

Abstract
The Secretin family of GPCRs are endocrine peptide hormone receptors that share a common genomic organization and are the subject of a wide variety of alternative splicing. All GPCRs contain a central seven transmembrane domain responsible for transducing signals from the outside of the cell as well as extracellular amino and intracellular carboxyl termini. Members of the Secretin receptor family have a relatively large N-terminus and a variety of lines of evidence support a common mode of ligand binding and a common ligand binding fold. These receptors are best characterized as coupling to intracellular signalling pathways via Gαs and Gαq but are also reported to couple to a multitude of other signalling pathways. The intracellular loops are implicated in regulating the interaction between the receptor and heterotrimeric G protein complexes. Alternative splicing of exons encoding both the extracellular N-terminal domain as well as the extracellular loops of some family members has been reported and as expected these splice variants display altered ligand affinity as well as differential activation by endogenous ligands. Various forms of alternative splicing have also been reported to alter intracellular loops 1 and 3 as well as the C-terminus and as one might expect these display differences in signalling bias towards downstream effectors. These diverse pharmacologies require that the physiological role of these splice variants be addressed but should provide unique opportunities for drug design and development.
LINKED ARTICLES
This article is part of a themed section on Secretin Family (Class B) G Protein-Coupled Receptors. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.166.issue-1
Keywords: GPCR, Secretin family, family B, alternative splicing, receptor variant
Introduction
GPCRs are the largest gene family in eukaryotes comprising between 2% and 4% of the genome. GPCRs are responsible for transducing a wide range of extracellular stimuli including light, odours, hormones and neurotransmitters. Although the primary amino acid sequence homology is extremely low across GPCRs, they all share the same topology of an extracellular N-terminus, a seven transmembrane (7TM) helix bundle and an intracellular C-terminus.
A subset of GPCRs is the family B, or Secretin family. In humans, these are a group of 15 peptide hormone receptors that form a distinct clade based on primary amino acid sequence (Fredriksson et al., 2003). In some nomenclatures family B GPCRs include the 33 receptors (in humans) that are related to the Adhesion receptor so for clarity we will avoid using the term family B. There are two more bases for placing these receptors in a distinct family. First, they share a common genomic organization with four to six exons encoding the N-terminal domain and eight to nine exons encoding the 7TM bundle and C-terminus (see Figure 1), an arrangement that differs markedly from the genomic arrangement found in the related Adhesion, Glutamate and Frizzled receptor families. Second, the N-terminal domain of these receptors is very likely to form a common fold. The sequence homology in the N-terminus across this family is very low (Figure 2); however, the solution and crystal structures for isolated N-termini of corticotropin releasing factor receptor 2β (CRF2β) (Grace et al., 2004), pituitary adenylate cyclase activating peptide receptor (PAC1) (Sun et al., 2007), glucose-dependent insulinotropic polypeptide receptor (GIP) (Parthier et al., 2007), glucagon-like peptide receptor 1 (GLP-1) (Runge et al., 2008), corticotropin releasing factor receptor 1 (CRF1) (Pioszak et al., 2008), parathyroid hormone receptor 1 (PTH1) (Pioszak and Xu, 2008), CRF2α (Pal et al., 2010) and calcitonin gene related peptide receptor [CGRP, a dimer between calcitonin-like receptor (CLR) and receptor activity modifying protein 1 (RAMP1) (Haar et al., 2010)] demonstrate a high conservation of secondary structure and a common mode of ligand binding (discussed below).
Figure 1.
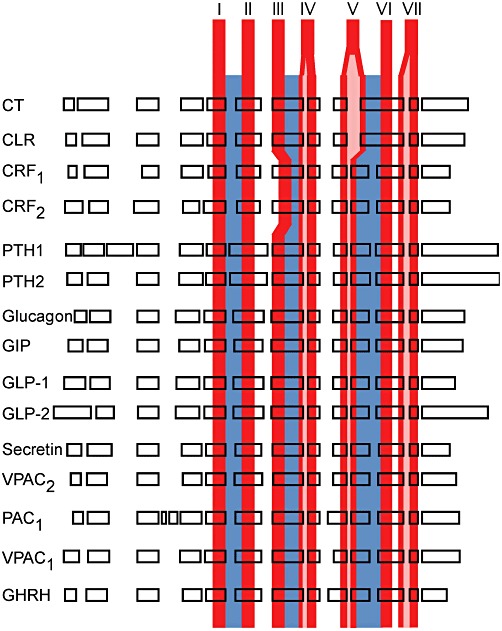
Schematic of the genomic organization of Secretin family GPCRs. Boxes indicate coding exons and are scaled according to amino acid length coded by each exon. The relative location of exons with respect to the overall receptor fold is depicted. The location of transmembrane helices is indicated in red (exon spanning helices shown with pink insert) and ICLs in blue.
Figure 2.

Alignment of the extracellular N-termini of Secretin family GPCRs. Amino acids encoded by alternative exons are shown in alternating black and blue text with amino acids encoded by two exons shown in red text. Predicted signal peptides are highlighted as italicized, underlined text. The six conserved cysteine residues are highlighted by red background and have the cysteine number indicated below the alignment. The disulphide bonding pattern for all receptors is 1–4, 2–5 and 3–6. The other conserved residues are highlighted in purple. Above the alignment alpha helices are indicated with H, beta strands with B and beta turns with T.
The Secretin family of GPCRs all respond to paracrine or endocrine peptide hormones that are typically in the range of 30–40 amino acids. These receptors are targets for existing drugs that treat osteoporosis [PTH and calcitonin receptor (CT)], hypercalcaemia (CT), Paget's disease (CT), type II diabetes (GLP-1, glucagon receptor) and are being actively pursued as targets for migraine (CGRP), depression and anxiety (CRF1) and pancreatic diagnostics (Secretin receptor). Many of these receptors have multiple endogenous ligands and at least for the GLP-1, signalling bias of these endogenous ligands has been demonstrated (Koole et al., 2010). Activation of these receptors regulates a wide variety of cellular physiology including cell cycle, differentiation, proliferation and release of other endocrine hormones. Their activation is most closely coupled to adenylate cyclase via Gαs and to a lesser extent to PLC and intracellular calcium mobilization via Gαq, although their signalling is not confined to these pathways.
As has been mentioned above, the structure of the N-terminal extracellular domain of these receptors almost certainly forms a common fold. This common fold contains a small number of conserved residues (Figure 2) being aspartic acid, tryptophan, proline, glycine and tryptophan as well as six conserved cysteine residues, which form three conserved disulphide bridges (1–4, 2–5 and 3–6) (Grauschopf et al., 2000; Perrin et al., 2001; 2003; Bazarsuren et al., 2002; Grace et al., 2004; 2007; Lisenbee et al., 2005; Parthier et al., 2007; Sun et al., 2007; Pioszak and Xu, 2008; Pioszak et al., 2008; Runge et al., 2008; Haar et al., 2010). This structural information supports not only a common fold but also a common mode of binding, in which the carboxy end of the ligand interacts with the N-terminal domain of the receptor. Experiments using chimeric receptors and ligands for CT/glucagon receptor (Stroop et al., 1995), Secretin/vasoactive intestinal peptide receptor 1 (VPAC1) (Holtmann et al., 1995), CT/PTH1 (Bergwitz et al., 1996), GIP/GLP-1 (Gelling et al., 1997), glucagon/GLP-1 (Runge et al., 2003) support a model in which the amino end of the ligand interacts with the juxtamembrane region and extracellular loops (ECL) and are consistent with the structural data. Progressive truncation of the N-terminus of Secretin receptor family ligands progressively converts ligands for VIP (Turner et al., 1986), PTH1 (Goldman et al., 1988), CGRP (Wang et al., 1991), CT (Feyen et al., 1992), GIP (Tseng et al., 1996) and GLP-1 (Montrose-Rafizadeh et al., 1997) from agonists into antagonists demonstrating that although, in many cases, the C-terminus is required for high affinity binding, it is the N-terminus that is competent to stabilize the active conformation of these receptors. Secretin family ligands therefore interact in a bivalent mode with the receptor, the N-terminal receptor domain providing a high affinity site for the peptide carboxy terminus that constrains the lower affinity interaction of the amino terminus of the peptide with the juxtamembrane domain and ECLs to stabilize the active receptor conformation. The active conformation of the receptor then acts as a guanine nucleotide exchange factor for the Gα subunit of the trimeric G protein complex. There is no direct evidence that addresses the regions of the cytoplasmic face of GPCRS that are responsible for interactions with the trimeric G protein complex. A large amount of mutational data as well as inferences made from crystal structures of both family A GPCRs and G proteins predict that intracellular loops (ICL) 2 and 3 as well as the C-terminal tail are involved in this interaction (reviewed in Huang and Tesmer, 2011).
