

Abstract
Glucagon-like peptide-1(7-36)amide (GLP-1) is a 30-residue peptide hormone released from intestinal L cells following nutrient consumption. It potentiates the glucose-induced secretion of insulin from pancreatic beta cells, increases insulin expression, inhibits beta-cell apoptosis, promotes beta-cell neogenesis, reduces glucagon secretion, delays gastric emptying, promotes satiety and increases peripheral glucose disposal. These multiple effects have generated a great deal of interest in the discovery of long-lasting agonists of the GLP-1 receptor (GLP-1R) in order to treat type 2 diabetes. This review article summarizes the literature regarding the discovery of GLP-1 and its physiological functions. The structure, function and sequence–activity relationships of the hormone and its natural analogue exendin-4 (Ex4) are reviewed in detail. The current knowledge of the structure of GLP-1R, a Family B GPCR, is summarized and discussed, before its known interactions with the principle peptide ligands are described and summarized. Finally, progress in discovering non-peptide ligands of GLP-1R is reviewed. GLP-1 is clearly an important hormone linking nutrient consumption with blood sugar control, and therefore knowledge of its structure, function and mechanism of action is of great importance.
LINKED ARTICLES
This article is part of a themed section on Secretin Family (Class B) G Protein-Coupled Receptors. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.166.issue-1
Keywords: GLP-1, GPCR, exendin-4, exenatide, liraglutide, diabetes, glucagon, insulin
Introduction
The incretin effect describes the increased secretory response of beta cells, above that of glycaemia itself, which results from the actions of gut-derived factors (McIntyre et al., 1964). Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are the hormones responsible for the incretin effect, and together they account for up to 60% of postprandial insulin release (Nauck et al., 1986). However, despite their equivalent roles in potentiating glucose-induced insulin release, therapeutic strategies for the treatment of type 2 diabetes have focused primarily on GLP-1 since, unlike GIP, it continues to elicit a response in type 2 diabetic patients (Nauck et al., 1993).
GLP-1 is expressed in intestinal L cells and is derived from the cell-specific post-translational processing of the preproglucagon gene (Mojsov et al., 1986). Initially, the peptide GLP-1(1-37) was identified from this processing, but it was the two N-terminally truncated products, GLP-1(7-37) and GLP-1(7-36)amide, that were found to recognize the pancreatic receptor and which were determined to be the active species in vivo (Drucker et al., 1987; Holst et al., 1987; Mojsov et al., 1987; Ørskov and Nielsen, 1988; Weir et al., 1989). These two shorter peptides are equipotent (Ørskov et al., 1993) and are the major physiological incretin in humans (Edwards et al., 1999); hence, these N-terminally truncated peptides are the forms implied by the generic term ‘GLP-1’ throughout this review.
The mechanisms of action of GLP-1 (reviewed extensively by Doyle and Egan, 2007) go beyond just the augmentation of glucose-induced insulin secretion and include increasing insulin expression (Drucker et al., 1987), inhibiting beta-cell apoptosis and promoting beta-cell neogenesis (Perfetti and Hui, 2004; Cornu and Thorens, 2009); reducing glucagon secretion (Kreymann et al., 1987), delaying gastric emptying (Wettergren et al., 1993; Nauck et al., 1997), promoting satiety (Turton et al., 1996; Flint et al., 1998) and increasing peripheral glucose disposal (D'Alessio et al., 1994; 1995; Egan et al., 2002). It is clear from these varied actions that GLP-1 plays a central role in controlling postprandial blood sugar levels, and hence, the enhancement of GLP-1 action has become an important area for the development of new therapies to treat type 2 diabetes (e.g. Ahrén, 2011). GLP-1 raises insulin and lowers glucagon in type 2 diabetic patients (Nathan et al., 1992; Nauck et al., 1993), and repeated s.c. injections were found to normalize plasma glucose levels (Nauck et al., 1996). However, the half-life of GLP-1 in vivo is short, and it is rapidly degraded to GLP-1(9-36)amide by DPPIV (Deacon et al., 1995; Knudsen and Pridal, 1996; Hansen et al., 1999), and hence, there are continuing attempts to find long-lasting peptide agonists with glucoregulatory properties (e.g. Gong et al., 2011). Indeed, current therapeutic approaches have utilized DPPIV-resistant mimetics of GLP-1, most notably exenatide (Iltz et al., 2006) and liraglutide (Neumiller and Campbell, 2009), both of which are in current therapeutic use. Exenatide is the synthetic form of the naturally occurring peptide exendin-4 (Ex4), originally isolated from the saliva of the Gila monster Heloderma suspectum (Eng et al., 1992). It is 53% identical to GLP-1 but is resistant to DPPIV cleavage, largely due to the substitution of Ala2 by Gly (Figure 1). Liraglutide, on the other hand, is a synthetic analogue of GLP-1 itself, with minor sequence alterations and a covalently linked C-16 acyl chain.
Figure 1.

A sequence alignment of GLP-1, Ex4 and several analogues mentioned at various stages in the text. All peptides are C-terminally amidated. Note that the first residue of GLP-1 is His7, while that of Ex4 is His1. Residues changed from the original sequence are underlined.
The receptor for GLP-1 (GLP-1R) was first cloned from a cDNA library derived from rat pancreatic islets (Thorens, 1992), and the following year, the human receptor was cloned (Dillon et al., 1993; Graziano et al., 1993; Thorens et al., 1993). The cloning revealed a receptor sequence of 463 residues that resembled the receptors for secretin, parathyroid hormone and calcitonin. These receptors formed a new branch of the GPCR superfamily, named ‘Family B’ (or ‘Class B’, or ‘secretin receptor-like’), which to date includes 15 members (Hoare, 2005). These receptors possess a unique extracellular N-terminal domain (NTD) of 100–150 residues, which is connected to the integral membrane core domain (or J domain) that is typical of all GPCRs.
Family B GPCRs bind their peptide ligands via a common mechanism known as the ‘two-domain model’ (Bergwitz et al., 1996; Hoare, 2005) in which the NTD first binds to the C-terminal helical region of the ligand, thereby enabling a second interaction between the N-terminal region of the ligand and the core domain of the receptor. The latter interaction is essential for enabling agonist-induced receptor activation. An alternative model to explain Family B GPCR activation has been proposed, in which the ligand-binding event induces a conformational change that exposes an ‘endogenous agonist’, which then interacts with the receptor core domain to initiate activity (Dong et al., 2006; Dong et al., 2008).
The importance of the ligand's α-helical secondary structure in the two-domain model for mediating the initial interaction with the NTD has been reviewed (Parthier et al., 2009), while a common helix-capping model to explain how the various ligands of Family B GPCRs might activate their receptors has been proposed (Neumann et al., 2008). An example of how knowledge of the two-domain model can be used to design and synthesize novel ligands has recently been described in an elegant study by Devigny et al. (2011), who produced high potency probes with in vivo activity. An azide-modified peptide based upon the C-terminus of the ligand, corticotropin releasing factor, was used as a carrier protein and fused to an acetylene-tagged peptide library from which the activating mimetic of the ligand's N-terminus was derived.
This article will review the structure and function of GLP-1R with particular emphasis upon how it interacts with its peptide ligands. In addition, the sequence– and structure–activity properties of the peptide ligands will be considered, as will the few non-peptide ligands that have been discovered to date.
Peptide ligands
GLP-1
Using perfused rat pancreas, Suzuki et al. (1989) demonstrated that the free N-terminal His7 was important for GLP-1's insulinotropic activity. While GLP-1(7-37) elicited a clear response at 0.1–1 nM, its N-terminally truncated analogue GLP-1(8-37) did not. Moreover, since GLP-1(1-37) and GLP-1(6-37) also failed to initiate activity, it suggested that His7 was required as the N-terminal residue of the agonist peptide. In contrast, truncation of the two extreme C-terminal residues (Arg36 and Gly37) resulted in a less dramatic effect, although potency was nevertheless reduced. These sequence–activity data were in agreement with the findings of Mojsov (1992), using insulin-secreting RIN1046-38 cells. Radioligand binding demonstrated that the removal of C-terminal residues resulted in a 5- to 10-fold reduction in affinity, whereas removal of the N-terminal His7[GLP-1(8-37)], or inclusion of the full N-terminal sequence [GLP-1(1-37)], resulted in greater than 300-fold reduced affinity. Removal of the first 14 residues of GLP-1 yielded a peptide with 1500-fold lower affinity than GLP-1 and with antagonist properties, while removal of the C-terminal 11 residues resulted in undetectable binding and activity (Gallwitz et al., 1990).
A residue-by-residue substitution of the GLP-1 sequence was carried out by Novo Nordisk in 1994 (Adelhorst et al., 1994). Each residue was individually substituted by Ala, unless it was already Ala in which case it was substituted by its equivalent in glucagon (Ala25 was not studied in this series, presumably because it is also Ala in glucagon). Adenylyl cyclase activity was used to assess agonist potency and intrinsic activity using membranes from RIN-2A18 cells, while competition binding assays were used to assess ligand affinity using membranes derived from Chinese hamster lung cells expressing recombinant rat GLP-1R (rGLP-1R). The binding assays suggested that the N-terminal positions 7, 10, 12, 13 and 15 were important for GLP-1 affinity at rGLP-1R since the substitutions resulted in a 41- to 219-fold increase in IC50. A similar Ala-scan study in the same year, using RINm5F cells, identified the same five residues in the N-terminal region as being important for affinity (132- to 425-fold reduced affinity; Gallwitz et al., 1994). The His7 substitution by Ala resulted in a 111- to 374-fold reduction in affinity in these two studies, in contrast with only a 10-fold reduction at recombinant human GLP-1R (hGLP-1R) caused by replacing His7 by Tyr (Xiao et al., 2001). In another study using rGLP-1R expressed in Chinese hamster lymphoblast cells, His7 to Ala resulted in a 397-fold reduction in affinity, compared with 174-fold reduction by the Tyr substitution (Hareter et al., 1997). Furthermore, the His7 to Ala substitution resulted in a peptide that displayed no detectable adenylyl cyclase activity using 10 µM peptide, whereas the Tyr substituted peptide had a potency of 34 nM (GLP-1 control EC50 was 2.6 nM; Adelhorst et al., 1994; Xiao et al., 2001).
The substitution of Ala8 with D-Ala resulted in less than a twofold effect on affinity and less than a 10-fold reduction in potency at hGLP1-R (Xiao et al., 2001), while substitution by Ser resulted in a ninefold reduction in affinity at rGLP-1R, with no effect upon potency (Adelhorst et al., 1994). Therefore, Ala8 appears to be less important to the peptide's function compared with His7. Truncation of both these residues to give GLP-1(9-36)amide (the natural DPPIV cleavage product) yielded a peptide with partial agonist activities. A study using transfected CHO cells expressing recombinant rGLP-1R demonstrated that GLP-1(9-36)amide displayed a 94-fold reduced affinity compared with GLP-1 and produced approximately 50% of the cAMP levels at 1 µM compared with that produced by 10 nM GLP-1 (Montrose-Rafizadeh et al., 1997). This partial agonist is present in the circulation at four to five times the level of GLP-1 (Egan et al., 2002), but initial studies did not demonstrate that it produced any biological activity (Vahl et al., 2003). However, it has since been suggested that GLP-1(9-36)amide is indeed insulinotropic, but that its actions are masked by the more potent effects of GLP-1 itself (Elahi et al., 2008).
