

Abstract
BACKGROUND AND PURPOSE
IL-13 is a pleiotropic Th2 cytokine considered likely to play a pivotal role in asthma. Here we describe the preclinical in vitro and in vivo characterization of CAT-354, an IL-13-neutralizing IgG4 monoclonal antibody (mAb), currently in clinical development.
EXPERIMENTAL APPROACH
In vitro the potency, specificity and species selectivity of CAT-354 was assayed in TF-1 cells, human umbilical vein endothelial cells and HDLM-2 cells. The ability of CAT-354 to modulate disease-relevant mechanisms was tested in human cells measuring bronchial smooth muscle calcium flux induced by histamine, eotaxin generation by normal lung fibroblasts, CD23 upregulation in peripheral blood mononuclear cells and IgE production by B cells. In vivo CAT-354 was tested on human IL-13-induced air pouch inflammation in mice, ovalbumin-sensitization and challenge in IL-13 humanized mice and antigen challenge in cynomolgus monkeys.
KEY RESULTS
CAT-354 has a 165 pM affinity for human IL-13 and functionally neutralized human, human variant associated with asthma and atopy (R130Q) and cynomolgus monkey, but not mouse, IL-13. CAT-354 did not neutralize human IL-4. In vitro CAT-354 functionally inhibited IL-13-induced eotaxin production, an analogue of smooth muscle airways hyperresponsiveness, CD23 upregulation and IgE production. In vivo in humanized mouse and cynomolgus monkey antigen challenge models CAT-354 inhibited airways hyperresponsiveness and bronchoalveolar lavage eosinophilia.
CONCLUSIONS AND IMPLICATIONS
CAT-354 is a potent and selective IL-13-neutralizing IgG4 mAb. The preclinical data presented here support the trialling of this mAb in patients with moderate to severe uncontrolled asthma.
Keywords: CAT-354, IL-13, IgG4, preclinical development for asthma, uncontrolled asthma, therapeutic antibody, asthma treatment
Introduction
Asthma is a complex, persistent, inflammatory disease characterized by airways hyperresponsiveness (AHR) in association with airway inflammation. Moderate to severe asthma is primarily and generally treated with inhaled corticosteroids and β2-adrenoceptor agonists which act to give the patient ‘control’ or a relatively normal life not restricted by their asthma. Control of asthma manifestations is the key goal of treatment (GINA Guidelines, 2010). Although current therapy controls asthma for the majority of patients it is recognized that a significant proportion of asthmatics are inadequately controlled even with maximal steroid treatment (Peters et al., 2006). Despite the 2003 approval of omalizumab (Xolair, Genentech, South San Francisco, CA, USA/Novartis, East Hanover, NJ, USA), for the treatment of moderate to severe persistent allergic asthma in patients whose symptoms are inadequately controlled by inhaled corticosteroids, there still remains a significant clinical need for alternative treatments in the wider population of poorly controlled asthmatic patients.
IL-13 is a cytokine which is secreted predominantly by CD4+ T helper type 2 (Th2) cells. It shares receptor components and many biological properties with IL-4. IL-13 receptors (heterodimers of IL-13Rα1 and IL-4Rα) are expressed on mast cells, leukocyte subsets (eosinophils, monocytes, B cells) and structural cells (epithelial cells, fibroblasts, smooth muscle) all of which are considered relevant in asthma. The action of IL-13 on these cells is to stimulate processes that promote AHR, inflammation and underlying airway structural changes including fibrosis and mucus hypersecretion that are cardinal features of asthma (Hershey, 2003).
IL-13 mRNA and protein levels are elevated in bronchial biopsies, sputum and bronchoalveolar lavage (BAL) fluid from asthmatics compared with control subjects (Kotsimbos et al., 1996; Humbert et al., 1997; Naseer et al., 1997; Komai-Koma et al., 2001) and further increased in BAL samples from allergen challenged asthmatics (Huang et al., 1995; Kroegel et al., 1996). IL-13 protein expression in the lung is also correlated with both asthma severity and control. Mild steroid-naïve asthmatics have higher levels of sputum IL-13 than normals as do severe asthmatics who have poor control despite standard of care up to, and including, oral corticosteroids. Moderate asthmatics, who are controlled by inhaled corticosteroids, do not have upregulated sputum IL-13 expression compared to normals (Saha et al., 2008).
Genetic polymorphisms in the IL-13 pathway are also associated with both atopy and asthma. The variant IL-13, R130Q, has been associated with asthma, atopy and raised serum IgE (Heinzmann et al., 2000; Liu et al., 2000; Kauppi et al., 2001) while activating the signalling IL-13 receptor (Rα1) and downstream functions more efficiently, and binding to the decoy receptor (Rα2) less efficiently, than WT IL-13 (Vladich et al., 2005). Similar polymorphisms associated with risk of atopy and asthma have been observed in the IL-13 promoter (Howard et al., 2001 and van der Pouw Kraan et al., 1999), IL-4Rα (Howard et al., 2002) and in the key signalling molecule downstream of the IL-13 receptor – STAT6 (Tamura et al., 2003).
Animal model data from models mimicking aspects of human asthma support the hypothesis that IL-13 is a key mediator in the development and maintenance of the asthma phenotype. Administration of recombinant IL-13 to the airway of allergen-naïve mice results in AHR, airway inflammation and mucus production (Grunig et al., 1998; Wills-Karp et al., 1998). Similar pathologies are seen in transgenic mice in which IL-13 is selectively overexpressed in the lung although longer-term exposure to IL-13 also results in fibrosis (Zhu et al., 1999). The same ‘asthmatic’ phenotype occurs following allergen challenge in sensitized mice and is also associated with IL-13. Soluble IL-13Rα2, a potent IL-13 neutralizer, has been shown to inhibit AHR, mucus hypersecretion and pulmonary inflammation (Grunig et al., 1998; Wills-Karp et al., 1998). Non-human primate segmental antigen challenge studies have also shown an anti-inflammatory effect for an IL-13 neutralizing antibody (Bree et al., 2007).
Given the range of cells involved in asthma on which IL-13 is known to act and the pathogenic functions ascribed to this cytokine a significant role for IL-13 in asthma can be expected. Coupled with the evidence of a relationship between IL-13 expression and disease severity in patients (Saha et al., 2008) neutralization of IL-13 is a credible approach for the treatment of asthma. Based on this rationale we have generated an IL-13 neutralizing IgG4 monoclonal antibody (mAb), CAT-354, which we are advancing to clinical development. This paper describes the preclinical in vitro and in vivo characterization of CAT-354 necessary to provide confidence that the mAb would affect all the key disease mechanisms associated with human asthma and drive a transition to clinical development.
Methods
Generation of CAT-354
CAT-354 (BAK 1.1) was isolated from a phage library and generated as a human IgG4 as described (Thom et al., 2006).
TF-1 cell proliferation assay
This assay was performed as described (Thom et al., 2006). In brief antibodies were added to various forms of IL-13 or other cytokines at a cytokine concentration mediating approximately 80% maximal effect and preincubated for 30 min at room temperature before adding to TF-1 cells (RnD Systems, Oxford, UK) and incubating at 37°C for 72 h. [3H]-thymidine (10µL; 50 mCi·mL−1; NEN) was added and the cells incubated for a further 4 h. Cells were harvested on glass fibre filter plates (PerkinElmer, Shelton, CT, USA) and thymidine incorporation determined using a Packard TopCount microplate liquid scintillation counter.
HDLM-2 cell proliferation assay
HDLM-2 cells (Deutsche SammLung von Mikroorganismen und Zellkulturen, Braunschweig, Germany) were maintained according to supplied protocols in RPMI-1640 with GLUTAMAX I (Invitrogen, Paisley, UK) and 20% fetal bovine serum. Washed cells (50 µL; 2 × 105 cells·mL−1 media) were added to each triplicate assay point in a 96-well assay plate and 50 µL test antibody added before incubating at 37°C under 5% CO2 for 72 h. [3H]-thymidine (25µL;10 µCi·mL−1) was then added to each well and the assay incubated for a further 4 h before harvesting on to glass fibre filter plates (PerkinElmer) using a cell harvester. Thymidine incorporation was determined using a Packard TopCount (Waltham, MA, USA) microplate liquid scintillation counter.
Human umbilical vein endothelial cell (HUVEC) vascular cell adhesion molecule (VCAM)-1 upregulation assay
HUVECs (Biowhittaker, Workingham, UK) were maintained according to supplied protocols in EGM-2 media (Biowhittaker). Test solutions of antibody (in triplicate) were diluted in EGM-2. Recombinant cytokines (IL-13, IL-4 or IL-1β) were added at concentrations inducing approximately 80% maximal VCAM-1 upregulation in a total well volume of 100 µL. All samples were preincubated for 30 min at room temperature. Assay samples were then added to HUVEC that had been pre-seeded and grown to near confluence in 96-well assay plates and the assay incubated for 16–20 h at 37°C under 5% CO2. Assay media was aspirated and replaced with blocking solution (PBS plus 4% Marvel v/v) and incubated for 1 h at room temperature. Plates were washed three times with PBST (PBS/0.1% Tween-20 v/v [PBST] ) before 100 µL biotinylated anti-VCAM-1 antibody (AbD Serotec, Oxford, UK, 1:500 dilution in PBST/1% Marvel) was added to each well and incubated at room temperature for 1 h, washed three times with Delfia™ wash buffer (PerkinElmer) and 100 µL europium-labelled Streptavidin anti-murine IgG1 (1:1000 dilution in Delfia™ assay buffer PerkinElmer) added to each well. Assay plates were then incubated at room temperature for 1 h, washed seven times with Delfia™ wash buffer (PerkinElmer). Finally 100 µL of enhancement solution (PerkinElmer) was added to each well and fluorescence intensity determined.
Normal human lung fibroblast (NHLF) eotaxin-1 release assay
NHLFs (Biowhittaker) were maintained according to supplied protocols in fibroblast growth medium-2 (Biowhittaker). Test solutions of antibody (in triplicate) were diluted in assay media with 0.8 nM recombinant bacterially derived human IL-13 (Peprotech), which induced approximately 80% maximal eotaxin-1 release, in a total well volume of 120 µL and pre-incubated for 30 min at room temperature. Assay samples were then added to pre-seeded and near confluence NHLFs in 96-well assay plates before being incubated for 16–20 h at 37°C under 5% CO2. Assay plates were centrifuged at 300 g for 5 min to pellet detached cells. Supernatant eotaxin levels were determined by elisa (enzyme-linked immunosorbent assay) using reagents and methods described by the manufacturer (R&D Systems).
Human eosinophil shape change assay
NHLFs were grown in media supplemented with 9.6 nM IL-13, 286 pM TNFα and 160 pM TGF-β1 for 48 h at 37°C under 5% CO2. The conditioned media was aspirated, assayed for eotaxin-1 content and stored at −80°C until ready to be used in the eosinophil shape change assay.
Eosinophil shape change was carried out essentially as described previously (Sabroe et al., 1999). Blood (50 mL) from normal healthy human volunteers was taken into EDTA and erythrocytes removed by dextran sedimentation. Polymorphonuclear leukocytes were then separated from mononuclear cells using discontinuous plasma-Percoll gradients and resuspended in buffer (PBS containing 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM d-glucose, and 0.1% BSA, pH 7.3) at a concentration of 5 × 106 cells·mL−1 and allowed to rest for 30 min at 37°C. Aliquots of cells (100 µL) were then supplemented with various experimental treatments to a total volume of 400 µL. Tubes were returned to 37°C, incubated for 8.5 min and then transferred to an ice-water bath and 25 µL 10 × Cellfix buffer (BD, Oxford, UK) added. Samples were analysed immediately. Eosinophils were identified on the flow cytometer (FACSCalibur, BD) by their FL2 (fluorescence channel 2, 564–606 nm wavelength, yellow/orange) autofluorescence; non FL2-autofluorescent granulocytes were considered neutrophils. Shape change was calculated as a percentage of the forward scatter (FSC) shape change caused by buffer alone.
