

Abstract
During evolution, mammals have developed remarkably similar molecular mechanisms to respond to external challenges and maintain survival. Critical regulators of these mechanisms are the family of ‘stress’-peptides that consists of the corticotropin-releasing hormone (CRH) and urocortins (Ucns). These neuropeptides ‘fine-tune’ integration of an intricate series of physiological responses involving the autonomic, endocrine, immune, cardiovascular and reproductive systems, which induce a spectrum of behavioural and homeostatic changes. CRH and Ucns exert their actions by activating two types of CRH receptors (CRH-R), CRH-R1 and CRH-R2, which belong to the class-B1 family of GPCRs. The CRH-Rs exhibit signalling promiscuity facilitated by their ability to couple to multiple G-proteins and regulate diverse intracellular networks that involve intracellular effectors such as cAMP and an array of PKs in an agonist and tissue-specific manner, a property that allows them to exert unique roles in the integration of homeostatic mechanisms. We only now begin to unravel the plethora of CRH-R biological actions and the transcriptional and post-translational mechanisms such as alternative mRNA splicing or phosphorylation-mediated desensitization developed to tightly control CRH-Rs biological activity and regulate their physiological actions. This review summarizes the current understanding of CRH-R signalling complexity and regulatory mechanisms that underpin cellular responses to CRH and Ucns.
LINKED ARTICLES
This article is part of a themed section on Secretin Family (Class B) G Protein-Coupled Receptors. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.166.issue-1
Keywords: alternative splicing, class-B1 GPCRs, CRH, CRH-R, MAPK, protein kinases, signalling, urocortin
Introduction
Mammalian adaptive responses to stress involve activation of the hypothalamo-pituitary-adrenal axis, predominantly via the hypothalamic hormone, corticotropin releasing hormone (CRH), which regulates the secretion of adrenocorticotrophin (ACTH) from the anterior pituitary (Bale and Vale, 2004). CRH also exerts multiple central and peripheral actions, which underpin its critical role in integrating the activity of diverse physiological mechanisms. This complex process of stress adaptation is fine-tuned by other CRH-like peptides, such as the family of urocortins (Ucn1, Ucn2 and Ucn3) that also exerts complementary or sometimes distinct actions (Suda et al., 2004). CRH and Ucns actions in target cells involve activation of specific receptors expressed on the cell membrane of target cells. Mammalian tissues express two types of specific CRH receptors (CRH-R), termed R1 and R2, which belong to the class B1 of GPCRs (receptors for ‘brain-gut’ neuropeptides).
The cognate agonists of class B1 GPCRs regulate important physiological functions such as exocrine and endocrine secretions, feeding behaviour, metabolism, growth, and neuro- and immuno-modulations. Members of this GPCR family are encoded by 15 genes in humans and the ligands for these receptors are polypeptide hormones that most often act in a paracrine or autocrine manner, including secretin, glucagon, vasoactive intestinal peptide, CRH and parathyroid hormone. Evolution studies suggest that the class B1 GPCRs could have descended from a subgroup of adhesion GPCRs (Nordstrom et al., 2009). They have a complex genomic architecture consisting of multiple exons and introns in their open reading frame, a feature that permits generation of multiple mRNA splice variants. Interestingly, a number of well-conserved splice sites have been identified across the B1 GPCRs which are also present in adhesion GPCRs. Amino acid frequency comparisons, relative to their position in the GPCR structure, across all class B1 GPCRs demonstrated that exons encoding the distal N-terminal segment and C-terminal tail of these receptors exhibit extensive amino acid hypervariability (<10% of amino acids conserved in two-thirds of GPCRs) consistent with a crucial role in ligand discrimination and signalling specificity respectively (Markovic and Grammatopoulos, 2009).
CRH receptors are also found in non-mammalian vertebrate species (Chang and Hsu, 2004). The CRH family of peptides exhibit significant sequence identity and structure similarity with the diuretic hormones in insects (Audsley et al., 1995). Their actions are also mediated via GPCR homologous to the mammalian CRH receptors. A clear evolutionary trail for the origin of the CRH/CRH receptor system from invertebrates to vertebrates has emerged, where the ancestors of the diuretic hormone and diuretic hormone receptors co-evolved and gave rise to diuretic hormone/diuretic hormone receptor in insects and CRH/CRH receptor in vertebrates (Chang and Hsu, 2004). Although the vertebrate CRH/CRH receptor signalling could have originated as a paracrine signalling system important for osmoregulation, during early evolution of vertebrates, the ancestor CRH/CRH receptor signalling pathway assumed additional functions that included the regulation of stress responses to environmental stimuli. It is likely that the presence of multiple highly conserved CRH-like peptides and receptors in vertebrates conferred an evolutionary advantage. Interestingly, in amphibian species, CRH has been shown to regulate metamorphosis in response to pond drying (Denver, 1997), suggesting a dual role as an osmoregulator and stress transducer between environmental and physiological responses of an organism. Thus, the CRH family peptides and their receptors are phylogenetically ancient developmental signalling molecules that allow developing organisms to coordinate physiological responses in a changing environment.
Structural determinants of agonist – CRH-R interactions and receptor activation
Structurally, the types 1 and 2 CRH-R are approximately 70% identical at the amino acid level, but exhibit considerable divergence at the N-terminal extracellular domain (ECD) (approximately 47%), consistent with their distinct pharmacological properties and agonist selectivity, a characteristic important for their unique physiological roles, when natively co-expressed in specific tissues. CRH-R1 binds CRH as well as Ucn1, but not Ucn2 or Ucn3, with equivalent high affinity. In contrast, the CRH-R2 exhibits ligand selectivity and binds all the Ucns with significantly higher binding affinity than CRH, suggesting that these peptides may be its natural ligands (Chen et al., 1993; Lovenberg et al., 1995; Vaughan et al., 1995; Hsu and Hsueh, 2001; Lewis et al., 2001; Reyes et al., 2001).
Within the CRH-R ligands amino acid sequence (Figure 1), a number of residues have been identified as important for determining binding specificity (Jahn et al., 2004; Mazur et al., 2004). A proline residue at position 11 (the numbering of residues is based on human CRH sequence) is found only in CRH-R2 selective ligands Ucn2 and Ucn3, and substitution of Pro11 in the Ucn2 sequence with corresponding amino acids, found in the CRH-R non-selective agonists, decreased binding potency to CRH-R2 while increasing CRH-R1 activity. In contrast, introduction of Pro11 in the amino acid sequence of CRH decreases α-helicity and impairs binding to CRH-R1. Furthermore, CRH-R2 selective peptides contain alanine residues at positions 35 and 39, while CRH-R non-selective peptides contain an invariant arginine at position 35 and an acidic amino acid at position 39. Introduction of proline at position 11 and alanine at positions 35 and 39 increases CRH-R2 selectivity in CRH-R non-selective peptides, mainly through the loss of CRH-R1 potency (Mazur et al., 2004). Interestingly, unlike Ucn1, this substitution in the CRH sequence results in a loss of potency at both the CRH-R1 and R2, indicating that CRH requires additional substitutions to achieve recognition by CRH-R2.
Figure 1.

Amino acid sequence alignment of CRH and CRH-related peptides. CRH-R1 binds CRH as well as Ucn1, but not Ucn2 or Ucn3, whereas the CRH-R2 binds Ucns with significantly higher binding affinity than CRH. A proline residue at position 11 (coloured red), is found only in CRH-R2 selective ligands Ucn2 and Ucn3. CRH-R2 selective peptides also contain alanine residues at positions 35 and 39 (coloured red), while CRH-R non-selective peptides contain an invariant arginine at position 35 and an acidic amino acid at position 39 (coloured blue).
The N-terminus ECD
The CRH-Rs N-terminal ECD comprises the major ligand binding site and serves to dock peptide ligand via its C-terminal segment along the hydrophobic face of their helices. Early mutation studies identified a number of regions such as amino acids 43–50, 76–84 and 68–109, within the N-terminal ECD as crucial for binding of CRH-R1 receptor agonists and peptide antagonists (Wille et al., 1999). Subsequent studies employing NMR and X-ray crystallography (Grace et al., 2004; 2007; 2010; Pioszak et al., 2008; Pal et al., 2010) identified that both CRH-R1 and R2 ECDs contain a short consensus repeat fold, a characteristic of the ECD of class B1 GPCRs. Agonist-receptor molecular interaction positions the N-terminal segment of the agonist in close proximity to interact with the receptor's juxtamembrane domains to activate the receptor and induces a helix formation towards the N terminus of the ligand to generate a conformational active state (the two-step model for ligand-binding and signalling of class B1 GPCRs). The CRH-R2 ECD peptide-binding site also contains a unique α-helix formed by its pseudo signal peptide. Comparison of the electrostatic surface potentials of the ECD suggests that ligand discrimination is achieved through a charge compatibility mechanism involving a single amino acid difference in the receptors (CRH-R1 Glu104/CRH-R2 Pro-100) at a site proximal to peptide residue 35 (Arg in CRH/Ucn1, Ala in Ucn2/3). CRH-R1 Glu-104 acts as a selectivity filter preventing Ucn2/3 binding because the non-polar Ala-35 is incompatible with the negatively charged Glu-104 (Pal et al., 2010).