The N-terminal region also contains other important residues and, indeed, the further truncation of GLP-1, to form GLP-1(15-36)amide, resulted in a 367-fold reduction in affinity (rGLP-1R in CHO cells) and no detectable activity at 1 µM peptide (Montrose-Rafizadeh et al., 1997). Although Glu-9 was not identified as being of particular importance by Adelhorst et al. (1994), Xiao et al. (2001) demonstrated that its substitution resulted in an 81-fold reduction in affinity and a 30-fold reduction in potency. Adelhorst et al. (1994) detected no adenylyl cyclase activity for GLP-1 peptides with either Gly10 or Asp15 substituted by Ala, while the Gly10 to Ala substitution by Xiao et al. (2001) resulted in more than 100-fold reduced affinity and greatly reduced potency. Watanabe et al. (1994) also studied position 10 and found that substitution by Ala resulted in a 221-fold reduction in affinity, while the D-Ala and β-Ala substitutions had less effect. Siegel et al. (1999) demonstrated that substitution of Ser14 and Asp15 resulted in significant decrease in affinity and activity at RINm5f cells.
An N-terminal fragment of GLP-1, corresponding to GLP-1(7-17) but with Val16 and Ser17 substituted with biphenylalanine derivatives, displayed potency that was only 206-fold lower than GLP-1 itself (Mapelli et al., 2009). Further modification by replacing Ala2 (by aminoisobutyric acid) and Phe6 (by α-methyl-phenylalanine derivative) led to the discovery of an 11-mer with potency that was only two to threefold lower than GLP-1. Hence the region of GLP-1 that is essential for activity must be located within positions 7–17. Therefore, although the two domain model suggests that the activating N-terminal region of the peptide agonists must be ‘delivered’ to the core domain via binding of the C-terminus to the NTD, this 11-mer agonist suggests that it is nevertheless possible to obtain high potency from the secondary interaction.
Adelhorst et al. (1994) and Gallwitz et al. (1994) also identified both Phe28 and Ile29 as being important for GLP-1's activity, probably due to their role in maintaining the helical structure of the peptide as assayed by far-UV CD spectroscopy (Adelhorst et al., 1994). However, many residues in GLP-1 appear not to be critical for affinity or activity, and indeed the discovery and analysis of a GLP-1-like peptide from Xenopus reveals that, despite 9 residue substitutions (Phe12–Tyr, Ser14–Asn, Ser17–Thr, Ser18–Glu, Gly22–Glu, Gln23–Lys, Ala30–Glu, Val33–Ile, Arg36–Lys), its affinity and potency at hGLP-1R were nevertheless slightly better than GLP-1 itself (Irwin et al., 1997). The non-involvement of these residues in receptor affinity and efficacy is reflected in the results of the Ala scan of Adelhorst et al. (1994) and Gallwitz et al. (1994) with the possible exception of Phe-12 (substitution to Ala resulted in 133-fold reduced affinity and 13-fold reduced potency), although the larger Tyr side chain in the Xenopus sequence is likely to be a more acceptable substitution compared with Ala.
Valuable insights into GLP-1 structure and function, as well as potential anti-obesity compounds, have come from studying the features that govern the selectivity between GLP-1 and glucagon at their related receptors. For example, the replacement of the C-terminal sequence ‘VKGR’ of GLP-1 with the corresponding ‘MNT’ of glucagon resulted in a 475-fold decrease in affinity. However, the substitution of residues in the N-terminal half of GLP-1 for those of glucagon had much smaller effects (Hjorth et al., 1994; Hjorth and Schwartz, 1996). Runge et al. (2003a,b) demonstrated, using chimaeric receptors, that the selectivity of GLP-1R for GLP-1 over glucagon is due to the NTD recognising the C-terminus of the peptide, while the toleration of GLP-1R for the glucagon N-terminal sequence results from the core domain. Understanding the specificity requirements for GLP-1 and glucagon at GLP-1R and the glucagon receptor has enabled the design of a co-agonist peptide that eliminates obesity in a rodent model (Day et al., 2009; Day et al., 2010). Full potency was obtained at GLP-1R using a peptide corresponding to glucagon but with changes at just three side chains (Ser16-Glu, Arg18-Ala, Gln20-Lys) and amidation of the C-terminus.
Exendin-4
The study of GLP-1R has been greatly enhanced by the discovery of the peptide agonist exendin-4 (Ex4) from the saliva of the Gila monster H suspectum (Eng et al., 1992). Although useful for elucidating aspects of GLP-1R's structure-function, Ex4 is not simply a research tool since, in its synthetic form, it constitutes the drug exenatide (Byetta; Iltz et al., 2006). It was actually the related peptide exendin-3, discovered in H horridum, which was discovered first and found to have efficacy in dispersed pancreatic acini (Eng et al., 1990). However, despite only differing by 2 of the 39 residues (Gly2, Glu3 in Ex4, compared with Ser2, Asp3 in exendin-3), Ex4 was found to have distinct pharmacological properties and does not act at VIP or secretin receptors. The N-terminal truncation of the first eight residues of exendin-3 yielded an antagonist that was initially used as a tool to elucidate the pharmacology of exendin-3 (Raufman et al., 1991). However, since exendin-3 and Ex4 are identical from residues 4–39, this antagonist, Ex4(9-39), has become a valuable tool for studying GLP-1R. Ex4 was found to share a similar pharmacological profile to GLP-1 (Raufman et al., 1992) being a potent GLP-1R agonist, while Ex4(9-39) is an antagonist (Göke et al., 1993; Thorens et al., 1993; Schepp et al., 1994; Kolligs et al., 1995). However, while usually described as an antagonist, Ex4(9-39) has also been described as an inverse agonist at the murine GLP-1R (Serre et al., 1998).
Despite their related sequences and similar pharmacological profile, sequence–activity studies using Ex4 have highlighted some clear differences with GLP-1. While the removal of the first two N-terminal residues of GLP-1 yielded a partial agonist with a 100-fold reduction in affinity, Ex4(3-39) has been reported to be a high-affinity antagonist (Montrose-Rafizadeh et al., 1997). Furthermore, while the further truncation of GLP-1 to remove the first eight residues resulted in a peptide with 370-fold reduced affinity [GLP-1(15-36)amide], Ex4(9-39) retains high affinity for GLP-1R. Hence, it is clear that, despite sharing eight of the nine residues at the N-terminus, these regions of the two peptides play a subtly different role in receptor interactions. For example, while the Gly2-Ala substitution in Ex4 is tolerated (Al-Sabah and Donnelly, 2003a; 2004; Patterson et al., 2011a), the converse Ala2-Gly change in GLP-1 reduces affinity by three- to fivefold (Deacon et al., 1998; Burcelin et al., 1999; Patterson et al., 2011a). Therefore, the interaction between the NTD and the C-terminal helix of the ligand can result in a different interaction between the N-terminus and the core domain (Al-Sabah and Donnelly, 2004). Indeed, substitution of Glu22, Glu27 and Ala30 in GLP-1, with Gly, Leu and Glu from Ex4, enabled Gly2 tolerance (Patterson et al., 2011a).
Interestingly, while Ex4(2-39) retains high affinity and full activity at 10 nM, the replacement of Asp9 with Glu resulted in the complete loss of efficacy, despite no effect upon affinity (Montrose-Rafizadeh et al., 1997), suggesting that Asp9 plays an important role in activity.
A large number of GLP-1/Ex4 chimaeric peptides have been utilized in order to understand the determinants that enable Ex4(9-39) to retain potent antagonist activity, while GLP-1(15-36) remains an agonist (Patterson et al., 2011b). Glu16, Val19 and Arg20 were determined to be the essential determinants of Ex4(9-39)'s antagonism, in agreement with earlier chimaeric studies that demonstrated that the ‘E16EAVRL’ sequence within the N-terminally truncated Ex4 was the determinant of its greater affinity over that of GLP-1(15-36)amide.
Updated data – a new view!
Recent data from the author's laboratory and in Patterson et al. (2011b) challenge some of the commonly held views about GLP-1 and Ex4 efficacy. The data reviewed in the sections above clearly point to the N-terminal region of GLP-1 as being essential for activity. However, using recombinant HEK-293 cell lines, it is possible to obtain robust activity for a number of peptide ligands previously believed to be devoid of efficacy (e.g. [His7-Ala]GLP-1; [Gly10-Ala]GLP-1; GLP-1(15-36)amide; Ex4(3-39); Figure 2; Patterson et al., 2011b). It is likely that the receptor reserve in the HEK-293 expression system enables the detection of agonists with low efficacy and therefore provides a useful environment in which to distinguish affinity-generating determinants from those responsible for creating efficacy.
Figure 2.
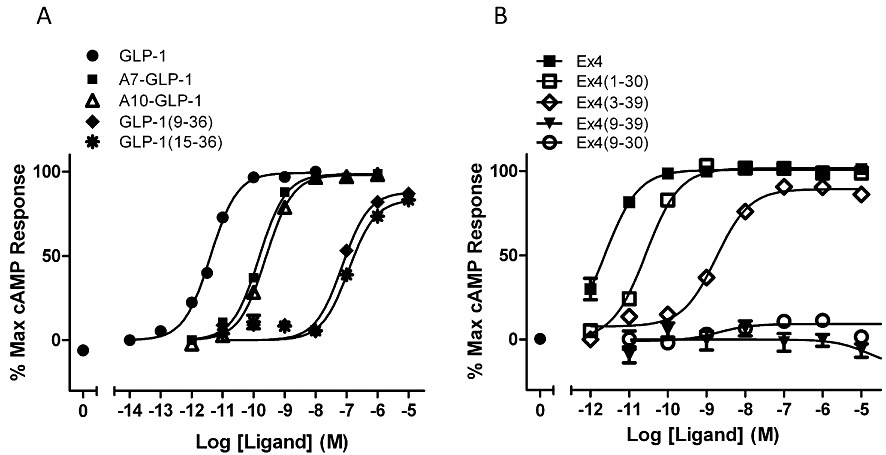
Concentration–response curves (LANCE cAMP assay) for various peptides at hGLP-1R, expressed in HEK-293 cells, using the same cell line as described previously by Mann et al. (2010a,b). pEC50 values, averaged over three independent experiments, were (A) GLP-1, 11.17 ± 0.12; A7-GLP-1,9.64 ± 0.09; A10-GLP-1, 9.45 ± 0.08; GLP-1(9-36), 6.97 ± 0.09; GLP-1(15-36), 6.72 ± 0.13. (B) Ex4, 12.14 ± 0.06; Ex4(1-30), 11.42 ± 0.03; Ex4(3-39), 8.87 ± 0.06; Ex(9-39) and Ex4(9-30) gave no response (Nasr, Wishart and Donnelly, unpubl.). Note that several peptides [A7-GLP-1, A10-GLP-1, GLP-1(15-36), Ex4(3-39)], believed to be inactive in other expression systems, display robust activity in HEK-293 cells.
Peptide structures
NMR analysis of GLP-1 in dodecylphosphocholine micelles revealed that, while the N-terminal residues 7–13 are unstructured, the rest of the peptide forms two helices from positions 13–20 and 24–35, separated by a linker region formed around Gly22 (Thornton and Gorenstein, 1994). Parker et al. (1998) utilized a number of cross-linked analogues of GLP-1 in order to trap an active conformation. The structures were largely compatible with the two-helix description of Thornton and Gorenstein. In contrast, Ex4 (which has Glu-16 in the equivalent position to Gly22 in GLP-1) forms a single helix spanning residues 11–27 (Neidigh et al., 2001). A recent NMR analysis of an analogue of Ex4 with enhanced activity (β-Ala replaces Glu-3, while Tyr replaces Gln-3) has shown a longer helical region that extends towards the N-terminus and that may be responsible for its apparent increase in activity (Wang et al., 2010). The NMR structures of GLP-1 and Ex4 are dependent upon the medium in which they reside (Andersen et al., 2002; Neidigh and Andersen, 2002). In the micelle-bound state, the C-terminal extension of Ex4 (residues 31–39) is disordered. However, in aqueous TFE, this region forms a distinct fold called a Tryptophan-cage (Trp-cage), which has been the seed-corn for parallel studies in the field of protein design and folding (e.g. Neidigh et al., 2002). The literature regarding the role of the putative Trp-cage and/or the C-terminal extension of Ex4 has appeared to be contradictory (Doyle et al., 2003; Al-Sabah and Donnelly, 2003a; Runge et al., 2007). However, this has recently been clarified (Mann et al., 2010b) and will be discussed in more detail later in this review.
