

Abstract
BACKGROUND AND PURPOSE
Action potential (AP) recordings in ex vivo heart preparations constitute an important component of the preclinical cardiac safety assessment according to the ICH S7B guideline. Most AP measurement models are sensitive, predictive and informative but suffer from a low throughput. Here, effects of selected anti-arrhythmics (flecainide, quinidine, atenolol, sotalol, dofetilide, nifedipine, verapamil) on field/action potentials (FP/AP) of guinea pig and rabbit ventricular slices are presented and compared with data from established in vitro and in vivo models.
EXPERIMENTAL APPROACH
Data from measurements of membrane currents (hERG, INa), AP/FP (guinea pig and rabbit ventricular slices), AP (rabbit Purkinje fibre), haemodynamic/ECG parameters (conscious, telemetered dog) were collected, compared and correlated to complementary published data (focused literature search).
KEY RESULTS
The selected anti-arrhythmics, flecainide, quinidine, atenolol, sotalol, dofetilide, nifedipine and verapamil, influenced the shape of AP/FP of guinea pig and rabbit ventricular slices in a manner similar to that observed for rabbit PF. The findings obtained from slice preparations are in line with measurements of membrane currents in vitro, papillary muscle AP in vitro and haemodynamic/ECG parameters from conscious dogs in vivo, and were also corroborated by published data.
CONCLUSION AND IMPLICATIONS
FP and AP recordings from heart slices correlated well with established in vitro and in vivo models in terms of pharmacology and predictability. Heart slice preparations yield similar results as papillary muscle but offer enhanced throughput for mechanistic investigations and may substantially reduce the use of laboratory animals.
Keywords: action potential, field potential, conscious dog, QT interval, cardiac repolarization, hERG potassium current, hNav1.5 sodium current, ICH S7B, in vitro electrophysiological methods, papillary muscle, Purkinje fibre, rabbit Langendorff heart, heart slice
Introduction
Although action potential (AP) recordings in ex vivo heart preparations are not a requirement within the framework of the ICH S7B guideline for the cardiac safety of drugs (Anonymous, 2005), they still constitute an important component of the preclinical cardiac safety assessment of new chemical entities, because AP recordings provide the electrophysiologist with a wealth of information. While most AP measurement models (e.g. papillary muscle, Purkinje fibre, ventricular strips, etc.) are sensitive, predictive and informative, they usually suffer from a low throughput. One promising approach to overcome this limitation constitutes the synchronous recording of bioelectrical signals from multiple cardiac tissue slices, a technique that has been recently optimized (Bussek et al., 2009). Here, effects of selected class 1 to 4 anti-arrhythmics on field potential (FP) and AP of guinea pig and rabbit ventricular slices are presented and compared with established in vitro and in vivo models.
Tissue slices from brain, kidney, liver, lung and pancreas are well-established models for electrophysiological, biochemical and toxicological studies (Edwards et al., 1989; Parrish et al., 1995; Vickers and Fisher, 2004; Colbert, 2006). Compared with isolated cells (e.g. cardiomyocytes), tissue slices offer the advantages of preserved tissue structure, no enzymatic digestion, no selection of cells during the isolation procedure, no exposure to alien growth factors or serum and viability for considerable periods of time when maintained under appropriate conditions. While isolated cardiomyocytes allow numerous measurements in cells from a single heart, the lack of intercellular contacts or cell surface damage by enzymatic digestion may lead to erroneous conclusions about drug actions. On the other hand, pharmacological and electrophysiological experiments in Langendorff hearts, papillary muscle or Purkinje fibres provide evidence of drug action under conditions more or less close to in vivo physiology but are time-consuming and expensive, because only a limited number of drugs or drug concentrations can be tested in the heart of one animal. Heart slices combine the advantages of whole organ and isolated cells, because they exhibit intact tissue structure and cellular contacts, yet a large number of preparations can be obtained from a single heart. Furthermore, the slice thickness (350 µm) ensures maintained tissue oxygenation, which may become limiting in other in vitro heart preparations like papillary muscles (Barclay, 2005).
In this paper, we present integrated data on the electrophysiological effects of selected anti-arrhythmic drugs (flecainide, quinidine, atenolol, sotalol, dofetilide, nifedipine, verapamil) using complementary in vitro and in vivo approaches. The experimental techniques range from measurement of membrane currents (hERG, INa, ICa.L), to AP/FP in slices, Purkinje fibre APs and haemodynamic/ECG parameters in conscious, telemetered Beagle dogs and also include published data. The main result of this complementary approach is that FP/AP recordings from heart slices correlate well with established in vitro and in vivo models in terms of pharmacology and predictability. Heart slice preparations yield similar results as papillary muscles but offer enhanced throughput for mechanistic investigations and may substantially reduce the use of laboratory animals. This work has been presented previously at the annual meeting of the Safety Pharmacology Society (Sept. 2010, Boston, MA).
Methods
Drugs
Chemicals and drugs [flecainide acetate (Flecainid-Isis® tablets), quinidine hydrochloride monohydrate, atenolol, dofetilide, d,l-sotalol hydrochloride (Sotalol ratiopharm® tablets), nifedipine, verapamil hydrochloride] were provided by Bayer-Schering Pharma (Berlin, Germany) or obtained from commercial sources (Sigma-Aldrich, Taufkirchen, Germany; RBI, Natick, MA, USA; local pharmacy) and stored at room temperature or as appropriate. On each of the experimental days, drug solutions were freshly prepared using frozen stock solutions (in dimethyl sulfoxide) for in vitro studies. For in vivo studies (oral administration of gelatine capsules, 0.25 mL·kg−1), drugs were formulated in water (verapamil), aqueous Tylose MH300 (0.5%; atenolol, dofetilide), ethanol/polyethylene glycol 400 (PEG400, 10:90; quinidine), PEG400 (nifedipine) or as pulverized tablets in gelatine capsules (flecainide, d,l-sotalol). Solutions and formulations were stored at room temperature.
Drug/molecular target nomenclature (e.g. receptors, ion channels, etc.) follows and conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011).
In vitro electrophysiology studies: hERG K+ current
HEK 293 cells stably transfected with cDNA encoding the hERG (human ether-a-go-go-related gene) K+ channel (Zhou et al., 1998) were cultured as previously described (Himmel, 2007). The single-electrode whole-cell voltage clamp method was applied using an EPC-9 amplifier and TIDA 5 software (HEKA Elektronik, Lambrecht, Germany) at room temperature as previously reported (Himmel, 2007).
The clamp protocol consisted of stepping the command voltage to +20 mV (duration 1000 ms) followed by a hyperpolarizing step to −120 mV (500 ms) and a step back to the holding potential of −80 mV (cycle length 12 s). The inward tail current elicited by stepping from +20 to −120 mV was used to quantify hERG K+ current.
The standard extracellular solution was composed of (in mM) NaCl 146.0, KCl 4.0, CaCl2 2.0, MgCl2 2.0, HEPES 10.0 (pH 7.4; 300–310 mosmol·L−1?). The intracellular (electrode filling) solution was composed of (in mM) KCl 135.0, MgATP 2.0, HEPES 10.0, EGTA 10.0 (pH 7.4; 290–300 mosmol·L−1).
The concentration-dependence of effects was modelled with a standard four-parameter logistic equation: effect = min + (max / (1 + 10[(logIC50 − logX) × nH])), with minimal and maximal effects (min, max), half-maximal inhibitory drug concentration (IC50), drug concentration (X) and Hill slope (nH). Minimal and maximal effects were usually treated as constants (max = 100 and min = 0), and IC50 and nH as variables (GraphPad Prism 3, GraphPad Software Inc., La Jolla, CA, USA).
In vitro electrophysiology studies: hNav1.5 Na+ current
HEK 293 cells stably transfected with cDNA encoding the hNav1.5 Na+ channel were cultured under standard conditions (section 2.2; Himmel and Hoffmann, 2010). The clamp protocol (whole-cell voltage clamp, 22°C) consisted of stepping the command voltage to −120 mV (duration 500 ms) followed by a depolarizing step to −35 mV (20 ms) and a step back to the holding potential of −80 mV (cycle length 2 s). The peak inward current elicited by stepping from −120 to −35 mV was used to quantify hNav1.5 Na+ current. The bath solution consisted of (mM) NaCl 150, KCl 2, MgCl2 1, CaCl2 1.5, HEPES 10, glucose 5 (pH 7.4); the pipette solution contained (mM) KCl 35, CsF 105, EGTA 10, HEPES 10 (pH 7.4).
In vitro electrophysiology studies: rabbit Purkinje fibre AP
Purkinje fibres were carefully dissected from the ventricles of female rabbit hearts (strain: New Zealand White and Chinchilla; age 4–14 months; body weight 2–5 kg) and stored in high-K+ Tyrode solution at 37°C as previously described (Vormberge et al., 2006; Himmel, 2007). Purkinje fibres were mounted in a horizontal organ bath that was perfused in a non-circulating manner with oxygenated (95% O2, 5% CO2) standard recording Tyrode solution (3 mL·min−1; 37°C). The recording Tyrode solution was composed of (in mM) NaCl 127.0, KCl 3.0, CaCl2 1.8, MgCl2 0.5, NaH2PO4 0.36, NaHCO3 22.0, d(+)-glucose 5.5 (pH 7.4 ± 0.1 at 37 ± 1°C following equilibration with 95% O2, 5% CO2). The preparations were stimulated electrically by means of square wave pulses (duration: 2 ms; 50% above threshold) at a standard stimulation rate of 1 Hz (software ISO-2, MFK, Niedernhausen, Germany). Conventional borosilicate glass microelectrodes with tip resistances of 20–40 MΩ when filled with 3 M KCl were used in order to measure APs intracellularly with a microelectrode amplifier (model BA-1S, npi electronic, Tamm, Germany).
Each preparation was equilibrated in the bath for approximately 60 min. When the impalement was stable, the bath was perfused with drug solution at cumulatively increasing concentrations (30 min per concentration) followed by washout. The frequency-dependence of effects was assessed by varying the stimulation frequency (0.2, 1.0 and 2.5 Hz).
Digitally recorded (10 kHz) APs were analysed for resting membrane potential (RMP), AP amplitude (APA), maximal upstroke velocity (Vmax), plateau potential at 35 ms after the upstroke and AP duration at 50% and 90% of repolarization (APD50, APD90). Furthermore, the ratio of APD50 divided by APD90 was calculated in order to quantify the potential AP triangulation.
In vitro electrophysiology studies: guinea pig papillary muscle AP
Papillary muscles were dissected from the ventricles of female guinea pig hearts (strain: Dunkin Hartley; body weight 200–250 g). Papillary muscles were mounted in a horizontal organ bath that was perfused in a non-circulating manner with oxygenated (95% O2, 5% CO2) Tyrode solution (35°C). Conventional borosilicate glass microelectrodes (20–40 MΩ, 3 M KCl) were used in order to measure APs at 1 Hz (software ISO-2, MFK) with a microelectrode amplifier (model BA-1S, npi electronic).