The role of extracellular loops (ECLs) and G-protein coupling for ligand high affinity binding
The N-terminus ECD–agonist interaction alone is not sufficient to stimulate coupling of the receptor to G proteins and elicit a signalling response. An additional interaction is required between the juxta-membrane domain of the receptor (the transmembrane helices and intervening loops, the J-domain) and the N-terminal segment of the peptide, which penetrates into the transmembrane segments of the receptor. Different contributions of the J-domain in ligand binding have been described for the R1 and R2 CRH-R: at the G-protein uncoupled state (or R state), the J-domain of the CRH-R2 receptor binds ligands more strongly than the J-domain of the CRH-R1. Data from a number of studies (Liaw et al., 1997a,b; Perrin et al., 1998; Ruhmann et al., 1998; Sydow et al., 1999; Wille et al., 1999; Assil et al., 2001; Rivier et al., 2002; Hoare et al., 2004) suggest that agonist interaction with the CRH-R1 J-domain weakly stabilizes agonist binding, whereas G-protein binding strongly stabilizes agonist–J-domain interaction and agonist high affinity via an allosteric effect (Hoare et al., 2004). Non-peptide antagonist affinity and the full antagonist effect are provided predominantly by the J-domain. Moreover, when expressed independently, the N-terminus ECD of the receptor binds peptide agonist ligands with affinity similar to that seen in the low-affinity, R state of the whole receptor. In contrast, the isolated J-domain mediates full high-affinity binding of non-peptide antagonists and nearly full efficacy receptor activation by peptide agonists, as demonstrated by activation of the J-domain by a tethered N-terminal CRH fragment (Nielsen et al., 2000). In agreement with this, CRH-R1 agonists like CRH and receptor antagonists require interactions with the ECLs and transmembrane domain (TMD) for high affinity binding (Figure 2). Within the J-domain of the CRH-R1, a number of regions have been identified as important for optimal agonist binding: regions within the ECL1 domain, amino acids 175–178 and His189 at the junction of ECL2 and TMD3, and the junction of ECL2 and TMD5 that involves three amino acid residues, Val266, Tyr267 and Thr268. In addition, specific amino acids with ECL2, Trp259 and Phe260 are also involved in receptor interactions with the agonist N-terminus, but do not participate in antagonist binding (Gkountelias et al., 2009). The ECL3 loop is also involved in ligand binding, especially the cassette Tyr346-Asn348 located in close proximity with TMD7 (Sydow et al., 1999). Also, experiments utilizing allosteric modulators with varying effect on CRH binding (inhibition or enhancement) suggest a CRH-R1 activation model in which the receptor exists in three predominant states: an inactive state, a weakly active state and a CRH-bound fully active state; allosteric inverse agonists stabilize the inactive state, and allosteric agonists stabilize the weakly active state; and antagonism of CRH signalling results from destabilization of the fully active state (Hoare et al., 2008). Non-peptide CRH-R1 antagonists, which bind the J-domain, are able to block peptide agonist binding and receptor–G-protein interaction (R-G coupling), whereas the binding of peptide antagonists, predominantly to the N-domain, was unaffected by R-G coupling. A naturally occurring model of these structural/functional relationships is a CRH-R1 variant isoform, termed R1β, which contains a 29-amino acid insert, and has reduced binding affinity for CRH-R1 agonists although its N- and J-domains are structurally intact (Xiong et al., 1995). Impaired R-G coupling due to the presence of the 29-amino acid insert in the ICL1 probably results in reduced ligand affinity of the CRH-R1.
Figure 2.

Amino acid regions, present in the N-terminus EC loops and TMD, important for different agonist (CRH and Ucn1) and antagonist (NBI) binding to CRH-R1. CRH-R1 ligand discrimination is achieved through a mechanism involving a single amino acid Glu-104, which acts as a selectivity filter preventing Ucn2/3 binding because the non-polar Ala-35 is incompatible with the negatively charged Glu-104. Although the CRH-R1 N-terminus is crucial for agonist binding, coupling of the receptor to G proteins requires an additional interaction the juxta-membrane domain of the receptor (the transmembrane helices and intervening loops, the J-domain) and the N-terminal segment of the peptide, which penetrates into the transmembrane segments of the receptor.
The CRH-R2 J-domain also appears to interact with agonists and/or antagonists. Studies (Hoare et al., 2005) using the subtype 2α of the CRH-R2 revealed that the juxta-membrane receptor domain determines the selectivity of antisauvagine-30 (a synthetic CRH-R2 antagonist), whereas the N-terminal-ECD contributes to selectivity of Ucn3, and both domains contribute to selectivity of Ucn2 and astressin2-B [a CRH-R2 antagonist (Rivier et al., 2002)]. Therefore, ligands differ in the contribution of receptor domains to their selectivity, and CRH-R2-selective antagonists can bind the J-domain. Unlike the CRH-R1 receptor, the CRH-R2 J-domain stabilizes receptor structure in a high affinity state for agonists like Ucn2 by about 30-fold and might act as a CRH-R2/CRH-R1 selectivity determinant. Only a weak increase in CRH-R2 affinity for Ucn2 is achieved by R-G coupling at the R-G state (Hoare et al., 2005).
CRH-R G-protein activation and signalling characteristics
G-protein coupling
The CRH-R, like all members of the GPCR family, has the intrinsic ability to couple to heterotrimeric GDP/GTP-bound proteins. Interestingly, studies in native tissues (mouse cortex) and overexpression in HEK293 cells demonstrated that the CRH-Rs are highly promiscuous and can activate multiple Gα subunits, including Gαs, Gαo, Gαq/11, Gαi1/2 and Gαz (Grammatopoulos et al., 1999; 2001). Similar evidence was provided by heterologous expression of CRH-R1α or CRH-R2β in the fission yeast Schizosaccharomyces pombe ectopically expressing yeast–mammalian chimaeric Gα proteins (Ladds et al., 2003).
There is little information regarding CRH-R structural domains important for G-protein interaction. Studies on the molecular mechanisms regulating CRH-R1–G protein interaction suggest that CRH-R1 agonists activate the receptor by binding to at least two J-domain configurations, which can couple to either Gs or Gi. The presence of G-protein specifc J-domain active conformations is supported by studies employed biased agonists generated by amino acid modifications of Ucn1 signalling domain (aminoacid positions 6–15) (Beyermann et al., 2007). Although these agonists retained abilty to activate Gs, they exhibited characteristics of competitive antagonism to Gi activation. Moreover, non-peptide antagonists can antagonize CRH-R1 coupling to Gs competitively but that to Gi allosterically (Berger et al., 2006). The identification of specific CRH-R1 variants with aberrant structure due to exonic sequence modificiations offers some clues about the structural domains important for G-protein coupling. For example, studies on the signalling properties of the CRH-R1d variant (which lacks 14 amino acids from TMD7) suggest that an intact conformation and/or orientation of the C-tail is crucial for receptor interaction with G-proteins, and that distortion of C-tail profoundly impairs G-protein activation (Grammatopoulos et al., 1999). Furthermore, amino acids within ICL3, especially Ser301, appear to be important for efficient CRH-R1 coupling to Gαs-, Gαo-, Gαq/11- and Gαi1/2 (Papadopoulou et al., 2004). The nature of the residue present in position 301 plays a significant role in determining the G-protein activation efficiency of the CRH-R1, either through stabilization of the amphipathic α-helix of the ICL3 or direct amino acid-to-amino acid interactions between CRH-R1 and G-proteins. Gαo-proteins appeared to be the most sensitive G-protein to amino acid substitution, whereas the weakly activated Gαi was found to be the least sensitive. This study also suggested a preference of Gαs-protein for acidic, but not charged, amino acid residues at position 301 in order to achieve full activation through the interaction with the receptor.
The cAMP/PKA signalling pathway
Pro-opiomelanocortin (POMC) release from pituitary corticotrophs by CRH is mediated through stimulation of the cAMP/PKA pathway (Reisine et al., 1985). Indeed, most physiological functions of CRH and Ucns in the CNS and the periphery involve coupling of CRH-R to Gαs-proteins. There are some notable exceptions to this, and CRH-R expressed in placenta or testis is unable to activate Gαs-proteins but rather employs alternative signalling cascades such as activation of MAPK or intracellular calcium (Ca2+)/PKC pathways (Ulisse et al., 1989; Karteris et al., 2000). The physiological rationale for this signalling selectivity is unknown.
The cAMP/PKA pathway initiates intracellular events both in the cytoplasm, resulting in the acute post-translational modification of target proteins by PKA, and in the nucleus at the level of gene transcription regulation by cAMP response element-binding proteins (Shaywitz and Greenberg, 1999). Moreover, in various native cellular models, the effects of CRH-R1-driven cAMP/PKA pathway can diverge through activation of other downstream signalling molecules such as membrane guanylyl cyclase, the NF-κB transcription factor, glycogen synthase kinase-3 and the Wnt/β-catenin pathway, K(+) current [I(sAHP)], ERK1/2 and tyrosine hydroxylase (Haug and Storm, 2000; Aggelidou et al., 2002; Kovalovsky et al., 2002; Zhao and Karalis, 2002; Facci et al., 2003; Nemoto et al., 2005; Khattak et al., 2010). Recently, cAMP-dependent but PKA-independent pathways potentially involving guanine nucleotide exchange proteins directly activated by cAMP (EPACs) have been identified as important mediators of CRH-R1 in AtT20 cells and CRH-R2 overexpressed in HEK293 cells (Van Kolen et al., 2010; Markovic et al., 2011) (Figure 3).