GLP-1 receptor
The cloning of GLP-1R (Thorens, 1992; Dillon et al., 1993; Graziano et al., 1993; Thorens et al., 1993) identified it as part of the Family B GPCR family (Segre and Goldring, 1993) and hence intimated that its structure would resemble the two-domain fold common to the sub-group. As with many integral membrane proteins, the experimentally determined structure of a representative Family B GPCR has yet to be achieved, although some progress has been made in expressing, purifying and characterizing the full-length hGLP-1R in an Escherichia coli expression system (Schröder-Tittmann et al., 2010). Based upon its amino acid sequence, GLP-1R is expected to have the seven transmembrane (7TM) bundle typical of all GPCRs (e.g. Palczewski et al., 2000), with the seven α-helices (TM1-TM7) separated by three intracellular loops (ICL1–ICL3) and three extracellular loops (ECL1–ECL3). However, the sequence of Family B GPCRs is devoid of the consensus sequences and motifs that define Family A receptors, with the possible exception of the disulphide bond between ECL1 and ECL2 (Mann et al., 2010a). However, using approaches that were shown to be reasonably successful at predicting the helical arrangements in Family A GPCRs (Donnelly et al., 1989; 1994; Baldwin, 1993), some studies have attempted to predict the features of the core domain of family B GPCRs (Donnelly, 1997; Tams et al., 1998; Frimurer and Bywater, 1999).
A complicating factor in understanding and determining the structure of family B GPCRs may be their tendency to form oligomers. For example, a predicted lipid-exposed face (Donnelly, 1997) of TM4 of the secretin receptor has been implicated in functionally relevant oligomerization (Harikumar et al., 2007). Disruption of this interface using either peptides derived from the sequence of TM4, or site-directed mutagenesis of Gly243 and Ile244, demonstrated that the oligomerization mediated by the lipid-exposed face of TM4 was important for the full potency of secretin. Disruption of the interface resulted in an 18- to 28-fold reduction in potency, despite no effect upon ligand affinity. A similar interface has been identified for the calcitonin receptor (Harikumar et al., 2010).
The cloning of GLP-1R suggested the presence of a signal peptide at the extreme N-terminus, and this was shown to be required for the receptor's synthesis in HEK-293 cells (Huang et al., 2010). Cleavage of the signal peptide was essential for correct processing and trafficking, such that only the mature and fully glycosylated receptor reached the plasma membrane (Huang et al., 2010). The N-glycosylation of GLP-1R had previously been demonstrated in RINm5F cells (Göke et al., 1994), with glycopeptidase F reducing the apparent molecular weight from 63 to 51 kDa and tunicamycin reducing detectable receptor expression but not ligand affinity. In recombinant CHO cells, mutation of any two of the three N-glycosylation sites resulted in disruption of receptor trafficking to the plasma membrane (Chen et al., 2010a). Therefore, it appears that GLP-1R is a glycoprotein, and that the glycosylation, together with the cleavage of the signal peptide, is an essential process in the correct trafficking and processing of the receptor.
GLP-1R signalling
As with family B GPCRs in general, GLP-1R predominantly signals through the Gs G-protein to raise intracellular cAMP levels (Nauck et al., 1993; Coopman et al., 2010), although coupling to other G-proteins has been observed (Bahekar et al., 2007; Doyle and Egan, 2007). The receptor couples to G-proteins via its intracellular loops, and there is some evidence that demonstrates that different regions and residues couple the receptor to different G-proteins. For example, using synthetic peptides to directly activate G-proteins, Hällbrink et al. (2001) have shown that the N-terminal half of ICL3 is responsible for Gs coupling, while the C-terminal half mediates signalling via Gi/o.
Nevertheless, it is the cAMP production mediated by GLP-1R activation that constitutes the principle driver of insulin secretion. Transfection of HIT-T15 cells with GLP-1R enabled GLP-1-mediated insulin secretion in contrast to the transfection of two deletion mutants that were known to be uncoupled from Gs (Salapatek et al., 1999). Wheeler's group have scanned the intracellular loops using deletion- and site-directed mutagenesis (Takhar et al., 1995; Mathi et al., 1997) and have shown that Lys334, along with the two following residues (Leu and Lys), is required for full receptor activity. In addition, Val327, Ile328 and Val331, at the TM5/ICL3 junction, play an important part in coupling the receptor and may contact Gs. ECL2 does not play a significant part in G-protein coupling, but mutation of Arg176 on ECL1 to Ala reduced receptor activity to about 25% of normal. Heller et al. (1996) also identified His180 as being important for agonist-induced coupling since its mutation to Arg resulted in a 20-fold decrease in affinity and a 50% decrease in cAMP production. In contrast to Takhar et al. (1995), Heller et al. (1996) also suggest that Arg348 (ECL3) is critical for receptor activity.
Following agonist-induced receptor activation, GLP-1R is internalized. This may be via a coated pit mechanism following phosphorylation by both homologous and heterologous mechanisms (Widmann et al., 1995; 1996a). Further investigation located the phosphorylation sites to Ser431/Ser432, Ser441/Ser442, Ser444/Ser445 and Ser451/Ser452 (Widmann et al., 1996b). Alternatively, GLP-1R internalization has been found to be arrestin-independent and associated with caveolin-1 (Syme et al., 2006).
N-terminal domain
The two-domain structure of Family B GPCRs lends itself to structural and functional analysis through the isolation of the NTD, an approach that has been extremely useful for studying GLP-1R (Wilmen et al., 1996; Xiao et al., 2000; Bazarsuren et al., 2002; López de Maturana et al., 2003; Al-Sabah and Donnelly, 2003a; Mann et al., 2007; 2010b; Runge et al., 2007; 2008; Underwood et al., 2010).
The isolated NTD was expressed and purified from mammalian COS-7 cells and was shown to cross-link to 125I-GLP-1. Using a 6xHis tag, the affinity of the NTD for GLP-1 was estimated to be 450 nM (Xiao et al., 2000). Prior to this, Wilmen et al. (1996) expressed the isolated NTD of rGLP-1R (residues 20–144) in E coli and purified the soluble fraction via a 6xHis tag. The purified protein was expressed at low levels but was shown to be functionally active since it competed with 125I-GLP-1 binding to cells expressing the full-length receptor and could also be cross-linked to 125I-GLP-1 with disuccinimidyl suberate (DSS). Disruption of the disulphide bonds within the NTD using β-mercaptoethanol resulted in its complete loss of ability to compete with 125I-GLP-1 binding to GLP-1R. Bazarsuren et al. (2002) also expressed the NTD of hGLP-1R in E coli, but, unlike Wilmen et al., they isolated the receptor domain from inclusion bodies from which the NTD was denatured, refolded and purified in quantities suitable for biophysical and structural analysis. Interaction with GLP-1 was demonstrated via cross-linking, surface plasmon resonance (SPR) and isothermal titration calorimetry, giving affinity constants for GLP-1 of 46–144 nM. Further analysis demonstrated the presence of three disulphide bonds linking residues 46–71, 62–104 and 85–126. Using this expression/refolding approach, López de Maturana et al. (2003) expressed and purified the NTD from rGLP-1R and demonstrated that, while GLP-1 binds with 400 nM affinity, the ligands Ex4, Ex4(3-39) and Ex4(9-39) bind with 1–3 nM affinity. The differential affinity of GLP-1 and Ex4 will be discussed in more detail in the next section.
The isolated NTD of hGLP-1R from the E coli inclusion body preparation was biophysically characterized by Runge et al. (2007), and the same protocol was used to produce a protein preparation which was incubated with Ex4(9-39), before being further purified, crystallized and analysed using X-ray diffraction (Runge et al., 2008). The resultant 2.2Å crystal structure resembled that of other Family B GPCR NTDs, displaying two regions of anti-parallel β-sheet, three disulphide bonds and an N-terminal α-helix (Parthier et al., 2009; Figure 3A). The core of the structure contains six conserved residues (Asp67, Trp72, Pro86, Arg102, Gly108 and Trp110), which are critical for the fold's stability and structure. A ligand-binding groove is present, which is lined by residues on the α-helix, turn 1, loop 2 and the C-terminal region of the NTD.
Figure 3.
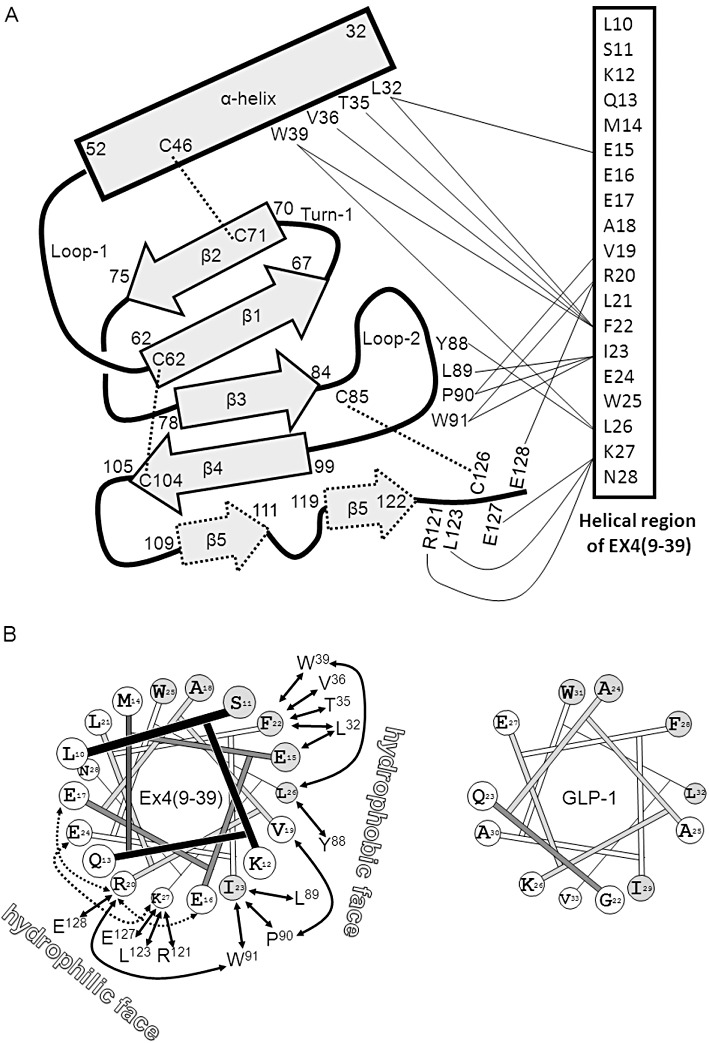
(A) A schematic representation of the structure of the NTD of hGLP-1R based upon the crystal structure of Runge et al. (2008). The residue numbers at the boundaries of the various secondary structural elements are shown. Disulphide bonds are depicted with dashed lines. On the right is the α-helical region of Ex4(9-39) with lines connecting to contacting residues on the NTD. (B) Left: Ideal helical wheel containing the residues in the helical region of Ex4(9-39). Residues conserved with GLP-1 are filled grey. Interactions with residues in the NTD are shown with solid arrows. Intra-helical interactions, which stabilize the helix, are shown with dotted arrows. The hydrophilic and hydrophobic faces are labelled. Right: The helical region of GLP-1 on the C-terminal side of the Gly-22 kink. Note that the helix-stabilizing interactions found in Ex4(9-39) are not formed in GLP-1 since the residues are not conserved.