Human bronchial smooth muscle cell (BSMC) calcium ion flux assay
Human BSMCs from one 18-year-old male Caucasian donor were grown in SmGM-2 growth media (both cells and media from Cambrex Biosciences, East Rutherford, NJ, USA) and maintained according to supplied protocols. BSMCs were plated at 2 × 104 cells per well in 96-well plates, allowed to attach for 24 h at 37°C and 5% CO2 before being refed and cultured for a further 24 h. Culture media was aspirated and replaced with SmGM-2 including either 4 nM IL-13 or 3.6 nM IL-4 ± 67 nM antibodies that had been preincubated together for 30 min and then cultured for a final 18–24 h.
Calcium flux was assessed using a fluorometric imaging plate reader (FLIPR, Molecular Devices, Sunnyvale, CA, USA). Cells were washed twice in SmBM basal media with 0.1% fetal calf serum (FCS, BioWhittaker) and loaded with dye for 45 min at 37°C under 5% CO2 by addition of 70 µL per well labelling mix (SmBM, 0.1% FCS, 2 µM Fluo 4-AM, 0.03% pluronic acid, 2 mM probenicid, 20 mM HEPES). Cells were washed once with SmBM and twice with FLIPR buffer (125 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 25 mM HEPES, 5 mM glucose, 1% FCS) before being incubated for 15 min at 37°C under 5% CO2 to equilibrate intra- and extracellular Ca++ prior to the FLIPR run.
BSMCs were challenged with a dose–response of histamine and the induced calcium ion flux followed out over 5 min at 37°C. FLIPR data was analysed using peak fluorescence after agonist addition with the average blank (background response) subtracted for each treatment. Histamine dose versus peak fluorescence dose–response curves were generated and the area under these curves analysed.
Peripheral blood mononuclear cell (PBMC) CD23 upregulation assay
PBMCs were generated as described above, under eosinophil shape change, and were stimulated with 80 pM IL-13 and/or 21.4 pM IL-4 in the absence or presence of 0.001–30 nM antibodies and cultured for 48 h. Subsequently PBMCs were co-stained with CD3, CD14, CD19 and CD23 to identify positive cell types by FACS analysis. CD23 expression was determined by comparing stimulated cells with unstimulated cells in terms of overall expression. Overall expression was the percentage of PBMCs expressing CD23 multiplied by the geometric mean fluorescence of this positive population and is reported as ‘CD23 score’.
B cell isotype switching assay
B cells from six individual donors were isolated from human buffy coats (National Blood Service, Watford, UK) by centrifugation over a density gradient followed by negative selection using magnetic beads. B cell purity was assessed by staining with anti-CD19-PE using standard flow cytometric methods. Following purification B cells were stimulated in 96-well format in Iscove's modified Dulbecco's medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10% FCS, 100 U·mL−1 penicillin, 100 U·mL−1 streptomycin and 20 µg·mL−1 human transferrin (Serologicals proteins Inc, USA). 5 × 104 B cells per well in a 24-well plate were treated with 2.4 nM IL-13, 0.7 nM IL-4 or IL-10 and 1 µg·mL−1 of the anti-CD40 mAb EA5 (Biosource, Camarillo, CA, USA) ± test antibodies in a total volume of 200 µL. Assay plates were incubated for 14 days at 37°C and 5% CO2 and the supernatants assessed for IgE content using an IgE elisa.
Fluoronunc immunosorb 96 well elisa plates were coated overnight at room temperature with 100 µL of 10 µg·mL−1 IgE-capture antibody (BD, Oxford, UK). Plates were then washed three times with PBS/0.1% (v/v) Tween-20 and blocked with 300 µL·well−1 of 1% BSA in PBS for 1 h at room temperature. Plates were washed three times with PBS/0.1% (v/v) Tween, 100 µL assay supernatant added and incubated for 90 min at room temperature. Plates were washed three times with PBS/0.1% Tween before 100 µL 0.1 µg·mL−1 in 1:10 superblock : PBS (Pierce, Rockford, IL, USA) biotinylated detection antibody (Bethyl Laboratories, Montgomery, TX, USA; A80-108A) was added to each well and incubated for 90 min at room temperature. Plates were washed five times with PBS/0.1% Tween and 100 µL per well Streptavidin-Europium (PerkinElmer) diluted 1:1000 in Delfia™ assay buffer was added and incubated for 60 min at room temperature. Plates were then washed 7 times in Delfia™ wash buffer before the addition of 100 µL·well−1 of enhancement solution (PerkinElmer). Plates were incubated for a final 15 min followed by reading at 615 nm. The detection limit of the elisa was 0.2 ng·mL−1.
Animals
All animal care and experimental procedures complied with the terms of the associated UK Home Office project licences or, for the work with cynomolgus monkeys, with Association for the Assessment and Accreditation of Laboratory Animal Care standards under the authority of the local Institutional Animal Care and Use Committee.
Mouse air pouch model
Female BALB/c mice (total 100; 17–21 g; Charles River, Manston, UK) were allowed to acclimatize to the animal house for at least 8 days before experimental use and allowed food and water ad libitum.
On day 0 mice were briefly anaesthetized with isofluorane and 2.5 mL sterile air (0.25 µm filtered) injected subcutaneously between the scapula to create a centrally positioned air pouch. On day 3 the injection with sterile air was repeated to re-inflate the air pouch. On day 6, 30 min before induction of inflammation by intrapouch (i.po.) injection of 0.75% carboxymethylcellulose in saline ± 2 µg rhuIL-13 (Peprotech) ± mAbs or PBS, animals were treated intravenously with antibody or PBS via the tail vein. Animals did not receive mAbs by both dosing routes. Twenty-four hours following induction of inflammation mice were killed and the air pouch lavaged with 1 mL heparinized PBS (5 U·mL−1). Total cells infiltrating the air pouch were counted on a Becton Dickinson FACSCalibur flow cytometer. Differential cell counts were determined by Wright–Giemsa staining of cytospun cells (Shandon, Runcorn, UK).
Generation of humanized IL-13 mice
Mice were humanized using homologous recombination and associated standard methods. In brief the mouse IL-13 gene, including introns, from start to stop codon was replaced with the human IL-13 gene from start to stop codon, including introns, such that the humanized mouse retained a mouse promoter and pA tail but had a human signal sequence.
This was achieved as follows: 5’- and 3’-mouse homology arms and the human gene from start to stop codon were generated by PCR utilizing primers with inbuilt restriction sites. Then mouse–human 5’-fusion and human–mouse 3’-fusion fragments and new mouse 5’- and 3’-homology regions were also generated by PCR using primers with inbuilt restriction sites for cloning. The 5’- and 3’- ends were then assembled from the four sections using overlapping PCR and cloned together into a vector containing a LoxP-flanked PGKneo cassette in the intron between exons 1 and 2. The entire vector was sequenced to confirm correct assembly.
The vector was linearized with AclI, which cut twice in the ampicillin gene, and electroporated into C57 BL/6 embryonic stem (ES) cells. Appropriate targeting at the 5’- and 3’-ends and single insertion was confirmed by Southern blotting. Two independently targeted ES cell lines were injected into BALB/c blastocysts to generate chimaeric mice which were then bred with wild type (WT) C57 BL/6 mice to generate heterozygote C57 BL/6 mice. The LoxP-flanked neomycin resistance cassette was removed by breeding the heterozygous mice onto C57 BL/6 Cre recombinase-expressing mice. The Cre recombinase was then removed by breeding the neomycin-cassette deleted heterozygous Cre recombinase-positive mice with C57 BL/6 mice and the resultant Cre-recombinase-negative neomycin-cassette-deleted heterozygous mice bred to homozygosity. Finally homozygote human IL-13 knock-in mice were transferred to a BALB/c background over five generations using speed congenics (as described by Wakeland et al., 1997).
Ovalbumin sensitization and challenge model
Humanized IL-13 BALB/c mice (see above; total 266), or WT BALB/c mice (total 96) were used at between 6 and 8 weeks of age and acclimatized for at least 1 week before initiation of procedures. The model of ovalbumin sensitization and challenge has been previously described (McMillan et al., 2002). In brief mice were sensitized intraperitoneally on days 0 and 12 with 10 µg ovalbumin (Sigma, Poole, UK) and 2 mg aluminium hydroxide (Serva, Heidelberg, UK) or 2 mg aluminium hydroxide alone before being challenged with a nebulized aerosol of 5% ovalbumin in saline for 20 min on days 19–24 inclusive. On day 25 mice were terminally anaesthetized, with 90 mg·kg−1 i.p. pentobarbital sodium (CEVA, Libourne, France), prior to either assessment of airflow limitation using forced manoeuvres (Buxco, Abington, UK) or AHR using Flexivent (Scireq, Montreal, Canada) and then a common step of exsanguination and BAL with PBS.
Cynomolgus monkey antigen challenge model
Twenty male and seven female adult, Ascaris suum-sensitive, cynomolgus monkeys were preselected on the basis of previously established AHR following nebulized antigen (Ascaris suum extract) challenge. The number of animals used was derived from a sample size calculation based on historical airway function data from two previous studies using the change in PC30 (the intravenous histamine dose required to generate a 30% increase in lung resistance [RL] above baseline) induced by double antigen challenge.
The study was run in two phases: phase I to establish the allergic response and phase II, in the presence of CAT-354, to investigate the effect of the drug on the allergic response. Primary endpoints were predetermined to be PC30 and BAL inflammation. All endpoints were defined as the within animal change between day 11 and day 1 in phase II compared with the change seen in the same animal between days 11 and day 1 in phase I. In other words the effect of CAT-354 on the double allergen challenge induced alterations in model phenotype on a per-animal basis. Statistical analysis of all study endpoints was performed using a one-sample t-test with the null hypothesis that the endpoint was not significantly different from zero.
In each phase airway function and lung inflammation was assessed by intravenous histamine challenge and bronchoalveolar lavage, respectively, on days 1 and 11. Histamine was administered at increasing doses, 0.1, 0.3, 1, 3, 10 and 30 µg·kg−1, until a strong positive response was seen (∼80% increase in RL and/or ∼50% decrease in dynamic compliance [CDYN], or until the highest histamine dose was administered). PC30 was determined from each histamine dose–response curve.
On days 9 and 10 animals were challenged with antigen via intermittent positive pressure breathing with a ventilator and an in-line antigen nebulizer delivering an individualized dose that historically elicited at least a 40% increase in RL and a 35% decrease in CDYN. The two antigen challenges caused AHR, as measured by a fall in PC30 at day 11 compared with day 1, in 21 of 27 animals. These 21 animals and 1 other were selected to enter phase II. BAL eosinophilia was also observed following antigen challenge. Following an 8-day rest phase II was run exactly as phase I except that all animals received an intravenous infusion of 30 mg·kg−1 CAT-354 in a volume of 3.2 mL·kg−1 over 20 min on days 1, 5 and 9 (70 min prior to antigen challenge).
Animals were anaesthetized on days 1, 9, 10 and 11 of each phase using ketamine HCl tranquilization (approximately 10 mg·kg−1, IM, to effect) followed by propofol (approximately 0.2 mg·kg·min−1, as necessary). For the day 9 and 10 challenge procedures animals were intubated and mechanically ventilated. BAL was performed on days 1 and 11 by guiding a paediatric bronchoscope past the carina to wedge in a major bronchus and the lung washed with three 20 mL aliquots of sterile saline. Following completion of the study animals were not killed but were returned to the holding colony.