Figure 3.

The central role of the cAMP pathway in amplifying and diversifying CRH-R signalling. In most tissues, both CRH-R1 and R2 receptor couple to and activate Gs-proteins and stimulate intracellular cAMP levels. Through activation of protein kinase A and EPACs, CRH-R agonists can activate a plethora of downstream intracellular effectors, such as GC, ERK1/2, NF-κB, ion channels, tyrosine hydroxylase phosphorylation and GSK-3β.
Activation of MAPKs
Members of the MAPK family (ERK1/2 and p38MAPK) have emerged as important effectors of CRH/Ucn central and peripheral effects; examples of their crucial role in mediating CRH-R biological actions include cardio-protection against ischaemia reperfusion injury (Brar et al., 2004a), behavioural and memory adaptation to stress (Sananbenesi et al., 2003), neuroprotection (Elliott-Hunt et al., 2002; Pedersen et al., 2002), positive or negative regulation of apoptosis depending on the cellular model (Dermitzaki et al., 2002; Radulovic et al., 2003), and regulation of IL-18 expression (Park et al., 2005). Both kinases can be stimulated by agonist-activated CRH-R1 or CRH-R2, although certain agonists exhibit cell type selectivity, most likely due to the different cell-specific signalling machinery employed to activate MAPK. In addition, distinct spatio-temporal characteristics of ERK1/2 activation have been identified between the R1 and R2 CRH-R when overexpressed in HEK293 cells (Punn et al., 2006; Markovic et al., 2008b): in contrast to a sustained ERK1/2 phosphorylation induced by the CRH-R1, ERK1/2 activation downstream of the CRH-R2 is transient and accumulates in both nuclear and cytoplasmic compartments.
The tissue-specific signalling characteristics of the CRH-R-ERK1/2 pathway have been extensively investigated over the past 10 years (Figure 4). In the hippocampus, neuronal cells or pituitary corticotrophs CRH-R induced ERK1/2 phosphoyrlation requires activation of the cAMP/PKA cascade (Elliott-Hunt et al., 2002; Kovalovsky et al., 2002). Interestingly, in other cellular models such as myometrium, endometrial adenocancinoma and breast cancer, CRH-R appears to employ the cAMP/PKA cascade in a different manner that leads to inhibition of ERK1/2 activation, highlighting a versatile use of this pathway to control ERK1/2 activity and downstream cell proliferation or differentiation (Grammatopoulos et al., 2000; Graziani et al., 2002; 2007). One potential inhibitory mechanism at the level of receptor–G-protein interaction might involve PKA-induced phosphorylation of the CRH-R1 ICL3 at position Ser301. This post-translational modification event prevents maximal coupling of overexpressed CRH-R1 with Gαq-protein and downstream activation of PKC-mediated ERK1/2 activation (Papadopoulou et al., 2004). Interestingly, a different functional relationship between the cAMP/PKA and ERK1/2 has been identified for the overexpressed R2 receptor as Ucn2/CRH-R2 activation of ERK1/2 is dependent on the cAMP/PKA cascade (Markovic et al., 2011).
Figure 4.

CRH-R regulation of ERK1/2 activation via the cAMP/PKA pathway. Examples of tissue/cell specific regulation of ERK1/2 activity by the CRH-Rs mediated via the cAMP/PKA pathway. In some tissues, CRH-activated PKA diminishes ERK1/2 phosphorylation and stimulation, whereas in others (right), ERK1/2 activation requires PKA.
The cAMP/PKA casade is not the only pathway, downstream of CRH-R, regulating MAPK phosphorylation. Overexpressed CRH-Rs are able to stimulate ERK1/2 also through PKA-independent mechanisms, involving various signalling cascades such as the Gαq-IP3-PKC pathway and Gβγ-dependent receptor tyrosine kinase transactivation (Punn et al., 2006). Other signalling molecules such as Gαi- and Gαo-proteins, PI3-K and its downstream effector PKB/Akt, MAPK kinase 1, Raf-1 kinase, and possibly intracellular Ca2+, have also been implicated in overexpressed CRH-R-induced ERK1/2 activation (Brar et al., 2004b; Punn et al., 2006). Recent studies, in both pituitary and CRH-R2 overexpresing HEK293 cells, suggested that ERK1/2 activation might also involve cAMP-activated EPAC signalling (Van Kolen et al., 2010; Markovic et al., 2011).
Regulation of NO and NF-κB signalling molecules
In various physiological systems, CRH and CRH-related agonists appear to be important regulators of the NO/cGMP pathway. In human placenta, myometrial smooth muscle and synoviocyte cells, activation of CRH-R1 stimulates expression and activity of the endothelial NO synthase (Aggelidou et al., 2002; Karteris et al., 2005; Ralph et al., 2007) through a Gαs-protein–PKA independent pathway (Aggelidou et al., 2002; Karteris et al., 2005). This interaction results in increased intracellular formation of NO, and the induction of intracellular events involving activation of the soluble form of GC and production of the second messenger cGMP. Interestingly, in other cellular systems such as the H5V murine endothelioma cells, CRH stimulation of CRH-R1 attenuates cytokine-stimulated inducible NO synthase (iNOS) protein expression (Cantarella et al., 2001), suggesting a more complex relationship and highlighting the concept of tissue-specific effects of CRH-R agonists. In contrast, CRH-R2 activation in human umbilical vein endothelial cells leads to potentiation of cytokine-induced iNOS expression (Cantarella et al., 2001).
It is increasingly recognized that CRH-R agonists exert potent immunomodulatory effects. This directed many studies towards investigation of the role of the transcription factor NF-κB as a target and mediator of CRH-R effects in various cells. In a number of cellular models, CRH activates NF-κB to regulate important biological actions; for example, in mouse thymocytes, CRH knockdown leads to impaired LPS-activated NF-κB response (Zhao and Karalis, 2002), whereas in rodent neonatal cardiomyocytes, Ucn1 and CRH-R2 activates NF-κB to induce IL-6 release, and in rodent models of atherosclerosis CRH and CRH-R1 enhance VCAM-1 expression through NF-κB activation, a molecular mechanism postulated to be important for CRH acceleration of atherosclerosis (Huang et al., 2009; Wu et al., 2009). An important role of NF-κB has been identified in corticotrophs in the regulation of pituitary POMC gene by CRH (Karalis et al., 2004); however, this effect appears to be tissue specific because in human immortalized epidermal (PIG1) melanocytes, CRH stimulation of POMC mRNA and ACTH peptide production involves inhibition of NF-κB activity (Zbytek et al., 2006). A similar inhibitory action of CRH on NF-κB transcriptional activity has been described in primary neurons and clonal hippocampal cells, which appears to be important for protection against oxidative cell death (Lezoualc'h et al., 2000).
Regulation of CRH-R signalling
The CRH-R activity is susceptible to intracellular mechanisms that rapidly attenuate signalling output and prevent cell overstimulation. Similar to most GPCRs, this mechanism involves three distinct cellular phenomena: receptor post-translational modification, usually phosphorylation by second messenger-activated PKs or G protein-coupled receptor kinases-(GRKs), followed by recruitment of specific adaptor proteins such as β-arrestins that induce receptor desensitization and uncoupling from G-proteins and subsequent receptor endocytosis.
CRH-R phosphorylation and desensitization
Although both CRH-R1 and R2 sequence contains a putative PKA phosphorylation site within ICL3, agonist-induced (homologous) desensitization of CRH-R1 appears to be a PKA-independent process (Hauger et al., 2000; Teli et al., 2005). Upon activation, PKA can indeed phosphorylate the CRH-R1 and this post-translational modification appears to be important for determining G-protein coupling and signalling direction (Papadopoulou et al., 2004). The CRH-R sequence also contains multiple PKC phosphorylation sites; however, current evidence suggests that PKC phosphorylation is not involved in homologous CRH-R1 desensitization (Hauger et al., 2003). Studies on the structural determinants of CRH-R1 coupling to Gs proteins and response to PKC phosphorylation identified Ser408 in the distal part of the CRH-R1 C-tail as a critical residue for receptor resistance to PKC-induced desensitization as replacement of Ser408 renders the CRH-R1 susceptible to PKC-induced desensitization and internalization (Teli et al., 2008). In addition, there is evidence to suggest that CRH-R activity is heterologously regulated by PKC; for example, in Y79 human neuroblastoma cells, PKC is involved in the heterologous desensitization of the CRH-induced cAMP response (Hauger et al., 2003), whereas in human pregnant term myometrium oxytocin activated, PKC diminishes CRH-R-induced activity of the Gs-protein-cAMP pathway (Grammatopoulos and Hillhouse, 1999).