This inclusion body/refolding approach has been extended to include the full-length receptor that has been obtained in an active form with distinct binding kinetics from those of the autonomously folded NTD (Schröder-Tittmann et al., 2010). However, to date, the structure of the full-length receptor remains unknown. An attempt was made to model GLP-1R and its ligand-binding sites using homology modelling with the templates being the crystal structures of the GIP receptor NTD and rhodopsin (Lin and Wang, 2009). More recent modelling (Miller et al., 2011) has utilized the X-ray structure of the GLP-1R NTD (Underwood et al., 2010) fused to 250 diverse helical bundle domain models from 25 transmembrane bundles, alongside experimentally derived photo-affinity contacts, in order to further understand the ligand–receptor interactions. Nevertheless, while it appears reasonable to assume that the core domain will interface with the NTD in the region close to the N-terminal ends of the helices of both the NTD and the ligand, the number of degrees of freedom remains very high and a structure of the full-length receptor will be required before we can fully understand the details of this interface. Further complexities have been proposed by a mutational analysis of the NTD of the related glucagon receptor using cysteine accessibility approaches (Prévost et al., 2010). A number of residues on one face of the NTD were found to be much less accessible to covalent modification than would have been expected from the GLP-1R NTD structures. However, these residues are on the opposite face of the NTD relative to the expected interface of the core domain and therefore may form an interface with another, as yet unidentified, protein.
Peptide–receptor interactions
Stage 1 interaction – binding of the ligand to the NTD
As described previously, the isolation of the NTD of GLP-1R as an independent soluble domain resulted in specific, though low affinity, binding of the natural ligand GLP-1. Since several mutations in the core domain of full-length GLP-1R resulted in a lowering of GLP-1 affinity, but not that of Ex4, it was hypothesized that Ex4 affinity was largely independent of the core domain (López de Maturana and Donnelly, 2002; Al-Sabah and Donnelly, 2003b). This was shown to be the case by demonstrating that the complete removal of the core domain has a minimal effect upon Ex4 affinity, compared with that of GLP-1, and that the interaction between Ex4 and the NTD was not dependent upon the N-terminal region of the ligand (López de Maturana et al., 2003). A model was proposed to explain the distinct pharmacological properties of the two peptide agonists at the isolated NTD, with the suggestion that Ex4 formed an additional interaction (termed the ‘Ex’ interaction), which was due to one or more of the unique structural features of Ex4, such as its increased helicity, its C-terminal extension, or residues that were not conserved with GLP-1 (López de Maturana et al., 2003). Runge et al. (2007) demonstrated that it was the increased helicity of Ex4, which accounted for its high affinity at the fully isolated NTD. However, when this domain was fused to the plasma membrane of HEK-293 cells via TM1, the large differential affinity observed at the isolated NTD of hGLP-1R was reduced to only eightfold (Mann et al., 2010a,b) and, furthermore, in the presence of Brij micelles, the affinity of GLP-1 and Ex4 at the isolated NTD was similar (Schröder-Tittmann et al., 2010).
The nine-residue C-terminal extension of Ex4 plays no significant role in the peptide's affinity at hGLP-1R (Runge et al., 2007; Mann et al., 2010a,b), which is in agreement with the X-ray structure of the isolated NTD bound with Ex4(9-39) (Runge et al., 2008). However, Ser32 in this extended region of the peptide is able to interact with Asp68 in the NTD of rGLP-1R, generating a >20-fold improvement in affinity (Mann et al., 2010a,b). The improved affinity of Ex4(9-39) at rGLP-1R, resulting from interaction between Ser32 of the ligand and Asp68 of the NTD, could be exchanged between hGLP-1 and rGLP-1R by swapping Glu and Asp at position 68. Hence, despite sharing similar physiochemical properties to Asp, Glu-68 does not appear to interact strongly with Ser32 of the ligand. Given that Glu-68 interacts with the C-terminus of GLP-1 (Day et al., 2010), it is possible that it preferentially interacts with residue 30 of Ex4(9-39), while the shorter Asp68 side chain in rGLP-1 interacts with Ser32. This would explain why the C-terminal extension on Ex4(9-39) improves the affinity for rGLP-1R but not hGLP-1R.
Photo-crosslinking of the C-terminal region of GLP-1 to GLP-1R demonstrated that residue 35 interacts with Glu-125, while residue 24 interacts with Glu-133 (Chen et al., 2009). On the other hand, Ex4(9-39) retains high affinity for the isolated rGLP-1R NTD consisting of residues Gln-26–Glu-127, suggesting that the NTD from Glu-128 onwards is not required for binding the C-terminal half of the ligand (Mann et al., 2007). The X-ray crystal structure of the hGLP-1R fragment Arg24–Tyr145, with either Ex4(9-39) (Runge et al., 2008) or GLP-1 (Underwood et al., 2010), reveal the details of the interaction of the C-terminal region of the peptides and the NTD of the receptor. Figure 3A summarizes the structure of the NTD and how it binds to Ex4(9-39). The antagonist forms a well-defined helix from residues Leu10–Asn28 and interacts with the NTD using residues within Glu15–Ser32. Although residues 9–14 do not interact with the NTD, residues 10–14 are nevertheless critical for high affinity binding (Runge et al., 2007), presumably because they are required to stabilize the helical structure of the ligand. The α-helix of Ex4(9-39) is amphipathic (Figure 3B) and interacts with the NTD via residues on both faces, although it is the hydrophobic face which appears most important, with Val19, Phe22, Ile23 and Leu26 being buried in the binding site.
GLP-1 also forms an α-helix when bound to the NTD (Underwood et al., 2010), but, in contrast to that in Ex4(9-39), it is kinked around Gly22 as observed in earlier NMR studies (Thornton and Gorenstein, 1994). Residues Ala24–Val33 interact with the NTD and, as with Ex4(9-39), the principal contacts are via a hydrophobic interface formed by residues Ala24, Ala25, Phe28, Ile29, Leu32 and Val33. Because the hydrophobic faces of the helices of Ex4(9-39) and GLP-1 are highly conserved (López de Maturana and Donnelly, 2002), their interactions with the NTD are very similar.
However, differences at the hydrophilic faces result in a degree of differential affinity. The ability of Arg20 of Ex4(9-39) to form intra-helical interactions with Glu16 and Glu17, and also for Lys27 to interact intra-helically with Glu24, results in stabilization of the α-helix (Figure 3B). However, three of these four charged sites are absent in GLP-1, and, moreover, Glu16 is replaced by Gly22, which reduces the helix stability and enables the distortion of this region. This hydrophilic surface may also be involved in a direct receptor interaction, since the crystal structures suggests that Arg20[Ex4(9-39)] and Lys26 (GLP-1) may interact with Glu128 of the NTD. However, truncation of the NTD to remove Glu128 did not remove high Ex4(9-39) affinity (Mann et al., 2007). On the other hand, Glu127, which appears to interact with the antagonist but not GLP-1, could not be truncated without loss of Ex4(9-39) affinity, suggesting that the observed interaction with Lys27 of the antagonist is important (Mann et al., 2007). Mutagenesis of Glu127 resulted in a sevenfold reduction of Ex4(9-39) affinity but with no effect upon GLP-1, suggesting it may be responsible for the differential affinity of these peptides at the NTD (Underwood et al., 2010). Given that their differential affinity is about eightfold at the membrane-tethered NTD (Mann et al., 2010b), the specific Glu127 interaction may indeed account for this. In addition, the mutation of Leu32, previously identified as a potential binding partner of Ex4(9-39) (Mann et al., 2007), also resulted in specific reduction of the antagonist's affinity, although the explanation for this is not obvious from the crystal structures (Underwood et al., 2010). It may be that, since Leu32 interacts with the ligand's helix close to the point at which GLP-1 kinks, it may have a differential interaction with the two ligands. Stabilization of the helix in this region of GLP-1, by substituting Ser18 by Lys and Gly22 by Glu, resulted in an eightfold increase in potency (Murage et al., 2008; 2010).
Further studies have shown important ligand–receptor interactions points on the NTD. A polymorphism of GLP-1R, Thr149–Met (Beinborn et al., 2005), resulted in a 60-fold reduction in affinity for GLP-1 but only fivefold reduction for Ex4, suggesting that this residue is involved in GLP-1 binding. However, the lack of a major effect on Ex4 affinity suggests that, either it does not interact with Thr149, or else it interacts with a region not essential for affinity, such as the N-terminus. The latter seems likely since the close-by residue Tyr145 has been labelled by a peptide probe containing benzoylphenylalanine (BPh) in place of Thr12 of GLP-1 (Chen et al., 2010b), and, furthermore, a similar ligand probe with BPh replacing Val16 was found to cross-link to Leu141 (Miller et al., 2011). The importance of helix stability in this region has been discussed by Neumann et al. (2008), and, indeed, Thr12 and Val16 would be predicted to be separated by one helical turn. These data therefore complement the study of Murage et al. (2008), who demonstrated that stabilization of helical structure between Thr11 and Gly22, using lactam bridges, improved activity. Therefore, the region linking the NTD to TM1 appears to interact with the N-terminal region of GLP-1, although, as would be expected if this was the case, this region is not required for the high affinity of Ex4(9-39) since the NTD can be truncated between Glu127 and Glu128 (Mann et al., 2007).
Stage 2 interaction – binding of the ligand to the core domain
Since there is no determined structure for the core domain of GLP-1R, our knowledge of the interaction between the peptide ligands and the core domain must be gleaned from more indirect approaches. The identification of mutations which reduce the affinity of GLP-1, but not that of GLP-1(15-36), have been useful for identifying residues in the core domain that interact with the N-terminus of GLP-1. For example, a double-alanine mutagenesis scan of ECL1 of rGLP-1R identified a residue pair Met204–Tyr205 as interacting with GLP-1's N-terminal 8 residues (López de Maturana et al., 2004). This approach has been validated by the identification of Tyr205 as the attachment point of a photoactive analogue of GLP-1 containing a BPh at the N-terminus (Chen et al., 2010b). While the scan of ECL1 suggested that it did not play a major role in ligand recognition (López de Maturana et al., 2004), a number of charged and polar residues have been identified as being involved in ligand binding (Xiao et al., 2000).
Further mutations with differential effects upon GLP-1 and GLP-1(15-36) affinity have been used to identify Asp198 (TM2/ECL1; López de Maturana and Donnelly, 2002) and Lys288 (TM4/ECL2; Al-Sabah and Donnelly, 2003b) as interaction points for the N-terminus of GLP-1. Furthermore, a double Ala-scan of ECL2 also identified a number of sites likely to be important in forming the binding site of the peptide's N-terminus (Table 1) and are also likely to be important for the agonist-induced activation of the receptor (Mann et al., 2010a,b). However, ECL2 may also contact the mid-region of the peptide since BPh at position 20 of GLP-1 was photo-crosslinked to Trp297 (Miller et al., 2011). The use of GLP-1/glucagon chimaeric ligands, coupled with site-directed mutagenesis of the glucagon receptor, has shown that the reduced activity of an analogue of glucagon, with Ala at position 2, could be rescued by mutating Asp385 to Glu, suggesting that the N-terminus binds close to the TM7/ECL3 boundary (Runge et al. (2003a,b).