Alternative splicing in Secretin family receptors
The genomic organization exhibited by the Secretin family of GPCRs engenders the potential for a wide variety of alternative splicing (AS). As it transpires, almost all combinations of AS that one might imagine appear to have evolved within this family (Table 1 and Figure 3).
Table 1.
Summary of splicing variants of Secretin family GPCRs that affect the coding region
| Receptor | Variation | Effect | Comment |
|---|---|---|---|
| N-termini | |||
| CRF1c | Coding exon 3 skipped | Severe reduction in ligand affinity | Supported |
| CRF2α | Alternative exon encoding 1st N-terminal alpha helix | Functional | Supported |
| CRF2β | Alternative exon encoding 1st N-terminal alpha helix | Functional | Supported |
| CRF2γ | Alternative exon encoding 1st N-terminal alpha helix | Functional | Supported |
| PAC13a | Exon insertion in N-terminal loop 1, between the first alpha helix and beta strand | Functional | Supported in rat, unsupported in human |
| Secretin receptor | Coding exon 3 skipped | Severe reduction in ligand affinity, dominant negative | Supported |
| CTRΔ47N | Apparent cryptic splice donor from 5′ UTR, skip coding exon 1 | No reduction of potency for calcitonins but reduced potency for amylin suggesting interaction with RAMP lost | Unsupported |
| Headless | |||
| CRF1e2 | Alternative first exon use of coding exon 5, receptor starts at TM1 | None reported | Unsupported |
| CRF1h2 | Cryptic first exon followed by coding exon 5 creating N-terminally truncated receptor | None reported | Unsupported |
| CTRheadless | Alternative first exon results in N-terminally truncated receptor | Severe reduction in ligand affinity, dominant negative | Supported in teleost fish but not orthologous to mammalian CT receptor genes |
| ECLs | |||
| CT receptor | Insertion of extra exon in ECL1 | Reduced affinity | Supported in rat, unsupported in human |
| ICLs | |||
| CT receptor | Insertion of extra exon in ICL1 | Signalling bias, reduced internalization | Supported |
| CTR7a | Cryptic additional exon 7 resulting in frame shift and stop codon. Predicted to generate N-terminus plus TM1 | None reported | Unsupported |
| CRF1β | Insertion of extra exon in ICL1 | Reduced affinity and potency, increased internalization | Supported |
| PAC1hip | Insertion of extra exon in ICL3 | Signalling bias | Supported |
| PAC1hop1 | Insertion of extra exon in ICL3 | Signalling bias | Supported |
| PAC1hop2 | Insertion of extra exon in ICL3 | Signalling bias | Supported in rat, proposed but unsupported in human |
| PAC1hiphop1 | Insertion of two extra exons in ICL3 | Signalling bias | Supported |
| Tailless | |||
| CRF1var | Coding exon 14 skipped, loss of half of TM7 then frame shift to stop codon equivalent to δ13e of CTR | No effect on affinity, impaired signalling | Unsupported |
| PTH1 | Coding exon 14 skipped, loss of half of TM7 then frame shift to stop codon equivalent to δ13e of CTR | Affinity not reported, impaired trafficking, insufficient signalling data to ascribe function | Unsupported |
| CTR δ13e | Coding exon 13 skipped, loss of half of TM7 then frame shift to stop codon | No effect on affinity, cAMP stimulation unaffected impaired signalling to IP3 | Reported in rabbit, unsupported in human |
Those splice variants listed as supported have supporting evidence from more than one group and/or organism as well as supporting EST data in the Ensembl/Havana merge of validated transcripts and/or ECgene. Those splice variants listed as unsupported do not have supporting EST data in the Ensembl/Havana merge of validated transcripts and/or ECgene.
Figure 3.
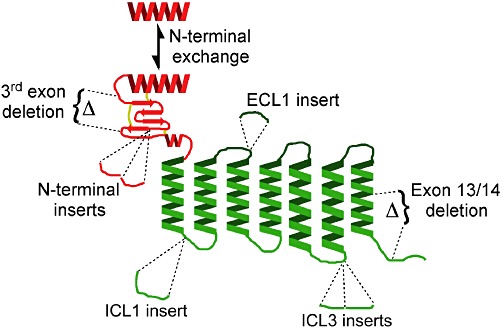
Cartoon depicting overall topology of Secretin family GPCRs. The N-terminal domain is shown in red with the seven transmembrane bundle in green. The relative locations of AS reported for the family are shown. N-terminal domain exchange is a feature of CRF2α, CRF2β and CRF2γ. CRF1, PAC1 and Secretin receptors have 3rd exon deletion variants. PAC1 has both N-terminal and ICL3 insert variants. CT receptor has variants with ICL1 insert, ECL1 insert and exon 13 deletion and CRF1 has an ICL1 insert variant.
The current nomenclature of AS within the Secretin family has a number of inconsistencies that make it difficult to follow. For example, the CT receptor has slice variants CTa and CTb, which in human refer to AS of the first ICL but in rat refer to AS in the first ECL. The PAC1 receptor has a wide variety of alternatively spliced variants with notations such as normal, short, very short, hip, hop and hiphop, which can be difficult for those unfamiliar with the literature to follow. We would recommend the adoption of a standardized nomenclature such as that suggest by Sammeth (Sammeth et al., 2008), although this has issues with the complexity of the nomenclature and its routine use.
In addition to AS of the Secretin family it should be noted that proteins directly interacting with this family, including RAMPs, Gα, Gβ, GPCR kinases and arrestins, are also subjected to AS of their coding regions potentially adding greatly to the possible diversity of receptor pharmacologies.
AS in the 5′ untranslated region
The 5′ untranslated region (UTR) of mRNA species is able to regulate their stability as well as the efficiency of translation initiation. Changes in the 5′ UTR can result in alternative coding regions and these will be discussed in the section on AS of the N-terminus.
The human Ensembl/Havana merge database of validated transcripts includes mRNA with alternative non-coding 5′ UTRs for CT, CLR, CRF2, PTH1 and PAC1. The alternative 5′ UTR of CT is the result of the use of an alternative osteoclast specific promoter and there is no evidence whether this alternative 5′ UTR confers any difference in stability or translational efficiency. The nature and function of the AS 5′ UTR in other human Secretin family receptors has yet to be determined.
AS in the N-terminus
In a bivalent ligand binding model, the receptor N-terminus serves as a high affinity bait for the carboxy terminus of the ligand. This then brings the amino terminus of the ligand in close proximity with the ECLs of the receptor where it is able to stabilize the receptor active state. In this model AS of the N-terminus of the receptor would enable new receptor variants to be generated. These could recognize alternative ligands that differed in their carboxy but not amino terminal end, alter the rank order of affinities a particular receptor had for multiple endogenous ligands or even generate alternative signalling bias from multiple endogenous ligands. Alternative N-termini have been reported for CRF1, CRF2, PAC1 and the Secretin receptor.