The major PK consistently identified in mammalian cells as regulator of CRH-R1 phosphorylation and desensitization is the family of GRKs. Studies in HEK293 and Y79 neuroblastoma cells have shown that multiple GRKs are involved in CRH-R1-phosphorylation and homologous desensitization (Dautzenberg et al., 2002; Teli et al., 2005): GRK3 appears to be the major ‘cytosolic’ isoform and requires Gβγ-subunits for its recruitment to the plasma membrane and association with the receptor. GRK6, which under basal conditions is localized in the plasma membrane, is also involved in receptor phosphorylation. However, GRK6 is able to interact with the CRH-R1 in a Gβγ-subunit independent manner, due to its constitutive association with the plasma membrane. Interestingly, in GRK3-deficient cellular systems, such as AtT-20 corticotroph cells, GRK2 participates in homologous CRH-R1 desensitization (Kageyama et al., 2006). The cAMP/PKA pathway may also have a role in this mechanism by activating GRK2 through phosphorylation. The putative GRK phospho-acceptor sites have been investigated within the tertiary structure of CRH-R1. The distal portion of the cytoplasmic tail of the CRH-R1 contains a cluster of Ser/Thr residues (Ser396-Ser405) and site-directed mutagenesis studies identified Thr399 as a GRK-phospho-acceptor residue (Teli et al., 2005). Further mutation studies localized the sites of agonist-induced CRH-R1 phosphorylation to Ser/Thr residues in both the IC3, which contains the putative GRK phosphorylation cassette Ser301-Thr306 and in the last 30 amino acids of the C-terminus, a segment that contains all putative GRK and PKC phosphorylation sites (Oakley et al., 2007). This study also suggested that initial phosphorylation of specific Ser/Thr residues in the receptor C-terminus may be necessary for subsequent phosphorylation of the Ser/Thr cluster in the IC3 loop (Figure 5A).
Figure 5.
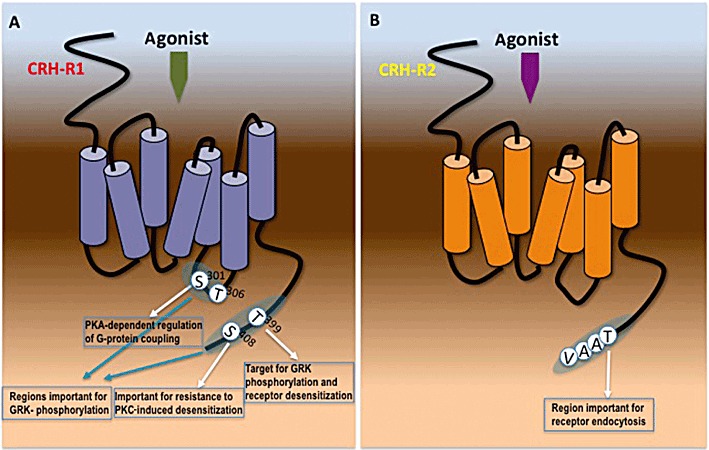
Structural determinants important for intracellular mechanisms regulating CRH-Rs activity. (A) Amino acid residues within the ICL3 and C-terminus of CRH-R1, identified as important phospho-acceptor sites of GRKs and second messenger PKs (PKA and PKC) and are involved in the intracellular mechanism that leads to β-arrestin recruitment and receptor endocytosis. (B) Amino acid casette within the C-terminus of CRH-R2, involved in the intracellular mechanism controlling rate of receptor endocytosis.
Agonist-induced receptor phosphorylation leads to translocation of β-arrestins to the plasma membrane where they interact with phosphorylated GPCRs. The binary protein complex formation initiates two important intracellular processes, receptor uncoupling and internalization, which are involved in both the desensitization and the recovery of cellular responses. Various studies have provided strong evidence that activation of both CRH-R1 and R2 induces rapid recruitment of β-arrestins to the plasma membrane and subsequent receptor interaction, an event leading to signalling desensitization (Rasmussen et al., 2004; Teli et al., 2005; Holmes et al., 2006; Markovic et al., 2006; 2008b; Oakley et al., 2007). Although it is generally accepted that CRH-R1 phosphorylation by GRK is required for efficient β-arrestin coupling, studies employing mutant CRH-R1 receptors resistance to GRK phosphorylation showed that these receptors were still able to recruit β-arrestin (Oakley et al., 2007). This suggests the presence of complex mechanisms that govern interactions between receptor (phosphorylated or not) and β-arrestins. This is supported further by the observation that β-arrestin recruitment to the plasma membrane is independent of receptor signalling, because the signalling impaired CRH-R1 variant, CRH-R1β, was able to efficiently induce β-arrestin recruitment to the plasma membrane (Markovic et al., 2006).
Most studies investigating the role of agonist-induced desensitization of CRH-R signalling have focused on the activation of AC, and important differences have been identified between the R1 and R2 subtypes as the CRH-R2 undergoes a more rapid rate of desensitization of cAMP responses following agonist activation (Teli et al., 2005; Markovic et al., 2008b). Moreover, it appears that not all CRH-R1-driven pathways are susceptible to desensitization; results from CRH-R1 over-expression systems suggest that homologous desensitization primarily attenuates Gs and Gq/11-driven signalling cascades (Wietfeld et al., 2004). In contrast, CRH-R1-Gi-protein interaction is not susceptible to desensitization, either because the important Ser/Thr is not accessible for phosphorylation or potential phosphorylation and β-arrestin binding do not interfere with Gi-protein-coupling. This might represent a regulatory mechanism that allows a more rapid decline of some CRH-R1 stimulatory effects. In fact, interaction between CRH-R1 and β-arrestin is involved in G-protein independent activation of ERK1/2 and p38MAPK (Punn et al., 2006), suggesting a crucial role of β-arrestin in terminating some G-protein-dependent signals such as cAMP, while stimulating others (MAPK) and thus controlling the overall direction of CRH-R1 intracellular effects. Lessons from other GPCRs suggest that β-arrestin binds to several components of the MAPK cascade, such as Raf and MEK1/2, assembling them in a multiprotein complex in close proximity, to facilitate signal propagation (Pierce et al., 2001). This role of β-arrestin appears to be specific for the CRH-R1 as CRH-R2-driven ERK1/2 activation was found to be exclusively G-protein dependent (Markovic et al., 2008b). In the latter cellular model, ERK/12 appears to finely tune β-arrestin1 actions, by modulating through phosphorylation, β-arrestin1 translocation to the plasma membrane and CRH-R2 desensitization/internalization kinetics. Loss of this ‘negative feedback’ mechanism through inhibition of the ERK1/2 activity results in significant attenuation of Ucn 2-induced cAMP response (Markovic et al., 2011). These findings provide another example of complex mechanisms regulating CRH-R functional responses and signal integration.
CRH-R internalization
Binding of β-arrestin to CRH-R initiates receptor endocytosis, a critical process for attenuating cellular responses to CRH and CRH-like agonists. At present, there are conflicting evidence about the fate of the CRH-R1-β-arrestin complex; in different experimental settings, the CRH-R1 appears to either form stable complexes with β-arrestin that internalize together as a unit into endocytic vesicles (Perry et al., 2005; Markovic et al., 2006) or dissociate from β-arrestin at or near the plasma membrane before internalization while β-arrestin remains in the membrane (Rasmussen et al., 2004; Holmes et al., 2006). In the former setting, β-arrestin might act as a scaffold to recruit other signalling molecules regulated by CRH-R1 such as phospho-ERK1/2 (Punn et al., 2006). The presence of distinct pathways leading to CRH-R1 internalization is supported by data suggesting that CRH-R1 internalization characteristics are ligand specific. Astressin, a neutral competitive antagonist, which binds with high affinity to the N-terminus of the CRH-R1, is capable of inducing CRH-R1 internalization through a pathway involving sequestration into intracellular compartments other than clathrin-coated pits and caveolae, despite being unable to stimulate CRH-R1 signalling, receptor phosphorylation or β-arrestin recruitment (Perry et al., 2005). Other studies have also suggested that CRH-R1 internalization appears to be independent on the degree of β-arrestin recruitment and/or receptor phosphorylation (Rasmussen et al., 2004), revealing a potential versatility of the CRH-R1 internalization mechanisms. These data raise the possibility that the two processes of CRH-R1 activation and signalling and receptor internalization might not be closely related, and demonstrate that under certain conditions (e.g. antagonist binding), the CRH-R1 can adopt distinct active conformations important for specific functions such as receptor internalization. Studies investigating the fate of the internalized receptor showed that in both HEK293 cells and cortical neurons, internalized CRH-R1 transited from Rab5-positive early endosomes to Rab4-positive recycling endosomes and was not targeted to lysosomes (Holmes et al., 2006). The CRH-R2 internalization characteristics have also been investigated (Markovic et al., 2008b); CRH-R2 activation induces transient β-arrestin1 and β-arrestin2, as well as clathrin, recruitment to the plasma membrane. This is followed by rapid receptor endocytosis in a mechanism that does not involve association with β-arrestin inside the cell. Site-directed mutagenesis studies on CRH-R2 C-terminus showed that the amino acid cassette TAAV at the end of the C-terminus is important for CRH-R2 rate of internalization because loss of a potential phospho-acceptor site of the cassette TAAV resulted in accelerated receptor internalization (Figure 5B).
Certainly, these intracellular mechanisms regulating CRH-R endocytosis are physiologically important for cellular adaptation to CRH actions as recently highlighted by a study, which identified gender-specific differences in rodent cortical CRH-R1 trafficking that could render CRH-receptive neurons of females more sensitive to low levels of CRH and less adaptable to high levels of CRH. Stress-induced CRH-R1 association with β-arrestin, an integral step in receptor internalization, was identified only in male rats. These differences might compromise stress adaptation in females and may underlie their increased vulnerability to develop stress-related pathology (Bangasser et al., 2010).