Table 1.
pIC50 values for GLP-1 and GLP-1(15-36) at wild-type rGLP-1R expressed in HEK-293 cells and at site-directed mutations targeted at pairs of residues in ECL2 (Al-Sabah and Donnelly, unpublished)
| GLP-1(7-36) pIC50 | GLP-1(15-36) pIC50 | |
|---|---|---|
| GLP-1R | 8.5 ± 0.07 | 6.4 ± 0.02 |
| W297A-T298A | 6.5 ± 0.06 | 6.7 ± 0.27 |
| R299A-N300A | 6.1 ± 0.06 | 6.3 ± 0.06 |
| Y305A-W306A | 6.0 ± 0.13 | 6.2 ± 0.05 |
| L307A-I308A | 6.1 ± 0.09 | 6.0 ± 0.05 |
| I309A-R310A | 6.8 ± 0.08 | 6.5 ± 0.02 |
pIC50 values were calculated from heterologous competition binding assays using 125I-Ex4(9-39) tracer. Methodology, cell lines and reagents are as described in Al-Sabah and Donnelly (2003b). The mean ± SE is shown for three independent experiments each performed in triplicate and shows that the mutations remove the sensitivity of the receptor to the presence of the N-terminal region of GLP-1.
A molecular modelling study, which incorporated the NTD X-ray studies (Runge et al., 2008; Underwood et al., 2010) with the outcome of several photo-crosslinking studies (Miller et al., 2011; Chen et al., 2009; Chen et al., 2010b), predicted a deep binding pocket for His7 of GLP-1, with Thr11 close to the receptor sites Tyr145 and Tyr205. Meanwhile Lys26 and Leu20 of GLP-1 were placed close to Glu133 and Tyr297 respectively (Miller et al., 2011). However, without knowledge of the structures of the GLP-1R core domain structure and the receptor-bound ligand, it remains difficult to fully understand the details of the stage 2 interaction.
Non-peptide ligands
Non-peptidic agonists of GLP-1R would be desirable as therapeutic agents since they have the potential to be orally active and hence circumvent the current requirement for self-injection by patients. However, the first non-peptide ligand at GLP-1R was in fact an antagonist, T-0632, which competes with 125I-Ex4(9-39) at hGLP-1R with an IC50 of 1.2 µM and was able to antagonize GLP-1-induced cAMP production in COS-7 cells (Tibaduiza et al., 2001). The compound was significantly less potent at rGLP-1R, and this species difference enabled the identification of Trp33 (Ser33 in rGLP-1R) as being the critical residue. This residue is located on the α-helix of the NTD but is not involved in peptide binding – presumably the non-peptidic binding site over-laps with that of the peptide.
The first two non-peptide agonists of GLP-1R were published in 2007 (Chen et al., 2007; Knudsen et al., 2007). Boc5 and its analogue S4P were discovered through a screen of more than 48 000 synthetic and natural compounds that yielded two hits, which actually turned out to be due to the dimeric versions of the original compounds that had formed spontaneously during their long-term storage in DMSO. Boc5 acted as a full agonist with 1.08 µM potency at rGLP-1R expressed in HEK-293 cells using a Luciferase-based cAMP reporter assay. Ex4(9-39) blocked the Boc5-induced receptor response and also competed with 125I-GLP-1 binding. Furthermore, Boc5 elicited a glucose-induced insulin response in isolated pancreatic islets from rat, inhibited food intake in rats and reduced HbA1c levels in db/db mice to non-diabetic levels. The GLP-1 mimetic properties of Boc5 were further characterized by Su et al. (2008).
The other class of non-peptidic agonist discovered in 2007 was ‘Compound 2’ (Cmp2; 6,7-dichloro-2methylsulfonyl-3-N-tert-butylaminoquinoxaline) and related structures (Knudsen et al., 2007; Teng et al., 2007). These compounds are significantly smaller than Boc5 and have distinct pharmacological properties. They are ago-allosteric modulators of hGLP-1R, since they can act both as independent agonists but also as allosteric enhancers of the activity of the natural agonist. In this case, the allosteric enhancement initially appeared to be limited to an increase in GLP-1 affinity (26-fold) rather than potency, probably due to a high degree of spare receptors. However, Cmp2 did potentiate the potency of GLP-1 in stable FlpIn-CHO cells expressing hGLP-1R (Koole et al., 2010).
The concentration–response curves for Cmp2 are bell-shaped since higher concentrations are associated with cell death (Coopman et al., 2010). Nevertheless, Cmp2 was able to stimulate glucose-dependent insulin secretion from normal mouse islets but not from those derived from GLP-1R knock-out mice (Knudsen et al., 2007). Related quinoxaline compounds have also been synthesized and shown to display anti-diabetic activity (Bahekar et al., 2007). A comparison of the effects of Cmp2 and GLP-1 in HEK-293 cells expressing hGLP-1R demonstrated that both agonists act via Gαs and that Cmp2 elicits 88% of the Emax of GLP-1 in cAMP assays (Coopman et al., 2010). Interestingly, Cmp2's cAMP response was enhanced by Ex4(9-39), and, furthermore, the antagonist inhibited Cmp2-mediated receptor internalization. Further in vivo studies using mice demonstrated that, while Cmp2 failed to prominently enhance glucose-mediated insulin release when compared with the more potent peptidic drugs exenatide and liraglutide, it did nevertheless significantly decrease the overall glucose excursion (Irwin et al., 2010). Ex4(9-39) did not impair Cmp2's glucoregulatory actions, consistent with this antagonist's inability to block Cmp2-induced cAMP activity in vitro (Knudsen et al., 2007; Coopman et al., 2010). Cmp2 also appears to possess peptide-dependent allosteric properties since it displayed clear enhancement of potency and affinity at GLP-1R for oxyntomodulin but not Ex4 (Koole et al., 2010).
An interesting non-peptide response at GLP-1R has been shown using the flavanoid quercetin which, despite not being able to activate GLP-R directly, enhanced the potency of the Ca2+ response mediated by GLP-1 and Ex4 (Koole et al., 2010). Using a range of flavanoid structures, the SAR between this pathway-selective behaviour and the flavanoid structure identified the 3-OH group as the key moiety of this activity (Wootten et al., 2011).
The pharmacology of a novel pyrimidine-based compound has recently been published by Sloop et al. (2010). ‘Compound B’ (CmpB) was discovered from screening a pharmacophore-based focused compound library, followed by rational modifications of the initial hit. It is a full agonist at hGLP-1R but displayed poor potency (0.7 µM) that is more than 1000-fold lower than GLP-1 itself. Nevertheless, CmpB evoked insulin secretion in rats and was able to normalize insulin secretion in islets derived from a human donor with type 2 diabetes. CmpB was able to activate a truncated version of GLP-1R, which lacks the NTD, demonstrating that this non-peptide acts via the core domain. Ex4(9-39) did not antagonize the CmpB-response, further demonstrating that its mode of binding is distinct from that of the peptide ligands.
Conclusion
The case for targeting GLP-1R for the treatment of type 2 diabetes is clear, and therefore, knowledge of the details of the peptide ligands, the receptor itself and their mode of interaction is important. Much is now known about the structure and function of GLP-1R and of its natural ligand GLP-1, while some initial progress has been made in the discovery of non-peptidic agonists. Many of the key recognition elements of the peptide are known and the sites to which they bind on the receptor have been partly deduced. Nevertheless, a great deal of work has yet to be carried out before our understanding of this important hormone–receptor interaction is complete and before the use of non-peptidic therapeutics becomes a reality.
Acknowledgments
I thank Dr Nasr Elsayed Nasr and Clare Wishart for carrying out the cAMP assays in Figure 2 and Dr Suleiman Al-Sabah for carrying out the competition binding assays in Table 1.
Glossary
- BPh
benzoylphenylalanine
- DPPIV
dipeptidyl peptidase IV
- DSS
disuccinimidyl suberate
- GIP
glucose-dependent insulinotropic polypeptide
- GLP-1
glucagon-like peptide-1
- GLP-1R
GLP-1 receptor
- hGLP-1R
human GLP-1R
- NTD
N-terminal domain
- rGLP-1R
rat GLP-1R
Conflict of interest
The author declares no conflict of interest.