Each preparation was equilibrated in the bath for approximately 60 min. When the impalement was stable, the bath was perfused with d,l-sotalol or quinidine at cumulatively increasing concentrations (30 min per concentration). The frequency-dependence of effects was assessed by varying the stimulation frequency (0.3, 1.0 and 3.0 Hz).
Digitally recorded APs were analysed for RMP, APA, Vmax and APD30, APD60, APD90.
In vitro electrophysiology studies: guinea pig and rabbit left ventricular slice AP and FP
Ventricular heart slices were obtained from male guinea pigs (about 300 g body weight; Charles River, Sulzfeld, Germany) and female rabbits (strain: Chinchilla; 2.0–2.5 kg body weight; Charles River) as described previously (Bussek et al., 2009). Briefly, animals were anaesthetized (guinea pig: 70% CO2, 30% O2; rabbit: ketamine/xylazine 50/12 mg·kg−1 i.m. and 10/5 mg·kg−1 i.v.), and their hearts were quickly removed and perfused on a Langendorff apparatus with oxygenated (5% CO2, 95% O2) Tyrode's solution (composition in mM: NaCl 126.7, NaH2PO4 0.4, NaHCO3 22, KCl 5.4, CaCl2 1.8, MgCl2 1.1, glucose 5, pH 7.4) for 1 min followed by a 1 min perfusion with high potassium (HK+) solution (composition in mM: NaCl 120, KCl 20, CaCl2 2, MgCl2 1, HEPES 10, glucose 10, pH 7.4) to inhibit electrical activity. Contractile activation was suppressed with 2,3-butanedione monoxime (BDM, 15 mM; Sellin and McArdle, 1994).
A tissue piece (10 × 4 mm) of the middle part of the left ventricle was glued directly to an agarose block with histoacryl tissue adhesive (Aesculap AG & Co. KG, Tuttlingen, Germany), with the tissue position corresponding to the slice direction. This procedure allows direct contact between the heart tissue and the superfusion solution in order to avoid the risk of inadequate oxygen supply. The block was then fixed to the cutting stage of a vibratome (Integraslice, Campden Instruments Ltd., Loughborough, UK). Vertical transmural slices (consisting of epi-, mid-myo- and endocardial layers) of 350 µm thickness were cut in cold (4°C) oxygenated HK+ solution containing 15 mM BDM. Cutting was done with a steel blade driven at a speed of 0.03 mm·s−1, amplitude of 1 mm and vibration frequency of 51 Hz. Freshly prepared slices were stored in oxygenated HK+ solution at room temperature, and fixed by a grid (‘slice holder’, SDH-27N/15, Harvard Apparatus, Holliston, MA).
To characterize the electrophysiological parameters of guinea pig and rabbit cardiac slices, we recorded extracellular FPs and intracellular APs in the mid-myocardium at the centre of the slices. Myocardial slices were transferred to a four-channel submerged type recording chamber [Lohmann Research Equipment (LRE), Castrop-Rauxel, Germany] and continuously superfused with oxygenated Tyrode's solution (2 mL·min−1) at 37°C. FPs were recorded simultaneously in up to four heart slices using the multiple slice evaluation system Synchroslice (LRE). Concentric bipolar stainless steel stimulation electrodes (LRE; diameter 1.5 mm), used in order to minimize effects of stimulation on neurotransmitter release, and tungsten platinum recording electrodes (Thomas Recording, Giessen, Germany) were advanced until contact with the slice surface using manually driven micromanipulators under visual control through a multiple CCD camera system. Data acquisition (sampling rate 10 kHz per channel, bandwidth 1 Hz–3 kHz), electrical stimulation (1 Hz) and application of drugs to the superfusion with an eight-channel Teflon valve system were controlled via automated software (SynchroHeart, LRE). The software was also used for offline analysis of single FP component amplitudes, slopes and latencies. In addition, the difference between the primary (positive or negative) and secondary (negative or positive) peak latencies, termed QNa, were analysed in order to detect changes in Na+ conductance.
Intracellular APs were recorded with conventional glass micropipettes (Bussek et al., 2009). Signals were accepted when the resting membrane potential was more negative than −75 mV. and the amplitude of the AP was larger than 115 mV. After 40 min under control conditions, drugs were cumulatively added (one concentration every 30 min) to the superfusion solution.
Cardiovascular function and ECG in conscious dogs in vivo
The animal care and experimental procedures were in accordance with the German Law on the Protection of Animals and were performed with the permission from the State Animal Welfare Committee. Furthermore, all dogs were examined before the experiments and found to be healthy. Beagle dogs of either sex (body weight: 11–20 kg, age: 2–8 years) were obtained from various commercial breeders. The dogs were identified by a tattooed ear number and a collar and housed in groups of two to three animals. Lights were on for 12 h per day from 6:00 h to 18:00 h, room temperature was 20–23°C and relative humidity 30–70%. The dogs were fed once daily with pelleted standard dog chow; drinking water was available ad libitum.
Briefly, under general anaesthesia, the dogs were implanted with a telemetry device [model TL11M2-D70-PCT; Data Science Inc. (DSI), St. Paul, MN] comprising a transmitter, a catheter and two electrodes. The body of the transmitter was inserted into a s.c. pouch made on the dog's flank. The catheter was passed s.c. to the femoral artery, inserted distal to the inguinal ligament and advanced approximately 20 cm upstream to measure blood pressure in the abdominal aorta. The two ECG electrodes were placed s.c. in a standard lead II configuration. After the implantation surgery, a post-operative recovery of at least 2 weeks was allowed.
For measurement of blood pressure and ECG, the dogs were separated into a cage equipped with two individual telemetry receivers (model RMC-1, DSI). The signals were captured using Ponemah P3 Plus software (V.4.10 including Dataquest OpenART, V.2.30, DSI). Collected data were averaged over a period of 15 min, and telemetry signals were analysed for systolic, diastolic and mean arterial blood pressure, heart rate and various ECG intervals (PQ, QRS, QT, QTc). QT intervals were corrected for heart rate according to Van de Water (QTcV; Van de Water et al., 1989).
Data collection began at least 90 min before administration of either of the test compounds. Following drug administration at 15 h 30 min, data were recorded overnight for a period of about 15 h. Drug effects were assessed as the changes versus pre-drug values as compared with those of the vehicle control group.
Determination of drug plasma concentrations in dogs
Blood samples for determination of drug plasma concentrations were taken from satellite animals of either sex without telemetry implant (n= 3–4 per group) via the jugular vein or cephalic vein into lithium-heparin-coated monovettes at 1, 3, 7 and 24 h post treatment. After centrifugation at ≤4°C, the plasma samples were stored at −20°C until analyis. Following protein precipitation with acetonitrile or acetonitrile/ammonium acetate including an appropriate internal standard, drug concentrations in plasma were determined by means of HPLC and tandem mass spectrometry detection. In the case of d,l-sotalol determination, plasma concentrations were measured after solid phase extraction using separation by HPLC and fluorescence detection. Lower limits of quantification were 0.25 µg·L−1 for flecainide, quinidine, dofetilide and verapamil; 0.5 µg·L−1 for nifedipine; 5 µg·L−1 for atenolol; and 12 µg·L−1 for d,l-sotalol.
Statistics
The data presented are rounded group mean values and corresponding SD of n experiments unless indicated otherwise. Statistical analysis was performed either as ordinary one-way or repeated-measures anova followed by Dunnett's multiple comparisons post hoc test or by applying an appropriate nonparametric test. Differences were considered statistically significant if P < 0.05. Statistical analysis and graphical presentation of data was done with GraphPad Prism (release 3) or Microsoft Excel.
Focused literature search
A literature search in PubMed (http://www.ncbi.nlm.nih.gov/pubmed/?) was conducted using the terms flecainide, quinidine, atenolol, sotalol, dofetilide, nifedipine, verapamil, anti-arrhythmic, hERG, calcium channel, sodium channel, AP, Purkinje fibre, papillary muscle, ECG, QT/QTc, QRS, plasma concentration either alone or in various combinations. Amongst the numerous retrieved articles, we focused on those presenting concentration–effect data in vitro and ex vivo, and dose-dependence of effects together with exposure data in vivo.
Results
Class 1 anti-arrhythmics flecainide and quinidine
As expected for class 1 anti-arrhythmics, both flecainide and quinidine were found to be inhibitors of the human cardiac sodium channel hNav1.5 with IC50 concentrations of 11 and 12 µM, respectively (Table 1, Figures 1B and 2B), and 20% inhibition at around 3 µM. At lower concentrations, however, quinidine and flecainide (IC50∼1.7 µM) also inhibited the hERG K+ channel (Table 1, Figures 1C and 2C) with an IC20 level at about 0.5 µM.
Table 1.
Effects of selected anti-arrhythmics on hERG K+ current, hNav1.5 Na+ current and cardiac L-type Ca++ current in vitro
| hERG K+ current | hNav1.5 Na+ current | L-type Ca++ current | |||
|---|---|---|---|---|---|
| Own IC50a (µM) | Literature IC50 (µM) | Own IC50a (µM) | Literature IC50 (µM) | Literature IC50 (µM) | |
| Flecainide | 1.7 | 0.7–3.9b | 11 | 2.5–7c | 20, 41, 63d |
| Quinidine | N/A | 0.41e | 12 | 20f | 10, 56g |
| Atenolol | >100 | >1000h | N/A | N/A | N/A |
| d,l-sotalol | 1200 | 69i | N/A | N/A | N/A |
| Dofetilide | 0.036 | 0.158j | N/A | N/A | N/A |
| Nifedipine | 315 | >50, 275k | N/A | N/A | 0.2–0.3l |
| Verapamil | 0.68 | 0.143m | N/A | N/A | 1.0n |
Largely unpublished IC50 values from the authors laboratory (d,l-sotalol IC50 from Vormberge et al., 2006); N/A, not available.
Estimated from Nitta et al., 1992; Ducroq et al., 2007.
Estimated from Ducroq et al., 2007.
Figure 1.