Splice variants in which the third coding exon is skipped (Δ3e) have been reported for CRF1[CRF1c (Ross et al., 1994)], PAC1[PAC1vs (Dautzenberg et al., 1999)] and Secretin (Figure 3 and Table 1) (Ding et al., 2002b). The third coding exon contains the 2nd, 3rd and 4th conserved cysteines and its deletion may lead to incorrect folding (see Figure 2). Consistent with a poorly functional N-terminus, CRF has very low potency at CRF1c compared with CRF1α and CRF1c did not bind CRF at concentrations tested (Ross et al., 1994), this is in spite of the fact that CRF1c does appear to traffic correctly to the cell surface (Zmijewski and Slominski, 2009b). The function of CRF1c has not been established although its expression is regulated by cell density (Zmijewski and Slominski, 2009a). CRF1 has been reported to form proximers1 (Kraetke et al., 2005) and also displays biphasic responses attributable to coupling to two different G protein pools (Wietfeld et al., 2004). This being the case, co-expression of CRF1c with CRF1α could alter the signalling profile, but this remains untested. In the case of the Secretin receptor, endogenous co-expression of full-length and Δ3e mRNA in pancreatic carcinoma cell lines results in at least a three order of magnitude reduction in Secretin potency (Ding et al., 2002a). This can be recovered by transfection of increasing amounts of wild-type receptor (Ding et al., 2002a). This pseudo dominant negative effect of the Δ3e Secretin receptor is very difficult to explain; Ding et al. (2002a) provide BRET data showing the wild-type and Δ3e receptors form a proximer alluding that interaction between the two receptor forms causes the observed loss of affinity (Ding et al., 2002b) and potency (Ding et al., 2002a). Elsewhere homo-dimerization of the Secretin receptor has been validated through combined disruption of BRET signal through mutation of the dimerization interface, disruption of BRET signal through the use of peptides that mimic the dimerization interface and cysteine disulphide bonding across TM regions of the receptor (Harikumar et al., 2007; 2008; Gao et al., 2009). Nonetheless, in the absence of direct data on the relative expression of the mature proteins at the cell surface as well as composition and stoichiometry of receptor proximers, interpretation of the severe loss of binding/signalling is extremely difficult. If we accept that endogenously all proximers contain both receptor variants, the implication is that the N-terminus of wild-type receptor provides strong cooperativity for ligand binding. Currently, the level of cooperativity reported across the Secretin dimer (Gao et al., 2009) appears insufficient to explain the magnitude of affinity and potency loss engendered by co-expression of the Δ3e variant. The PAC1 receptor is unusual in this family as the variant usually referred to as ‘normal’ contains six coding exons for the N-terminal domain (Figures 2 and 4). This is the result of the inclusion of two small exons between those corresponding to the 3rd and 4th of other family members (Figure 2 and PAC1n Figure 4). PAC1vs has exon 3 as well as the extra exons, with no equivalent in other family members, deleted (Figure 4) and is thus equivalent CRF1c and Δ3e Secretin receptors above. This variant displays a two order of magnitude reduction in affinity for its ligands pituitary adenylate cyclase activating peptide (PACAP) 38 (comprising 38 amino acids) and 27 (the same peptide with a C-terminal truncation) relative to the normal receptor variant (Dautzenberg et al., 1999), as well as a 20- to 100-fold decrease in potency for cAMP production (Dautzenberg et al., 1999; Lutz et al., 2006). The mRNA encoding this variant is co-expressed with full-length and short (see later, exon 3 present, unique exons absent) PAC1 in neuroblastoma cell lines SH-SY-5Y and Kelly cells (Lutz et al., 2006). The potency of PACAP38 to elicit a cAMP response in these cell lines is consistent with the potencies observed in heterologous systems for PAC1n and PAC1s (Lutz et al., 2006), suggesting that PAC1vs does not exert a dominant effect in this pathway.
Figure 4.
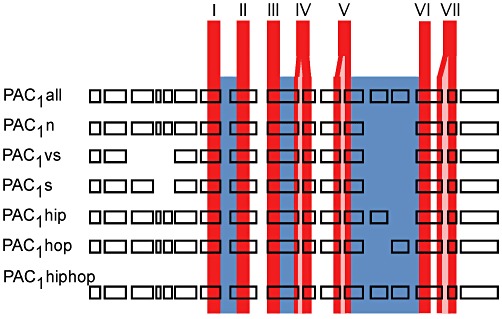
Schematic of the genomic organization of PAC1 receptor indicating the human splice variants discussed in this review. The top most exon pattern labelled all shows all coding exons for human PAC1; however, we are not aware of a splice variant that incorporates all these exons. Boxes indicate coding exons and are scaled according to amino acid length coded by each exon. The relative location of exons with respect to the overall receptor fold is depicted. The location of transmembrane helices is indicated in red (exon spanning helices shown with pink insert) and ICLs in blue.
In the above cases of CRF1c, Δ3e Secretin receptor and PAC1vs it is not clear what physiological relevance would be engendered by their pharmacology. In each case the N-terminus would be predicted to fold incorrectly and the effect on wild-type receptor signalling (if any) could be achieved simply by altering wild-type receptor expression. In comparison, the AS that gives rise to CRF2α, β and γ and PAC1n and PAC1s variants results in alternative N-termini that are all fully functional and elicit altered pharmacology. For CRF1, AS arises through the use of different promoters that drive expression of alternative first exons, with CRF2α containing a single first exon that is 5′ proximal to the first common exon and CRF2β containing two further upstream exons before splicing to the common first exon, CRF2γ contains a 3rd alternative first exon located between 5′αβ exons and again splices to the same common exon. The consequence of this AS is that the N-terminal sequence containing the first conserved cysteine differs between the three forms with protein lengths of 411, 438 and 397 amino acids respectively (Figure 3, N-terminal exchange). The crystal structure of CRF2α shows an alpha helix extending either side of the first conserved cysteine before the loop that connects it with the first beta strand (Pal et al., 2010). This contrasts with the NMR solution structure of CRF2β in which the corresponding structural element forms a disordered loop that is constrained by the disulphide bond and its link with the first beta strand (Grace et al., 2007). The N-terminus of CRF2γ has not been solved; however, the existing structural studies demonstrate that the interaction between the ligand and N-terminal domain occurs via the part of this fold opposite the AS sequence. In this light, the observation that these CRF2 variants have identical binding properties is unsurprising (Kostich et al., 1998; Ardati et al., 1999). These two studies have examined different downstream outputs from receptor activation. Kostich et al. (1998) measured cAMP accumulation and reported pEC50 values for CRF, sauvagine, urotensin and urocortin, which did not differ between CRF2α and γ but showed 10-fold higher potency at CRF2β. This is in contrast to the report of Ardati et al. (1999), who used a cAMP response element driven reporter assay and showed identical potencies of CRF2α and β with respect to CRF, sauvagine, urotensin and urocortin with pEC50 values consistent with the higher potency values established for CRF2β by Kostich et al. (1998). This difference may be due to differences in receptor reserve in the two assays. The β and γ variants show a more restricted pattern of tissue expression compared with the α variant; however, with the existing molecular pharmacology, it is unclear why the different isoforms exist. One possibility may be that CRF2 can form a complex with one or more RAMP isoforms. The crystal structure of the N-terminal domains of CLR in complex with RAMP1 shows extensive contacts between RAMP1 and the alpha helix of CLR (Haar et al., 2010), which corresponds to the AS region of the CRF2α, β and γ variants. AS splicing could, therefore, regulate the interaction between CRF2 and RAMPs thereby regulating ligand selectivity. As mentioned above, PAC1 is unusual among Secretin family members in that there is a common receptor variant (PAC1n) for which the N-terminus is encoded by six exons (Figure 3, C-terminal of the two N-terminal inserts and Figure 4). AS of this region to remove coding exons 4 and 5 results in a receptor, PAC1s (Figure 4), with exon organization that mimics the remainder of the family. PAC1, VPAC1 and VPAC2 all respond to physiologically relevant concentrations of PACAP; however, PAC1 is normally considered a type I PACAP receptor due to the low affinity and potency that VIP (vasoactive intestinal peptide) displays at this receptor. The first molecular pharmacological description comparing PAC1n and PAC1s was performed in HEK293 cells and indicated that VIP displays low affinity and potency for cAMP production only at the PAC1n variant and PAC1s does not show the same ligand selectivity (Dautzenberg et al., 1999). In contrast to these results a follow-up study performed in CHO cells showed that VIP had low affinity and potency for cAMP production at both PAC1n and PAC1s (Ushiyama et al., 2007). This, combined with the observation that RAMP2 selectively interacts with VPAC1 and alters its pharmacology (Christopoulos et al., 2003), suggests that AS of the N-terminal domain of PAC1 could yield receptors with significantly altered pharmacology by regulating the interaction with RAMP proteins. In rat testis an additional functional N-terminally spliced variant of PAC1 has been reported, PAC13a (Daniel et al., 2001). This variant has, in addition to the six exons that encode the N-terminus of PAC1n, an additional exon between coding exons 3 and 4 adding another 24 amino acids to the N-terminal domain (Figure 3, N-terminal of the two N-terminal inserts). At PAC13a PACAP27 displays equivalent affinity but slightly reduced potency for cAMP and IP3 production when compared with PAC1n (Daniel et al., 2001). PACAP38 displays higher affinity but significantly lower efficacy at PAC13a compared with PAC1n (Daniel et al., 2001) suggesting the higher affinity has been achieved in part by stronger G protein coupling. This variant has not been reported in humans.