Regulation of CRH-R biological activity through splicing
A growing number of CRH-R mRNA variants have been identified in humans and other species (Hillhouse and Grammatopoulos, 2006) generated via alternative pre-mRNA splicing, a mechanism that has emerged as an important regulatory mechanism of class-B1 GPCRs biological function. Our current understanding of the role of these receptor variants is limited and is based primarily on in vitro experiments. Certain receptor variants such as the soluble (s) CRH-R2α identified in mouse brain and rat oesophagus (Chen et al., 2005; Wu et al., 2007) where exon deletion results in a soluble receptor fragment protein. Overexpression studies suggest that these might modulate biological effects of CRH and CRH-related peptides, as they are able to inhibit cellular responses to CRH and Ucn1, possibly acting as extracellular ligand-binding proteins. Similar soluble variants have also been identified for the CRH-R1, namely isoforms e and h, in human skin and myometrium (Jin et al., 2007; Zmijewski and Slominski, 2009a) These variants are diffusely distributed in the cytoplasm or localized to the endoplasmic reticulum (ER) and additionally secreted in culture medium (Zmijewski and Slominski, 2009a). It has been suggested that these variants can exert a dual effect on the wild type (w.t) CRH-R1α cellular activity depending on their localization: when secreted, they can inhibit CRH activity, whereas their intracellular presence can lead to amplification of agonist-induced CRH-R1α intracellular effects (Zmijewski and Slominski, 2009b). Of particular interest is the CRH-R1d, a receptor variant mising exon 13, a modification that leads to the loss of 14 amino acids from the C-terminal end of the putative TMD7, resulting in a 6-TMD receptor containing a protein segment that fails to segregate into the membrane lipid bilayer and leads to an extracellular C-terminus (Grammatopoulos et al., 1999). However, only a small fraction of ‘TMD7-short’ CRH-R1d is localized to the plasma membrane and the majority of receptors are expressed as intracellular proteins possibly in the ER (Markovic et al., 2008a), suggesting that the absence of TMD7 could affect CRH-R1d folding or transport to the cell surface. Similar mRNA splice variants arising from the same exon excision have been described for other members of the B1 family of GPCRs, suggesting the presence of a highly conserved splicing ‘hot-spot’ (Markovic and Grammatopoulos, 2009). Whether or not this aberrant R1d mRNA transcript is significantly translated as protein in native tissues is unkwown. Although our current understanding of the mechanisms regulating CRH-R1 mRNA splicing is incomplete, emerging evidence points towards potentially important biological mechanisms dynamically regulating CRH-R mRNA splicing. For example, in human myometrial smooth muscle cells, progesterone and estradiol-17β exert antagonistic actions on exon 13 inclusion or skipping from the final CRH-R1 mRNA transcript (Karteris et al., 2010). Although the specific biological function of CRH-R1d remains to be elucidated, it appears that it might act as dominant negative (DN) regulator of CRH responses by interfering with w.t receptor cell membrane expression or activation by its cognate agonists. In particular, it has been suggested that CRH-R1d associate with w.t CRH-R1 to form hetero-oligomers that are trapped intracellularly. This leads to a reduction in the signalling response through inhibition of w.t receptor cell surface expression (Zmijewski and Slominski, 2009b). Interestingly, data suggest that CRH-R1d interaction with the CRH-R2 receptor ‘rescues’ plasma membrane expression of CRH-R1d (Markovic et al., 2008a); enhancement of cell surface expression allows CRH-R1d to act as a DN regulator and heterologously attenuate CRH-R2β signalling. This suggests that under certain conditions and suitable ‘partner’ proteins, biosynthetic trafficking pathways of ‘TMD7-short’ variants can be modified to transport these proteins to the plasma membrane. Therefore, it is conceivable that under certain conditions, increased expression of the aberrant R1d mRNA transcript at the expense of the w.t receptor might reduce expression levels of functional receptor and diminish cellular responsiveness.
DN roles have been described for other CRH-R variants; for example, an insertion variant (iv)-mCRH-R2β variant with a unique C-terminal tail has been isolated from the mouse heart (Sztainberg et al., 2009). When co-expressed with w.t mCRH-R2β, iv-mCRH-R2β significantly inhibits the w.t mCRH-R2β membrane expression and its functional signalling by ER-Golgi complex retention. Interestingly, induction of stress up-regulates (iv)-mCRH-R2β mRNA expression in the heart, suggesting a potential regulatory mechanism of the mCRH-R2β cardioprotective effects.
Conclusions
The multiplicity of CRH-like peptides and CRH-R subtypes underpins the actions of a modulatory system fundamental for species adaptation, development and survival. The CRH-Rs are multifaceted receptors with a high degree of versatility in transducing the biological actions of their cognate agonists in a tissue- and agonist-specific manner. Significant progress has been made towards understanding the plethora of intracellular mechanisms that fine-tune their signalling plasticity by influencing receptor expression, signalling direction and duration of signal (Figure 6). Although most of our current knowledge is based on studies from cellular models, either artificial overexpression systems or native cultured cells, use of in vivo models of disease have advanced our understanding of the physiological relevance of CRH-R signalling and dissection of the relative contribution of CRH-R1 and R2-driven pathways. Further elucidation of these mechanisms will allow us to characterize their sometimes distinct roles in human pathophysiology and exploit their potential as novel therapeutic targets.
Figure 6.

An overview of the regulatory mechanisms allowing cells to modulate CRH-R signalling potency and direction. Upon receptor activation by its cognate agonists, cells modify their responsiveness (either through signal termination of generation of alternative signals) through activation of mechanisms that lead to receptor desensitization and decrease of receptor number in the plasma membrane. These mechanisms also potentially involve receptor variants that act as DN regulators of receptor expression or signalling.
Acknowledgments
The work presented was supported by the Wellcome Trust. I would like to thank E. W. Hillhouse, M. A. Levine and H. Lehnert for their guidance, support and encouragement over the years.
Glossary
- ACTH
adrenocorticotropic hormone
- CRH
corticotropin-releasing hormone
- DN
dominant negative
- ECD
extracellular domain
- ECL
extracellular loop
- EPAC
exchange proteins directly activated by cAMP
- ER
endoplasmic reticulum
- GRK
G-protein receptor kinase
- ICL
intracellular loop
- mGC
membrane guanylyl cyclase
- PI3-K
phosphatidylinositol 3-kinase
- SCR
short concensus repeat
- Ucn
urocortin
- VCAM-1
vascular cell adhesion protein 1
- w.t
wild type
Conflict of interest
No conflict of interest to disclose.