Data analysis
All data were analysed with Microsoft Excel before being curve fitted with GraphPad Prism using the four-parameter non-linear regression variable slope equation to derive EC50 and IC50 values. Summary results are generally expressed as the geometric mean IC50, EC50 or IC50 : ligand concentration ratio with 95% confidence intervals in brackets.
Materials
Recombinant bacterially derived cytokines were purchased; human and mouse IL-13 from Peprotech (London, UK) and human IL-1β and IL-4 from RnD Systems (Oxford, UK). Recombinant baculovirus derived, C-terminally FLAG-His10 labelled, cytokines were cloned and expressed in Sf9 cells in-house before being purified to homogeneity on Ni-NTA columns; namely WT human, human variant (in which the arginine at position 130 is replaced with glutamine, R130Q) and cynomolgus non-human primate IL-13.
Results
CAT-354 is a potent and selective neutralizer of IL-13
The potency and species selectivity of CAT-354 was assessed using the TF-1 proliferation and HUVEC VCAM-1 upregulation assays. Both TF-1s (a human pre-myeloid erythroleukemic cell line) and HUVECs respond to human, cynomolgus monkey and mouse IL-13. CAT-354 potently neutralized human and cynomolgus monkey IL-13 with similar potency (Figure 1A) but did not functionally neutralize mouse IL-13 (Figure 1B). In the TF-1 assay CAT-354 had an IC50 of 1.1 (0.7, 1.6) nM against 2 nM human IL-13 with an IC50 : ligand ratio of 0.55 (0.35, 0.80) (n= 6). Against 4 nM cynomolgus IL-13 CAT-354 had an IC50 of 1.7 (1.2, 2.5) nM and an IC50 : ligand ratio of 0.43 (0.30, 0.63) (n= 6). In the same TF-1 assay system we tested the ability of CAT-354 to functionally inhibit IL-13 R130Q, the variant form associated with atopy and increased incidence of asthma (Heinzmann et al., 2000; Liu et al., 2000; Kauppi et al., 2001). CAT-354 potently inhibited 2 nM IL-13 R130Q with an IC50 of 1.6 (1.3, 2.0) nM and an IC50 : ligand ratio of 0.80 (0.65, 1.00) (n= 6; Figure 1A).
Figure 1.
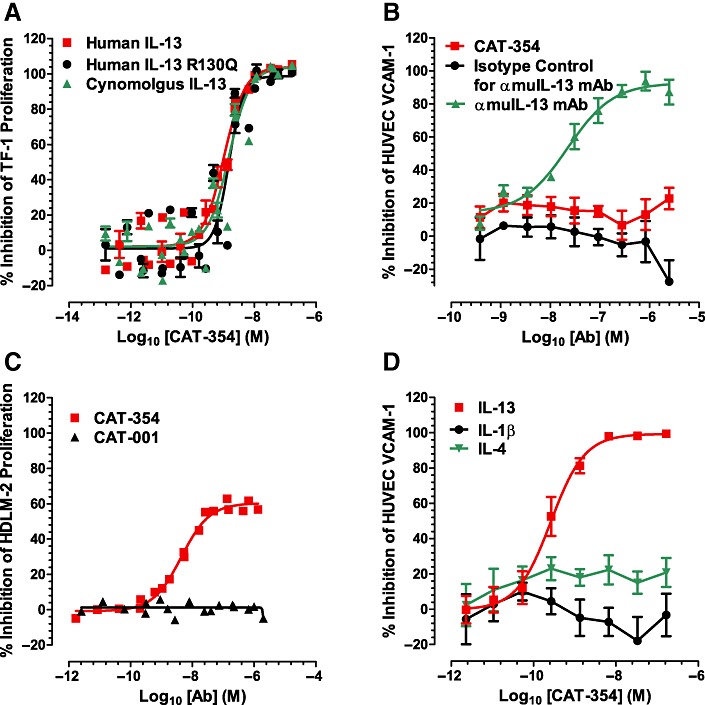
CAT-354 is a potent and selective neutralizer of IL-13. (A) Baculovirus-derived human, human R130Q and cynomolgus IL-13-induced TF-1 cell proliferation (n= 6). (B) Recombinant mouse IL-13-induced HUVEC VCAM-1 upregulation (n= 3). (C) Endogenous IL-13-driven HDLM-2 cell proliferation (n= 2). (D) Recombinant human IL-13, IL-4 and IL-1β-induced HUVEC VCAM-1 upregulation (n= 5, 3, 6 respectively). Data are shown as mean average points ± SEM from n independent experiments.
The Hodgkin's lymphoma-derived cell line HDLM-2 secretes IL-13 and is dependent upon it for growth (Skinnider et al., 2001). As it is critical for a therapeutic antibody to neutralize the endogenous target we tested the ability of CAT-354 to inhibit HDLM-2 proliferation. CAT-354, but not the isotype control antibody CAT-001, inhibited HDLM-2 proliferation with a 4.4 nM IC50 (n= 2; Figure 1C). The amount of IL-13 generated by the HDLM-2 cells was not measured so a mAb : ligand molar ratio is not reported.
A key feature of a therapeutic antibody is an ability to specifically neutralize the target without off-target effects. IL-4 is the most similar protein to IL-13 with 30% amino acid sequence identity (McKenzie et al., 1993). As shown in Figure 1D CAT-354 was not able to inhibit VCAM-1 upregulation induced by 160 pM IL-4 or 29 pM IL-1β but did inhibit that induced by 0.8 nM IL-13 with an IC50 of 368 (204, 664) pM and an IC50 : ligand ratio of 0.46 (0.26, 0.83) (n= 5).
CAT-354 inhibits key disease mechanisms in vitro
IL-13 is a pleiotropic Th2 cytokine which has been implicated in all the disease mechanisms considered important in asthma and we therefore tested the ability of CAT-354 to inhibit a variety of these key disease mechanisms.
Inflammatory cell recruitment and activation
Having established that CAT-354 prevented upregulation of the adhesion molecule VCAM-1 on endothelial cells we investigated chemokine generation and eosinophil recruitment. Normal human lung fibroblasts (NHLFs) produce eotaxin-1 in response to IL-13 stimulation (Terada et al., 2000). CAT-354, but not the isotype control antibody CAT-001, inhibited 0.8 nM IL-13-induced eotaxin-1 production with an IC50 of 267 (155, 459) pM and and IC50 : ligand ratio of 0.33 (0.19, 0.57) (n= 4; Figure 2A).
Figure 2.
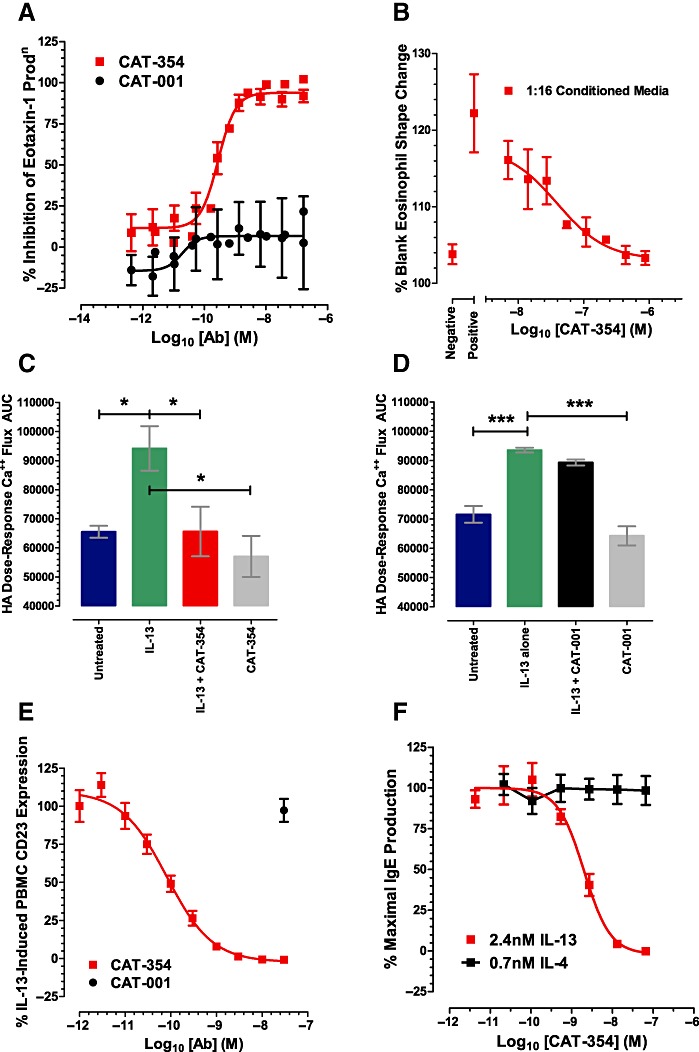
CAT-354 prevents IL-13-induced activation of a variety of cell types implicated in human asthma. Inflammatory cell recruitment and activation: (A) eotaxin-1 release from normal human lung fibroblasts (NHLF) (n= 4); (B) shape change in human eosinophils (n= 4). Activation of bronchial smooth muscle: (C and D) calcium flux in human bronchial smooth muscle cells (HBSM) (n = 3). Modulation of the IgE axis: (E) CD23 upregulation in human monocytes(n = 6); (F) isotype switching in B cells (n= 6). Data are shown as mean average points ± SEM from n independent experiments. In C and D differences in cell responses were compared using a one-way anova with Dunnett's multiple comparisons post-test. *P < 0.05; ***P < 0.001 significantly different from values with recombinant human IL-13.
Considering the important role eosinophils play in the asthmatic lung we decided to explore the potential consequences of IL-13 neutralization on eosinophil activation using a surrogate for chemotaxis, shape change. While IL-13 is not directly chemotactic it is found at high levels in the asthmatic lung (Saha et al., 2008) and can synergize with other asthma-relevant cytokines, such as TNFα (Terada et al., 2000) or TGF-β1 (Wenzel et al., 2002), to induce structural cells such as fibroblasts to synthesize eotaxin 1. Using factorial design we optimized the concentrations of TNFα, TGFβ1 and IL-13 to generate the most eotaxin-1 possible from NHLFs so enabling functional analysis.
Human eosinophil shape change assay
NHLFs stimulated for 48 h with 9.6 nM IL-13, 286 pM TNFα and 160 pM TGFβ1 secreted 9.6 nM eotaxin-1 into the culture media (conditioned media). In contrast NHLFs cultured with media alone secreted 0.1 nM eotaxin-1 into the culture media (control media). Dose-dependent inhibition of IL-13/TNFα/TGFβ1-stimulated eotaxin-1 production showed that the process was IL-13 dependent with an IC50 of 32.4 nM CAT-354 (data not shown).
Stimulated media was assessed for its leukocyte shape-changing properties as follows. A titration of conditioned media (no CAT-354) was performed using doubling dilutions. This experiment showed that shape change was proportional to media dilution and identified the linear range of the assay (data not shown). This led to testing the conditioned media at 1:16 (diluted in shape change buffer).
NHLF-conditioned media, but not control media, induced a clear eosinophil shape change of 122% versus 104% of buffer-alone levels, respectively (n= 4; Figure 2B). CAT-354 addition to costimulated media prior to NHLF culture resulted in a dose-dependent inhibition of eosinophil shape change with an IC50 of 14.0 (1.0, 199.5) nM and an IC50 : ligand ratio of 1.46 (0.10, 20.78) when assayed at 1:16 dilution (n= 4; Figure 2B).