Headless receptors
The expression of an N-terminally deleted, or headless, Secretin family GPCR in isolation would result in a non-functional receptor. If, on the other hand, a headless receptor was expressed that was competent to form homodimers with a full-length counterpart or heterodimers with other family members then altered pharmacology could result through, for example, loss of cooperativity.
In humans there are no examples of a headless Secretin family GPCR, nor is there evidence for such receptors through expressed sequence tag (EST) clustering in either Ensembl/Havana merge or ECgene. In spite of this, the existence of headless CRF1 variants has been proposed (Zmijewski and Slominski, 2010). The CT receptor, which has been shown to form a proximer (Harikumar et al., 2010), has been reported to have an AS variant in pufferfish that lacks the N-terminal domain (Nag et al., 2007a). These authors subsequently reported that this N-terminally truncated variant is able to act in a dominant negative manner (Nag et al., 2007b). These reports are unlikely to have any relevance to any mammalian Secretin family GPCR. Teleost fish are well documented to have undergone at least one whole-genome duplication subsequent to their divergence from the mammalian lineage (e.g. Jaillon et al., 2004). There appear to be four CT receptor genes in the teleost lineage (documented for zebrafish, medaka, pufferfish and stickleback) with two sharing similar genomic organization and total protein length when compared with the mammalian CT receptor. The CT receptor orthologue studied by this group has 23 exons and a predicted length of 794 amino acids and should not be considered as orthologous to human.
Although an attractive means to regulate receptor function we believe there is no evidence to support the existence of N-terminally truncated human Secretin family GPCRs.
Soluble N-termini
As has been discussed, the N-terminal domain is capable, in some cases, of high affinity ligand binding in its own right. Expression and secretion of soluble N-termini would therefore be predicted to sequester ligand and reduce its local effective concentration. If soluble N-terminal domains were stored in secretory vesicles and released in response to stimuli, including their own ligand, this would provide a means to provide additional temporal and spatial control over receptor signalling.
In humans the existence of an mRNA that may code for a soluble N-terminus of CRF1 has been reported (Pisarchik and Slominski, 2001); however, there is no supporting evidence for this variant in either Ensembl/Havana merge or ECgene. AS mRNA species predicted to code for soluble N-termini of both CRF1 and CRF2 have been reported in mouse (Pisarchik and Slominski, 2001; Chen et al., 2005). The data related to these forms are consistent with the possibility that they would be secreted and their action would be to reduce the effective concentration of available ligand (Perrin et al., 2001; Pisarchik and Slominski, 2001; Chen et al., 2005; Evans and Seasholtz, 2009; Zmijewski and Slominski, 2009a,b). At present we would regard the data related to these soluble N-termini as purely phenomenological.
AS of ECLs
The current model for Secretin family ligand–receptor interaction predicts the ECLs to be important in stabilizing the active state of the receptor–ligand complex. AS of these loops would therefore provide another means to alter pharmacology through altered ligand selectivity or altered signal transduction. It is evident from the genomic structure that insertions could be accommodated in all ECLs, with the exception of CT receptors and CL receptors that have the equivalent of coding exons 10 and 11 fused. Currently there is no evidence for such AS in humans through EST clustering in either Ensembl/Havana merge or ECgene.
In the rat brain, alternatively spliced variants of the CT receptor have been identified in which an extra exon is inserted between the 6th and 7th coding exon to create a receptor variant containing an additional 37 amino acids in ECL1 (ECL1+) (Figure 3) (Albrandt et al., 1993; Sexton et al., 1993). This variant displayed 50-fold lower affinity for the non-endogenous ligand salmon CT whereas rat CT showed no binding at the concentrations tested (Houssami et al., 1994). In a HEK293 background ligands displayed cAMP accumulation potencies consistent with binding affinities at both wild-type and ECL1+ receptors (Houssami et al., 1994). Contrastingly, in a Xenopus oocyte background, in which cystic fibrosis transmembrane conductance regulator transactivation was used as a measure of cAMP production, both receptor variants displayed equivalent responses to salmon CT but not human CT (Matsumoto et al., 1998). This variant has not been tested for interaction with RAMP proteins nor for its ability to act as a receptor for amylin.
AS of ICLs
The intracellular face of GPCRs provides the interaction surface for a wide range of proteins involved in signalling and regulation. Predictions based on mutagenesis studies would suggest that ICL2, ICL3 and the C-terminus would be involved in interactions with G proteins, while ICL1 may serve a regulatory role. Splice variants that alter ICLs could then provide a means of regulating receptor function. Additionally, GPCRs can be thought of as transducers of signals across the plasma membrane such that alterations on the intracellular face could result in differences in ligand affinity.