References
- Aggelidou E, Hillhouse EW, Grammatopoulos DK. Up-regulation of nitric oxide synthase and modulation of the guanylate cyclase activity by corticotropin-releasing hormone but not urocortin II or urocortin III in cultured human pregnant myometrial cells. Proc Natl Acad Sci USA. 2002;99:3300–3305. doi: 10.1073/pnas.052296399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assil IQ, Qi LJ, Arai M, Shomali M, Abou-Samra AB. Juxtamembrane region of the amino terminus of the corticotropin releasing factor receptor type 1 is important for ligand interaction. Biochemistry. 2001;40:1187–1195. doi: 10.1021/bi001758y. [DOI] [PubMed] [Google Scholar]
- Audsley N, Kay I, Hayes TK, Coast GM. Cross reactivity studies of CRF-related peptides on insect Malpighian tubules. Comp Biochem Physiol A Physiol. 1995;110:87–93. doi: 10.1016/0300-9629(94)00132-d. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Bangasser DA, Curtis A, Reyes BA, Bethea TT, Parastatidis I, Ischiropoulos H, et al. Sex differences in corticotropin-releasing factor receptor signaling and trafficking: potential role in female vulnerability to stress-related psychopathology. Mol Psychiatry. 2010;15:896–904. doi: 10.1038/mp.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger H, Heinrich N, Wietfeld D, Bienert M, Beyermann M. Evidence that corticotropin-releasing factor receptor type 1 couples to Gs- and Gi-proteins through different conformations of its J-domain. Br J Pharmacol. 2006;149:942–947. doi: 10.1038/sj.bjp.0706926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyermann M, Heinrich N, Fechner K, Furkert J, Zhang W, Kraetke O, et al. Achieving signalling selectivity of ligands for the corticotropin-releasing factor type 1 receptor by modifying the agonist's signalling domain. Br J Pharmacol. 2007;151:851–859. doi: 10.1038/sj.bjp.0707293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar BK, Jonassen AK, Egorina EM, Chen A, Negro A, Perrin MH, et al. Urocortin-II and urocortin-III are cardioprotective against ischemia reperfusion injury: an essential endogenous cardioprotective role for corticotropin releasing factor receptor type 2 in the murine heart. Endocrinology. 2004a;145:24–35. doi: 10.1210/en.2003-0689. [DOI] [PubMed] [Google Scholar]
- Brar BK, Chen A, Perrin MH, Vale W. Specificity and regulation of extracellularly regulated kinase1/2 phosphorylation through corticotropin-releasing factor (CRF) receptors 1 and 2beta by the CRF/urocortin family of peptides. Endocrinology. 2004b;145:1718–1729. doi: 10.1210/en.2003-1023. [DOI] [PubMed] [Google Scholar]
- Cantarella G, Lempereur L, Lombardo G, Chiarenza A, Pafumi C, Zappala G, et al. Divergent effects of corticotropin releasing hormone on endothelial cell nitric oxide synthase are associated with different expression of CRH type 1 and 2 receptors. Br J Pharmacol. 2001;134:837–844. doi: 10.1038/sj.bjp.0704322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Hsu SY. Ancient evolution of stress-regulating peptides in vertebrates. Peptides. 2004;25:1681–1688. doi: 10.1016/j.peptides.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Chen AM, Perrin MH, Digruccio MR, Vaughan JM, Brar BK, Arias CM, et al. A soluble mouse brain splice variant of type 2alpha corticotropin-releasing factor (CRF) receptor binds ligands and modulates their activity. Proc Natl Acad Sci USA. 2005;102:2620–2625. doi: 10.1073/pnas.0409583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Lewis KA, Perrin MH, Vale WW. Expression cloning of a human corticotropin-releasing-factor receptor. Proc Natl Acad Sci USA. 1993;90:8967–8971. doi: 10.1073/pnas.90.19.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautzenberg FM, Wille S, Braun S, Hauger RL. GRK3 regulation during CRF- and urocortin-induced CRF1 receptor desensitization. Biochem Biophys Res Commun. 2002;298:303–308. doi: 10.1016/s0006-291x(02)02463-4. [DOI] [PubMed] [Google Scholar]
- Denver RJ. Environmental stress as a developmental cue: corticotropin-releasing hormone is a proximate mediator of adaptive phenotypic plasticity in amphibian metamorphosis. Horm Behav. 1997;31:169–179. doi: 10.1006/hbeh.1997.1383. [DOI] [PubMed] [Google Scholar]
- Dermitzaki E, Tsatsanis C, Gravanis A, Margioris AN. Corticotropin-releasing hormone (CRH) induces Fas ligand production and apoptosis in PC12 cells via activation of p38 MAPK. J Biol Chem. 2002;277:12280–12287. doi: 10.1074/jbc.M111236200. [DOI] [PubMed] [Google Scholar]
- Elliott-Hunt CR, Kazlauskaite J, Wilde GJC, Grammatopoulos D, Hillhouse EW. Potential signalling pathways underlying corticotropin-releasing hormone's mediated neuroprotection from excitotoxicity in rat hippocampus. J Neurochem. 2002;80:416–425. doi: 10.1046/j.0022-3042.2001.00712.x. [DOI] [PubMed] [Google Scholar]
- Facci L, Stevens DA, Pangallo M, Franceschini D, Skaper SD, Strijbos PJ. Corticotropin-releasing factor (CRF) and related peptides confer neuroprotection via type 1 CRF receptors. Neuropharmacology. 2003;45:623–636. doi: 10.1016/s0028-3908(03)00211-9. [DOI] [PubMed] [Google Scholar]
- Gkountelias K, Tselios T, Venihaki M, Deraos G, Lazaridis I, Rassouli O, et al. Alanine scanning mutagenesis of the second extracellular loop of type 1 corticotropin-releasing factor receptor revealed residues critical for peptide binding. Mol Pharmacol. 2009;75:793–800. doi: 10.1124/mol.108.052423. [DOI] [PubMed] [Google Scholar]
- Grace CR, Perrin MH, DiGruccio MR, Miller CL, Rivier JE, Vale WW, et al. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc Natl Acad Sci USA. 2004;101:12836–12841. doi: 10.1073/pnas.0404702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace CR, Perrin MH, Gulyas J, Digruccio MR, Cantle JP, Rivier JE, et al. Structure of the N-terminal domain of a type B1 G protein-coupled receptor in complex with a peptide ligand. Proc Natl Acad Sci USA. 2007;104:4858–4863. doi: 10.1073/pnas.0700682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace CR, Perrin MH, Gulyas J, Rivier JE, Vale WW, Riek R. NMR structure of the first extracellular domain of corticotropin-releasing factor receptor 1 (ECD1-CRF-R1) complexed with a high affinity agonist. J Biol Chem. 2010;285:38580–38589. doi: 10.1074/jbc.M110.121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammatopoulos D, Hillhouse EW. Activation of protein kinase C by oxytocin inhibits the biological activity of the human myometrial CRH receptor at term. Endocrinology. 1999;140:585–594. doi: 10.1210/endo.140.2.6530. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos DK, Dai Y, Randeva HS, Levine MA, Karteris E, Easton AJ, et al. A novel spliced variant of the type 1 corticotropin-releasing hormone receptor with a deletion in the seventh transmembrane domain present in the human pregnant term myometrium and fetal membranes. Mol Endocrinol. 1999;13:2189–2202. doi: 10.1210/mend.13.12.0391. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos DK, Randeva HS, Levine MA, Katsanou ES, Hillhouse EW. Urocortin, but not corticotropin-releasing hormone (CRH), activates the mitogen-activated protein kinase signal transduction pathway in human pregnant myometrium: an effect mediated via R1alpha and R2beta CRH receptor subtypes and stimulation of Gq-proteins. Mol Endocrinol. 2000;14:2076–2091. doi: 10.1210/mend.14.12.0574. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos DK, Randeva HS, Levine MA, Kanellopoulou KA, Hillhouse EW. Rat cerebral cortex corticotropin-releasing hormone receptors: evidence for receptor coupling to multiple G-proteins. J Neurochem. 2001;76:509–519. doi: 10.1046/j.1471-4159.2001.00067.x. [DOI] [PubMed] [Google Scholar]
- Graziani G, Tentori L, Portarena I, Barbarino M, Tringali G, Pozzoli G, et al. CRH inhibits cell growth of human endometrial adenocarcinoma cells via CRH-receptor 1-mediated activation of cAMP-PKA pathway. Endocrinology. 2002;143:807–813. doi: 10.1210/endo.143.3.8694. [DOI] [PubMed] [Google Scholar]
- Graziani G, Tentori L, Muzi A, Vergati M, Tringali G, Pozzoli G, et al. Evidence that corticotropin-releasing hormone inhibits cell growth of human breast cancer cells via the activation of CRH-R1 receptor subtype. Mol Cell Endocrinol. 2007;264:44–49. doi: 10.1016/j.mce.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Haug T, Storm JF. Protein kinase A mediates the modulation of the slow Ca(2+)-dependent K(+) current, I(sAHP), by the neuropeptides CRF, VIP, and CGRP in hippocampal pyramidal neurons. J Neurophysiol. 2000;83:2071–2079. doi: 10.1152/jn.2000.83.4.2071. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Smith RD, Braun S, Dautzenberg FM, Catt KJ. Rapid agonist-induced phosphorylation of the human CRF receptor, type 1: a potential mechanism for homologous desensitization. Biochem Biophys Res Commun. 2000;268:572–576. doi: 10.1006/bbrc.2000.2183. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Olivares-Reyes JA, Braun S, Catt KJ, Dautzenberg FM. Mediation of corticotropin releasing factor type 1 receptor phosphorylation and desensitization by protein kinase C: a possible role in stress adaptation. J Pharmacol Exp Ther. 2003;306:794–803. doi: 10.1124/jpet.103.050088. [DOI] [PubMed] [Google Scholar]
- Hillhouse EW, Grammatopoulos DK. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev. 2006;27:260–286. doi: 10.1210/er.2005-0034. [DOI] [PubMed] [Google Scholar]