References
- Adelhorst K, Hedegaard BB, Knudsen LB, Kirk O. Structure-activity studies of glucagon-like peptide-1. J Biol Chem. 1994;269:6275–6278. [PubMed] [Google Scholar]
- Ahrén B. GLP-1 for type 2 diabetes. Exp Cell Res. 2011;317:1239–1245. doi: 10.1016/j.yexcr.2011.01.010. [DOI] [PubMed] [Google Scholar]
- Al-Sabah S, Donnelly D. A model for receptor-peptide binding at the glucagon-like peptide-1 (GLP-1) receptor through the analysis of truncated ligands and receptors. Br J Pharmacol. 2003a;140:339–346. doi: 10.1038/sj.bjp.0705453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sabah S, Donnelly D. The positive charge at Lys-288 of the glucagon-like peptide-1 (GLP-1) receptor is important for binding the N-terminus of peptide agonists. FEBS Lett. 2003b;553:342–346. doi: 10.1016/s0014-5793(03)01043-3. [DOI] [PubMed] [Google Scholar]
- Al-Sabah S, Donnelly D. The primary ligand-binding interaction at the GLP-1 receptor is via the putative helix of the peptide agonists. Protein Pept Lett. 2004;11:9–14. doi: 10.2174/0929866043478365. [DOI] [PubMed] [Google Scholar]
- Andersen NH, Brodsky Y, Neidigh JW, Prickett KS. Medium-dependence of the secondary structure of exendin-4 and glucagon-like-peptide-1. Bioorg Med Chem. 2002;10:79–85. doi: 10.1016/s0968-0896(01)00263-2. [DOI] [PubMed] [Google Scholar]
- Bahekar RH, Jain MR, Gupta AA, Goel A, Jadav PA, Patel DN, et al. Synthesis and antidiabetic activity of 3,6,7-trisubstituted-2-(1H-imidazol-2-ylsulfanyl)quinoxalines and quinoxalin-2-yl isothioureas. Arch Pharm (Weinheim) 2007;340:359–366. doi: 10.1002/ardp.200700024. [DOI] [PubMed] [Google Scholar]
- Baldwin JM. The probable arrangement of the helices in G protein-coupled receptors. EMBO J. 1993;12:1693–1703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazarsuren A, Grauschopf U, Wozny M, Reusch D, Hoffmann E, Schaefer W, et al. In vitro folding, functional characterization, and disulfide pattern of the extracellular domain of human GLP-1 receptor. Biophys Chem. 2002;96:305–318. doi: 10.1016/s0301-4622(02)00023-6. [DOI] [PubMed] [Google Scholar]
- Beinborn M, Worrall CI, McBride EW, Kopin AS. A human glucagon-like peptide-1 receptor polymorphism results in reduced agonist responsiveness. Regul Pept. 2005;130:1–6. doi: 10.1016/j.regpep.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Bergwitz C, Gardella TJ, Flannery MR, Potts JT, Kronenberg HM, Goldring SR, et al. Full activation of chimeric receptors by hybrids between parathyroid hormone and calcitonin. J Biol Chem. 1996;271:26469–26472. doi: 10.1074/jbc.271.43.26469. [DOI] [PubMed] [Google Scholar]
- Burcelin R, Dolci W, Thorens B. Long-lasting antidiabetic effect of a Dipeptidyl peptidase IV-resistant analog of glucagon-like peptide-1. Metabolism. 1999;48:252–258. doi: 10.1016/s0026-0495(99)90043-4. [DOI] [PubMed] [Google Scholar]
- Chen D, Liao J, Li N, Zhou C, Liu Q, Wang G, et al. A non-peptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proc Natl Acad Sci U S A. 2007;104:943–948. doi: 10.1073/pnas.0610173104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Pinon DI, Miller LJ, Dong M. Molecular basis for glucagon-like peptide 1 docking to its intact receptor studied with carboxyl-terminal photolabile probes. J Biol Chem. 2009;284:34135–34144. doi: 10.1074/jbc.M109.038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Miller LJ, Dong M. Role of N-linked glycosylation in biosynthesis, trafficking, and function of the human glucagon-like peptide 1 receptor. Am J Physiol Endocrinol Metab. 2010a;299:E62–E68. doi: 10.1152/ajpendo.00067.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Pinon DI, Miller LJ, Dong M. Spatial approximations between residues 6 and 12 in the amino-terminal region of glucagon-like peptide 1 and its receptor. J Biol Chem. 2010b;285:24508–24518. doi: 10.1074/jbc.M110.135749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coopman K, Huang Y, Johnston N, Bradley SJ, Wilkinson GF, Willars GB. Comparative effects of the endogenous agonist glucagon-like peptide-1 (GLP-1)-(7-36) amide and the small-molecule ago-allosteric agent ‘Compound 2’ at the GLP-1 receptor. J Pharmacol Exp Ther. 2010;334:795–808. doi: 10.1124/jpet.110.166009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornu M, Thorens B. GLP-1 protects beta-cells against apoptosis by enhancing the activity of an IGF-2/IGF1-receptor autocrine loop. Islets. 2009;1:280–282. doi: 10.4161/isl.1.3.9932. [DOI] [PubMed] [Google Scholar]
- D'Alessio DA, Kahn SE, Leusner CR, Ensinck JW. Glucagon-like peptide 1 enhances glucose tolerance both by stimulation of insulin release and by increasing insulin-independent glucose disposal. J Clin Invest. 1994;93:2263–2266. doi: 10.1172/JCI117225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessio DA, Prigeon RL, Ensinck JW. Enteral enhancement of glucose disposition by both insulin-dependant and insulin-independent processes. A physiological role of glucagon-like peptide I. Diabetes. 1995;44:1433–1437. doi: 10.2337/diab.44.12.1433. [DOI] [PubMed] [Google Scholar]
- Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5:749–757. doi: 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- Day JW, Li P, Patterson JT, Chabenne J, DiMarchi Chabenne M, Gelfanov VM, et al. Charge inversion at position 68 of the glucagon and glucagon-like peptide-1 receptors supports selectivity in hormone action. J Pept Sci. 2010;17:218–225. doi: 10.1002/psc.1317. [DOI] [PubMed] [Google Scholar]
- Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide01 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab. 1995;80:952–957. doi: 10.1210/jcem.80.3.7883856. [DOI] [PubMed] [Google Scholar]
- Deacon CF, Knudsen LB, Madsen K, Wiberg FC, Jacobsen O, Holst JJ. Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity. Diabetologia. 1998;41:271–278. doi: 10.1007/s001250050903. [DOI] [PubMed] [Google Scholar]
- Devigny C, Perez-Balderas F, Hoogeland B, Cuboni S, Wachtel R, Mauch CP, et al. Biomimetic screening of class-B G protein-coupled receptors. J Am Chem Soc. 2011;133:8927–8933. doi: 10.1021/ja200160s. [DOI] [PubMed] [Google Scholar]
- Dillon JS, Tanizawa Y, Wheeler MB, Leng X-H, Ligon BB, Rabin DU, et al. Cloning and functional expression of the human glucagon-like peptide-1 (GLP-1) receptor. Endocrinology. 1993;133:1907–1910. doi: 10.1210/endo.133.4.8404634. [DOI] [PubMed] [Google Scholar]
- Dong M, Pinon DI, Asmann YW, Miller LJ. Possible endogenous agonist mechanism for the activation of secretin family G protein-coupled receptors. Mol Pharm. 2006;70:206–213. doi: 10.1124/mol.105.021840. [DOI] [PubMed] [Google Scholar]
- Dong M, Gao F, Pinon DI, Miller LJ. Insights into the structural basis of endogenous agonist activation of family B G protein-coupled receptors. Mol Endocrinol. 2008;22:1489–1499. doi: 10.1210/me.2008-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly D. The arrangement of the transmembrane helices in the secretin receptor family of G-protein-coupled receptors. FEBS Lett. 1997;409:431–436. doi: 10.1016/s0014-5793(97)00546-2. [DOI] [PubMed] [Google Scholar]
- Donnelly D, Johnson MS, Blundell TL, Saunders J. An analysis of the periodicity of conserved residues in sequence alignments of G-protein coupled receptors. FEBS Lett. 1989;251:109–116. doi: 10.1016/0014-5793(89)81438-3. [DOI] [PubMed] [Google Scholar]
- Donnelly D, Findlay JBC, Blundell TL. The evolution and structure of aminergic G protein-coupled receptors. Receptors Channels. 1994;2:61–78. [PubMed] [Google Scholar]
- Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007;113:546–593. doi: 10.1016/j.pharmthera.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle ME, Theodorakis MJ, Holloway HW, Bernier M, Greig NH, Egan JM. The importance of the nine-amino acid C-terminal sequence of exendin-4 for binding to the GLP-1 receptor and for biological activity. Regul Pept. 2003;114:153–158. doi: 10.1016/s0167-0115(03)00120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci U S A. 1987;84:3434–3438. doi: 10.1073/pnas.84.10.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards CMB, Todd JF, Mahmoudi M, Wang Z, Wang RM, Ghatei MA, et al. Glucagon-like peptide 1 has a physiological role in the control of postprandial glucose in humans. Diabetes. 1999;48:86–93. doi: 10.2337/diabetes.48.1.86. [DOI] [PubMed] [Google Scholar]
- Egan JM, Meneilly GS, Habener JF, Elahi D. Glucagon-like peptide-1 augments insulin-mediated glucose uptake in the obese state. J Clin Endocrinol Metab. 2002;87:3768–3773. doi: 10.1210/jcem.87.8.8743. [DOI] [PubMed] [Google Scholar]
- Elahi D, Egan JM, Shannon RP, Meneilly GS, Khatri A, Habener JF, et al. GLP-1 (9-36) amide, cleavage product of GLP-1 (7-36) amide, is a glucoregulatory peptide. Obesity. 2008;16:1501–1509. doi: 10.1038/oby.2008.229. [DOI] [PubMed] [Google Scholar]
- Eng J, Andrews PC, Kleinman WA, Singh L, Raufman J-P. Purification and structure of exendin-3, a new pancreatic secretagogue isolated from Heloderma horridum venom. J Biol Chem. 1990;265:20259–20262. [PubMed] [Google Scholar]
- Eng J, Kleinman WA, Singh L, Singh G, Raufman J-P. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. J Biol Chem. 1992;267:7402–7405. [PubMed] [Google Scholar]
- Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101:515–520. doi: 10.1172/JCI990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frimurer TM, Bywater RP. Structure of the intergral membrane domain of the GLP1 receptor. Proteins. 1999;35:375–386. [PubMed] [Google Scholar]
- Gallwitz B, Schmidt WE, Conlon JM, Creutzfeldt W. Glucagon-like peptide-1 (7-36) amide: characterization of the domain responsible for binding to its receptor on rat insulinoma RINm5F cells. J Mol Endocrinol. 1990;5:33–39. doi: 10.1677/jme.0.0050033. [DOI] [PubMed] [Google Scholar]
- Gallwitz B, Witt M, Paetzold G, Morays-Wortmann C, Zimmermann B, Eckart K, et al. Structure/activity characterization of glucagon-like peptide-1. Eur J Biochem. 1994;225:1151–1156. doi: 10.1111/j.1432-1033.1994.1151b.x. [DOI] [PubMed] [Google Scholar]
- Göke R, Fehmann H-C, Linn T, Schmidt H, Krause M, Eng J, et al. Exendin-4 is a high potency agonist and truncated exendin-(9-39)-amide an antagonist at the glucagon-like peptide 1-(7-36)-amide receptor of insulin-secreting β-cells. J Biol Chem. 1993;268:19650–19655. [PubMed] [Google Scholar]
- Göke R, Just R, Lankat-Buttgereit B, Göke B. Glycosylation of the GLP-1 receptor is a prerequisite for regular receptor function. Peptides. 1994;15:675–681. doi: 10.1016/0196-9781(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Gong N, Ma A-N, Zhang L-J, Luo X-S, Zhang Y-H, Xu M, et al. Site-specific PEGylation of exenatide analogues markedly improved their glucoregulatory activity. Br J Pharmacol. 2011;163:399–412. doi: 10.1111/j.1476-5381.2011.01227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano MP, Hey PJ, Borkowski D, Chicchi GG, Strader CD. Cloning and functional expression of a human glucagon-like peptide-1 receptor. Biochem Biophys Res Commun. 1993;196:141–146. doi: 10.1006/bbrc.1993.2226. [DOI] [PubMed] [Google Scholar]
- Hällbrink M, Holmqvist T, Olsson M, Östenson C-G, Efendic S, Langel Ü. Different domains in the third intracellular loop of the GLP-1 receptor are responsible for Gαs and Gαi/Gαo activation. Biochim Biophys Acta. 2001;1546:79–86. doi: 10.1016/s0167-4838(00)00270-3. [DOI] [PubMed] [Google Scholar]