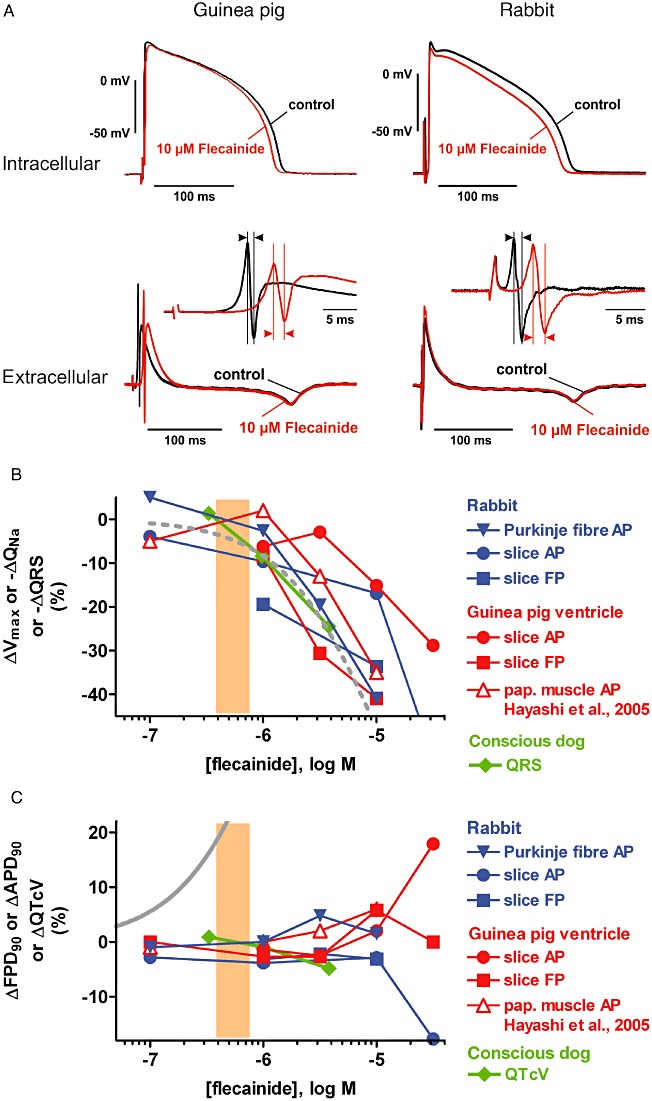
(A) Examples of intracellular (AP) and extracellular (FP) recordings from guinea pig (left) and rabbit (right) ventricular slices. Shown are superimposed tracings before (pre-drug control) and following flecainide exposure (10 µM). Insets: enlarged view of primary and secondary peak of the FP, the latency difference of which is a measure of Na+ conductance (QNa). (B) Concentration-dependence of effects of flecainide on ΔVmax, ΔQNa and ΔQRS in the following preparations: rabbit Purkinje fibre and ventricular slices, guinea pig ventricular slices and papillary muscle (data from Hayashi et al., 2005) and conscious dog. Also depicted is a concentration–response curve for inhibition of the hNav1.5 Na+ current (dashed grey line) and the range of therapeutically effective protein-unbound drug plasma concentrations in humans (orange area; data from Redfern et al., 2003). Please note that throughout all figures (i) solid symbols reflect data originating from the authors' laboratories, whereas data from the literature are depicted with unfilled symbols; and (ii) only mean values are depicted for clarity. (C) Concentration-dependence of effects of flecainide on ΔAPD90, ΔFPD90 and ΔQTcV in the following preparations: rabbit Purkinje fibre and ventricular slices, guinea pig ventricular slices and papillary muscle (data from Hayashi et al., 2005) and conscious dog. Also depicted is a concentration–response curve for inhibition of the hERG K+ current (solid grey line) and the range of therapeutically effective protein-unbound drug plasma concentrations in humans (orange area; data from Redfern et al., 2003).
Figure 2.
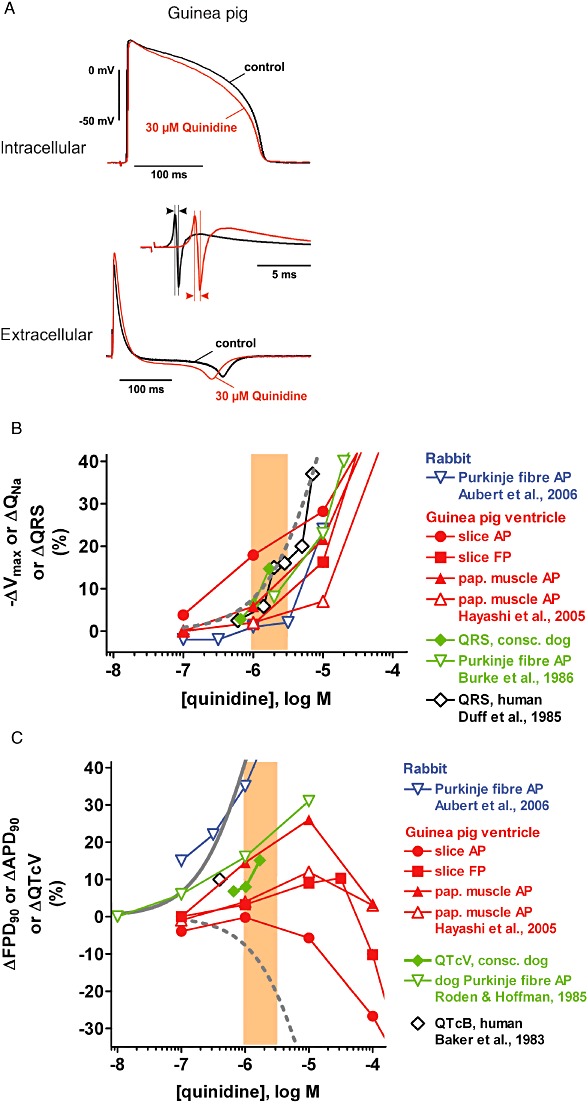
(A) Examples of intracellular (AP) and extracellular (FP) recordings from guinea pig ventricular slices. Shown are superimposed tracings before (pre-drug control) and following quinidine exposure (30 µM). Insets: enlarged view of primary and secondary peak of the FP, the latency difference of which is a measure of Na+ conductance (QNa). (B) Concentration-dependence of effects of quinidine on ΔVmax, ΔQNa and ΔQRS in the following preparations: rabbit Purkinje fibre (data from Aubert et al., 2006), guinea pig ventricular slices and papillary muscle (own data and data from Hayashi et al., 2005), conscious dog and dog Purkinje fibre (data from Burke et al., 1986) and human subjects (data from Duff et al., 1985). Also depicted is a concentration–response curve for inhibition of the hNav1.5 Na+ current (dashed grey line) and the range of therapeutically effective protein-unbound drug plasma concentrations in humans (orange area; data from Redfern et al., 2003). (C) Concentration-dependence of effects of quinidine on ΔAPD90, ΔFPD90 and ΔQTcV in the following preparations: rabbit Purkinje fibre (data from Aubert et al., 2006), guinea pig ventricular slices and papillary muscle (own data and data from Hayashi et al., 2005), conscious dog and dog Purkinje fibre (data from Roden and Hoffman, 1985) and human subjects (data from Baker et al., 1983). Also depicted are concentration–response curves for inhibition of the hERG K+ current (solid grey line) and the hNav1.5 Na+ current (dashed grey line), and the range of therapeutically effective protein-unbound drug plasma concentrations in humans (orange area; data from Redfern et al., 2003).
Because of this dual ion channel inhibition, flecainide and quinidine may interfere with both depolarization and repolarization, and hence the maximum rate of depolarization, Vmax, of the cardiac AP and its equivalent QNa in slices as well as APD and FPD are of particular interest. In guinea pig and rabbit ventricular slices (Table 3, Figures 1A,B and 2A,B), guinea pig papillary muscle (Table 2) and rabbit Purkinje fibre (Table 2, Figure 1B), Vmax was reduced and QNa was increased by both flecainide and quinidine with a concentration-dependence similar to that required for Na+ current inhibition. When recording ECGs in vivo, Na+ channel block manifests itself as slowing of conduction (i.e. widening of the QRS complex in the ECG). The corresponding effect was detected in conscious dogs (Table 4, Figures 1B and 2B), with a concentration-dependence and to an extent similar to those required for Na+ current inhibition.
Table 3.
Effects of selected anti-arrhythmics on AP and FP parameters from guinea pig (A) and rabbit (B) ventricular slice preparations in vitro§
| AP& | FP | |||||||
|---|---|---|---|---|---|---|---|---|
| Concentration (µM) | Vmax (V·s−1 or Δ%) | APD90 (ms or Δ%) | Triangulation (ms or Δ%) | −Vmax (V·s−1 or Δ%) | QNa (ms or Δ%) | FPD (ms or Δ%) | Triangulation (ms or Δ%) | |
| A: guinea pig | ||||||||
| Flecainide (NAP= 12/5, NFP= 8/2) | 0 | 162.7 | 180.2 | 36.8 | N/A | 0.9 | 190.8 | N/A |
| 1 | −6.2 | +1.4 | −0.3 | +8.3 | +2.7 | |||
| 3 | −2.9 | +2.5 | −0.5 | +30.6 | +2.7 | |||
| 10 | −15.1 | −2.0 | −3.3 | +41.0 | −5.8 | |||
| Quinidine (NAP= 6/5, NFP= 6/2) | 0 | 203.6 | 177.0 | 27.9 | N/A | 0.8 | 192.1 | N/A |
| 1 | −15.2 | +0.3 | +6.0 | +1.6 | +9.0 | |||
| 10 | −25.1 | −5.2 | +20.8 | +16.3 | +10.3 | |||
| 30 | +42.4 | −10.2 | ||||||
| 100 | −56.6 | −28.7 | +11.2 | +79.7 | −41.0 | |||
| Atenolol (NAP= 6/5, NFP= 6/2) | 0 | 222.6 | 169.3 | 26.3 | N/A | 0.8 | 197.8 | N/A |
| 1 | −14.7 | −11.3 | +5.0 | −1.2 | +1.9 | |||
| 10 | −21.7 | −15.9 | +21.8 | +7.8 | +6.0 | |||
| 50 | +3.3 | +5.2 | ||||||
| d,l-sotalol (NAP= 11/9, NFP= 9/2) | 0 | 173.6 | 187.5 | 40.3 | N/A | 0.8 | 193.1 | N/A |
| 1 | +2.2 | +3.6 | +1.6 | −2.3 | −0.7 | |||
| 3 | +0.7 | +5.3 | +3.6 | +5.9 | +3.5 | |||
| 10 | +3.5 | +12.1 | +14.3 | +4.9 | +12.0 | |||
| 30 | +3.4 | +18.2 | +28.4 | −6.2 | +22.7 | |||
| 100 | +12.5 | +19.3 | +48.7 | +25.0 | ||||
| Dofetilide (NAP= 7/4, NFP= 7/2) | 0 | 181.1 | 178.4 | 25.9 | N/A | 0.9 | 202.4 | N/A |
| 0.001 | +1.1 | +2.4 | +2.3 | +9.2 | ||||
| 0.01 | −0.4 | +13.1 | +17.3 | +10.2 | ||||
| 0.1 | −7.3 | +22.7 | +34.0 | −1.2 | +20.8 | |||
| 1 | −4.2 | +21.7 | +32.7 | +4.2 | +48.6 | |||
| Nifedipine (NAP= 8/4, NFP= 8/2) | 0 | 138.2 | 178.2 | 27.9 | N/A | 1.0 | 212.3 | N/A |
| 0.1 | −3.6 | −2.8 | −4.6 | −1.2 | −3.9 | |||
| 1 | −1.5 | −16.9 | −8.2 | +4.2 | −26.3 | |||
| 10 | +7.7 | −30.9 | −15.0 | +5.8 | −62.9 | |||
| Verapamil (NAP= 7/3, NFP= 7/2) | 0 | 220.4 | 173.2 | 25.9 | N/A | 0.9 | 193.9 | N/A |
| 0.1 | −15.3 | −3.5 | −1.3 | −5.3 | −5.6 | |||
| 1 | −6.3 | −8.7 | +2.1 | −17.3 | −6.9 | |||
| 10 | −7.0 | −23.1 | +11.7 | −11.1 | −22.4 | |||
| B: rabbit? | ||||||||
| Flecainide (NAP= 6/2, NFP= 4/2) | 0 | 170.9 | 182.0 | 52.3 | N/A | 0.9 | 183.9 | N/A |
| 0.1 | −3.9 | −2.8 | 0.2 | |||||
| 1 | −9.6 | −3.8 | 0.7 | 19.4 | −3.2 | |||
| 3 | 33.6 | −2.2 | ||||||
| 10 | −16.9 | −2.9 | 4.0 | 30.9 | −3.1 | |||
| 30 | −54.7 | −17.7 | −0.5 | |||||
| Dofetilide (NAP= 4/2, NFP= 4/2) | 0 | 139.0 | 215.8 | 46.4 | N/A | 0.7 | 192.6 | N/A |
| 0.001 | 0.0 | 6.5 | 2.5 | −5.0 | 4.1 | |||
| 0.01 | −6.6 | 45.1 | 40.8 | −5.3 | 18.4 | |||
| 0.1 | −5.8 | 201.3 | 88.6 | 2.3 | 23.1 | |||
| Nifedipine (NAP= 3/2, NFP= 4/2) | 0 | 141.7 | 215.7 | 36.3 | N/A | 0.9 | 211.7 | N/A |
| 0.1 | 11.7 | −4.1 | 4.9 | 5.1 | 0.4 | |||
| 1 | 3.1 | −9.3 | 7.0 | −3.4 | −14.4 | |||
| 10 | −1.7 | −19.6 | 42.5 | −0.7 | −43.4 | |||
Data are mean values from AP and FP measurements in guinea pig and rabbit ventricular slice preparations; NAP= i/j refers to the number of slices used for AP measurements with i slices from j animals (the same applies to FP measurements). Nomenclature analogous to Table 2; QNa, latency difference between primary and secondary peak of the FP as measure for Na+ conductance.