The ability of stimulants (IL-13, TNFα, TGFβ1) alone to induce eosinophil shape change was also investigated; 9.6 nM IL-13, 286 pM TNFα and 160 pM TGFβ1 did not induce a clear eosinophil shape change compared with media alone (102% versus 98%, n= 3, data not shown) suggesting that the eosinophil shape change-inducing ability of the conditioned media develops during culture and is not due to IL-13, TNFα and TGFβ1 in combination.
Activation of bronchial smooth muscle
IL-13 has been shown to directly modulate the contractility of airway smooth muscle. This effect is mediated, in part, through potentiation of contractile agonist-induced calcium (Ca++) mobilization from intracellular stores which is a prerequisite to smooth muscle contraction (Tliba et al., 2003). Pretreatment of primary human bronchial smooth muscle cells with either 4 nM IL-13 or 3.6 nM IL-4 significantly enhanced Ca++ signalling responses of the cells to the contractile agonist histamine. Dose–response curves of peak fluorescence after addition of histamine were constructed and the area under the curve (AUC) calculated for each treatment condition. 67 nM CAT-354, but not 67 nM isotype control antibody CAT-001, significantly inhibited the potentiation of Ca++ signalling responses induced by IL-13 but not IL-4 (n= 3, P < 0.05, Figures 2C and D, and data not shown). To exclude the possibility that differences in cell growth were responsible for the effects of cytokines on Ca++-signalling cell viability analysis was performed and revealed no significant differences in cell numbers between the pre-histamine challenge conditions (data not shown).
Modulation of the IgE axis
Induction of CD23 expression on monocytes
IL-13 and IL-4 induce cell surface expression of the low affinity IgE receptor, CD23 on PBMCs (Punnonen et al., 1993 and Defrance et al., 1987, respectively); 80 pM IL-13 and 21.4 pM IL-4 induced equivalent CD23 expression on PBMCs, B cells and monocytes as judged by CD23/CD19 and CD23/CD14 co-staining respectively (data not shown). CAT-354 dose-dependently inhibited 80 pM IL-13, but not 21 pM IL-4, induced CD23 expression on PBMCs with an IC50 of 120.2 (92.9, 155.4) pM and an IC50 : ligand ratio of 1.50 (1.16, 1.94) (n= 6, Figure 2E, and data not shown). 30 nM of the isotype control antibody CAT-001 did not inhibit either IL-13 or IL-4-induced PBMC CD23 expression (n= 6, Figure 2E and data not shown). Co-stimulation of PBMCs with IL-13 and IL-4 produced an additive CD23 response (data not shown). 30 nM CAT-354, but not 30 nM CAT-001, reduced CD23 expression levels to that of IL-4 stimulation alone (data not shown).
B cell isotype switching
IL-13 and IL-4 have both been shown to be switch factors for IgE production (Punnonen et al., 1993 and Pène et al., 1988 respectively). The therapeutic effect of omalizumab (αIgE) clearly demonstrates the relevance of IgE in asthma. B cells exhibited a dose-dependent IgE response induced by IL-13 (EC50= 0.32 (0.19, 0.56) nM) and IL-4 (EC50= 0.45 (0.38, 0.54) nM) but not IL-10 (data not shown) (all n = 6). CAT-354 was able to neutralize the response induced by 2.4 nM IL-13, but not 0.7 nM IL-4, in a dose-dependent fashion with an IC50 of 1.8 (1.4, 2.5) nM and an IC50 : ligand ratio of 0.75 (0.58, 1.04) (n= 6, Figure 2F). Treatment with the isotype control antibody CAT-001 had no effect on IL-13 or IL-4-induced IgE production (data not shown).
CAT-354 inhibits key disease mechanisms in vivo
As shown above CAT-354 is a potent, neutralizing, human- and cynomolgus-IL-13 specific, human monoclonal IgG4 antibody. The high specificity inherent in biological agents such as CAT-354 makes preclinical pharmacology and support for the clinic challenging. To address this we followed three approaches: use of recombinant human IL-13 to activate mouse IL-13 receptors and allergen challenge models in both humanized mice and cynomolgus monkeys.
Effect of CAT-354 on human IL-13-induced inflammation
As IL-13 plays a key role in promoting eosinophil recruitment to the lung through upregulation of the adhesion molecule VCAM-1 (Sironi et al., 1994) and release of the eosinophil chemoattractant eotaxin-1 from fibroblasts (Terada et al., 2000), smooth muscle cells (Moore et al., 2002) and epithelial cells (Matsukura et al., 2001) we decided to model this in a mouse air pouch model of inflammation. In this model a pouch is formed by subcutaneous injection of sterile air on the back of the mouse and inflammation induced by injection of 2 µg recombinant human IL-13 (rhuIL-13).
As shown in Figure 3 human IL-13 injected into the air pouch caused a significantly increased infiltration of total leukocytes (P < 0.001) and eosinophils (P < 0.001) at 24 h post-challenge compared with placebo. Locally administered (intrapouch) CAT-354 dose-dependently inhibited both total leukocyte and eosinophil infiltration into the air pouch caused by rhuIL-13. CAT-354 20 µg and 200 µg, but not 2 µg, significantly inhibited eosinophil influx (P < 0.01 and P < 0.001, respectively). CAT-354 200 µg also significantly inhibited total cell influx (P < 0.001). No inhibitory effects of the isotype control antibody CAT-251 were seen.
Figure 3.
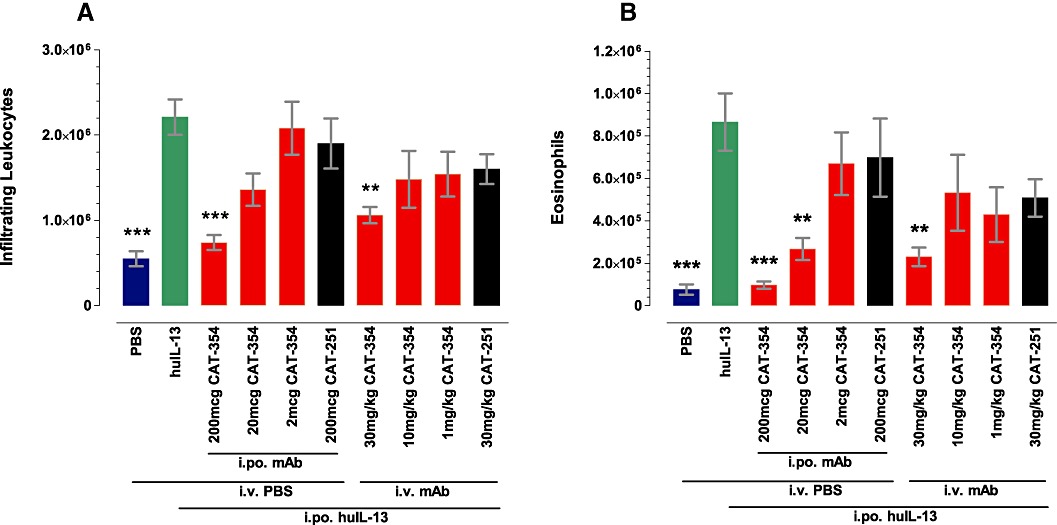
CAT-354 inhibits human IL-13-induced leukocytic inflammation in the mouse air pouch. (A) Total leukocytes and (B) eosinophils. Data are shown as mean number of infiltrating leukocytes ± SEM (n= 9–10 from two blocked experiments). **P < 0.01; ***P < 0.001, significantly different from recombinant human IL-13 (huIL-13) tested by one-way anova followed by Dunnett's multiple comparisons test.
Systemically administered (i.v.) CAT-354 also dose-dependently inhibited both total leukocyte and eosinophil infiltration into the air pouch caused by rhuIL-13. CAT-354 at 30 mg·kg−1, but not at 10 or 1 mg·kg−1, significantly inhibited eosinophil and total cell influx (both P < 0.01). No inhibitory effects of the control antibody, CAT-251, were seen.
Effect of CAT-354 in humanized mice
We generated a BALB/c knock-in mouse line where the human IL-13 genomic sequence replaced that of the mouse between the endogenous start and stop codons (huIL-13 KI mice). Appropriate targeting was ensured by sequencing the targeting vector and targeted locus as well as southern hybridization of targeted ES cells (data not shown). Homozygous mice were generated from two independent ES cell lines and found to be healthy, fertile and to express human, but not mouse, IL-13 mRNA and protein (data not shown). Their equivalence in terms of human IL-13 production and gross phenotype was confirmed in a previously reported model of ovalbumin sensitization and challenge (McMillan et al., 2002) (data not shown). A single BALB/c line, derived from the 5C11 ES clone, was then used in all subsequent experiments.
Ovalbumin sensitization and challenge, as outlined in Figure 4A, induced a clear allergic phenotype at day 25 characterized by eosinophilia (Figure 5E), AHR (Figure 5G), airflow limitation (Figure 5F) and increased BAL IL-13 (Figure 5H) when compared with mice who had not been sensitized but were challenged. Initial experiments using CAT-354 dosed daily during the challenge period (from days 18–24 inclusive, Figure 4B) demonstrated that 1, 3 or 10 mg·kg−1 i.p. CAT-354, but not 10 mg·kg−1 of the isotype control mAb CAT-251 (anti-doxorubicin human IgG4), abolished AHR (P < 0.001). Subsequent experiments giving CAT-354 every 3 days (i.p. on days 18, 21, 24) showed that 0.03 mg·kg−1 CAT-354 or 3 mg·kg−1 CAT-251 were ineffective but that 0.3 or 3 mg·kg−1 CAT-354 were as effective as twice-daily dexamathasone (1.5 mg·kg−1 s.c.) from days 19–24 and were able to inhibit AHR by approximately 50% (P < 0.05–0.01; Figure 4D).
Figure 4.