- Hansen L, Deacon CF, Ørskov C, Holst JJ. Glucagon-LikePeptide-1-(7-36) amide is transformed to glucagon-like peptide-1-(9-36) amide by Dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology. 1999;140:5356–5363. doi: 10.1210/endo.140.11.7143. [DOI] [PubMed] [Google Scholar]
- Hareter A, Hoffman E, Bode H-P, Göke B, Göke R. The positive charge of the imidazole side chain of Histidine7 is crucial for GLP-1 action. Endocr J. 1997;44:701–705. doi: 10.1507/endocrj.44.701. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Pinon DI, Miller LJ. Transmembrane segment IV contributes a functionally important interface for oligomerization of the class II G protein-coupled secretin receptor. J Biol Chem. 2007;282:30363–30372. doi: 10.1074/jbc.M702325200. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Ball AM, Sexton PM, Miller LJ. Importance of lipid-exposed residues in transmembrane segment four for family B calcitonin receptor homo-dimerization. Regul Pept. 2010;164:113–119. doi: 10.1016/j.regpep.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RS, Kieffer TJ, Habener JF. Point mutations in the first and third intracellular loops of the glucagon-like peptide-1 receptor alter intracellular signaling. Biochem Biophys Res Commun. 1996;223:624–632. doi: 10.1006/bbrc.1996.0945. [DOI] [PubMed] [Google Scholar]
- Hjorth SA, Schwartz TW. Glucagon and GLP-1 receptors: lessons from chimeric ligands and receptors. Acta Physiol Scand. 1996;157:343–345. doi: 10.1046/j.1365-201X.1996.37259000.x. [DOI] [PubMed] [Google Scholar]
- Hjorth SA, Adelhorst K, Brogaard Pedersen B, Kirk O, Schwartz TW. Glucagon and glucagon-like peptide 1: selective receptor recognition via distinct peptide epitopes. J Biol Chem. 1994;269:30121–30124. [PubMed] [Google Scholar]
- Hoare SRJ. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov Today. 2005;10:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- Holst JJ, Ørskov C, Vagn Nielsen O, Schwartz TW. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 1987;211:169–174. doi: 10.1016/0014-5793(87)81430-8. [DOI] [PubMed] [Google Scholar]
- Huang Y, Wilkinson GF, Willars GB. Role of the signal peptide in the synthesis and processing of the glucagon-like peptide-1 receptor. Br J Pharmacol. 2010;159:237–251. doi: 10.1111/j.1476-5381.2009.00517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iltz JL, Baker DE, Setter SM, Campbell RK. Exenatide: an incretin mimetic for the treatment of type 2 diabetes mellitus. Clin Ther. 2006;28:652–665. doi: 10.1016/j.clinthera.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Irwin DM, Satkunarajah M, Wen Y, Brubaker PL, Pederson RA, Wheeler MB. The Xenopus proglucagon gene encodes novel GLP-1-like peptides with insulinotropic properties. Proc Natl Acad Sci U S A. 1997;94:7915–7920. doi: 10.1073/pnas.94.15.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin N, Flatt PR, Patterson S, Green BD. Insulin-releasing and metabolic effects of small molecule GLP-1 receptor agonist 6, 7-dichloro-2-methylsulfonyl-3-N-tert-butylaminoquinoxaline. Eur J Pharmacol. 2010;628:268–273. doi: 10.1016/j.ejphar.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Knudsen LB, Pridal L. Glucagon-like peptide-1-(9-36) amide is a major metabolite of glucagon-like peptide-1-(7-36) amide after in vivo administration to dogs, and it acts as an antagonist on the pancreatic receptor. Eur J Pharmacol. 1996;318:429–435. doi: 10.1016/s0014-2999(96)00795-9. [DOI] [PubMed] [Google Scholar]
- Knudsen LB, Kiel D, Teng M, Behrens C, Bhumralkar D, Kodra JT, et al. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc Natl Acad Sci U S A. 2007;104:937–942. doi: 10.1073/pnas.0605701104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolligs F, Fehmann H-C, Göke R, Göke B. Reduction of the incretin effect in rats by the glucagon-like peptide 1 receptor antagonist exendin (9-36) amide. Diabetes. 1995;44:16–19. doi: 10.2337/diab.44.1.16. [DOI] [PubMed] [Google Scholar]
- Koole C, Wootten D, Simms J, Valant C, Sridhar R, Woodman OL, et al. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol Pharmacol. 2010;78:456–465. doi: 10.1124/mol.110.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet. 1987;330:1300–1304. doi: 10.1016/s0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]
- Lin F, Wang R. Molecular modelling of the three-dimensional structure of GLP-IR and its interactions with several agonists. J Mol Model. 2009;15:53–65. doi: 10.1007/s00894-008-0372-2. [DOI] [PubMed] [Google Scholar]
- López de Maturana R, Donnelly D. The glucagon-like peptide-1 receptor binding site for the N-terminus of GLP-1 requires polarity at Asp198 rather than negative charge. FEBS Lett. 2002;530:244–248. doi: 10.1016/s0014-5793(02)03492-0. [DOI] [PubMed] [Google Scholar]
- López de Maturana R, Willshaw A, Kuntzsch A, Rudolph R, Donnelly D. The isolated N-terminal domain of the Glucagon-like Peptide-1 (GLP-1) receptor binds exendin peptides with much higher affinity than GLP-1. J Biol Chem. 2003;278:10195–10200. doi: 10.1074/jbc.M212147200. [DOI] [PubMed] [Google Scholar]
- López de Maturana R, Treece-Birch J, Abidi F, Findlay JB, Donnelly D. Met-204 and Tyr-205 are together important for binding GLP-1 receptor agonists but not their N-terminally truncated analogues. Protein Pept Lett. 2004;11:15–22. doi: 10.2174/0929866043478491. [DOI] [PubMed] [Google Scholar]
- Mann R, Nasr N, Hadden D, Sinfield J, Abidi F, Al-Sabah S, et al. Peptide binding at the GLP-1 receptor. Biochem Soc Trans. 2007;35:713–716. doi: 10.1042/BST0350713. [DOI] [PubMed] [Google Scholar]
- Mann RJ, Al-Sabah S, López de Maturana R, Sinfield JK, Donnelly D. Functional coupling of Cys-226 and Cys-296 in the glucagon-like peptide-1 (GLP-1) receptor indicates a disulfide bond that is close to the activation pocket. Peptides. 2010a;31:2289–2293. doi: 10.1016/j.peptides.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Mann RJ, Nasr NE, Sinfield JK, Paci E, Donnelly D. The major determinant of exendin-4/glucagon-like peptide 1 differential affinity at the rat glucagon-like peptide 1 receptor N-terminal domain is a hydrogen bond from SER-32 of exendin-4. Br J Pharmacol. 2010b;160:1973–1984. doi: 10.1111/j.1476-5381.2010.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli C, Natarajan SI, Meyer JP, Bastos MM, Bernatowicz MS, Lee VG, et al. Eleven amino acid glucagon-like peptide-1 receptor agonists with antidiabetic activity. J Med Chem. 2009;52:7788–7799. doi: 10.1021/jm900752a. [DOI] [PubMed] [Google Scholar]
- Mathi SK, Chan Y, Li X, Wheeler MB. Scanning of the Glucagon-Like Peptide-1 Receptor localizes G protein-activating determinants primarily to the N terminus of the third intracellular loop. Mol Endocrinol. 1997;11:424–432. doi: 10.1210/mend.11.4.9913. [DOI] [PubMed] [Google Scholar]
- McIntyre N, Holdsworth CD, Turner DS. New interpretation of oral glucose tolerance. Lancet. 1964;284:20–21. doi: 10.1016/s0140-6736(64)90011-x. [DOI] [PubMed] [Google Scholar]
- Miller LJ, Chen Q, Lam PC-H, Pinon DI, Sexton PM, Abagyan R, et al. Refinement of glucagon-like peptide 1 docking to its intact receptor using mid-region photolabile probes and molecular modeling. J Biol Chem. 2011;286:15895–15907. doi: 10.1074/jbc.M110.217901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojsov S. Structural requirements for biological activity of glucagon-like peptide-I. Int J Peptide Protein Res. 1992;40:333–343. doi: 10.1111/j.1399-3011.1992.tb00309.x. [DOI] [PubMed] [Google Scholar]
- Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J Biol Chem. 1986;261:11880–11889. [PubMed] [Google Scholar]
- Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. 1987;79:616–619. doi: 10.1172/JCI112855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C, Yang H, Rodgers BD, Beday A, Pritchette LA, Eng J. High potency antagonists of the pancreatic glucagon-like peptide-1 receptor. J Biol Chem. 1997;272:21201–21206. doi: 10.1074/jbc.272.34.21201. [DOI] [PubMed] [Google Scholar]
- Murage EN, Schroeder JC, Beinborn M, Ahn J-M. Search for alpha-helical propensity in the receptor-bound conformation of glucagon-like peptide-1. Bioorg Med Chem. 2008;16:10106–10112. doi: 10.1016/j.bmc.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Murage EN, Gao G, Bisello A, Ahn J-M. Development of potent glucagon-like peptide-1 agonists with high enzyme stability via introduction of multiple lactam bridges. J Med Chem. 2010;53:6412–6420. doi: 10.1021/jm100602m. [DOI] [PubMed] [Google Scholar]
- Nathan DM, Schreiber E, Fogel H, Majsov S, Habener JF. Insuilinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care. 1992;15:270–276. doi: 10.2337/diacare.15.2.270. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Erbert R, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. 1986;63:492–498. doi: 10.1210/jcem-63-2-492. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Heimesaat MM, Ørskov C, Holst JJ, Erbert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993;91:301–307. doi: 10.1172/JCI116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauck MA, Wollschläger D, Werner J, Holst JJ, Ørskov C, Creutzfeldt W, et al. Effects of subcutaneous glucagon-like peptide 1 (GLP-1 [7-36 amide]) in patients with NIDDM. Diabetologia. 1996;39:1546–1553. doi: 10.1007/s001250050613. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Ørskov C, Ritzel R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273:E981–E988. doi: 10.1152/ajpendo.1997.273.5.E981. [DOI] [PubMed] [Google Scholar]
- Neidigh JW, Andersen NH. Peptide conformational changes induced by tryptophan-phosphocholine interactions in a micelle. Biopolymers. 2002;65:354–361. doi: 10.1002/bip.10272. [DOI] [PubMed] [Google Scholar]
- Neidigh JW, Fesinmeyer RW, Prickett KS, Andersen NH. Exendin-4 and glucagon-like-peptide-1: NMR structural comparisons in the solution and micelle-associated states. Biochemistry. 2001;40:13188–13200. doi: 10.1021/bi010902s. [DOI] [PubMed] [Google Scholar]
- Neidigh JW, Fesinmeyer RM, Andersen NH. Designing a 20-residue protein. Nat Struct Biol. 2002;9:425–430. doi: 10.1038/nsb798. [DOI] [PubMed] [Google Scholar]
- Neumann J-M, Couvineau A, Murail S, Lacapère J-J, Jamin N, Laburthe M. Class-B GPCR activation: is ligand helix-capping the key? Trends Biochem Sci. 2008;33:314–319. doi: 10.1016/j.tibs.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Neumiller JJ, Campbell RK. Liraglutide: a once-daily incretin mimetic for the treatment of type 2 diabetes mellitus. Ann Pharmacother. 2009;43:1433–1444. doi: 10.1345/aph.1M134. [DOI] [PubMed] [Google Scholar]
- Ørskov C, Nielsen JH. Truncated glucagon-like peptide-1 (proglucagon 78-107 amide), an intestinal insulin-releasing peptide, has specific receptors on rat insulinoma cells (RIN 5AH) FEBS Lett. 1988;229:175–178. doi: 10.1016/0014-5793(88)80821-4. [DOI] [PubMed] [Google Scholar]
- Ørskov C, Wettergren A, Holst JJ. Biological effects and metabolic rates of glucagon-like peptide-1 7-36 amide and glucagon-like peptide-1 7-37 in healthy subjects are indistinguishable. Diabetes. 1993;42:658–661. doi: 10.2337/diab.42.5.658. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fo BA, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Parker JC, Andrews KM, Rescek DM, Massefski W, Andrews GC, Contillo LG, et al. Structure-function analysis of a series of glucagon-like peptide-1 analogs. J Pept Res. 1998;52:398–409. doi: 10.1111/j.1399-3011.1998.tb00664.x. [DOI] [PubMed] [Google Scholar]