Pre-drug control resting membrane potential in guinea pig and rabbit ventricular slices. Guinea pig: flecainide, −82.4 ± 1.5 mV (mean ± SEM, n= 12); quinidine, −88.3 ± 2.5 mV (n= 6); atenolol, −87.2 ± 2.5 mV (n= 6); d,l-sotalol, −86.9 ± 1.3 mV (n= 11); dofetilide, −88.4 ± 1.1 mV (n= 7); nifedipine, −88.2 ± 2.9 mV (n= 8); verapamil, −87.4 ± 1.8 mV (n= 7). Rabbit: flecainide, −84.9 ± 1.4 mV (n= 6); dofetilide, −91.7 ± 1.3 mV (n= 5); nifedipine, −90.2 ± 2.8 mV (n= 3). No major changes (i.e. >±3 mV) of resting membrane potential occurred throughout the experiments.
Table 2.
Effects of selected anti-arrhythmics on rabbit cardiac Purkinje fibre and guinea pig papillary muscle AP parameters in vitro§
| Rabbit Purkinje fibre AP& | Guinea pig papillary muscle AP | |||||||
|---|---|---|---|---|---|---|---|---|
| Concentration (µM) | Vmax (V·s−1 or Δ%) | APD90 (ms or Δ%) | Triangulation (ms or Δ%) | −Vmax (V·s−1 or Δ%) | Vmax (V·s−1 or Δ%) | APD90 (ms or Δ%) | Triangulation (ms or Δ%) | |
| Flecainide (n= 4–6) | 0 | 393.0 | 445.3 | 124.0 | −0.783 | |||
| 0.1 | +5.0 | +1.0 | −2.9 | −2.1 | ||||
| 1 | −2.6 | 0.0 | +44.5 | −36.7 | ||||
| 3 | −19.6 | −4.8 | +164.0 | −65.7 | ||||
| 10 | −40.9 | −1.5 | +257.1 | −78.2 | ||||
| Quinidine (n= 6) | 0 | 153.2 | 158.8 | 58.7 | ||||
| 1 | −5.8 | +14.5 | +1.7 | |||||
| 10 | −21.6 | +26.0 | +21.0 | |||||
| 100 | −59.0 | +3.5 | +12.4 | |||||
| Atenolol (n= 5–6) | 0 | 259.4 | 475.7 | 135.5 | −0.682 | |||
| 1 | +17.3 | +0.1 | +4.1 | −6.3 | ||||
| 10 | +4.3 | −1.6 | +4.7 | −6.7 | ||||
| 100 | +8.5 | −1.5 | −0.8 | −3.3 | ||||
| d,l-sotalol (n= 5–6) | 0 | 273.4 | 331.8 | 116.2 | −0.775 | 170.9 | 163.3 | 62.8 |
| 1 | −5.8 | +11.5 | −4.7 | |||||
| 3 | −21.4 | +6.1 | −5.0 | −4.9 | ||||
| 10 | −23.4 | +41.8 | +15.5 | −18.2 | −7.8 | +24.9 | +1.0 | |
| 30 | +2.9 | +119.0 | +127.9 | −29.0 | −7.4 | +29.6 | +36.6 | |
| 100 | −8.0 | +45.5 | +44.9 | |||||
| Dofetilide (n= 3–6) | 0 | 194.8 | 415.8 | 145.9 | −0.772 | |||
| 0.3 | −7.5 | +12.1 | +16.7 | −12.2 | ||||
| 1 | −12.5 | +13.6 | +11.5 | −15.0 | ||||
| 3 | −24.3 | +66.5 | +81.1 | −42.7 | ||||
| 10 | −9.7 | +105.4 | +98.4 | −57.7 | ||||
| Nifedipine (n= 5–7) | 0 | 318.0 | 447.3 | 77.4 | −0.810 | |||
| 0.1 | +17.1 | −3.8 | −10.8 | −0.9 | ||||
| 1 | −2.3 | −8.9 | +19.8 | +8.5 | ||||
| 10 | +7.1 | −36.3 | +2.3 | +5.4 | ||||
| Verapamil (n= 4–6) | 0 | 305.8 | 397.3 | 80.3 | −0.689 | |||
| 0.1 | −1.7 | −2.1 | +12.8 | −13.3 | ||||
| 1 | +5.7 | +11.5 | +31.1 | −25.5 | ||||
| 10 | +10.0 | +61.7 | +236.7 | −61.7 | ||||
Data are mean values from AP measurements in rabbit Purkinje fibres and guinea pig papillary muscle; n refers to the number of preparations investigated (usually one preparation per animal). Pre-drug control values (concentration = 0) are absolute values in V·s−1 or ms, whereas all other values are expressed as % change versus pre-drug control (Δ%). Vmax, maximal depolarization velocity; triangulation, difference between APD90 and APD50 (Purkinje fibre) or APD90 and APD30 (papillary muscle); −Vmax, maximal repolarization velocity.
Pre-drug control resting membrane potential in rabbit Purkinje fibres: flecainide, −88.7 ± 0.6 mV (mean ± SEM, n= 6) (depolarization to −80.1 ± 3.4 mV at 10 µM); quinidine, N/A; atenolol, −89.4 ± 0.5 mV (n= 5); d,l-sotalol, −91.1 ± 1.4 mV (n= 6); dofetilide, −90.5 ± 0.5 mV (n= 6); nifedipine, −87.6 ± 0.4 mV (n= 9); verapamil, −89.4 ± 0.5 mV (n= 8). No major changes (i.e. >±3 mV) of resting membrane potential occurred throughout the experiments.
Table 4.