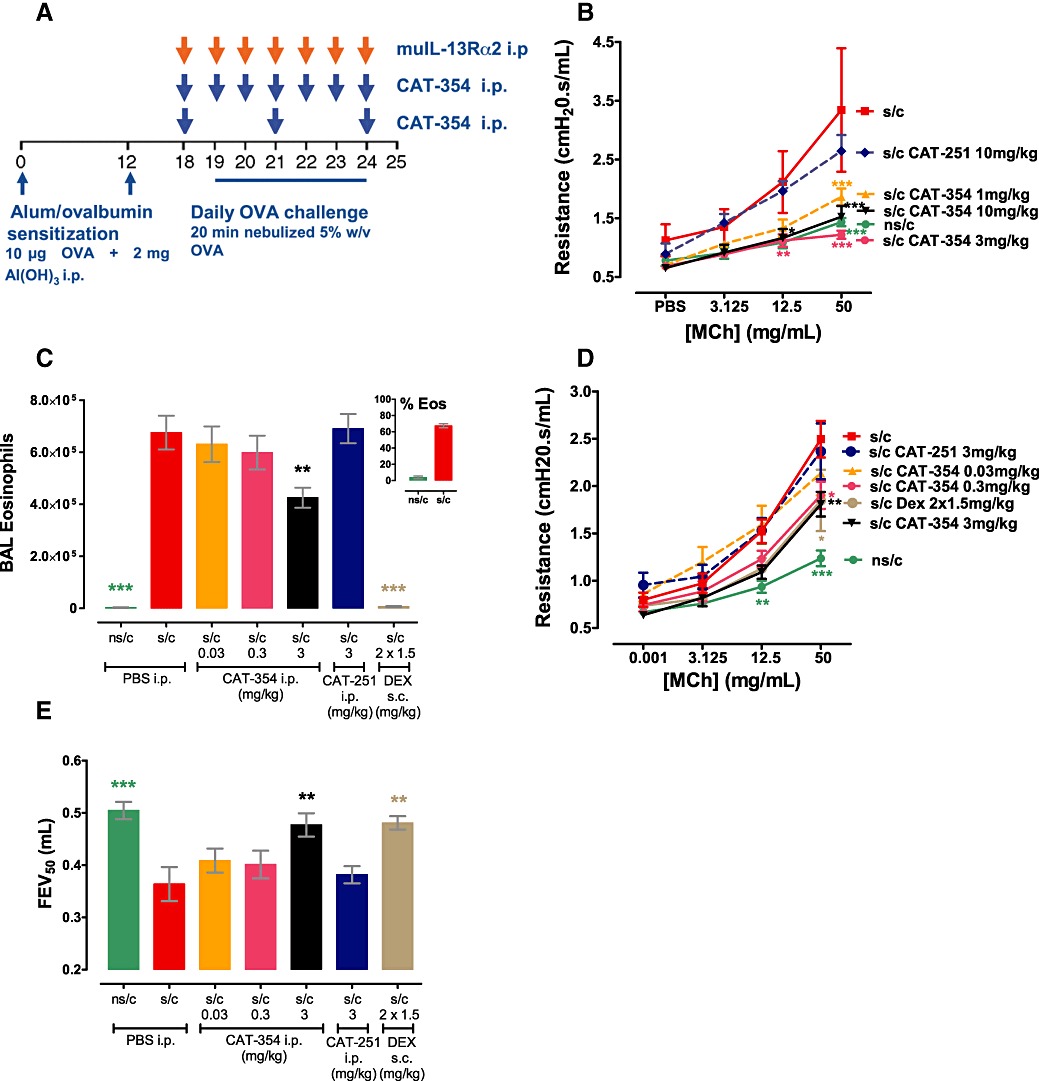
Effects of CAT-354 on the allergic phenotype in humanized BALB/c mice. (A) Model schematic. Daily mAb dosing. (B) Airway hyperresponsiveness (n= 4–8). 72h mAb dosing. (C) BAL eosinophilia with inset % eosinophils (n= 20–25); (D) Airway hyperresponsiveness (n= 14–18 except dex n= 8); (E) Forced expiratory volume (n= 6–8). Data are shown as mean average ± SEM from 1 (B) or 2 (C–E) blocked experiments. *P < 0.05; **P < 0.01; ***P < 0.001 versus s/c group using one-way (two-way for AHR) anova followed by Dunnett's (Bonferroni's for AHR) multiple comparisons test using s/c as the control group.
Figure 5.
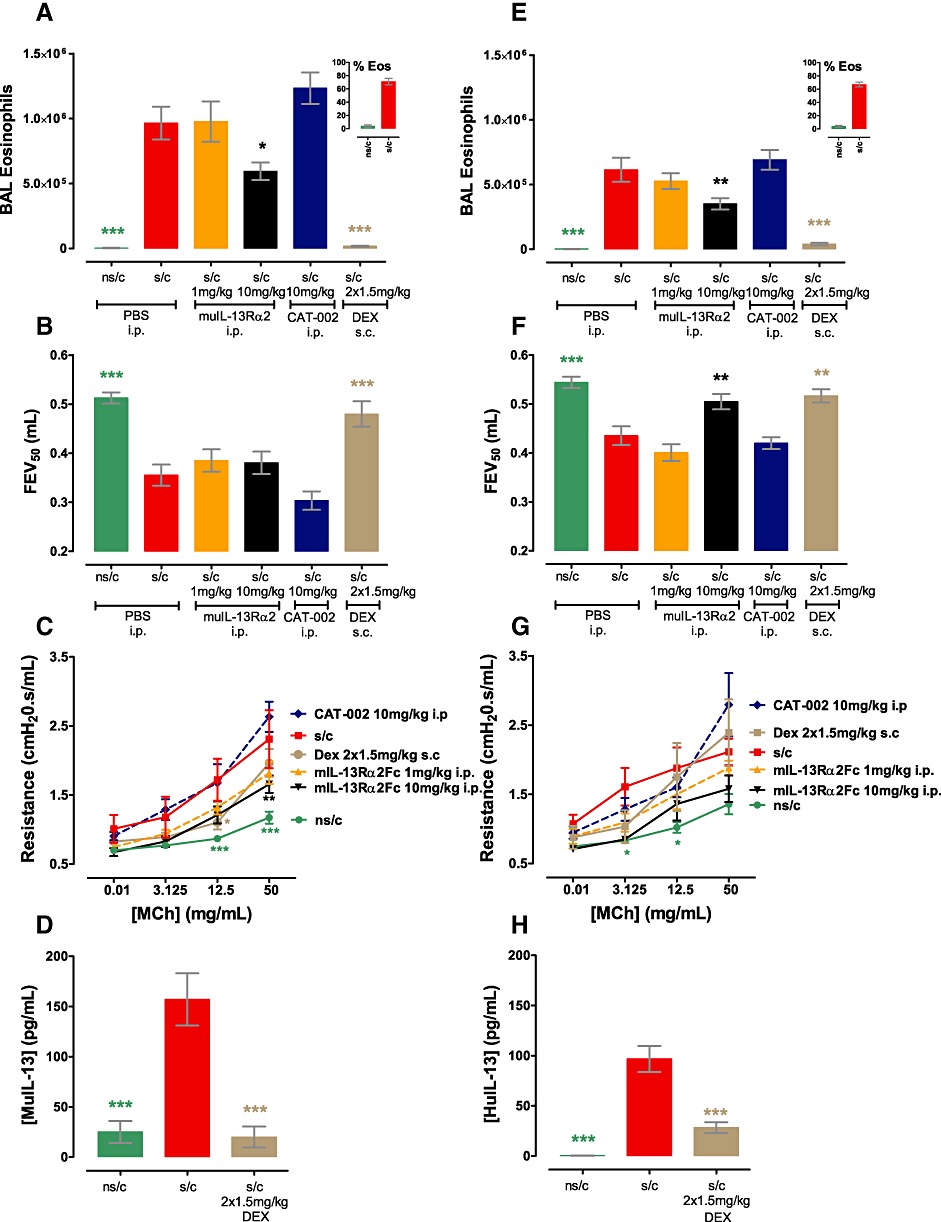
Differential effects of modulating the IL-13 axis on the allergic phenotype in WT BALB/c (A–D) and huIL-13 knock-in BALB/c (E-H) mice. (A and E) BAL eosinophilia with inset % eosinophils (n= 14–16); (B and F) FEV (n= 8); (C and G) AHR (n= 4–8); (D) BAL muIL-13 and (H) BAL huIL-13 (n= 15–16). Data are shown as mean average ± SEM from two blocked experiments. *P < 0.05; **P < 0.01; ***P < 0.001, significantly different from values in the sensitization and challenge (s/c) group; one-way (two-way for AHR) anova followed by Dunnett's (Bonferroni's for AHR) multiple comparisons test.
When given i.p. only 3 mg·kg−1 CAT-354, but not lower CAT-354 doses or 3 mg·kg−1 CAT-251, was able to inhibit BAL eosinophilia reducing it by 35% (Figures 4C, P < 0.01). In contrast twice daily 1.5 mg·kg−1 s.c. dexamethasone totally prevented eosinophilia (P < 0.001). Ovalbumin sensitization and challenge also induced a clear airflow limitation as measured by a decreased FEV50 (forced expiratory volume in 50 ms, which is considered analogous to FEV1 in humans); 3 mg·kg−1 i.p. CAT-354 or twice-daily 1.5 mg·kg−1 s.c. dexamethasone, but not lower CAT-354 doses or 3 mg·kg−1 i.p. CAT-251, reversed this airflow limitation (P < 0.01, Figure 4E).
Parallel studies using the mouse protein muIL-13Rα2-Fc, which functionally neutralizes both human and mouse IL-13, were undertaken in WT and huIL-13 KI mouse ovalbumin models to investigate whether humanization of IL-13 had altered the biology of the axis. Ovalbumin sensitization and challenge induced mouse IL-13 production in WT mice (Figure 5D) and production of human IL-13 in huIL-13KI mice (Figure 5H) which was significantly inhibited by twice daily s.c. 1.5 mg·kg−1 dexamethasone given from days 18 to 24 (P < 0.001 in WT and huIL-13KI). Ovalbumin sensitization and challenge also induced a similar degree of eosinophilia in both wild type (Figure 5A) and humanized mice (Figure 5E) which was significantly inhibited by 10 mg·kg−1 i.p. muIL-13Rα2-Fc (P < 0.05 WT, P < 0.01 KI), but not 10 mg·kg−1 i.p. of the isotype control mAb CAT-002, dosed daily from days 18 to 24; twice daily s.c. 1.5 mg·kg−1 dexamethasone abolished eosinophilia (P < 0.001 WT and KI). AHR was also induced to a similar extent in WT (Figure 5C) and KI (Figure 5G) mice which was significantly inhibited by daily 10 mg·kg−1 i.p. muIL-13Rα2 in WT mice (P < 0.01); there was a non-significant inhibition in the KI mice; 10 mg·kg−1 i.p. CAT-002 failed to affect AHR in either WT or huIL-13KI mice.
The key difference between WT and huIL-13KI mice came when examining airflow limitation. Ovalbumin sensitization and challenge induced a significant reduction in FEV50 compared with animals which had not been sensitized but were challenged in both WT and huIL-13KI mice (Figure 5B and F, respectively – both P < 0.001). In WT mice airflow limitation could not be reversed with 10 mg·kg−1 i.p. muIL-13Rα2 or 10 mg·kg−1 i.p. CAT-002 but only with twice daily s.c. 1.5 mg·kg−1 dexamethasone (P < 0.001, Figure 5B). In contrast the airflow limitation in huIL-13 KI mice was IL-13 sensitive as it was significantly inhibited by 10 mg·kg−1 i.p. muIL-13Rα2 (P < 0.01) or twice daily s.c. 1.5 mg·kg−1 dexamethasone (P < 0.01) but not 10 mg·kg−1 i.p. CAT-002 (Figure 5F).
Taken together this study confirmed that humanization of IL-13 had altered the biology of the IL-13 axis in mice. While the AHR and eosinophilia phenotypes were IL-13 dependent in both strains of mice, airflow limitation was dependent on IL-13 only in the humanized, but not WT, mice.
Cynomolgus monkey antigen challenge model
To further understand CAT-354 pharmacology we investigated the effect of three 30 mg·kg−1 doses of CAT-354 on the phenotype of a lung antigen challenge model using cynomolgus monkeys which were allergic to Ascaris suum, an intestinal parasite to which many cynomolgus monkeys are intrinsically atopic (Turner et al., 1996). In each of the two phases of the allergen challenge study cynomolgus monkeys were challenged on subsequent days with nebulized Ascaris suum which induces an AHR and lung eosinophilia (Figure 6A).
Figure 6.