- Parthier C, Reedtz-Runge S, Rudoloh R, Stubbs MT. Passing the baton in class B GPCRs: peptide hormone activation via helix induction? Trends Biochem Sci. 2009;34:303–310. doi: 10.1016/j.tibs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Patterson JT, Day JW, Gelfanov VM, DiMarchi RD. Functional association of the N-terminal residues with the central region in glucagon-related peptides. J Pept Sci. 2011a;17:659–666. doi: 10.1002/psc.1385. [DOI] [PubMed] [Google Scholar]
- Patterson JT, Ottaway N, Gelfanov VM, Smiley DL, Perez-Tilve D, Pfluger PT, et al. A novel human-based receptor antagonist of sustained action reveals body weight control by endogenous GLP-1. ACS Chem Biol. 2011b;6:135–145. doi: 10.1021/cb1002015. [DOI] [PubMed] [Google Scholar]
- Perfetti R, Hui H. The role of GLP-1 in the life and death of pancreatic beta cells. Horm Metab Res. 2004;36:804–810. doi: 10.1055/s-2004-826167. [DOI] [PubMed] [Google Scholar]
- Prévost M, Vertongen P, Raussens V, Roberts DJ, Cnuddle J, Perret J, et al. Mutational and cysteine scanning analysis of the glucagon receptor N-terminal domain. J Biol Chem. 2010;285:30951–30598. doi: 10.1074/jbc.M110.102814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raufman J-P, Singh L, Eng J. Exendin-3, a novel peptide from Heloderma horridum venom, interacts with vasoactive intestinal peptide receptors and a newly described receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1991;266:2897–2902. [PubMed] [Google Scholar]
- Raufman J-P, Singh L, Singh G, Eng J. Truncated glucagon-like peptide-1 interacts with exendin receptors on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267:21432–21437. [PubMed] [Google Scholar]
- Runge A, Schimmer S, Oschmann J, Schiødt CB, Knudsen SM, Jeppesen CB, et al. Differential structural properties of GLP-1 and exendin-4 determine their relative affinity for the GLP-1 receptor N-terminal extracellular domain. Biochemistry. 2007;46:5830–5840. doi: 10.1021/bi062309m. [DOI] [PubMed] [Google Scholar]
- Runge S, Gram C, Bräuner-Osborne H, Madsen K, Knudsen LB, Wulff BS. Three distinct epitopes on the extracellular face of the glucagon receptor determine specificity for the glucagon amino terminus. J Biol Chem. 2003a;278:28005–28010. doi: 10.1074/jbc.M301085200. [DOI] [PubMed] [Google Scholar]
- Runge S, Wulff BS, Madsen K, Bräuner-Osborne H, Knudsen LB. Different domains of the glucagon and glucagon-like peptide-1 receptors provide the critical determinants of ligand selectivity. Br J Pharmacol. 2003b;138:787–794. doi: 10.1038/sj.bjp.0705120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge S, Thøgersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- Salapatek A-MF, MacDonald PE, Gaisano HY, Wheeler MB. Mutations to the Third Cytoplasmic Domain of the Glucagon-Like Peptide 1 (GLP-1) Receptor Can Functionally Uncouple GLP-1-Stimulated Insulin Secretion in HIT-T15 Cells. Mol Endocrinol. 1999;13:1305–1317. doi: 10.1210/mend.13.8.0321. [DOI] [PubMed] [Google Scholar]
- Schepp W, Schmidtler J, Riedel T, Dehne K, Schusdziarra V, Holst JJ, et al. Exendin-4 and exendin-(9-39)-amide: agonist and antagonist, respectively at the rat parietal cell receptor for glucagon-like peptide-1-(7-36)-amide. Eur J Pharmacol. 1994;269:183–191. doi: 10.1016/0922-4106(94)90085-x. [DOI] [PubMed] [Google Scholar]
- Schröder-Tittmann K, Bosse-Doenecke E, Reedtz-Runge S, Ihling C, Sinz A, Tittmann K, et al. Recombinant expression, in vitro refolding, and biophysical characterization of the human glucagon-like peptide-1 receptor. Biochemistry. 2010;49:7956–7965. doi: 10.1021/bi101159s. [DOI] [PubMed] [Google Scholar]
- Segre GV, Goldring SR. Receptors for secretin, calcitonin, parathyroid hormone (PTH)/PTH-related peptide, vasoactive intestinal peptide, glucagonlike peptide 1, growth hormone-releasing hormone, and glucagon belong to a newly discovered G-protein-linked receptor family. Trends Endocrinol Metab. 1993;4:309–314. doi: 10.1016/1043-2760(93)90071-l. [DOI] [PubMed] [Google Scholar]
- Serre V, Dolci W, Schaerer E, Scrocchi L, Drucker D, Efrat S, et al. Exendin-(9-39) is an inverse agonist of the murine glucagon-like peptide-1 receptor: implications for basal intracellular cyclic adenosine 3′, 5′- monophosphate levels and β-cell glucose competence. Endocrinology. 1998;139:4448–4454. doi: 10.1210/endo.139.11.6295. [DOI] [PubMed] [Google Scholar]
- Siegel EG, Gallwitz B, Scharf G, Mentlein R, Morys-Wortmann C, Fölsch UR, et al. Biological activity of GLP-1-analogues with N-terminal modifications. Regul Pept. 1999;79:93–102. doi: 10.1016/s0167-0115(98)00155-4. [DOI] [PubMed] [Google Scholar]
- Sloop KW, Willard FS, Brenner MB, Ficorilli J, Valasek K, Showalter AD, et al. Novel small molecule glucagon-like peptide-1 agonist stimulates insulin secretion in rodents and from human islets. Diabetes. 2010;59:3099–3107. doi: 10.2337/db10-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, He M, Li H, Liu Q, Wang J, Wang Y, et al. Boc5, a non-peptidic glucagon-like peptide-1 receptor agonist, invokes sustained glycemic control and weight loss in diabetic mice. PLoS ONE. 2008;3:1–11. doi: 10.1371/journal.pone.0002892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Kawai K, Ohashi S, Mukai H, Yamashita K. Comparison of the effects of various C-terminal and N-terminal fragment peptides of glucagon-like peptide-1 on insulin and glucagon release from the isolated perfused rat pancreas. Endocrinology. 1989;125:3109–3114. doi: 10.1210/endo-125-6-3109. [DOI] [PubMed] [Google Scholar]
- Syme CA, Zhang L, Bisello A. Caveolin-1 regulates cellular trafficking and function in the glucagon-like peptide 1 receptor. Mol Endocrinol. 2006;20:3400–3411. doi: 10.1210/me.2006-0178. [DOI] [PubMed] [Google Scholar]
- Takhar A, Gyomorey S, Su R-C, Mathi SK, Li X, Wheeler MB. The third cytoplasmic domain of the GLP-1[7-36 amide] receptor is required for coupling to the adenylyl cyclase system. Endocrinology. 1995;137:2175–2178. doi: 10.1210/endo.137.5.8612565. [DOI] [PubMed] [Google Scholar]
- Tams JW, Knudsen SM, Fahrenkrug J. Proposed arrangement of the seven transmembrane helices in the secretin receptor family. Receptors Channels. 1998;5:79–90. [PubMed] [Google Scholar]
- Teng M, Johnson MD, Thomas C, Kiel D, Lakis JN, Kercher T, et al. Small molecule ago-allosteric modulators of the human glucagon-like peptide-1 (hGLP-1) receptor. Bioorg Med Chem Lett. 2007;17:5472–5478. doi: 10.1016/j.bmcl.2007.06.086. [DOI] [PubMed] [Google Scholar]
- Thorens B. Expression cloning of the pancreatic β cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc Natl Acad Sci U S A. 1992;89:8641–8645. doi: 10.1073/pnas.89.18.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B, Porett A, Bühler L, Deng SP, Morel P, Widmann C. Cloning and functional expression of the human islet GLP-1 receptor. Demonstration that exendin-4 is an agonist and exendin-(9-39) an antagonist of the receptor. Diabetes. 1993;42:1678–1682. doi: 10.2337/diab.42.11.1678. [DOI] [PubMed] [Google Scholar]
- Thornton K, Gorenstein DG. Structure of glucagon-like peptide (7-36) amide in a dodecylphosphocholine micelle as determined by 2D NMR. Biochemistry. 1994;33:3532–3539. doi: 10.1021/bi00178a009. [DOI] [PubMed] [Google Scholar]
- Tibaduiza EC, Chen C, Beinborn M. A small molecule ligand of the glucagon-like peptide 1 receptor targets its amino-terminal hormone binding domain. J Biol Chem. 2001;276:37787–37793. doi: 10.1074/jbc.M106692200. [DOI] [PubMed] [Google Scholar]
- Turton MD, O'Shea D, Gunn I, Beak SA, Edwards CMB, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. 1996;379:69–72. doi: 10.1038/379069a0. [DOI] [PubMed] [Google Scholar]
- Underwood CR, Garibay P, Knudsen LB, Hastrup S, Peters GH, Rudolph R, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem. 2010;285:723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahl TP, Paty BW, Fuller BD, Prigeon RL, D'Alessio DA. Effects of GLP-1-(7-36) amide, GLP-1-(7-37), and GLP-1-(9-36) amide on intravenous glucose tolerance and glucose-induced insulin secretion in healthy humans. J Clin Endocrinol Metab. 2003;88:1772–1779. doi: 10.1210/jc.2002-021479. [DOI] [PubMed] [Google Scholar]
- Wang S, Yu J, Li W, Li F. Structural study of an active analog of EX-4 in solution and micelle associated states. Biopolymers (Peptide Science) 2010;96:348–357. doi: 10.1002/bip.21566. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Kawai K, Oshashi S, Yokota C, Suzuki S, Yamashita K. Structure-activity relationships of glucagon-like peptide-1 (7-36) amide: insulinotropic activities in perfused rat pancreases, and receptor binding and cyclic AMP production in RINm5F cells. J Endocrinol. 1994;140:45–52. doi: 10.1677/joe.0.1400045. [DOI] [PubMed] [Google Scholar]
- Weir GC, Mojsov S, Hendrick GK, Habener JF. Glucagon-like peptide I (7-37) actions on endocrine pancreas. Diabetes. 1989;38:338–342. doi: 10.2337/diab.38.3.338. [DOI] [PubMed] [Google Scholar]
- Wettergren A, Schjoldager B, Mortensen PE, Myhre J, Christiansen J, Holst JJ. Truncated GLP-1 (proglucagon 78-107-amide) inhibits gastric and pancreatic functions in man. Dig Dis Sci. 1993;38:665–673. doi: 10.1007/BF01316798. [DOI] [PubMed] [Google Scholar]
- Widmann C, Dolci W, Thorens B. Agonist-induced internalization and recycling of the glucagon-like peptide-1 receptor in transfected fibroblasts and in insulinomas. Biochem J. 1995;310:203–214. doi: 10.1042/bj3100203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Dolci W, Thorens B. Desensitization and phosphorylation of the glucagon-like peptide-1 (GLP-1) receptor by GLP-1 and 4-Phorbol 12-Myristate 13-Acetate. Mol Endocrinol. 1996a;10:62–75. doi: 10.1210/mend.10.1.8838146. [DOI] [PubMed] [Google Scholar]
- Widmann C, Dolci W, Thorens B. Heterologous desensitization of the glucagon-like peptide-1 receptor by phorbol esters requires phosphorylation of the cytoplasmic tail at four different sites. J Biol Chem. 1996b;271:19957–19963. doi: 10.1074/jbc.271.33.19957. [DOI] [PubMed] [Google Scholar]
- Wilmen A, Göke B, Göke R. The isolated N-terminal extracellular domain of the glucagon-like peptide-1 (GLP)-1 receptor has intrinsic binding activity. FEBS Lett. 1996;398:43–47. doi: 10.1016/s0014-5793(96)01214-8. [DOI] [PubMed] [Google Scholar]
- Wootten D, Simms J, Koole C, Woodman OL, Summers RJ, Christopoulos A, et al. Modulation of the glucagon-like peptide-1 receptor signaling by naturally occurring and synthetic flavonoids. J Pharmacol Exp Ther. 2011;336:540–550. doi: 10.1124/jpet.110.176362. [DOI] [PubMed] [Google Scholar]
- Xiao Q, Jeng W, Wheeler MB. Characterization of glucagon-like peptide-1 receptor binding determinants. J Mol Endocrinol. 2000;25:321–335. doi: 10.1677/jme.0.0250321. [DOI] [PubMed] [Google Scholar]
- Xiao Q, Giguere J, Parisen M, Jeng WS, Pierre PL, Brubaker PL, et al. Biological activities of glucagon-like peptide-1 analogues in vitro and in vivo. Biochemistry. 2001;40:2860–2869. doi: 10.1021/bi0014498. [DOI] [PubMed] [Google Scholar]