- Hoare SR, Sullivan SK, Schwarz DA, Ling N, Vale WW, Crowe PD, et al. Ligand affinity for amino-terminal and juxtamembrane domains of the corticotropin releasing factor type I receptor: regulation by G-protein and nonpeptide antagonists. Biochemistry. 2004;43:3996–4011. doi: 10.1021/bi036110a. [DOI] [PubMed] [Google Scholar]
- Hoare SR, Sullivan SK, Fan J, Khongsaly K, Grigoriadis DE. Peptide ligand binding properties of the corticotropin-releasing factor (CRF) type 2 receptor: pharmacology of endogenously expressed receptors, G-protein-coupling sensitivity and determinants of CRF2 receptor selectivity. Peptides. 2005;26:457–470. doi: 10.1016/j.peptides.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Hoare SR, Fleck BA, Gross RS, Crowe PD, Williams JP, Grigoriadis DE. Allosteric ligands for the corticotropin releasing factor type 1 receptor modulate conformational states involved in receptor activation. Mol Pharmacol. 2008;73:1371–1380. doi: 10.1124/mol.107.042978. [DOI] [PubMed] [Google Scholar]
- Holmes KD, Babwah AV, Dale LB, Poulter MO, Ferguson SS. Differential regulation of corticotropin releasing factor 1alpha receptor endocytosis and trafficking by beta-arrestins and Rab GTPases. J Neurochem. 2006;96:934–949. doi: 10.1111/j.1471-4159.2005.03603.x. [DOI] [PubMed] [Google Scholar]
- Hsu SY, Hsueh AJ. Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nat Med. 2001;7:605–611. doi: 10.1038/87936. [DOI] [PubMed] [Google Scholar]
- Huang M, Kempuraj D, Papadopoulou N, Kourelis T, Donelan J, Manola A, et al. Urocortin induces interleukin-6 release from rat cardiomyocytes through p38 MAP kinase, ERK and NF-kappaB activation. J Mol Endocrinol. 2009;42:397–405. doi: 10.1677/JME-08-0120. [DOI] [PubMed] [Google Scholar]
- Jahn O, Tezval H, van Werven L, Eckart K, Spiess J. Three-amino acid motifs of urocortin II and III determine their CRF receptor subtype selectivity. Neuropharmacology. 2004;47:233–242. doi: 10.1016/j.neuropharm.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Jin D, He P, You X, Zhu X, Dai L, He Q, et al. Expression of corticotropin-releasing hormone receptor type 1 and type 2 in human pregnant myometrium. Reprod Sci. 2007;14:568–577. doi: 10.1177/1933719107307821. [DOI] [PubMed] [Google Scholar]
- Kageyama K, Hanada K, Moriyama T, Nigawara T, Sakihara S, Suda T. G protein-coupled receptor kinase 2 involvement in desensitization of corticotropin-releasing factor (CRF) receptor type 1 by CRF in murine corticotrophs. Endocrinology. 2006;147:441–450. doi: 10.1210/en.2005-0376. [DOI] [PubMed] [Google Scholar]
- Karalis KP, Venihaki M, Zhao J, van Vlerken LE, Chandras C. NF-kappaB participates in the corticotropin-releasing, hormone-induced regulation of the pituitary proopiomelanocortin gene. J Biol Chem. 2004;279:10837–10840. doi: 10.1074/jbc.M313063200. [DOI] [PubMed] [Google Scholar]
- Karteris E, Grammatopoulos D, Randeva H, Hillhouse EW. Signal transduction characteristics of the corticotropin-releasing hormone receptors in the feto-placental unit. J Clin Endocrinol Metab. 2000;85:1989–1996. doi: 10.1210/jcem.85.5.6590. [DOI] [PubMed] [Google Scholar]
- Karteris E, Vatish M, Hillhouse EW, Grammatopoulos DK. Preeclampsia is associated with impaired regulation of the placental nitric oxide-cyclic guanosine monophosphate pathway by corticotropin-releasing hormone (CRH) and CRH-related peptides. J Clin Endocrinol Metab. 2005;90:3680–3687. doi: 10.1210/jc.2004-2210. [DOI] [PubMed] [Google Scholar]
- Karteris E, Markovic D, Chen J, Hillhouse EW, Grammatopoulos DK. Identification of a novel corticotropin-releasing hormone type 1beta-like receptor variant lacking exon 13 in human pregnant myometrium regulated by estradiol-17beta and progesterone. Endocrinology. 2010;151:4959–4968. doi: 10.1210/en.2010-0622. [DOI] [PubMed] [Google Scholar]
- Khattak MN, Buchfelder M, Kleindienst A, Schöfl C, Kremenevskaja N. CRH and SRIF have opposite effects on the Wnt/β-catenin signalling pathway through PKA/GSK-3β in corticotroph pituitary cells. Cancer Invest. 2010;28:797–805. doi: 10.3109/07357907.2010.494318. [DOI] [PubMed] [Google Scholar]
- Kovalovsky D, Refojo D, Liberman AC, Hochbaum D, Pereda MP, Coso OA, et al. Activation and induction of NUR77/NURR1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol Endocrinol. 2002;16:1638–1651. doi: 10.1210/mend.16.7.0863. [DOI] [PubMed] [Google Scholar]
- Ladds G, Davis K, Hillhouse EW, Davey J. Modified yeast cells to investigate the coupling of G protein-coupled receptors to specific G proteins. Mol Microbiol. 2003;47:781–792. doi: 10.1046/j.1365-2958.2003.03336.x. [DOI] [PubMed] [Google Scholar]
- Lewis K, Li C, Perrin MH, Blount A, Kunitake K, Donaldson C, et al. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc Natl Acad Sci USA. 2001;98:7570–7555. doi: 10.1073/pnas.121165198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezoualc'h F, Engert S, Berning B, Behl C. Corticotropin-releasing hormone-mediated neuroprotection against oxidative stress is associated with the increased release of non-amyloidogenic amyloid beta precursor protein and with the suppression of nuclear factor-kappaB. Mol Endocrinol. 2000;14:147–159. doi: 10.1210/mend.14.1.0403. [DOI] [PubMed] [Google Scholar]
- Liaw CW, Grigoriadis DE, Lorang MT, De Souza EB, Maki RA. Localization of agonist- and antagonist-binding domains of human corticotropin-releasing factor receptors. Mol Endocrinol. 1997a;11:2048–2053. doi: 10.1210/mend.11.13.0034. [DOI] [PubMed] [Google Scholar]
- Liaw CW, Grigoriadis DE, Lovenberg TW, De Souza EB, Maki RA. Localization of ligand-binding domains of human corticotropin-releasing factor receptor: a chimeric receptor approach. Mol Endocrinol. 1997b;11:980–985. doi: 10.1210/mend.11.7.9946. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Liaw CW, Grigoriadis DE, Clevenger W, Chalmers DT, De Souza EB, et al. Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proc Natl Acad Sci USA. 1995;92:836–840. doi: 10.1073/pnas.92.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic D, Grammatopoulos DK. Focus on the splicing of secretin GPCRs transmembrane-domain 7. Trends Biochem Sci. 2009;34:443–452. doi: 10.1016/j.tibs.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Markovic D, Papadopoulou N, Teli T, Randeva H, Levine MA, Hillhouse EW, et al. Differential responses of corticotropin-releasing hormone receptor type 1 variants to protein kinase C phosphorylation. J Pharmacol Exp Ther. 2006;319:1032–1042. doi: 10.1124/jpet.106.107441. [DOI] [PubMed] [Google Scholar]
- Markovic D, Lehnert H, Levine MA, Grammatopoulos DK. Structural determinants critical for localization and signaling within the seventh transmembrane domain of the type 1 corticotropin releasing hormone receptor: lessons from the receptor variant R1d. Mol Endocrinol. 2008a;22:2505–2519. doi: 10.1210/me.2008-0177. [DOI] [PubMed] [Google Scholar]
- Markovic D, Punn A, Lehnert H, Grammatopoulos DK. Intracellular mechanisms regulating corticotropin-releasing hormone receptor-2beta endocytosis and interaction with extracellularly regulated kinase 1/2 and p38 mitogen-activated protein kinase signaling cascades. Mol Endocrinol. 2008b;22:689–706. doi: 10.1210/me.2007-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic D, Punn A, Lehnert H, Grammatopoulos DK. Molecular determinants and feedback circuits regulating type 2 CRH receptor signal integration. Biochim Biophys Acta Mol Cell Res. 2011;1813:896–907. doi: 10.1016/j.bbamcr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Mazur AW, Wang F, Tcheiner M, Donnelly E, Isfort RJ. Determinants of corticotropin releasing factor. Receptor selectivity of corticotropin releasing factor related peptides. J Med Chem. 2004;47:3450–3454. doi: 10.1021/jm049883l. [DOI] [PubMed] [Google Scholar]
- Nemoto T, Mano-Otagiri A, Shibasaki T. Urocortin 2 induces tyrosine hydroxylase phosphorylation in PC12 cells. Biochem Biophys Res Commun. 2005;330:821–831. doi: 10.1016/j.bbrc.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Nielsen SM, Nielsen LZ, Hjorth SA, Perrin MH, Vale WW. Constitutive activation of tethered-peptide/corticotropin-releasing factor receptor chimeras. Proc Natl Acad Sci USA. 2000;97:10277–10281. doi: 10.1073/pnas.97.18.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstrom KJ, Lagerstrom MC, Waller LM, Fredriksson R, Schioth HB. The secretin GPCRs descended from the family of adhesion GPCRs. Mol Biol Evol. 2009;26:71–84. doi: 10.1093/molbev/msn228. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Olivares-Reyes JA, Hudson CC, Flores-Vega F, Dautzenberg FM, Hauger RL. Carboxyl-terminal and intracellular loop sites for CRF1 receptor phosphorylation and beta-arrestin-2 recruitment: a mechanism regulating stress and anxiety responses. Am J Physiol Regul Integr Comp Physiol. 2007;293:R209–R222. doi: 10.1152/ajpregu.00099.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal K, Swaminathan K, Xu HE, Pioszak AA. Structural basis for hormone recognition by the human CRFR2{alpha} G protein-coupled receptor. J Biol Chem. 2010;285:40351–40361. doi: 10.1074/jbc.M110.186072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulou N, Chen J, Randeva H, Levine MA, Hillhouse EW, Grammatopoulos D. Protein kinase A-induced negative regulation of the corticotropin-releasing hormone (CRH) R1α receptor-ERK signal transduction pathway: the critical role of Ser301 for signalling switch and selectivity. Mol Endocrinol. 2004;18:624–639. doi: 10.1210/me.2003-0365. [DOI] [PubMed] [Google Scholar]