Alternative splicing of ICL1 has been reported for both CT and CRF1 receptors (Figure 3). In both cases the AS spliced version results from inclusion of an extra exon that encodes 16 [CT (Gorn et al., 1992)] or 29 [CRF1 (Chen et al., 1993)] additional amino acids. The CT ICL1+ receptor displayed no difference when compared with ICL1− in affinity for its ligands (Moore et al., 1995; Nussenzveig et al., 1995) but displayed impaired coupling to Gαs as assessed by an approximate 100-fold decrease in potency of CT to stimulate cAMP production (Moore et al., 1995; Nussenzveig et al., 1995).The ICL1+ variant of the CT receptor was not able to couple to intracellular calcium release (presumably via Gαq) and displayed impaired internalization when compared with the ICL1− variant (Moore et al., 1995). This study was performed in the baby hamster kidney cell background. Experiments in a HEK293 background showed a loss of coupling to intracellular calcium release and cAMP accumulation for the ICL1+ CT receptor variant (Raggatt et al., 2000). Although cell background-dependent variations in responses are observed, unpublished data from our laboratory confirmed impaired coupling of the CT receptor ICL1+ variant in stable transfectants of HEK293, mouse fibroblast (3T3) and African green monkey kidney (Cos7) cell lines suggesting the observed change in cAMP potency is an intrinsic property of this receptor variant. In contrast studies on the CRF1 ICL1+ are not as straightforward to interpret. The initial report comparing CRF1 ICL1+/− in a transiently transfected Cos7 system reported a twofold reduction in affinity of CRF for the ICL1+ receptor and an approximate 100-fold reduction in potency (Xiong et al., 1995). A subsequent study by the same group of stable transfectants of ICL+ and – variants in LLCPK-1 cells (pig kidney) showed a fourfold to fivefold decrease in affinity and an approximate 10-fold decrease in both potency and efficacy of CRF for cAMP accumulation at the ICL1+ variant (Nabhan et al., 1995). In radioligand binding on isolated membranes, GTPγS caused an apparent decrease in the affinity of CRF1 ICL− but not ICL+ for CRF (Nabhan et al., 1995), providing direct support for decreased G protein coupling of the ICL1+ variant. Two recent reports from a different group reported a fourfold to fivefold reduction in affinity of the CRF1 ICL1+ variant [transient transfection CHO, transient and stable HEK293 (Markovic et al., 2006; Teli et al., 2008)] with similar receptor levels with no significant difference in potency of CRF for cAMP stimulation but rather a difference in efficacy (Markovic et al., 2006; Teli et al., 2008). These studies also conclude that the ICL1+ variant of CRF1 is more sensitive to desensitization and internalization in a PKC-dependent fashion, in contrast to the increased resistance to internalization of the ICL+ CT receptor variant (Moore et al., 1995). Both CT and CRF1 ICL1+ variants display a more restricted tissue expression compared with their ICL1- counterparts; however, there is currently no data that demonstrate whether individual cells express only single or both receptor variants. Both CT and CRF1 receptors have been shown to form higher order complexes but without data on AS expression in native tissues it is not possible to speculate as to the physiological role of the ICL1+ receptors. Clearly there is a need for experiments on native tissues.
PAC1 is subject to AS of ICL3 (Figures 3 and 4). ICL3 inserts generated by AS of two additional exons were originally identified in rat (Spengler et al., 1993), then subsequently in human (Pisegna and Wank, 1996). The 5′ of these extra exons has been termed hip (Figure 4) and encodes an extra 28 amino acids (Spengler et al., 1993; Pisegna and Wank, 1996). The 3′ exon has been termed hop (Figure 4) and in humans also encodes an extra 28 amino acids (Pisegna and Wank, 1996), although in rat has been shown to encode 28 (hop1) or 27 (hop2) amino acids through the use of AS acceptor sites (Journot et al., 1995). In rat and mouse PAC1 ICL3 variants significant differences in pathway coupling are observed. No PAC1 variants studied were able to alter IP3 concentration when stimulated by PACAP27 whereas PACAP38 can stimulate IP3 equally via PAC1n and PAC1hop, to a lesser extent PAC1hiphop and not at all through PAC1hip (Spengler et al., 1993; Journot et al., 1995; Ushiyama et al., 2007). In contrast human PAC1 ICL3 isoforms show little or no difference in their abilities to stimulate cAMP or IP3 production in response to either PACAP38 or 27 (Pisegna and Wank, 1996; Pisegna et al., 1996; Lutz et al., 2006). The amino acid sequence encoded by rat and human hop1 exons is identical, whereas the sequence encoded by hip differs by two non-conservative substitutions of alanine (rat) to threonine and proline (rat) to leucine. The substantial signalling differences reported between rat and human PAC1 ICL3 variants could therefore result simply from these substitutions or may be a result of cell background effects as has been discussed above. In addition to the reports describing coupling of rat and human receptors via effectors to stimulate cAMP and IP3 production, there have been two reports from the same group describing coupling of bovine PAC1 to voltage gated calcium channels (VGCCs) (Mustafa et al., 2007; 2010). In this case both PACAP38 and 27 show similar affinities and potencies for cAMP accumulation via PAC1hop and PAC1n; however, only PAC1hop is able to couple to VGCCs (Mustafa et al., 2007). In addition, it is coupling via VGCCs that is required for PACAP-stimulated catecholamine release, implicating PAC1hop as the physiologically important isoform in acute adrenal stress response. These data have yet to be reported for human PAC1 variants; however, the bovine hop cassette encodes the identical amino acid sequence to the human cassette.
Tailless receptors
The 7th transmembrane domain of Secretin family members is encoded by two exons. The second of these exons encodes the last 14 amino acids of this transmembrane domain (Figures 1 and 3). Skipping of this exon in humans has been proposed for CRF1 (Markovic et al., 2008) and PTH1 (Alonso et al., 2011). For both receptors this is proposed to result in a 6TM receptor that displays impaired trafficking (Alonso et al., 2011) with CRF1 displaying similar ligand affinity to the full-length CRF variants but impaired signalling (Markovic et al., 2008). An equivalent CT receptor has been reported in rabbits (termed δ13e). This CT receptor variant displays similar ligand affinity to full-length CT with less than a twofold reduction in its ability to stimulate cAMP production in response to hCT but complete loss of ability to stimulate IP3 accumulation. Currently insufficient data exist regarding this type of variant to make any conclusion about pharmacological or physiological relevance.
Discussion
A wide array of AS events have been reported for the Secretin family of GPCRs. These splice variants do, indeed, display a range of altered pharmacologies and there is little doubt that the variants identified in this family engender diverse alternative receptor phenotypes, which warrant careful examination. It is clear that AS is capable of generating new receptor types that are able to distinguish different endogenous ligands as well as couple to alternative intracellular second messenger pathways and/or alter receptor regulation. This is likely to be extremely important physiologically for different cell types to respond appropriately to the same endocrine ligand. Moreover, understanding the location and nature of signalling from alternatively spliced receptor variants provides the opportunity to design more effectively targeted pharmaceuticals.
In spite of this very positive view there are a number of limitations in almost all the literature published on the molecular pharmacology of these variants. The literature relating to expression of AS Secretin family receptors almost exclusively examines expression at the tissue and mRNA level. Many of the studies cited, and splice variants reported are proposed to exert alternative pharmacology through their interaction with other splice variants of the same receptor. There are very few instances, however, in which co-expression of both variants has been convincingly demonstrated. There is also an underlying assumption that presence, or even quantitation, of various AS mRNA species corresponds with expression of mature protein at the cell surface. In this regard there is an urgent need for both high-quality pan splice variant specific antibodies as well as antibodies generated capable of distinguishing between splice variants. This is especially true in the case of examining transformed or primary cell lines that endogenously express more than one variant of a particular receptor. Presently the research in this field takes the form of identification of AS mRNA species followed by heterologous expression in model cell lines to examine the resulting molecular pharmacology. This is an entirely reasonable approach; however, a number of theoretical AS variants have been published for which little or no supporting evidence exists (these have been omitted from this review). Having established the pharmacology by the above method studies do need to be extended. Experiments in transformed cell lines that endogenously express receptor splice variants need to be performed to assess cell background-dependent changes in receptor activity. In addition experiments on primary cells harbouring endogenous receptors as well as endogenous combinations of splice variants are necessary if the molecular pharmacology is to be related to receptor physiology. Indeed, the physiological relevance of even the best characterized splice variants is unknown simply because there are effectively no data on which splice variants are endogenously expressed in particular cell types.
Acknowledgments
This work was funded by Program Grant 519461 and Project Grant 436780 from the National Health and Medical Research Council (NHMRC) of Australia. AC is a Senior, and PMS a Principal Research Fellow of the NHMRC.