Effects of selected anti-arrhythmics on haemodynamic and ECG parameters in conscious, telemetry device-implanted Beagle dogs in vivo§
| BPS | BPD | HR | QRS | PQ | QT | QTcV | Cmax | Cmax.u | ||
|---|---|---|---|---|---|---|---|---|---|---|
| mmHg Δ% | mmHg Δ% | Bpm Δ% | ms Δ% | ms Δ% | ms Δ% | ms Δ% | µg·L−1 | µM | ||
| Flecainide (mg·kg−1) | BL | 129 | 76 | 76 | 45 | 114 | 234 | 253 | – | – |
| 3 | +1.1 | +3.9 | +13.0 | −1.4 | −2.8 | −2.9 | +0.9 | 238 | 0.33 | |
| 10 | −8.4 | −1.0 | +20.6 | +8.4 | +4.0 | −5.1 | −0.9 | 693 | 0.97 | |
| 30 | +6.9 | +23.7 | +49.3 | +24.5 | +15.5 | −8.7 | −4.8 | 2700 | 3.78 | |
| Quinidine (mg·kg−1) | BL | 127 | 75 | 78 | 46 | 116 | 234 | 253 | – | – |
| 5 | −14.4 | −15.9 | +24.9 | +2.8 | −9.9 | +4.1 | +6.8 | 1631 | 0.65 | |
| 15 | −15.5 | −13.8 | +39.3 | +6.4 | −10.8 | +2.7 | +8.0 | 2538 | 1.02 | |
| 45 | −21.5 | −18.4 | +58.4 | +14.7 | −11.7 | +2.6 | +15.1 | 4229 | 1.69 | |
| Atenolol (mg·kg−1) | BL | 129 | 75 | 76 | 49 | 117 | 237 | 254 | – | – |
| 0.3 | −7.3 | −9.3 | −5.9 | −0.4 | +10.2 | +0.4 | −1.3 | 144 | 0.52 | |
| 1.5 | −6.2 | −6.1 | −11.2 | +1.3 | +17.1 | +5.3 | +1.6 | 594 | 2.16 | |
| 5 | −2.5 | −5.1 | −12.6 | −1.5 | +18.1 | +1.7 | −2.0 | 2388 | 8.70 | |
| 15 | −6.9 | −8.6 | −10.1 | +3.8 | +17.1 | +4.2 | +0.9 | 8069 | 29.39 | |
| d,l-sotalol (mg·kg−1) | BL | 134 | 83 | 85 | 51 | 110 | 234 | 259 | – | – |
| 5 | −4.5 | −8.2 | −12.6 | +1.3 | +14.9 | +13.2 | +10.7 | 3073 | 11.28 | |
| 10 | −10.3 | −9.9 | +3.8 | +3.2 | +15.9 | +4.2 | +11.4 | 5533 | 20.31 | |
| 25 | −18.9 | −24.9 | −10.9 | +2.5 | +18.8 | +17.5 | +17.2 | 12767 | 46.87 | |
| 50 | −18.2 | −15.5 | +3.6 | +2.1 | +14.2 | +16.3 | +19.1 | 26302 | 96.56 | |
| Dofetilide (mg·kg−1) | BL | 133 | 78 | 88 | 48 | 108 | 227 | 255 | – | – |
| 0.01 | +0.1 | −3.1 | +1.4 | +1.7 | −2.5 | +4.9 | +4.6 | 1.55 | 0.0016 | |
| 0.03 | +9.8 | +4.9 | −13.5 | +2.0 | +3.2 | +13.6 | +8.6 | 4.24 | 0.0044 | |
| 0.1 | −2.9 | −2.1 | −6.7 | −0.3 | +11.6 | +16.5 | +12.8 | 16.90 | 0.0176 | |
| Nifedipine (mg·kg−1) | BL | 130 | 78 | 85 | 43 | 112 | 228 | 253 | – | – |
| 0.3 | −6.6 | −9.8 | −4.0 | +2.6 | +4.9 | +1.4 | +0.3 | 16 | 0.0002 | |
| 1 | −15.7 | −16.9 | +36.1 | +1.7 | −6.5 | −8.5 | −1.0 | 53 | 0.0006 | |
| 3 | −20.6 | −24.0 | +90.4 | −6.2 | −16.2 | −18.2 | −3.5 | 177 | 0.0020 | |
| Verapamil (mg·kg−1) | BL | 127 | 78 | 82 | 48 | 117 | 233 | 255 | – | – |
| 3 | −3.4 | −4.3 | +8.9 | +3.3 | +5.5 | −0.1 | +0.7 | 16 | 0.005 | |
| 10 | −4.4 | −2.6 | +17.4 | +1.7 | +20.8 | −3.7 | −1.3 | 149 | 0.049 | |
| 20 | −12.5 | −10.5 | +36.1 | −3.3 | +30.8 | −5.9 | +0.5 | 872 | 0.288 |
Data are mean values (n= 5–6 per dose) of the following haemodynamic and ECG parameters: BPD, diastolic blood pressure; BPS, systolic blood pressure; HR, heart rate; QTcV, QT interval corrected for heart rate according to Van de Water. Pre-drug baseline values (BL) are absolute values in mmHg or bpm or ms, whereas all other values are expressed as % change versus pre-drug baseline levels (Δ%). Maximal drug plasma concentrations (Cmax) were determined in satellite animals (n= 3–4 per dose) and corrected for protein binding (Cmax.u). The following data (MW, molecular weight; fu, protein-unbound fraction) were used to calculate Cmax.u values: flecainide: MW 414.3, fu 58% (human: Zordan et al., 1993); quinidine: MW 324.4, fu 13% (dog: Rakhit et al., 1984); atenolol: MW 266.3, fu 97% (human: Drug Information); d,l-sotalol: MW 272.4, fu 100% (dog: Schnelle and Garrett, 1973?); dofetilide: MW 441.6, fu 46% (dog: Smith et al., 1992); nifedipine: MW 346.3, fu 4% (dog: Bayer internal data); verapamil: MW 454.6, fu 15% (dog: Belpaire et al., 1989).
Although flecainide is a potent inhibitor of hERG (Table 1), it did not substantially alter APD90 and/or FPD90 in guinea pig and rabbit ventricular slices (Table 3, Figure 1C), guinea pig papillary muscle and rabbit Purkinje fibre (Table 2, Figure 1C), unless at concentrations ≥10 µM. Also, QTcV in conscious dogs in vivo remained essentially unchanged (Table 4, Figure 1C) at concentrations overlapping and exceeding the human therapeutic range. It should be pointed out, however, that flecainide dramatically altered the shape of APs, particularly in rabbit Purkinje fibres, where the maximal repolarization velocity was reduced and significant triangulation was observed (Table 2).
The results situation was more complex with quinidine, since there was considerable variation between types of tissue preparation and species. Concentration-dependent APD90 prolongation was only observed in rabbit Purkinje fibres (Lu et al., 2001; Aubert et al., 2006; Ducroq et al., 2007), at concentrations inhibiting hERG K+ current (Figure 2C). In guinea pig papillary muscle, APD90 was prolonged at ≤10 µM, while at ≥10 µM APD90 returned to baseline (Table 2, Figure 2C; Hayashi et al., 2005). In guinea pig ventricular slices, FPD90 was prolonged at ≤30 µM and shortened at higher concentrations, whereas APD90 was only shortened at ≥10 µM (Table 3, Figure 2C). In conscious dogs, the application of quinidine was associated with QTcV prolongation (Table 4, Figure 2C).
Class 2 anti-arrhythmic atenolol
The class 2 anti-arrhythmic drug atenolol lacks any relevant interactions with cardiac ion channels (e.g. the hERG K+ channel) (Table 1), and this was also reflected by the fact that atenolol hardly alters the shape of cardiac action or FPs. This was demonstrated in guinea pig ventricular slices (Figure 3, Table 3) and in rabbit Purkinje fibre (Table 2, Figure 3). In the former preparation, however, the application of atenolol was associated with a concentration-dependent shortening of the APD90 in the range of 5–15%. Finally, in vivo, atenolol prolonged the PQ interval (lowered heart rate) but was without effect on the duration of the QTc interval in conscious dogs (Table 4, Figure 3B) at concentrations overlapping and exceeding the human therapeutic range (Figure 3B).
Figure 3.
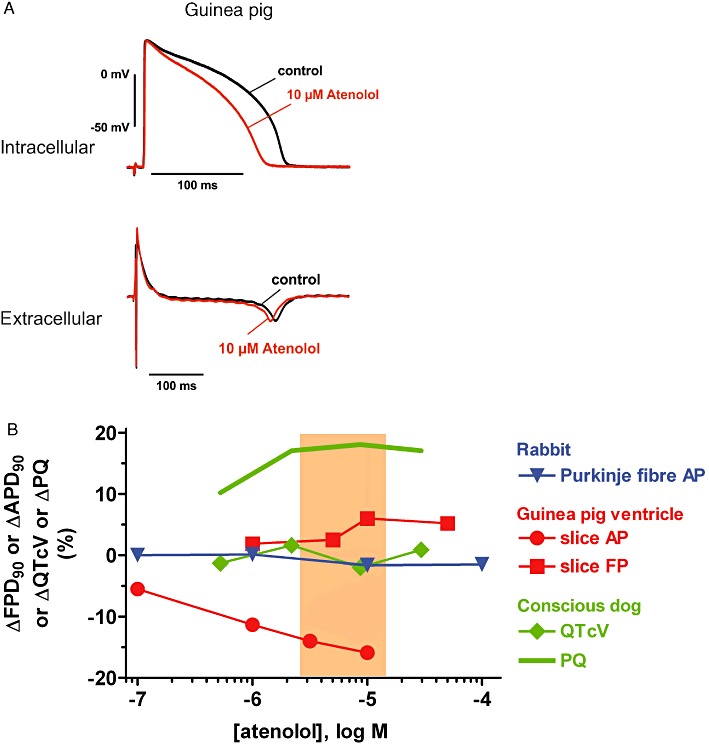
(A) Examples of intracellular (AP) and extracellular (FP) recordings from guinea pig ventricular slices. Shown are superimposed tracings before (pre-drug control) and following atenolol exposure (10 µM). (B) Concentration-dependence of effects of atenolol on ΔAPD90, ΔFPD90, ΔQTcV and ΔPQ in the following preparations: rabbit Purkinje fibre, guinea pig ventricular slices and conscious dog. Also depicted is the range of therapeutically effective protein-unbound drug plasma concentrations in humans (orange area; data from Redfern et al., 2003).
Class 3 anti-arrhythmics d,l -sotalol and dofetilide
The characteristic feature of class 3 anti-arrhythmics is their ability to delay cardiac repolarization as for instance by inhibition of the hERG K+ channel by d,l-sotalol and dofetilide, with IC20 concentrations of about 20 µM and 10 nM respectively (Table 1, Figures 4B and 5B). In guinea pig ventricular slice preparations, the effect of d,l-sotalol was characterized by a concentration-dependent drug-mediated prolongation of APD90 and FPD90, the extent of which was approximately 20% at 30 µM (Table 3, Figure 4). A similar concentration-dependence of APD90 prolongation was observed in guinea pig papillary muscle (Table 2; Figure 4B), whereas rabbit Purkinje fibres were more sensitive to d,l-sotalol-mediated APD prolongation (Table 2; Figure 4B). In vivo, d,l-sotalol prolonged the QT interval, as demonstrated in conscious and anaesthetized dogs at plasma concentrations, and to an extent that was similar to that observed in guinea pig ventricular slices and to hERG K+ current inhibition (Table 4, Figure 4B).
Figure 4.
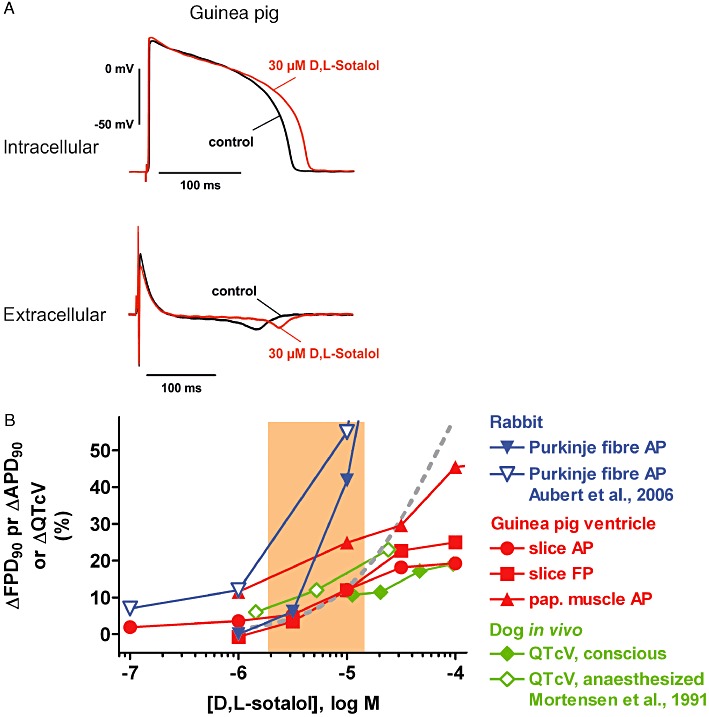
(A) Examples of intracellular (AP) and extracellular (FP) recordings from guinea pig ventricular slices. Shown are superimposed tracings before (pre-drug control) and following d,l-sotalol exposure (30 µM). (B) Concentration dependence of effects of d,l-sotalol on ΔAPD90, ΔFPD90 and ΔQTcV in the following preparations: rabbit Purkinje fibre (own data and data from Aubert et al., 2006), guinea pig ventricular slices and papillary muscle, conscious dog and anaesthetized dog (data from Mortensen et al., 1991). Also depicted is a concentration–response curve for inhibition of the hERG K+ current (dashed grey line; data from Ducroq et al., 2007) and the range of therapeutically effective protein-unbound drug plasma concentrations in humans (orange area; data from Redfern et al., 2003).