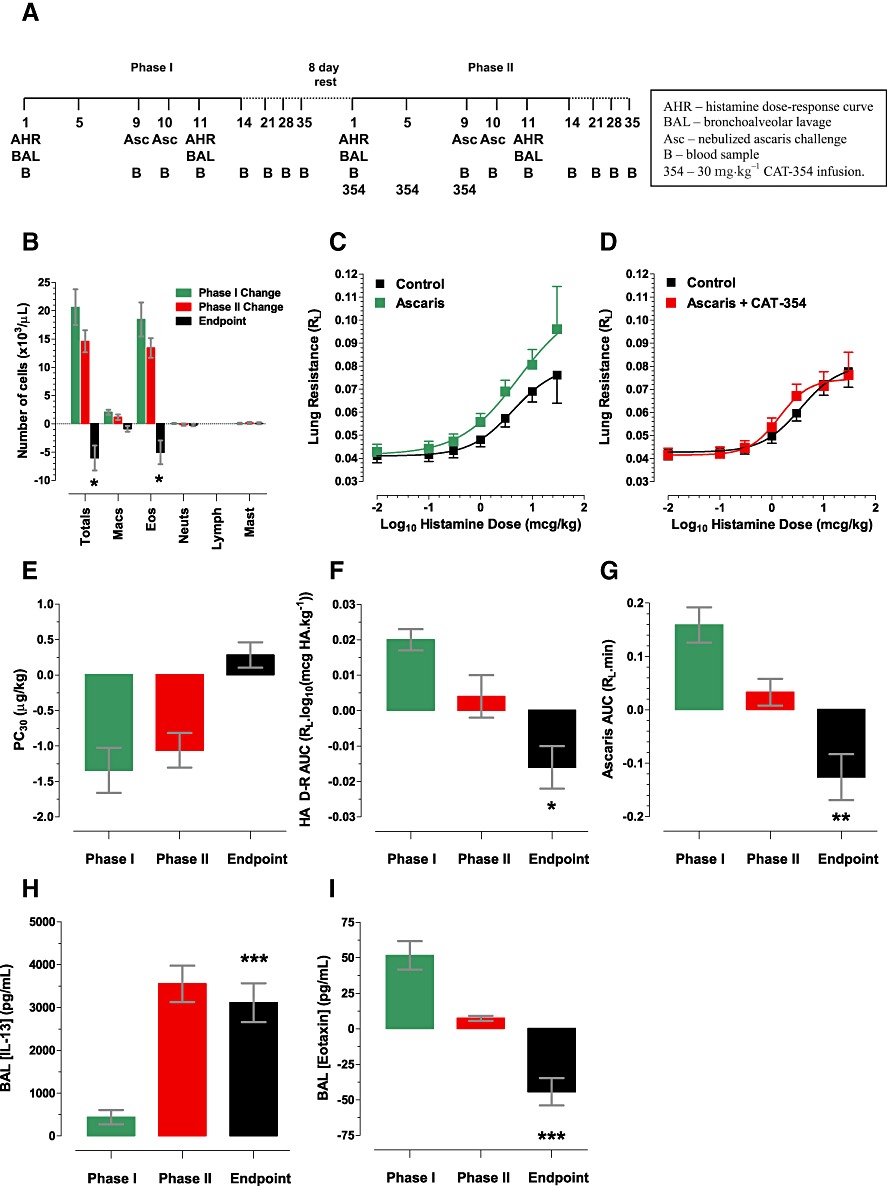
CAT-354 decreases AHR and eosinophilia in a cynomolgus model of antigen challenge. (A) Model schematic. (B) BAL inflammation (n= 21). (C) Histamine AHR in phase I (n= 18) and (D) Phase II (n= 18). (E) Histamine PC30 (n= 18). (F) Histamine AHR by RL AUC (n= 14). (G) Antigen priming (n= 20). (H) BAL IL-13 (n= 22). (I) BAL eotaxin (n= 22). Data displayed as the arithmetic mean ± SEM of individual animal changes at day 11 compared with day 1 for the phase I and II changes, respectively and phase II compared with phase I in the endpoint column. Statistical testing was performed only on the endpoint and was a two-way Student's t-test with the null hypothesis that the endpoint contained the number zero. *P < 0.05; **P < 0.01; ***P < 0.001.
This study was of serial design rather than a more usual parallel design. This was considered appropriate as cynomolgus monkeys are an outbred population with a natural genetic diversity and hence a potentially heterogeneous model response. Hence the most appropriate measure of change is not across groups but between individuals over time; the change induced by allergen challenge in the control phase (phase I) compared with change induced after allergen challenge in the treatment phase (phase II). Historical data suggested that AHR, BAL eosinophilia and antigen priming (observed phenomenon in which the bronchoconstrictive response to antigen is greater following the second, day 10, challenge as compared with the first, day 9, challenge and shown by the increased area under the day 10 versus day 9 pre- and post-challenge RL-time curve) were unaltered between two serial phases of a study (data not shown) giving confidence that any changes seen would be due to the agent tested and not time-based alterations in phenotype. As this was a proof of concept study a high dose level of CAT-354 was chosen to ensure target saturation at the supposed site of action, the lung.
The difference between the phases was examined in the ‘endpoint’ for BAL inflammation, antigen priming and histamine-induced AHR. The ‘endpoint’ was the difference between the performance of an individual animal in phase II compared with phase I (endpoint = change in phase II – change in phase I; a negative value would mean an inhibition, a positive value a stimulation).
Double allergen challenge induced eosinophilia (Figure 6B) and AHR (Figure 6C) in phase I (the control phase). In Phase II (active phase) CAT-354 inhibited AHR (Figure 6D) as measured by PC30 (theoretical dose of intravenous histamine required to increase baseline lung resistance by 30%) but this did not reach statistical significance (Figure 6E). CAT-354 did significantly inhibit AHR as measured by RL AUC (area under the histamine challenge lung resistance – time curve) (Figure 6F, P < 0.05). These measures are not fully comparable because not every animal that showed a decrease in PC30 after double allergen challenge also showed an associated increase in histamine-induced RL AUC and vice versa. Hence it is likely that they represent different forms of, or mechanisms within, AHR and may result from differences between hypersensitivity (left shift of the histamine dose-response curve) and hyperreactivity (increased maxima, or slope, of the histamine dose-response curve). When taken together the data do suggest activity for CAT-354 against the AHR that develops in response to allergen challenge.
CAT-354 also significantly inhibited both antigen priming (Figure 6G, P < 0.01) and BAL inflammation (Figure 6B). CAT-354 significantly inhibited total cell (P < 0.05) and eosinophil (P < 0.05) but not macrophage, lymphocyte or mast cell influx into the BAL. CAT-354 also inhibited BAL eotaxin (Figure 6I, P < 0.001) but increased lung BAL IL-13 levels (Figure 6H, P < 0.001). This apparent increase likely represents ‘biologically inactive’ IL-13:CAT-354 complexes, because when BAL was concentrated, on average 3.5-fold, and profiled in a CAT-354-based elisa system no IL-13 could be detected (data not shown). Thus, taken together, in a cynomolgus lung allergen challenge model CAT-354 significantly improved both lung function and lung inflammation.
Discussion and conclusions
A growing body of preclinical and clinical evidence implicates IL-13 as a key player in the development and maintenance of the asthma phenotype. In the clinic evidence for the importance of IL-13 in asthma comes from the IL-13 neutralizing antibody IMA-638 which has been shown to inhibit the early and late airway responses to allergen challenge in mild atopic asthmatics (Gauvreau et al., 2011). Additional encouragement comes from studies examining the effect of IL-4Rα neutralization which mediates functional inhibition of both IL-4 and IL-13. Aerovant, an IL-4 mutein, has been shown to block both the late phase reaction to antigen challenge in mild atopic asthmatics (Wenzel et al., 2007) as well as exacerbations in moderate to severe asthmatics (blood eosinophils >0.35/mm3) precipitated by the withdrawal of long acting β-agonists and inhaled corticosteroids (Wenzel et al., 2010). AMG-317, an IL-4Rα blocking IgG2, showed trends towards improved FEV1, reduced use of β-agonists and decreased exacerbation incidence in moderate atopic asthmatics in the worst tertile of disease control (Corren et al., 2010). Taken together these data suggest both a definite role for IL-13 in human asthma and a likely requirement for a patient-selection strategy.
In this paper we present evidence that agrees with, and supplements, published results and provides support for the current programme of clinical testing to determine if CAT-354 may be developed as a novel therapeutic agent that, when added to current standard of care, may enhance asthma control in patients.
CAT-354 is a highly potent and specific IL-13 neutralizing antibody with a 165 pM affinity for human IL-13 that neutralized human and cynomolgus IL-13 with near equivalent potency but did not neutralize mouse IL-13 nor the most homologous cytokine to human IL-13, human IL-4. CAT-354 also neutralized the variant human IL-13, R130Q, which has been associated with atopy and asthma as well as endogenous (mammalian glycosylated) human IL-13 (Figure 1). To build on this basic package and provide enough confidence to advance CAT-354 to clinical development, we undertook a broad panel of in vitro (Figure 2) and in vivo (Figures 3, 4 and 6) studies encompassing the range of effects reported to be mediated by IL-13. In addition we worked with the Institute of Lung Health to understand disease expression showing that IL-13 was expressed not only by mild patients controlled by β-agonists alone (GINA1) but also those severe patients (GINA4-5) that are currently insufficiently controlled by standard of care (Saha et al., 2008).
At the outset of this project we considered the most important, asthma relevant, effect of IL-13 to be development and maintenance of AHR and airflow limitation suggested by observations including IL-13-induced modulation of airway smooth muscle contractility (Laporte et al., 2001; Grunstein et al., 2002), direct action of IL-13 on smooth muscle potentiating the effect of contractile agonists (Tliba et al., 2003; Deshpande et al., 2004) and decreased relaxation induced by β-agonists (Grunstein et al., 2002). Our data support these observations as CAT-354 prevented the potentiation of bronchial smooth muscle Ca++ signalling responses induced by IL-13 (Figure 2C). In addition CAT-354 in humanized mice and muIL-13Rα2-Fc in WT mice, dosed during antigen challenge, prevented the development of AHR (Figures 4B and 5C). Finally CAT-354 also prevented the recapitulation of AHR in atopic cynomolgus monkeys that underwent antigen challenge (Figure 6D–F). Taken together these data fully support published reports that IL-13 is a key driver of AHR.
The only new treatment of the last decade to help severe atopic asthmatics regain control of their disease is omalizumab (Xolair – Genentech/Novartis) which works by reducing the levels of serum IgE below a critical level of 10 IU·mL−1. Among many functions IL-13, like IL-4, is known to induce B cell proliferation and is a potent mediator of IgE synthesis by human B cells in vitro (Punnonen et al., 1993) in concert with CD40 cross-linking, which can be induced by anti-CD40 antibodies (Jabara et al., 1990). Although responses to IL-4 and IL-13 are similar the levels of proliferation and IgE produced in response to IL-13 are generally found to be several-fold lower than those induced by IL-4 (Punnonen and de Vries, 1994). As omalizumab offers clinical benefit to a proportion of eligible atopic asthmatics we were interested in the ability of CAT-354 to influence the IgE axis. Previous data showed that mast cell proliferation and FcεRI, the high affinity receptor for IgE, expression can be induced by IL-13 and mediate increased histamine release; both effects are inhibited by CAT-354 (Kaur et al., 2006). While we found that CAT-354 could prevent IL-13, but not IL-4, mediated IgE production from B cells (Figure 2F) and CD23 upregulation on PBMCs (Figure 2E) we did not see any clear evidence for total IgE modulation by CAT-354 in a cynomolgus monkey single allergen challenge model (data not shown). This is consistent with previous mouse data suggesting that IL-13, via IL-13Rα1, is involved in baseline IgE maintenance but not IgE responses to T cell-dependent antigens (Munitz et al., 2008). These data are also consistent with data from a single allergen challenge cynomolgus monkey model which showed that a single intravenous administration of 10 mg·kg−1 IL-13 neutralizing mAb did not affect total IgE immediately after challenge but did reduce Ascaris-specific IgE by 8 weeks post-challenge (Kasaian et al., 2008). Taken together these data suggest that CAT-354 is unlikely to dramatically reduce serum IgE levels in humans.