Glossary
- AS
alternative splicing/alternative splice
- CGRP
calcitonin gene related peptide (and receptor)
- CLR
calcitonin-like receptor
- CRF1
corticotropin releasing factor receptor 1
- CRF2
corticotropin releasing factor receptor 2
- CT
calcitonin receptor or calcitonin
- ECL
extracellular loop
- EST
expressed sequence tag
- GIP
gastric inhibitory polypeptide receptor or glucose-dependent insulinotropic polypeptide receptor
- GLP-1
glucagon-like peptide receptor 1
- GLP-2
glucagon-like peptide receptor 2
- ICL
intracellular loop
- PAC1
pituitary adenylate cyclase activating peptide receptor
- PACAP
pituitary adenylate cyclase activating peptide
- PTH1
parathyroid hormone receptor 1
- PTH2
parathyroid hormone receptor 2
- RAMP
receptor activity modifying protein
- UTR
untranslated region
- VPAC1
vasoactive intestinal peptide receptor 1
- VPAC2
vasoactive intestinal peptide receptor 2
Footnotes
We have chosen to use the term proximer to denote the higher order complex whose existence is demonstrated by resonance energy transfer methods such as BRET and FRET. The distance over which energy resonance transfer operates is 1–10 nm. For comparison the height of the lipid bilayer is approximately 4 nm and the width of a GPCR is about 3.8 nm, thus resonance energy transfer could conceivably occur between two GPCRs separated by two to three intervening GPCRs or similarly sized proteins.
Conflict of interest
None declared.
References
- Albrandt K, Mull E, Brady EM, Herich J, Moore CX, Beaumont K. Molecular cloning of two receptors from rat brain with high affinity for salmon calcitonin. FEBS Lett. 1993;325:225–232. doi: 10.1016/0014-5793(93)81078-e. [DOI] [PubMed] [Google Scholar]
- Alonso V, Ardura JA, Wang B, Sneddon WB, Friedman PA. A naturally occurring isoform inhibits parathyroid hormone receptor trafficking and signaling. J Bone Miner Res. 2011;26:143–155. doi: 10.1002/jbmr.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardati A, Goetschy V, Gottowick J, Henriot S, Valdenaire O, Deuschle U, et al. Human CRF2 alpha and beta splice variants: pharmacological characterization using radioligand binding and a luciferase gene expression assay. Neuropharmacology. 1999;38:441–448. doi: 10.1016/s0028-3908(98)00201-9. [DOI] [PubMed] [Google Scholar]
- Bazarsuren A, Grauschopf U, Wozny M, Reusch D, Hoffmann E, Schaefer W, et al. In vitro folding, functional characterization, and disulfide pattern of the extracellular domain of human GLP-1 receptor. Biophys Chem. 2002;96:305–318. doi: 10.1016/s0301-4622(02)00023-6. [DOI] [PubMed] [Google Scholar]
- Bergwitz C, Gardella TJ, Flannery MR, Potts JT, Kronenberg HM, Goldring SR, et al. Full activation of chimeric receptors by hybrids between parathyroid hormone and calcitonin. Evidence for a common pattern of ligand-receptor interaction. J Biol Chem. 1996;271:26469–26472. doi: 10.1074/jbc.271.43.26469. [DOI] [PubMed] [Google Scholar]
- Chen R, Lewis KA, Perrin MH, Vale WW. Expression cloning of a human corticotropin-releasing-factor receptor. Proc Natl Acad Sci USA. 1993;90:8967–8971. doi: 10.1073/pnas.90.19.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AM, Perrin MH, Digruccio MR, Vaughan JM, Brar BK, Arias CM, et al. A soluble mouse brain splice variant of type 2alpha corticotropin-releasing factor (CRF) receptor binds ligands and modulates their activity. Proc Natl Acad Sci USA. 2005;102:2620–2625. doi: 10.1073/pnas.0409583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Christopoulos G, Morfis M, Udawela M, Laburthe M, Couvineau A, et al. Novel receptor partners and function of receptor activity-modifying proteins. J Biol Chem. 2003;278:3293–3297. doi: 10.1074/jbc.C200629200. [DOI] [PubMed] [Google Scholar]
- Daniel PB, Kieffer TJ, Leech CA, Habener JF. Novel alternatively spliced exon in the extracellular ligand-binding domain of the pituitary adenylate cyclase-activating polypeptide (PACAP) type 1 receptor (PAC1R) selectively increases ligand affinity and alters signal transduction coupling during spermatogenesis. J Biol Chem. 2001;276:12938–12944. doi: 10.1074/jbc.M009941200. [DOI] [PubMed] [Google Scholar]
- Dautzenberg FM, Mevenkamp G, Wille S, Hauger RL. N-terminal splice variants of the type I PACAP receptor: isolation, characterization and ligand binding/selectivity determinants. J Neuroendocrinol. 1999;11:941–949. doi: 10.1046/j.1365-2826.1999.00411.x. [DOI] [PubMed] [Google Scholar]
- Ding W-Q, Cheng Z-J, McElhiney J, Kuntz SM, Miller LJ. Silencing of secretin receptor function by dimerization with a misspliced variant secretin receptor in ductal pancreatic adenocarcinoma. Cancer Res. 2002a;62:5223–5229. [PubMed] [Google Scholar]
- Ding WQ, Kuntz S, Böhmig M, Wiedenmann B, Miller LJ. Dominant negative action of an abnormal secretin receptor arising from mRNA missplicing in a gastrinoma*. Gastroenterology. 2002b;122:500–511. doi: 10.1053/gast.2002.31039. [DOI] [PubMed] [Google Scholar]
- Evans RT, Seasholtz AF. Soluble corticotropin-releasing hormone receptor 2alpha splice variant is efficiently translated but not trafficked for secretion. Endocrinology. 2009;150:4191–4202. doi: 10.1210/en.2009-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyen JH, Cardinaux F, Gamse R, Bruns C, Azria M, Trechsel U. N-terminal truncation of salmon calcitonin leads to calcitonin antagonists. Structure activity relationship of N-terminally truncated salmon calcitonin fragments in vitro and in vivo. Biochem Biophys Res Commun. 1992;187:8–13. doi: 10.1016/s0006-291x(05)81450-0. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin L-G, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Gao F, Harikumar KG, Dong M, Lam PC-H, Sexton PM, Christopoulos A, et al. Functional importance of a structurally distinct homodimeric complex of the family B G protein-coupled secretin receptor. Mol Pharmacol. 2009;76:264–274. doi: 10.1124/mol.109.055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelling RW, Wheeler MB, Xue J, Gyomorey S, Nian C, Pederson RA, et al. Localization of the domains involved in ligand binding and activation of the glucose-dependent insulinotropic polypeptide receptor. Endocrinology. 1997;138:2640–2643. doi: 10.1210/endo.138.6.9104. [DOI] [PubMed] [Google Scholar]
- Goldman ME, McKee RL, Caulfield MP, Reagan JE, Levy JJ, Gay CT, et al. A new highly potent parathyroid hormone antagonist: [D-Trp12,Tyr34]bPTH-(7-34)NH2. Endocrinology. 1988;123:2597–2599. doi: 10.1210/endo-123-5-2597. [DOI] [PubMed] [Google Scholar]
- Gorn AH, Lin HY, Yamin M, Auron PE, Flannery MR, Tapp DR, et al. Cloning, characterization, and expression of a human calcitonin receptor from an ovarian carcinoma cell line. J Clin Invest. 1992;90:1726–1735. doi: 10.1172/JCI116046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace CRR, Perrin MH, DiGruccio MR, Miller CL, Rivier JE, Vale WW, et al. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc Natl Acad Sci USA. 2004;101:12836–12841. doi: 10.1073/pnas.0404702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace CRR, Perrin MH, Gulyas J, Digruccio MR, Cantle JP, Rivier JE, et al. Structure of the N-terminal domain of a type B1 G protein-coupled receptor in complex with a peptide ligand. Proc Natl Acad Sci USA. 2007;104:4858–4863. doi: 10.1073/pnas.0700682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauschopf U, Lilie H, Honold K, Wozny M, Reusch D, Esswein A, et al. The N-terminal fragment of human parathyroid hormone receptor 1 constitutes a hormone binding domain and reveals a distinct disulfide pattern. Biochemistry. 2000;39:8878–8887. doi: 10.1021/bi0001426. [DOI] [PubMed] [Google Scholar]