Figure 5.
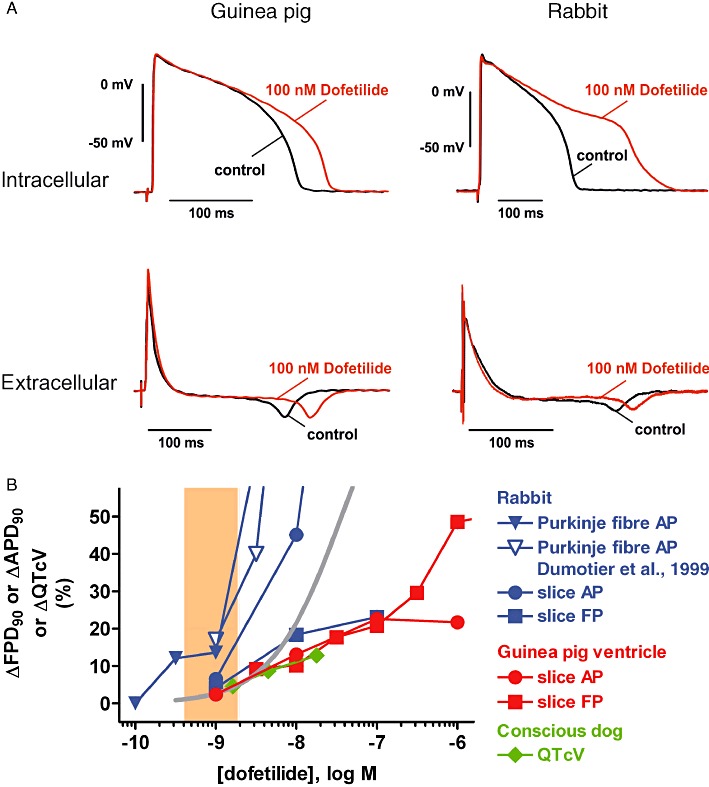
(A) Examples of intracellular (AP) and extracellular (FP) recordings from guinea pig (left) and rabbit (right) ventricular slices. Shown are superimposed tracings before (pre-drug control) and following dofetilide exposure (100 nM). (B) Concentration-dependence of effects of dofetilide on ΔAPD90, ΔFPD90 and ΔQTcV in the following preparations: rabbit Purkinje fibre (own data and data from Dumotier et al., 1999) and ventricular slices, guinea pig ventricular slices and conscious dog. Also depicted are a concentration–response curve for inhibition of the hERG K+ current (solid grey line) and the range of therapeutically effective protein-unbound drug plasma concentrations in humans (orange area; data from Redfern et al., 2003).
In a similar manner, the effect of dofetilide in both guinea pig and rabbit ventricular slice preparations was characterized by a concentration-dependent drug-mediated prolongation of APD90 and FPD90, the extent of which was approximately 20% at 10–30 nM (Table 3, Figure 5). Also here, rabbit Purkinje fibres were more sensitive to dofetilide-mediated APD or FPD prolongation (Table 2; Figure 5B). In vivo, dofetilide prolonged the QT interval in conscious dogs at plasma concentrations and to an extent that was similar to the findings in ventricular slice preparations and to hERG K+ current inhibition (Table 4, Figure 5B).
Class 4 anti-arrhythmics nifedipine and verapamil
The class 4 anti-arrhythmics nifedipine and verapamil inhibited L-type calcium channels at submicromolar concentrations (Table 1, Figures 6B and 7B). In addition, both drugs were inhibitors of the hERG K+ channel (Table 1), albeit with one important difference. Nifedipine had very low potency as an hERG blocker displaying an IC50 of about three orders of magnitude higher than that for calcium channel inhibition, whereas verapamil inhibited both hERG K+ and L-type calcium channel at almost identical concentrations (Table 1, Figures 6B and 7B).
Figure 6.
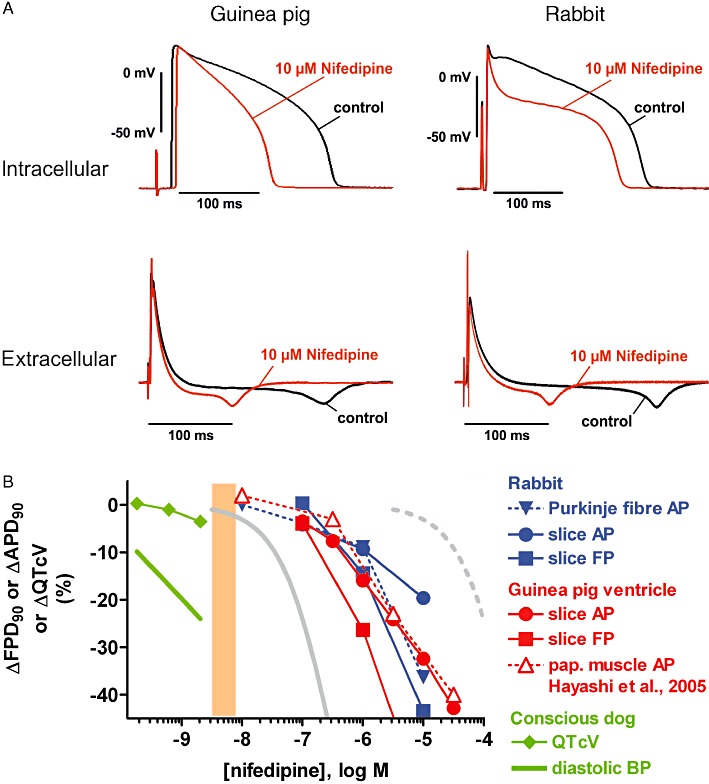
(A) Examples of intracellular (AP) and extracellular (FP) recordings from guinea pig (left) and rabbit (right) ventricular slices. Shown are superimposed tracings before (pre-drug control) and following nifedipine exposure (10 µM). (B) Concentration-dependence of effects of nifedipine on ΔAPD90, ΔFPD90 and ΔQTcV in the following preparations: rabbit Purkinje fibre and ventricular slices, guinea pig ventricular slices and papillary muscle (data from Hayashi et al., 2005) and conscious dog. Also depicted are concentration–response curves for inhibition of the hERG K+ current (dashed grey line) and the L-type Ca2+ current (solid grey line; from Uehara and Hume, 1985) and the range of therapeutically effective protein-unbound drug plasma concentrations in humans (orange area; data from Redfern et al., 2003). The decrease in diastolic blood pressure (BP) in conscious dogs is also shown.
Figure 7.
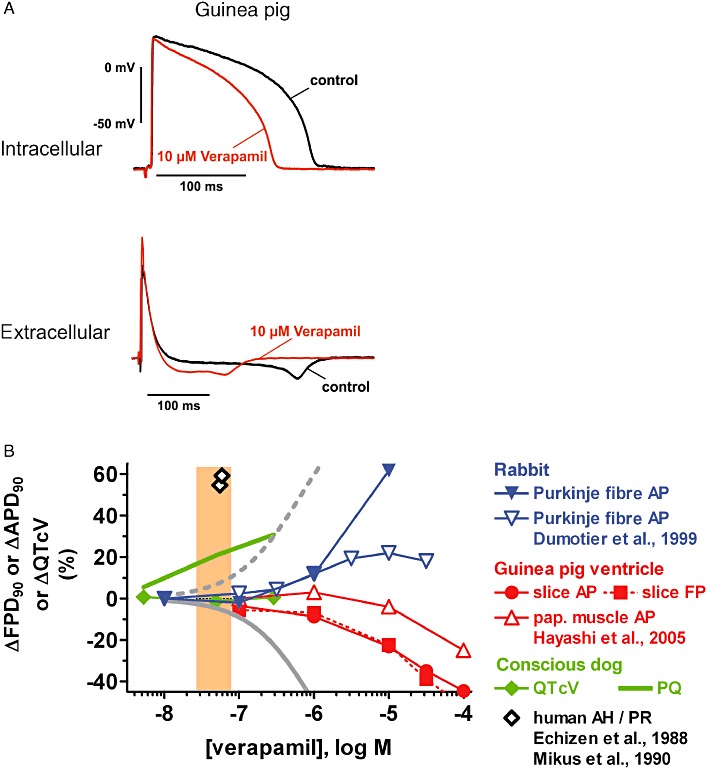
(A) Examples of intracellular (AP) and extracellular (FP) recordings from guinea pig ventricular slices. Shown are superimposed tracings before (pre-drug control) and following verapamil exposure (10 µM). (B) Concentration-dependence of effects of verapamil on ΔAPD90, ΔFPD90 and ΔQTcV in the following preparations: rabbit Purkinje fibre (own data and data from Dumotier et al., 1999), guinea pig ventricular slices and papillary muscle (data from Hayashi et al., 2005), conscious dog and human subjects (Echizen et al., 1988; Mikus et al., 1990). Also depicted are concentration–response curves for inhibition of the hERG K+ current (dashed grey line) and the L-type Ca2+ current (from Zhang et al., 1999) and the range of therapeutically effective protein-unbound drug plasma concentrations in humans (orange area; data from Redfern et al., 2003).
The effects of nifedipine on the shape of the cardiac AP and FP were characterized by concentration-dependent shortening of the APD90 and/or FPD90 in guinea pig and rabbit ventricular slices (Figure 6; Table 3), and rabbit Purkinje fibres (Table 2, Figure 6B) and a decrease in the AP plateau potential (Figure 6A); AP upstroke or QNa was unaffected (Tables 2 and 3). The extent of APD/FPD shortening was similar in guinea pig slices, rabbit slices and rabbit Purkinje fibres, and amounted to approximately 10–25% at 1 µM. In contrast, the diastolic blood pressure in conscious dogs (Table 4, Figure 6B) was decreased to a similar extent at concentrations approximately three orders of magnitude lower; at these low single-digit nanomolar concentrations, the QTcV interval in conscious dogs was not prolonged but rather slightly shortened (Table 4, Figure 6B).
In guinea pig ventricular slice preparations, the effect of verapamil was characterized by a concentration-dependent decrease in APD90 and FPD90 (Figure 7; Tables 2 and 3); this decrease was approximately 20% at 10 µM. In contrast, concentration-dependent verapamil-mediated APD90 prolongation together with pronounced triangulation prevailed in rabbit Purkinje fibres (Table 2, Figure 7B). In conscious dogs, verapamil prolonged the PQ interval at concentrations that were about one order of magnitude lower than those associated with AP/FP duration changes in vitro, while the QTc interval duration was hardly affected within the same concentration range (Table 4, Figure 7B).