To understand whether functionally neutralizing IL-13 with CAT-354 had the potential to modulate the over-exuberant inflammatory response seen in asthma we examined a number of cellular mechanisms considered relevant for this disease phenotype in simple in vitro systems including eotaxin generation, eosinophil activation via eosinophil shape change and endothelial upregulation of adhesion molecules (Figures 1 and 2). While IL-13 is not directly chemotactic it is found at high levels in the lungs of severe asthmatics (Saha et al., 2008) and can synergize with other cytokines found in the asthmatic lung, such as TNF-α (Terada et al., 2000; Obase et al., 2001) or TGF-β1 (Chu et al., 2000; Wenzel et al., 2002) to induce structural cells such as fibroblasts to synthesize eosinophil chemoattractants such as eotaxin-1. We optimized the concentrations of TNF-α, TGF-β1 and IL-13 to generate the most eotaxin-1 possible to enable functional analysis and in so doing were able to demonstrate that using CAT-354 to neutralize IL-13 we could prevent eotaxin-1 production and also eosinophils undergoing the first stage of chemotaxis, shape change (Figure 2B). Taken together with CAT-354-mediated inhibition of IL-13-induced endothelial VCAM-1 upregulation (Figure 1D), IL-13-induced air pouch eosinophilia (Figure 3B) and intratracheally instilled IL-13-induced lung eosinophilia (Blanchard et al., 2005) this evidence was consistent with CAT-354 being likely to have an anti-eosinophilic effect in the lung.
There are mixed results about the role of IL-13 in driving airway eosinophilia in more complex in vivo models. In genetically modified mouse models transgenic lung IL-13-overexpression alone induces strong eosinophilia (Zhu et al., 1999) whereas IL-13-deficient mice mount a normal eosinophillic response to ovalbumin sensitization and challenge (OSAC) (Webb et al., 2000). Use of IL-13-neutralizing IL-13Rα2-Fc in the challenge phase of a mouse OSAC model strongly inhibited (Grunig et al., 1998) or failed to affect (Wills-Karp et al., 1998) BAL eosinophilia. The data from non-human primate models is also variable; one group report reductions of eosinophilia of 70–80% in a single Ascaris challenge model with different IL-13 neutralizing mAbs dosed once intravenously at 10 mg·kg−1 (Kasaian et al., 2008 and Bree et al., 2007 respectively). In contrast a second group found no effect on lung eosinophilia following a single Ascaris challenge (Martin et al., 2008).
We found that IL-13 was not a key driver of lung eosinophilia in any of the models tested; whether it was the weak effect of muIL-13Rα2-Fc (which functionally neutralizes both mouse and human IL-13) on BAL eosinophilia in the OSAC model in WT or IL-13 humanized mice (Figure 5A and E) or whether it was CAT-354 in the OSAC IL-13 humanized mouse model (Figure 4C) or in the cynomolgus monkey double Ascaris challenge model (Figure 6B). In each case IL-13 neutralization only decreased eosinophilia by approximately 30%. Taken together these data suggest that while IL-13 drives mechanisms mediating eosinophilic inflammation these mechanisms are not dominant in the multiple allergen challenge models we tested in vivo.
Early in the preclinical development of CAT-354 it became clear that mouse cross-reactivity was unachievable and that pharmacologically relevant species would be limited to man and cynomolgus monkey. A novel feature of this study is the route we chose to provide additional pharmacological data with CAT-354; generation of humanized mice. This was achieved by the targeted replacement of the mouse gene with the human gene, from start to stop codon, retaining a mouse promoter and pA tail. This was considered a rational approach as human IL-13 can drive mouse hybridoma B9 cell proliferation in a similar fashion to mouse IL-13 (Bouteiller et al., 1995). In addition human IL-13 has been shown to mimic the effects of mouse IL-13 in the mouse lung driving eosinophilia and AHR (Blanchard et al., 2005). Initial characterization provided the expected results of appropriate replacement of mouse with human IL-13 and a WT-approximate phenotype in the OSAC model of AHR, eosinophilia, airflow limitation and human (not mouse) BAL IL-13. Parallel ovalbumin sensitization and challenge studies using muIL-13Ra2-Fc, which neutralizes both human and mouse IL-13, in WT and humanized mice confirmed the validity of the humanized model (Figure 5). Consistent with previously published studies IL-13 neutralization mediated inhibition of AHR with weak effects on BAL eosinophilia (Wills-Karp et al., 1998) in both huIL-13KI and WT mice. However OSAC-induced airflow limitation was only IL-13 dependent in humanized, but not in WT, mice. Based on these data we believe that the inhibition of airflow limitation by either CAT-354 or muIL-13Ra2-Fc in the IL-13 humanized mice is an artefact and suggest that while humanization of gene loci to enable pharmacological characterization of clinical candidate drugs is a rational and viable approach significant care must be exercised in defining the biology of the resultant mice to avoid erroneous conclusions.
In summary we have characterized CAT-354, a potent and specific IL-13 neutralizing mAb, in mechanistic in vitro and complex in vivo model systems to demonstrate beneficial effects on AHR and eosinophilic inflammation. Taken together, with compelling disease expression data linking IL-13 to severe uncontrolled asthma, these data support investigation of CAT-354 in the clinic with the key aim of improving asthma control for those patients who remain uncontrolled despite standard of care.
Acknowledgments
We thank Tamara Baker, Ann Traher, Sue Pearce, Helen Brant, Tracey Myers, Karen Balch, Rob Orvis and Chris Traher for their expert technical advice and support in the generation of the mouse data and Margaret Blundy and the tissue culture team for the care and provision of cell cultures.
MedImmune generated CAT-354 and provided financial support for all aspects of this work.
Glossary
- AAALAC
Association for the Assessment and Accreditation of Laboratory Animal Care, International
- AHR
airways hyperresponsiveness
- BAL
bronchoalveolar lavage
- BSMC
bronchial smooth muscle cell
- CMC
carboxymethylcellulose
- HUVEC
human umbilical vein endothelial cell
- IACUC
Institutional Animal Care and Use Committee
- NHLF
normal human lung fibroblast
- PBMC
peripheral blood mononuclear cell
- RL
lung resistance, PC30, the intravenous histamine dose required to generate a 30% increase in lung resistance above baseline
Conflicts of interest
RDM1, PDM2, ESC1, DM2, FD2, ND1, AJD1, DJC1, JW1, CJ-C1, LC2, NB2, JP2, IKA1 are all1, or were2, employees of MedImmune Ltd which owns and is developing CAT-354.
ECM is an employee of Charles River Laboratories which provided contract research services to MedImmune.
RW is a statistical consultant contracted to MedImmune.
References
- Blanchard C, Mishra A, Saito-Akei H, Monk P, Anderson I, Rothenberg ME. Inhibition of human interleukin-13-induced respiratory and oesophageal inflammation by anti-human-interleukin-13 antibody (CAT-354) Clin Exp Allergy. 2005;35:1096–1103. doi: 10.1111/j.1365-2222.2005.02299.x. [DOI] [PubMed] [Google Scholar]
- Bouteiller CL, Astruc R, Minty A, Ferrara P, Lupker JH. Isolation of an IL-13-dependent subclone of the B9 cell line useful for the estimation of human IL-13 bioactivity. J Immunol Methods. 1995;181:29–36. doi: 10.1016/0022-1759(94)00323-o. [DOI] [PubMed] [Google Scholar]
- Bree A, Schlerman FJ, Wadanoli M, Tchistiakova L, Marquette K, Tan X-Y, et al. IL-13 blockade reduces lung inflammation after Ascaris suum challenge in cynomolgus monkeys. J Allergy Clin Immunol. 2007;119:1251–1257. doi: 10.1016/j.jaci.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Chu HW, Trudeau JB, Balzar S, Wenzel S. Peripheral blood and airway tissue expression of transforming growth factor beta by neutrophils in asthmatic subjects and normal control subjects. J Allergy Clin Immunol. 2000;106:1115–1123. doi: 10.1067/mai.2000.110556. [DOI] [PubMed] [Google Scholar]
- Corren J, Busse W, Meltzer EO, Mansfield L, Bensch G, Fahrenholz J, et al. A randomized, controlled, phase 2 study of AMG 317, an IL-4Rα antagonist, in patients with asthma. Am J Respir Crit Care Med. 2010;181:788–796. doi: 10.1164/rccm.200909-1448OC. [DOI] [PubMed] [Google Scholar]
- Defrance T, Aubry JP, Rousset F, Vanbervliet B, Bonnefoy JY, Arai N, et al. Human recombinant interleukin 4 induces Fc epsilon receptors (CD23) on normal human B lymphocytes. J Exp Med. 1987;165:1459–1467. doi: 10.1084/jem.165.6.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande DA, Dogan S, Walseth TF, Miller SM, Amrani Y, Panettieri RA, et al. Modulation of calcium signalling by IL-13 in human airway smooth muscle: role of CD38/cADPR pathway. Am J Respir Cell Mol Biol. 2004;31:36–42. doi: 10.1165/rcmb.2003-0313OC. [DOI] [PubMed] [Google Scholar]
- Gauvreau GM, Boulet LP, Cockcroft DW, Fitzgerald JM, Carlsten C, Davis BE, et al. The effects of IL-13 blockade on allergen-induced airway responses in mild atopic asthma. Am J Respir Crit Care Med. 2011;183:1007–1014. doi: 10.1164/rccm.201008-1210OC. [DOI] [PubMed] [Google Scholar]
- Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein MM, Hakonarson H, Leiter J, Chen M, Whelen R, Grunstein JS, et al. IL-13 dependent autocrine signalling mediates altered responsiveness of IgE-sensitised airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2002;282:L520–L528. doi: 10.1152/ajplung.00343.2001. [DOI] [PubMed] [Google Scholar]
- GINA Guidelines. GINA report, Global strategy for asthma management and prevention. 2010. http://www.ginasthma.org/pdf/GINA_report_2010.pdf.