- Park HJ, Kim HJ, Lee JH, Lee JY, Cho BK, Kang JS, et al. Corticotropin-releasing hormone (CRH) downregulates interleukin-18 expression in human HaCaT keratinocytes by activation of p38 mitogen-activated protein kinase (MAPK) pathway. J Invest Dermatol. 2005;124:751–755. doi: 10.1111/j.0022-202X.2005.23656.x. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Wan R, Zhang P, Mattson MP. Urocortin, but not urocortin II, protects cultured hippocampal neurons from oxidative and excitotoxic cell death via corticotropin-releasing hormone receptor type I. J Neurosci. 2002;22:404–412. doi: 10.1523/JNEUROSCI.22-02-00404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin MH, Sutton S, Bain DL, Berggren WT, Vale WW. The first extracellular domain of corticotropin releasing factor-R1 contains major binding determinants for urocortin and astressin. Endocrinology. 1998;13:566–570. doi: 10.1210/endo.139.2.5757. [DOI] [PubMed] [Google Scholar]
- Perry SJ, Junger S, Kohout TA, Hoare SRJ, Struthers RS, Grigoriadis DE, et al. Distinct conformations of the corticotropin releasing factor type 1 receptor adopted following agonist and antagonist binding are differentially regulated. J Biol Chem. 2005;280:11560–11568. doi: 10.1074/jbc.M412914200. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Luttrell LM, Lefkowitz RJ. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene. 2001;20:1532–1539. doi: 10.1038/sj.onc.1204184. [DOI] [PubMed] [Google Scholar]
- Pioszak AA, Parker NR, Suino-Powell K, Xu HE. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J Biol Chem. 2008;283:32900–32912. doi: 10.1074/jbc.M805749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punn A, Levine MA, Grammatopoulos DK. Identification of signaling molecules mediating corticotropin-releasing hormone-R1alpha-mitogen-activated protein kinase (MAPK) interactions: the critical role of phosphatidylinositol 3-kinase in regulating ERK1/2 but not p38 MAPK activation. Mol Endocrinol. 2006;20:3179–3195. doi: 10.1210/me.2006-0255. [DOI] [PubMed] [Google Scholar]
- Radulovic M, Hippel C, Spiess J. Corticotropin-releasing factor (CRF) rapidly suppresses apoptosis by acting upstream of the activation of caspases. J Neurochem. 2003;84:1074–1085. doi: 10.1046/j.1471-4159.2003.01594.x. [DOI] [PubMed] [Google Scholar]
- Ralph JA, Zocco D, Bresnihan B, Fitzgerald O, McEvoy AN, Murphy EP. A role for type 1alpha corticotropin-releasing hormone receptors in mediating local changes in chronically inflamed tissue. Am J Pathol. 2007;170:1121–1133. doi: 10.2353/ajpath.2007.061000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen TN, Novak I, Nielsen SM. Internalization of the human CRF receptor 1 is independent of classical phosphorylation sites and of beta-arrestin 1 recruitment. Eur J Biochem. 2004;271:4366–4374. doi: 10.1111/j.1432-1033.2004.04371.x. [DOI] [PubMed] [Google Scholar]
- Reisine T, Rougon G, Barbet J, Affolter HU. Corticotropin-releasing factor-induced adrenocorticotropin hormone release and synthesis is blocked by incorporation of the inhibitor of cyclic AMP-dependent protein kinase into anterior pituitary tumor cells by liposomes. Proc Natl Acad Sci USA. 1985;82:8261–8265. doi: 10.1073/pnas.82.23.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes TM, Lewis K, Perrin MH, Kunitake KS, Vaughan J, Arias CA, et al. Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc Natl Acad Sci USA. 2001;98:2843–2848. doi: 10.1073/pnas.051626398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivier J, Gulyas J, Kirby D, Low W, Perrin MH, Kunitake K, et al. Potent and long-acting corticotropin releasing factor (CRF) receptor 2 selective peptide competitive antagonists. J Med Chem. 2002;45:4737–4747. doi: 10.1021/jm0202122. [DOI] [PubMed] [Google Scholar]
- Ruhmann A, Bonk I, Lin CR, Rosenfeld MG, Spiess J. Structural requirements for peptidic antagonists of the corticotropin-releasing factor receptor (CRFR): development of CRFR2beta-selective antisauvagine-30. Proc Natl Acad Sci USA. 1998;95:15264–15269. doi: 10.1073/pnas.95.26.15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Mitogen-activated protein kinase signaling in the hippocampus and its modulation by corticotropin-releasing factor receptor 2: a possible link between stress and fear memory. J Neurosci. 2003;23:11436–11443. doi: 10.1523/JNEUROSCI.23-36-11436.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Suda T, Kageyama K, Sakihara S, Nigawara T. Physiological roles of urocortins, human homologues of fish urotensin I, and their receptors. Peptides. 2004;25:1689–1701. doi: 10.1016/j.peptides.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Sydow S, Flaccus A, Fischer A, Spiess J. The role of the fourth extracellular domain of the rat corticotropin-releasing factor receptor type 1 in ligand binding. Eur J Biochem. 1999;259:55–62. doi: 10.1046/j.1432-1327.1999.00007.x. [DOI] [PubMed] [Google Scholar]
- Sztainberg Y, Kuperman Y, Issler O, Gil S, Vaughan J, Rivier J, et al. A novel corticotropin-releasing factor receptor splice variant exhibits dominant negative activity: a putative link to stress-induced heart disease. FASEB J. 2009;23:2186–2196. doi: 10.1096/fj.08-128066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teli T, Markovic D, Levine MA, Hillhouse EW, Grammatopoulos DK. Regulation of corticotropin-releasing hormone receptor type 1alpha signaling: structural determinants for G protein-coupled receptor kinase-mediated phosphorylation and agonist-mediated desensitization. Mol Endocrinol. 2005;19:474–490. doi: 10.1210/me.2004-0275. [DOI] [PubMed] [Google Scholar]
- Teli T, Markovic D, Hewitt ME, Levine MA, Hillhouse EW, Grammatopoulos DK. Structural domains determining signalling characteristics of the CRH-receptor type 1 variant R1beta and response to PKC phosphorylation. Cell Signal. 2008;20:40–49. doi: 10.1016/j.cellsig.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Ulisse S, Fabbri A, Dufau ML. Corticotropin-releasing factor receptors and actions in rat Leydig cells. J Biol Chem. 1989;264:2156–2163. [PubMed] [Google Scholar]
- Van Kolen K, Dautzenberg FM, Verstraeten K, Royaux I, De Hoogt R, Gutknecht E, et al. Corticotropin releasing factor-induced ERK phosphorylation in AtT20 cells occurs via a cAMP-dependent mechanism requiring EPAC2. Neuropharmacology. 2010;58:135–144. doi: 10.1016/j.neuropharm.2009.06.022. [DOI] [PubMed] [Google Scholar]
- Vaughan J, Donaldson C, Bittencourt J, Perrin MH, Lewis K, Sutton S, et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature. 1995;378:287–292. doi: 10.1038/378287a0. [DOI] [PubMed] [Google Scholar]
- Wietfeld D, Heinrich N, Furkert J, Fechner K, Beyermann M, Bienert M, et al. Regulation of the coupling to different G proteins of rat corticotropin-releasing factor receptor type 1 in human embryonic kidney 293 cells. J Biol Chem. 2004;279:38386–38394. doi: 10.1074/jbc.M405335200. [DOI] [PubMed] [Google Scholar]
- Wille S, Sydow S, Palchaudhuri MR, Spiess J, Dautzenberg FM. Identification of amino acids in the N-terminal domain of corticotropin-releasing factor receptor 1 that are important determinants of high-affinity ligand binding. J Neurochem. 1999;72:388–395. doi: 10.1046/j.1471-4159.1999.0720388.x. [DOI] [PubMed] [Google Scholar]
- Wu SV, Yuan PQ, Wang L, Peng YL, Chen CY, Taché Y. Identification and characterization of multiple corticotropin-releasing factor type 2 receptor isoforms in the rat esophagus. Endocrinology. 2007;148:1675–1687. doi: 10.1210/en.2006-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhang R, Zhou C, Xu Y, Guan X, Hu J, et al. Enhanced expression of vascular cell adhesion molecule-1 by corticotrophin-releasing hormone contributes to progression of atherosclerosis in LDL receptor-deficient mice. Atherosclerosis. 2009;203:360–370. doi: 10.1016/j.atherosclerosis.2008.05.059. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Xie LY, Abou-Samra AB. Signaling properties of mouse and human corticotropin-releasing factor (CRF) receptors: decreased coupling efficiency of human type II CRF receptor. Endocrinology. 1995;136:1828–1834. doi: 10.1210/endo.136.5.7720627. [DOI] [PubMed] [Google Scholar]
- Zbytek B, Pfeffer LM, Slominski AT. CRH inhibits NF-kappa B signaling in human melanocytes. Peptides. 2006;27:3276–3283. doi: 10.1016/j.peptides.2006.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Karalis KP. Regulation of nuclear factor-kappaB by corticotropin-releasing hormone in mouse thymocytes. Mol Endocrinol. 2002;16:2561–2570. doi: 10.1210/me.2001-0334. [DOI] [PubMed] [Google Scholar]
- Zmijewski MA, Slominski AT. CRF1 receptor splicing in epidermal keratinocytes: potential biological role and environmental regulations. J Cell Physiol. 2009a;218:593–602. doi: 10.1002/jcp.21632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmijewski MA, Slominski AT. Modulation of corticotropin releasing factor (CRF) signaling through receptor splicing in mouse pituitary cell line AtT-20–emerging role of soluble isoforms. J Physiol Pharmacol. 2009b;60:39–46. [PMC free article] [PubMed] [Google Scholar]