Discussion
Class 1 anti-arrhythmics flecainide and quinidine
Quinidine and flecainide have been used in past decades to suppress/prevent ventricular tachyarrhythmias in humans [quinidine (Halkin et al., 1979; Carliner et al., 1980; Rosen and Wit, 1983; Duff et al., 1985), flecainide (Duff et al., 1981; Hodges et al., 1982; Lui et al., 1982; Estes et al., 1984) ]. Later, the pro-arrhythmic potential of these class 1 anti-arrhythmic drugs came more into focus (Morganroth and Horowitz, 1984) and led to investigations of the underlying electrophysiological mechanism(s).
Both flecainide and quinidine are inhibitors of the human cardiac sodium channel hNav1.5 as demonstrated here (Table 1, Figures 1B and 2B) and as expected from the literature (Hondeghem and Katzung, 1977; Konzen et al., 1990; Snyders and Hondeghem, 1990; Nitta et al., 1992; Ducroq et al., 2007). At slightly lower concentrations, however, quinidine and flecainide also inhibit the hERG K+ channel (Table 1, Figures 1C and 2C; Paul et al., 2002; Ducroq et al., 2007), and at higher concentrations, both drugs interfere with other K+ channels (Slawsky and Castle, 1994) and with L-type Ca2+ channels (Table 1).
As expected for hNav1.5 blockers, the maximum rate of depolarization, Vmax, of the cardiac AP was reduced by flecainide and quinidine with a concentration-dependence similar to that required for Na+ current inhibition. This has been demonstrated in numerous ex vivo AP measurements, for example, in preparations of rabbit Purkinje fibre (Table 2, Figures 1B and 2B; Konzen et al., 1990; Aubert et al., 2006), dog Purkinje fibre (Burke et al., 1986), guinea pig papillary muscle (Table 2, Figure 1B; Hayashi et al., 2005) and guinea pig ventricular slices (Table 3, Figures 1B and 2B). In an in vivo situation, Na+ channel block manifests itself as a slowing of conduction (i.e. widening of the QRS complex in the ECG). The respective effect was detected in dogs (Table 4, Figures 1B and 2B; Tashibu et al., 2005; Toyoshima et al., 2005) and in humans [flecainide (Duff et al., 1981; Hodges et al., 1982; Lui et al., 1982; Conrad and Ober, 1984; Estes et al., 1984), quinidine (Henning and Nyberg, 1973; Halkin et al., 1979; Carliner et al., 1980; Duff et al., 1985)] in the same range of drug concentrations that cause hNav1.5 inhibition and Vmax decrease, and that overlap with the range of therapeutically effective protein-unbound drug plasma concentrations in humans (Redfern et al., 2003). As an equivalent to a prolonged QRS complex in the ECG following Na+ channel blockade, the increase in the latency difference between the primary and the secondary peak of the FP (QNa) indicates reduced Na+ conductance in cardiac slices (Table 3; Figures 1A,B and 2A,B). In fact, both flecainide and quinidine increased QNa at concentrations that induced QRS widening in vivo (compare Tables 3 and 4).
Drugs that are highly selective blockers of the hERG K+ channel are expected to prolong the duration of AP/FP and the QT interval, whereas with mixed ion channel blockers such as flecainide and quinidine the net effect on APD, FPD and QT is more difficult to predict. Despite being a potent inhibitor of hERG (Table 1, Figure 1C), flecainide did not substantially alter APD90/FPD90 in rabbit Purkinje fibre (Table 2, Figure 1C), guinea pig and rabbit ventricular slices (Table 3, Figure 1A,C) and papillary muscle (Hayashi et al., 2005) unless at concentrations ≥10 µM (Figure 1C; Ducroq et al., 2007). Then, flecainide prolonged APD in guinea pig ventricular slices but shortened it in rabbit ventricular slices; this might be explained by a differential contribution of L-type Ca2+ channels to the shape of the ventricular slice AP, contributing more current in guinea pig and less in rabbit. Also, QTcV in conscious dogs in vivo remained essentially unchanged (Table 4, Figure 1C) at concentrations overlapping and exceeding the human therapeutic range (Redfern et al., 2003). Finally, in rabbit Purkinje fibres, flecainide reduced the maximal repolarization velocity resulting in significant triangulation (Table 2), whereas in ventricular preparations, such a dramatic alteration of the shape of APs was not observed. The most likely reason for this difference is that the Purkinje fibre AP plateau depends to a greater extent on Na+ and Ca2+ influx than that of ventricular muscle.
Quinidine presents us with a more complex picture, since there was considerable variation between types of tissue preparation and species. Concentration-dependent APD90 prolongation was only observed in rabbit Purkinje fibres (Lu et al., 2001; Aubert et al., 2006; Ducroq et al., 2007), at concentrations inhibiting hERG K+ current (Figure 2C). In guinea pig papillary muscle, APD90 was prolonged at ≤10 µM, while at ≥10 µM, APD90 returned to baseline (Table 2, Figure 2C; Jurevicious et al., 1991?; Hayashi et al., 2005). In guinea pig ventricular slices, FPD90 was prolonged at ≤30 µM and shortened at higher concentrations, whereas APD90 was only shortened at ≥10 µM (Table 3, Figure 2C). Overall, the prolongation of APD and FPD at lower quinidine concentrations in guinea pig ventricular preparations was consistent with the inhibition of repolarizing hERG K+ currents, while at higher quinidine concentrations, the additional inhibition of depolarizing L-type Ca2+ currents (Table 1) shifted the balance towards an APD/FPD shortening. In conscious dogs, the application of quinidine was associated with QTcV prolongation (Table 4, Figure 2C) occurring at a similar magnitude as APD/FPD increases in the range of therapeutically effective protein-unbound drug plasma concentrations in humans (Figure 2C; Baker et al., 1983; Redfern et al., 2003). In dog Purkinje fibres, however, quinidine has been reported to mediate concentration-dependent APD90 prolongation (Figure 2C; Roden and Hoffman, 1985; Davidenko et al., 1989; Nemeth et al., 1997; Lu et al., 2001) as well as APD90 shortening (Figure 2C; Burke et al., 1986) and even a biphasic effect (Wyse et al., 1993). These latter results cannot be explained based on differential inhibition of depolarizing versus repolarizing ion channels but may be due to differences in experimental protocols.
In general, the effects of multichannel blockers like flecainide or quinidine on AP shape are difficult to predict. For flecainide, blocking effects on IKr, IKur, Ito and ICa have been reported. Since the contribution of individual channels to a certain AP shape shows large variations depending on species and location within the heart of one species, the effects of drugs like flecainide and quinidine will depend on the predominating conductance(s) during the plateau or repolarization phase and how they are affected by overlapping active concentration ranges (Dumotier et al., 1999), thus resulting in APD prolongation as well as shortening.
Class 2 anti-arrhythmic atenolol
For several decades, atenolol and other class 2 anti-arrhythmics have been amongst the mainstay drugs for treatment of heart failure and hypertension, and are known to possess few adverse effects, particularly with regard to pro-arrhythmia, and to improve life expectancy. The rather benign profile of atenolol is based on the lack of relevant interactions with cardiac ion channels, including the hERG K+ channel (Table 1; Kawakami et al., 2006), and is also reflected by the fact that atenolol hardly alters the shape of cardiac APs. This has been demonstrated in rabbit Purkinje fibre (Table 2, Figure 3B) and papillary muscle (Manley et al., 1986), and in guinea pig ventricular slices (Figure 3, Table 3). In the latter preparation, however, atenolol was associated with a concentration-dependent shortening of the APD90, which was most likely as a result of ‘rundown’ of the preparation. Finally, in the in vivo situation, atenolol prolonged the PQ interval (lowered heart rate) but was without effect on the duration of the QTc interval in conscious dogs (Table 4, Figure 3B; McAinsh and Holmes, 1983; Davies and McAinsh, 1986; Coppi et al., 1987; Kvetina et al., 1999) and human volunteers and patients (Coppi et al., 1987; Way et al., 1988; Demolis et al., 1997) and even in case of human intoxication (Snook et al., 2000).
Class 3 anti-arrhythmics d,l -sotalol and dofetilide
Class 3 anti-arrhythmics had been developed with the specific aim to prolong the refractory period by prolonging the AP duration, this being the main distinguishing feature compared to class 1 anti-arrhythmics. The electrophysiological basis of the delayed cardiac repolarization is inhibition of the hERG K+ channel by d,l-sotalol and dofetilide (Table 1, Figures 4 and 5; Numaguchi et al., 2000; Davie et al., 2004; Ducroq et al., 2007). This is in line with concentration-dependent drug-mediated prolongation of APD90 and FPD90 in the following ex vivo preparations: rabbit Purkinje fibre (Table 2, Figures 4B and 5B; Abrahamsson et al., 1993; Dumotier et al., 1999; Lu et al., 2001; 2002; Aubert et al., 2006; Ducroq et al., 2007) and papillary muscle (Manley et al., 1986; Abrahamsson et al., 1993), dog Purkinje fibre (Gwilt et al., 1991; Knilans et al., 1991; Wyse et al., 1993; Lee et al., 1996; Nemeth et al., 1997; Lu et al., 2001) and papillary muscle (Nemeth et al., 1997), guinea pig papillary muscle (Table 2, Figure 4B; Tande et al., 1990; Davie et al., 2004), and guinea pig ventricular slices (Table 3, Figures 4 and 5). In vivo, both d,l-sotalol and dofetilide prolonged the QT interval as demonstrated in conscious (Table 4, Figures 4B and 5B; Schneider et al., 2005) and anaesthetized dogs (Mortensen et al., 1991; Tashibu et al., 2005; Vormberge et al., 2006) as well as in human patients (Wang et al., 1986; Way et al., 1988; Redfern et al., 2003) or intoxications (Elonen et al., 1979; Neuvonen et al., 1981; Wang et al., 1986; Edvardsson and Varnauskas, 1987?).