- Heinzmann A, Mao XQ, Akaiwa M, Kreomer RT, Gao PS, Ohshima K, et al. Genetic variants of IL-13 signalling and human asthma and atopy. Hum Mol Genet. 2000;9:549–559. doi: 10.1093/hmg/9.4.549. [DOI] [PubMed] [Google Scholar]
- Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol. 2003;111:677–690. doi: 10.1067/mai.2003.1333. [DOI] [PubMed] [Google Scholar]
- Howard TD, Whittaker PA, Zaiman AL, Koppelman GH, Xu J, Hanley MT, et al. Identification and association of polymorphisms in the interleukin-13 gene with asthma and atopy in a Dutch population. Am J Respir Cell Mol Biol. 2001;25:377–384. doi: 10.1165/ajrcmb.25.3.4483. [DOI] [PubMed] [Google Scholar]
- Howard TD, Koppelman GH, Xu J, Zheng SL, Postma DS, Meyers DA, et al. Gene-gene interaction in asthma: IL4RA and IL13 in a Dutch population with asthma. Am J Hum Genet. 2002;70:230–236. doi: 10.1086/338242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SK, Xiao HQ, Kleine-Tebbe J, Paciotti G, Marsh DG, Lichtenstein LM, et al. IL-13 expression at the sites of allergen challenge in patients with asthma. J Immunol. 1995;155:2688–2694. [PubMed] [Google Scholar]
- Humbert M, Durham SR, Kimmitt P, Powell N, Assoufi B, Pfister R, et al. Elevated expression of messenger ribonucleic acid encoding IL-13 in the bronchial mucosa of atopic and nonatopic subjects with asthma. J Allergy Clin Immunol. 1997;99:657–665. doi: 10.1016/s0091-6749(97)70028-9. [DOI] [PubMed] [Google Scholar]
- Jabara HH, Fu SM, Geha RS, Vercelli D. CD40 and IgE: synergism between anti-CD40 monoclonal antibody and interleukin 4 in the induction of IgE synthesis by highly purified human B cells. J Exp Med. 1990;172:1861–1864. doi: 10.1084/jem.172.6.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasaian MT, Tan XY, Jin M, Fitz L, Marquette K, Wood N, et al. Interleukin-13 neutralization by two distinct receptor blocking mechanisms reduces immunoglobulin E responses and lung inflammation in cynomolgus monkeys. J Pharmacol Exp Ther. 2008;325:882–892. doi: 10.1124/jpet.108.136515. [DOI] [PubMed] [Google Scholar]
- Kauppi P, Lindblad-Toh K, Sevon P, Toivonen HT, Rioux JD, Villapakkam A, et al. A second-generation association study of the 5q31 cytokine gene cluster and the interleukin-4 receptor in asthma. Genomics. 2001;77:35–42. doi: 10.1006/geno.2001.6613. [DOI] [PubMed] [Google Scholar]
- Kaur D, Hollins F, Woodman L, Yang W, Monk P, May R, et al. Mast cells express IL-13Rα1: IL-13 promotes human lung mast cell proliferation and FcεRI expression. Allergy. 2006;61:1047–1053. doi: 10.1111/j.1398-9995.2006.01139.x. [DOI] [PubMed] [Google Scholar]
- Komai-Koma M, McKay A, Thomson L, McSharry C, Chalmers GW, Liew FY, et al. Immuno-regulatory cytokines in asthma: IL-15 and IL-13 in induced sputum. Clin Exp Allergy. 2001;31:1441–1448. doi: 10.1046/j.1365-2222.2001.01174.x. [DOI] [PubMed] [Google Scholar]
- Kotsimbos TC, Ernst P, Hamid QA. Interleukin-13 and interleukin-4 are coexpressed in atopic asthma. Proc Assoc Am Phys. 1996;108:368–373. [PubMed] [Google Scholar]
- Kroegel C, Julius P, Matthys H, Virchow JC, Jr, Luttmann W. Endobronchial secretion of interleukin-13 following local allergen challenge in atopic asthma: relationship to interleukin-4 and eosinophil counts. Eur Respir J. 1996;9:899–904. doi: 10.1183/09031936.96.09050899. [DOI] [PubMed] [Google Scholar]
- Laporte JC, Moore PE, Baraldo S, Jouvin M, Church TL, Shwartzman IN, et al. Direct effects of interleukin-13 on signalling pathways for physiological responses in cultured human airway smooth muscle cells. Am J Respir Crit Care Med. 2001;164:141–148. doi: 10.1164/ajrccm.164.1.2008060. [DOI] [PubMed] [Google Scholar]
- Liu X, Nickel R, Beyer K, Wahn U, Ehrlich E, Freidhoff LR, et al. An IL13 coding region variant is associated with a high total serum IgE level and atopic dermatitis in the German multicenter atopy study (MAS-90) J Allergy Clin Immunol. 2000;106:167–170. doi: 10.1067/mai.2000.107935. [DOI] [PubMed] [Google Scholar]
- McKenzie AN, Li X, Largaespada DA, Sato A, Kaneda A, Zurawski SM, et al. Structural comparison and chromosomal localization of the human and mouse IL-13 genes. J Immunol. 1993;150:5436–5444. [PubMed] [Google Scholar]
- McMillan SJ, Bishop B, Townsend MJ, McKenzie AN, Lloyd CM. The absence of interleukin 9 does not affect the development of allergen-induced pulmonary inflammation nor airway hyperreactivity. J Exp Med. 2002;195:51–57. doi: 10.1084/jem.20011732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PL, Fisher D, Glass W, O'Neil K, Das A, Martin EC, et al. Preclinical safety and pharmacology of an anti-human interleukin-13 monoclonal antibody in normal macaques and in macaques with allergic asthma. Int J Toxicol. 2008;27:351–358. doi: 10.1080/10915810802430509. [DOI] [PubMed] [Google Scholar]
- Matsukura S, Stellato C, Georas SN, Casolaro V, Plitt JR, Miura K, et al. Interleukin-13 upregulates eotaxin expression in airway epithelial cells by a STAT6-dependent mechanism. Am J Respir Cell Mol Biol. 2001;24:755–761. doi: 10.1165/ajrcmb.24.6.4351. [DOI] [PubMed] [Google Scholar]
- Moore PE, Church TL, Chism DD, Panettieri RA, Jr, Shore SA. IL-13 and IL-4 cause eotaxin release in human airway smooth muscle cells: a role for ERK. Am J Physiol Lung Cell Mol Physiol. 2002;282:L847–L853. doi: 10.1152/ajplung.00245.2001. [DOI] [PubMed] [Google Scholar]
- Munitz A, Brandt EB, Mingler M, Finkelman FD, Rothenberg ME. Distinct roles for IL-13 and IL-4 via IL-13 receptor α1 and the type II IL-4 receptor in asthma pathogenesis. Proc Natl Acad Sci U S A. 2008;105:7240–7245. doi: 10.1073/pnas.0802465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseer T, Minshall EM, Leung DY, Laberge S, Ernst P, Martin RJ, et al. Expression of IL-12 and IL-13 mRNA in asthma and their modulation in response to steroid therapy. Am J Respir Crit Care Med. 1997;155:845–851. doi: 10.1164/ajrccm.155.3.9117015. [DOI] [PubMed] [Google Scholar]
- Obase Y, Shimoda T, Mitsuta K, Matsuo N, Matsuse H, Kohno S. Correlation between airway hyperresponsiveness and airway inflammation in a young adult population: eosinophil, ECP, and cytokine levels in induced sputum. Ann Allergy Asthma Immunol. 2001;86:304–310. doi: 10.1016/S1081-1206(10)63303-0. [DOI] [PubMed] [Google Scholar]
- Pène J, Rousset F, Brière F, Chrétien I, Bonnefoy JY, Spits H, et al. IgE production by normal human lymphocytes is induced by interleukin 4 and suppressed by interferons gamma and alpha and prostaglandin E2. Proc Natl Acad Sci U S A. 1988;85:6880–6884. doi: 10.1073/pnas.85.18.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters SP, Ferguson G, Deniz Y, Reisner C. Uncontrolled asthma: a review of the prevalence, disease burden and options for treatment. Respir Med. 2006;100:1139–1151. doi: 10.1016/j.rmed.2006.03.031. [DOI] [PubMed] [Google Scholar]
- van der Pouw Kraan TC, van Veen A, Boeije LC, van Tuyl SA, de Groot ER, Stapel SO, et al. An IL-13 promoter polymorphism associated with increased risk of allergic asthma. Genes Immun. 1999;1:61–65. doi: 10.1038/sj.gene.6363630. [DOI] [PubMed] [Google Scholar]
- Punnonen J, de Vries JE. IL-13 induces proliferation, Ig isotype switching, and Ig synthesis by immature human fetal B cells. J Immunol. 1994;152:1094–1102. [PubMed] [Google Scholar]
- Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G, et al. Interleukin 13 induces interleukin -4 independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci U S A. 1993;90:3730–3734. doi: 10.1073/pnas.90.8.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabroe I, Hartnell A, Jopling LA, Bel S, Ponath PD, Pease JE, et al. Differential regulation of eosinophil chemokine signalling via CCR3 and non-CCR3 pathways. J Immunol. 1999;162:2946–2955. [PubMed] [Google Scholar]
- Saha SK, Berry MA, Parker D, Siddiqui S, Morgan A, May R, et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol. 2008;121:685–691. doi: 10.1016/j.jaci.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi M, Sciacca FL, Matteucci C, Conni M, Vecchi A, Bernasconi S, et al. Regulation of endothelial and mesothelial cell function by interleukin-13: selective induction of vascular cell adhesion molecule-1 and amplification of interleukin-6 production. Blood. 1994;84:1913–1921. [PubMed] [Google Scholar]
- Skinnider BF, Kapp U, Mak TW. Interleukin 13: a growth factor in Hodgkin lymphoma. Int Arch Allergy Immunol. 2001;126:267–276. doi: 10.1159/000049523. [DOI] [PubMed] [Google Scholar]
- Tamura K, Suzuki M, Arakawa H, Tokuyama K, Morikawa A. Linkage and association studies of STAT6 gene polymorphisms and allergic diseases. Int Arch Allergy Immunol. 2003;131:33–38. doi: 10.1159/000070432. [DOI] [PubMed] [Google Scholar]
- Terada N, Hamano N, Nomura T, Numata T, Hirai K, Nakajima T, et al. Interleukin-13 and tumour necrosis factor-alpha synergistically induce eotaxin production in human nasal fibroblasts. Clin Exp Allergy. 2000;30:348–355. doi: 10.1046/j.1365-2222.2000.00750.x. [DOI] [PubMed] [Google Scholar]
- Thom G, Cockroft AC, Buchanan AG, Candotti CJ, Cohen ES, Lowne D, et al. Probing a protein-protein interaction by in vitro evolution. Proc Natl Acad Sci U S A. 2006;103:7619–7624. doi: 10.1073/pnas.0602341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tliba O, Deshpande D, Chen H, van Beisien C, Kannan M, Panettieri RA, et al. IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br J Pharmacol. 2003;140:1159–1162. doi: 10.1038/sj.bjp.0705558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CR, Andresen CJ, Smith WB, Watson JW. Characterization of a primate model of asthma using anti-allergy/anti-asthma agents. Inflamm Res. 1996;45:239–245. doi: 10.1007/BF02259610. [DOI] [PubMed] [Google Scholar]
- Vladich FD, Brazille SM, Stern D, Peck ML, Ghittoni R, Vercelli D. IL-13 R130Q, a common variant associated with allergy and asthma, enhances effector mechanisms essential for human allergic inflammation. J Clin Invest. 2005;115:747–754. doi: 10.1172/JCI22818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeland E, Morel L, Achey K, Yui M, Longmate J. Speed congenics: a classic technique in the fast lane (relatively speaking) Immunol Today. 1997;10:472–477. doi: 10.1016/s0167-5699(97)01126-2. [DOI] [PubMed] [Google Scholar]
- Webb DC, McKenzie ANJ, Koskinen AML, Yang M, Mattes J, Foster PS. Integrated signals between IL-13, IL-4, and IL-5 regulate airways hyperreactivity. J Immunol. 2000;165:108–113. doi: 10.4049/jimmunol.165.1.108. [DOI] [PubMed] [Google Scholar]
- Wenzel SE, Trudeau JB, Barnes S, Zhou X, Cundall M, Wescott J, et al. TGF-β and IL-13 synergistivally increase eotaxin-1 production in human airway fibroblasts. J Immunol. 2002;169:4613–4619. doi: 10.4049/jimmunol.169.8.4613. [DOI] [PubMed] [Google Scholar]
- Wenzel S, Wilbraham D, Fuller R, Burmeister Getz E, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- Wenzel SE, Ind PW, Otulana BA, Bleecker ER, Kuna P, Yen YP. Inhaled pitrakinra, an IL-4/IL-13 antagonist, reduced exacerbations in patients with eosinophilic asthma. P3980, 714s, European Respiratory Society 2010 Meeting. 2010. http://www.ersnet.org/learning_resources_player/abstract_print_10/main_frameset.htm.
- Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]