- Haar ET, Koth CM, Abdul-Manan N, Swenson L, Coll JT, Lippke JA, et al. Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure. 2010;18:1083–1093. doi: 10.1016/j.str.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Pinon DI, Miller LJ. Transmembrane segment IV contributes a functionally important interface for oligomerization of the Class II G protein-coupled secretin receptor. J Biol Chem. 2007;282:30363–30372. doi: 10.1074/jbc.M702325200. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Happs RM, Miller LJ. Dimerization in the absence of higher-order oligomerization of the G protein-coupled secretin receptor. Biochim Biophys Acta. 2008;1778:2555–2563. doi: 10.1016/j.bbamem.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikumar KG, Ball AM, Sexton PM, Miller LJ. Importance of lipid-exposed residues in transmembrane segment four for family B calcitonin receptor homo-dimerization. Regul Pept. 2010;164:113–119. doi: 10.1016/j.regpep.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmann MH, Hadac EM, Miller LJ. Critical contributions of amino-terminal extracellular domains in agonist binding and activation of secretin and vasoactive intestinal polypeptide receptors. Studies of chimeric receptors. J Biol Chem. 1995;270:14394–14398. doi: 10.1074/jbc.270.24.14394. [DOI] [PubMed] [Google Scholar]
- Houssami S, Findlay DM, Brady CL, Myers DE, Martin TJ, Sexton PM. Isoforms of the rat calcitonin receptor: consequences for ligand binding and signal transduction. Endocrinology. 1994;135:183–190. doi: 10.1210/endo.135.1.8013352. [DOI] [PubMed] [Google Scholar]
- Huang C-C, Tesmer JJG. Recognition in the face of diversity: interactions of heterotrimeric G proteins and G protein-coupled receptor (GPCR) kinases with activated GPCRs. J Biol Chem. 2011;286:7715–7721. doi: 10.1074/jbc.R109.051847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Aury J-M, Brunet F, Petit J-L, Stange-Thomann N, Mauceli E, et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004;431:946–957. doi: 10.1038/nature03025. [DOI] [PubMed] [Google Scholar]
- Journot L, Waeber C, Pantaloni C, Holsboer F, Seeburg PH, Bockaert J, et al. Differential signal transduction by six splice variants of the pituitary adenylate cyclase-activating peptide (PACAP) receptor. Biochem Soc Trans. 1995;23:133–137. doi: 10.1042/bst0230133. [DOI] [PubMed] [Google Scholar]
- Koole C, Wootten D, Simms J, Valant C, Sridhar R, Woodman OL, et al. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol Pharmacol. 2010;78:456–465. doi: 10.1124/mol.110.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostich WA, Chen A, Sperle K, Largent BL. Molecular identification and analysis of a novel human corticotropin-releasing factor (CRF) receptor: the CRF2gamma receptor. Mol Endocrinol. 1998;12:1077–1085. doi: 10.1210/mend.12.8.0145. [DOI] [PubMed] [Google Scholar]
- Kraetke O, Wiesner B, Eichhorst J, Furkert J, Bienert M, Beyermann M. Dimerization of corticotropin-releasing factor receptor type 1 is not coupled to ligand binding. J Recept Signal Transduct Res. 2005;25:251–276. doi: 10.1080/10799890500468838. [DOI] [PubMed] [Google Scholar]
- Lisenbee CS, Dong M, Miller LJ. Paired cysteine mutagenesis to establish the pattern of disulfide bonds in the functional intact secretin receptor. J Biol Chem. 2005;280:12330–12338. doi: 10.1074/jbc.M414016200. [DOI] [PubMed] [Google Scholar]
- Lutz EM, Ronaldson E, Shaw P, Johnson MS, Holland PJ, Mitchell R. Characterization of novel splice variants of the PAC1 receptor in human neuroblastoma cells: consequences for signaling by VIP and PACAP. Mol Cell Neurosci. 2006;31:193–209. doi: 10.1016/j.mcn.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Markovic D, Papadopoulou N, Teli T, Randeva H, Levine MA, Hillhouse EW, et al. Differential responses of corticotropin-releasing hormone receptor type 1 variants to protein kinase C phosphorylation. J Pharmacol Exp Ther. 2006;319:1032–1042. doi: 10.1124/jpet.106.107441. [DOI] [PubMed] [Google Scholar]
- Markovic D, Lehnert H, Levine MA, Grammatopoulos DK. Structural determinants critical for localization and signaling within the seventh transmembrane domain of the type 1 corticotropin releasing hormone receptor: lessons from the receptor variant R1d. Mol Endocrinol. 2008;22:2505–2519. doi: 10.1210/me.2008-0177. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Kaibara M, Uezono Y, Izumi F, Sumikawa K, Sexton PM, et al. Function of the rat calcitonin receptors, C1a and C1b, expressed in Xenopus oocytes. Biochem Biophys Res Commun. 1998;242:484–491. doi: 10.1006/bbrc.1997.7991. [DOI] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C, Yang H, Rodgers BD, Beday A, Pritchette LA, Eng J. High potency antagonists of the pancreatic glucagon-like peptide-1 receptor. J Biol Chem. 1997;272:21201–21206. doi: 10.1074/jbc.272.34.21201. [DOI] [PubMed] [Google Scholar]
- Moore EE, Kuestner RE, Stroop SD, Grant FJ, Matthewes SL, Brady CL, et al. Functionally different isoforms of the human calcitonin receptor result from alternative splicing of the gene transcript. Mol Endocrinol. 1995;9:959–968. doi: 10.1210/mend.9.8.7476993. [DOI] [PubMed] [Google Scholar]
- Mustafa T, Grimaldi M, Eiden LE. The hop cassette of the PAC1 receptor confers coupling to Ca2+ elevation required for pituitary adenylate cyclase-activating polypeptide-evoked neurosecretion. J Biol Chem. 2007;282:8079–8091. doi: 10.1074/jbc.M609638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa T, Walsh J, Grimaldi M, Eiden LE. PAC1hop receptor activation facilitates catecholamine secretion selectively through 2-APB-sensitive Ca(2+) channels in PC12 cells. Cell Signal. 2010;22:1420–1426. doi: 10.1016/j.cellsig.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabhan C, Xiong Y, Xie LY, Abou-Samra AB. The alternatively spliced type II corticotropin-releasing factor receptor, stably expressed in LLCPK-1 cells, is not well coupled to the G protein(s) Biochem Biophys Res Commun. 1995;212:1015–1021. doi: 10.1006/bbrc.1995.2071. [DOI] [PubMed] [Google Scholar]
- Nag K, Kato A, Sultana N, Ogoshi M, Takei Y, Hirose S. Fish calcitonin receptor has novel features. Gen Comp Endocrinol. 2007a;154:48–58. doi: 10.1016/j.ygcen.2007.06.017. [DOI] [PubMed] [Google Scholar]
- Nag K, Sultana N, Kato A, Hirose S. Headless splice variant acting as dominant negative calcitonin receptor. Biochem Biophys Res Commun. 2007b;362:1037–1043. doi: 10.1016/j.bbrc.2007.08.107. [DOI] [PubMed] [Google Scholar]
- Nussenzveig DR, Mathew S, Gershengorn MC. Alternative splicing of a 48-nucleotide exon generates two isoforms of the human calcitonin receptor. Endocrinology. 1995;136:2047–2051. doi: 10.1210/endo.136.5.7720653. [DOI] [PubMed] [Google Scholar]
- Pal K, Swaminathan K, Xu HE, Pioszak AA. Structural basis for hormone recognition by the human CRFR2{alpha} G protein-coupled receptor. J Biol Chem. 2010;285:40351–40361. doi: 10.1074/jbc.M110.186072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthier C, Kleinschmidt M, Neumann P, Rudolph R, Manhart S, Schlenzig D, et al. Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc Natl Acad Sci USA. 2007;104:13942–13947. doi: 10.1073/pnas.0706404104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin MH, Fischer WH, Kunitake KS, Craig AG, Koerber SC, Cervini LA, et al. Expression, purification, and characterization of a soluble form of the first extracellular domain of the human type 1 corticotropin releasing factor receptor. J Biol Chem. 2001;276:31528–31534. doi: 10.1074/jbc.M101838200. [DOI] [PubMed] [Google Scholar]
- Perrin MH, DiGruccio MR, Koerber SC, Rivier JE, Kunitake KS, Bain DL, et al. A soluble form of the first extracellular domain of mouse type 2beta corticotropin-releasing factor receptor reveals differential ligand specificity. J Biol Chem. 2003;278:15595–15600. doi: 10.1074/jbc.M210476200. [DOI] [PubMed] [Google Scholar]
- Pioszak AA, Xu HE. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc Natl Acad Sci USA. 2008;105:5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak AA, Parker NR, Suino-Powell K, Xu HE. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J Biol Chem. 2008;283:32900–32912. doi: 10.1074/jbc.M805749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarchik A, Slominski AT. Alternative splicing of CRH-R1 receptors in human and mouse skin: identification of new variants and their differential expression. FASEB J. 2001;15:2754–2756. doi: 10.1096/fj.01-0487fje. [DOI] [PubMed] [Google Scholar]
- Pisegna JR, Wank SA. Cloning and characterization of the signal transduction of four splice variants of the human pituitary adenylate cyclase activating polypeptide receptor. Evidence for dual coupling to adenylate cyclase and phospholipase C. J Biol Chem. 1996;271:17267–17274. doi: 10.1074/jbc.271.29.17267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisegna JR, Moody TW, Wank SA. Differential signaling and immediate-early gene activation by four splice variants of the human pituitary adenylate cyclase-activating polypeptide receptor (hPACAP-R) Ann NY Acad Sci. 1996;805:54–64. doi: 10.1111/j.1749-6632.1996.tb17473.x. discussion 64–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raggatt LJ, Evdokiou A, Findlay DM. Sustained activation of Erk1/2 MAPK and cell growth suppression by the insert-negative, but not the insert-positive isoform of the human calcitonin receptor. J Endocrinol. 2000;167:93–105. doi: 10.1677/joe.0.1670093. [DOI] [PubMed] [Google Scholar]
- Ross PC, Kostas CM, Ramabhadran TV. A variant of the human corticotropin-releasing factor (CRF) receptor: cloning, expression and pharmacology. Biochem Biophys Res Commun. 1994;205:1836–1842. doi: 10.1006/bbrc.1994.2884. [DOI] [PubMed] [Google Scholar]
- Runge S, Wulff BS, Madsen K, Bräuner-Osborne H, Knudsen LB. Different domains of the glucagon and glucagon-like peptide-1 receptors provide the critical determinants of ligand selectivity. Br J Pharmacol. 2003;138:787–794. doi: 10.1038/sj.bjp.0705120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge S, Thøgersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- Sammeth M, Foissac S, Guigo R. A general definition and nomenclature for alternative splicing events. PLoS Comput Biol. 2008;4:e1000147. doi: 10.1371/journal.pcbi.1000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton PM, Houssami S, Hilton JM, O'Keeffe LM, Center RJ, Gillespie MT, et al. Identification of brain isoforms of the rat calcitonin receptor. Mol Endocrinol. 1993;7:815–821. doi: 10.1210/mend.7.6.8395656. [DOI] [PubMed] [Google Scholar]
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg P, et al. Differential signal transduction by five splice variants of the PACAP receptor. 1993;365:170–175. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]
- Stroop SD, Kuestner RE, Serwold TF, Chen L, Moore EE. Chimeric human calcitonin and glucagon receptors reveal two dissociable calcitonin interaction sites. Biochemistry. 1995;34:1050–1057. doi: 10.1021/bi00003a040. [DOI] [PubMed] [Google Scholar]
- Sun C, Song D, Davis-Taber RA, Barrett LW, Scott VE, Richardson PL, et al. Solution structure and mutational analysis of pituitary adenylate cyclase-activating polypeptide binding to the extracellular domain of PAC1-RS. Proc Natl Acad Sci USA. 2007;104:7875–7880. doi: 10.1073/pnas.0611397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teli T, Markovic D, Hewitt ME, Levine MA, Hillhouse EW, Grammatopoulos DK. Structural domains determining signalling characteristics of the CRH-receptor type 1 variant R1beta and response to PKC phosphorylation. Cell Signal. 2008;20:40–49. doi: 10.1016/j.cellsig.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Tseng CC, Kieffer TJ, Jarboe LA, Usdin TB, Wolfe MM. Postprandial stimulation of insulin release by glucose-dependent insulinotropic polypeptide (GIP). Effect of a specific glucose-dependent insulinotropic polypeptide receptor antagonist in the rat. J Clin Invest. 1996;98:2440–2445. doi: 10.1172/JCI119060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JT, Jones SB, Bylund DB. A fragment of vasoactive intestinal peptide, VIP(10-28), is an antagonist of VIP in the colon carcinoma cell line, HT29. Peptides. 1986;7:849–854. doi: 10.1016/0196-9781(86)90105-1. [DOI] [PubMed] [Google Scholar]
- Ushiyama M, Ikeda R, Sugawara H, Yoshida M, Mori K, Kangawa K, et al. Differential intracellular signaling through PAC1 isoforms as a result of alternative splicing in the first extracellular domain and the third intracellular loop. Mol Pharmacol. 2007;72:103. doi: 10.1124/mol.107.035477. [DOI] [PubMed] [Google Scholar]
- Wang MW, Young AA, Rink TJ, Cooper GJ. 8-37h-CGRP antagonizes actions of amylin on carbohydrate metabolism in vitro and in vivo. FEBS Lett. 1991;291:195–198. doi: 10.1016/0014-5793(91)81282-d. [DOI] [PubMed] [Google Scholar]
- Wietfeld D, Heinrich N, Furkert J, Fechner K, Beyermann M, Bienert M, et al. Regulation of the coupling to different G proteins of rat corticotropin-releasing factor receptor type 1 in human embryonic kidney 293 cells. J Biol Chem. 2004;279:38386–38394. doi: 10.1074/jbc.M405335200. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Xie LY, Abou-Samra AB. Signaling properties of mouse and human corticotropin-releasing factor (CRF) receptors: decreased coupling efficiency of human type II CRF receptor. Endocrinology. 1995;136:1828–1834. doi: 10.1210/endo.136.5.7720627. [DOI] [PubMed] [Google Scholar]
- Zmijewski MA, Slominski AT. CRF1 receptor splicing in epidermal keratinocytes: potential biological role and environmental regulations. J Cell Physiol. 2009a;218:593–602. doi: 10.1002/jcp.21632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmijewski MA, Slominski AT. Modulation of corticotropin releasing factor (CRF) signaling through receptor splicing in mouse pituitary cell line AtT-20–emerging role of soluble isoforms. J Physiol Pharmacol. 2009b;60(Suppl. 4):39–46. [PMC free article] [PubMed] [Google Scholar]
- Zmijewski MA, Slominski AT. Emerging role of alternative splicing of CRF1 receptor in CRF signaling. Acta Biochim Pol. 2010;57:1–13. [PMC free article] [PubMed] [Google Scholar]