Although the direction of drug-induced changes was consistent throughout the various models in general, there was considerable variation in terms of sensitivity when individual models and species were compared. The magnitude of effects as well as the d,l-sotalol sensitivity of models was comparable for inhibition of the hERG K+ current, prolongation of APD90/FPD90 in guinea pig papillary muscle and ventricular slices and prolongation of the QTc interval in anaesthetized and conscious dogs (Figure 4B); there, the concentrations producing a 20% effect were mostly between 10 and 100 µM (Figure 4B). In contrast, both the magnitude of the effect and sotalol sensitivity appeared to be greater in rabbit and dog Purkinje fibres and in humans, since concentration–response curves were steeper and shifted to approximately 10-fold lower concentrations (Figure 4B). The picture was similar for dofetilide (Figure 5B) where 20% prolongation of APD90 in rabbit and dog Purkinje fibres occurred in the single-digit nanomolar range and overlapped with human therapeutically effective protein-unbound plasma concentrations. However, for hERG K+ current inhibition, APD90/FPD90 prolongation in rabbit, dog and guinea pig ventricular muscle, and QTcV prolongation in conscious dogs, double digit nanomolar dofetilide concentrations were required to reach the 20% effect level. In addition, rabbit appears to be the most and guinea pig the least sensitive species, regardless of whether this rank order was established for Purkinje fibre or for ventricular muscle preparations. Literature reports point to the same direction with regard to (i) a differential species sensitivity of Purkinje fibres towards delayed repolarization (e.g. Lu et al., 2001) and (ii) a higher sensitivity of Purkinje fibres versus ventricular muscle towards the APD shortening or prolonging effects of sodium channel or hERG K+ channel blockers [e.g. Abrahamsson et al., 1996 (rabbit); Lathrop, 1985 (dog) ] because of quantitative differences of the underlying ionic currents [e.g. Cordeiro et al., 1998 (rabbit) ]. Another important point of discussion concerns a potential bias introduced by sex-specific differences in ventricular repolarization (for review, see Cheng, 2006). Females are more sensitive towards APD prolongation than males, particularly at extremely low stimulation rates, as demonstrated in rabbit Purkinje fibre (Lu et al., 2000) and Langendorff heart (Liu et al., 1998), and in human patients and volunteers (Lehmann et al., 1999; Somberg et al., 2011), whereas this issue remains controversial in guinea pig ventricle (Brouillette et al., 2007; Hreiche et al., 2009) and in canine ventricle (Xiao et al., 2006). Also, inward currents vary with sex and cardiac region as shown for the L-type calcium current (Sims et al., 2008).
Class 4 anti-arrhythmics nifedipine and verapamil
Both nifedipine and verapamil share the common feature of class 4 anti-arrhythmics; that is, they inhibit L-type calcium channels at submicromolar concentrations (Table 1, Figures 6B and 7B; Uehara and Hume, 1985; Charnet et al., 1987; Zhang et al., 1999; Shen et al., 2000). However, since the potency of nifedipine is markedly increased at depolarized potentials, nifedipine targets predominantly the L-type calcium channels modulating vascular smooth muscle tone, whereas the main therapeutic effect of verapamil is to delay atrioventricular conduction. In addition, both drugs are inhibitors of the hERG K+ channel. However, nifedipine had a very low potency of about three orders of magnitude beyond that of calcium channel inhibition, thus physiologically irrelevant, whereas verapamil inhibited both hERG K+ and L-type calcium channel at almost identical concentrations (Table 1, Figures 6B and 7B; Zhang et al., 1999; Zhabyeyev et al., 2000?).
Since nifedipine is a fairly selective calcium channel blocker, its effects on the shape of cardiac APs were straightforward and characterized by concentration-dependent shortening of the APD90/FPD90 in rabbit Purkinje fibres (Table 2, Figure 6B; Dumotier et al., 1999), dog Purkinje fibres (Lee et al., 1996), cat and guinea pig papillary muscle (Bayer et al., 1977; Jurevicious et al., 1991; Hayashi et al., 2005) and guinea pig and rabbit ventricular slices (Table 3, Figure 6). This APD90 shortening was observed at concentrations approximately two to three orders of magnitude higher than those required to decrease the diastolic blood pressure in conscious dogs (Table 4, Figure 6B) or the range of therapeutically effective protein-unbound drug plasma concentrations in man (Redfern et al., 2003). Due to the functional antagonism of hERG K+ and L-type calcium channel block and consistent with the in vitro/ex vivo findings, the QTcV interval in conscious and anaesthetized dogs was not prolonged but rather slightly shortened (Table 4, Figure 6B; Amlie et al., 1979; Tashibu et al., 2005). Whenever nifedipine-induced QTc prolongation is reported, the actual consensus opinion attributes this to an inadequate heart rate correction of the QT interval (e.g. Toyoshima et al., 2005).
The well-balanced mixed ion channel blocker verapamil presented a decidedly different picture, since the effect on APD90/FPD90 in ex vivo preparations ranged from prolongation to shortening, obviously depending on the type of preparation (Tables 2 and 3, Figure 7B). Concentration-dependent verapamil-mediated APD90 prolongation prevailed in canine (Cranefield et al., 1974; Rosen et al., 1974) and rabbit Purkinje fibres (Table 2, Figure 7B; Dumotier et al., 1999), whereas in guinea pig papillary muscle (Zhang et al., 1997; Hayashi et al., 2005) or ventricular slices (Table 3, Figure 7), APD90 shortening was observed. The desired effect of PQ interval prolongation occurred in conscious and anaesthetized dogs within the same concentration range as the therapeutic effect in man (Table 4, Figure 7B; Echizen et al., 1988; Mikus et al., 1990; Shiina et al., 2000; Redfern et al., 2003), while the QTc interval duration was hardly affected, adequate heart rate correction was provided (Table 4, Figure 7B; Shiina et al., 2000; Fossa et al., 2002; Schneider et al., 2005; Tashibu et al., 2005; Toyoshima et al., 2005).
Limitations and opportunities
Similar to other multicellular preparations, it is conceivable that neurotransmitters are also released due to field stimulation in cardiac tissue slices. In our preparation, we used a small concentric stimulation electrode, which is expected to minimize the effect of electrical stimulation on neurotransmitter release. This problem, however, has not yet been addressed systematically by inhibiting neurotransmitter release or by blocking neurotransmitter effects.
In cardiac slice preparations, it might reasonably be expected that drug actions on fibroblasts could be an area of considerable interest or matter of concern (Porter and Turner, 2009). Fibroblasts within cardiac tissue are assumed to play an important role in bridging electrical activity between myocytes. Since fibroblasts are coupled to myocytes by connexins (MacCannell et al., 2007), drug interaction with connexins but also interactions with fibroblast ion channels might influence drug effects on the cardiac AP. Investigating such an interaction was beyond the scope of our present work, and we are also not aware of publications addressing this question. We believe that these putative interactions have to be studied on isolated fibroblasts and in co-culture with cardiac myocytes. Although the tissue architecture in cardiac slice preparations is in principle not different from that of other multicellular preparations, cardiac slices may be more readily accessible for the study of specific modulation of fibroblast characteristics by gene transfer and/or knock-down methods.
Conclusion
In this paper, we have presented integrated data regarding electrophysiological effects of selected anti-arrhythmic drugs (flecainide, quinidine, atenolol, sotalol, dofetilide, nifedipine, verapamil) using complementary in vitro and in vivo study types. These complementary approaches range from measurement of membrane currents (hERG, INa, ICa.L), to AP/FP in slices, Purkinje fibre AP and haemodynamic/ECG parameters (conscious dog), and also include published data. The main result of this complementary approach is that FP and AP recordings from heart slices correlate well with established in vitro and in vivo models in terms of pharmacology and predictability. Heart slice preparations yield similar results as papillary muscle but offer enhanced throughput for mechanistic investigations and reduce the use of laboratory animals.
Acknowledgments
The expert technical assistance of Manuela Weisflog and Waldemar Hink (heart slice AP), Susanne Herbold (voltage clamp), Marcus Deitermann (Purkinje fibre AP) and Thomas Vormberge (conscious dog model) is gratefully acknowledged. This work was supported by the German Federal Ministry for Economy and Technology BMWi (PRO INNO II program, grant KF0682501 SB8 to AB, EW, MS and HL). Finally, the authors would like to thank Michael Kayser and Dr Frank Thorsten Hafner (Bioanalytics, Bayer Pharma) for measuring drug plasma concentrations.
Glossary
- AP
action potential
- APA
AP amplitude
- APD
AP duration
- BDM
2,3-butanedione monoxime
- FP
field potential
- FPD
FP duration
- hERG
human ether-a-go-go-related gene
- HK+
solution, high potassium solution
- ICa.L
L-type Ca2+ current
- INa
Na+ current
- PM
papillary muscle
- QNa
latency difference between primary and secondary peak of the FP as measure for Na+ conductance
- QTcV
QT interval corrected for heart rate according to Van de Water
- RMP
resting membrane potential
- Vmax
maximal AP upstroke velocity
Author contributions
Major contributions by the authors to this work were as follows: (1) conception and design (HMH, HL, EW); (2) collection and assembly of data: voltage clamp experiments (HMH), rabbit Purkinje fibre AP recordings (HMH), guinea pig papillary muscle AP recordings (RB), ventricular slice AP and FP recordings (AB, EW, MS, HL), studies in conscious dogs (MH) and literature search (HMH); (3) data analysis and interpretation (HMH, AB, EW, MS, HL, MH, RB); and (4) manuscript writing (HMH).
Conflict of interest
The authors have no conflicts of interest that could inappropriately influence this work.
References
- Abrahamsson C, Duker G, Lundberg C, Carlsson L. Electrophysiological and inotropic effects of H-234/09 (almokalant) in vitro– a comparison with 2 other novel IK blocking drugs, UK-68,798 (dofetilide) and E-4031. Cardiovasc Res. 1993;27:861–867. doi: 10.1093/cvr/27.5.861. [DOI] [PubMed] [Google Scholar]
- Abrahamsson C, Carlsson L, Duker G. Lidocaine and nisoldipine attenuate almokalant-induced dispersion of repolarisation and early afterdepolarisations in vitro. J Cardiovasc Electrophysiol. 1996;7:1074–1081. doi: 10.1111/j.1540-8167.1996.tb00483.x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlie JP, Refsum H, Landmark K. The effect of nifedipine on the monophasic action potential and refractoriness of the right ventricle of the dog heart in situ after beta-adrenergic receptor blockade. Acta Pharmacol Toxicol (Copenh) 1979;44:185–190. doi: 10.1111/j.1600-0773.1979.tb02315.x. [DOI] [PubMed] [Google Scholar]
- Anonymous. ICH S7B: the nonclinical evaluation of the potential for delayed ventricular repolarisation (QT interval prolongation) by human pharmaceuticals. 2005. Availabble at: http://www.ich.org/products/guidelines/safety/article/safety-guidelines.html (accessed 12 June 2011) [PubMed]
- Aubert M, Osterwalder R, Wagner B, Parrilla I, Cavero I, Doessegger L, et al. Evaluation of the rabbit Purkinje fiber assay as an in vitro tool for assessing the risk of drug-induced Torsades de Pointes in humans. Drug Saf. 2006;29:237–254. doi: 10.2165/00002018-200629030-00007. [DOI] [PubMed] [Google Scholar]
- Baker BJ, Gammill J, Massengill J, Schubert E, Karin A, Doherty JE. Concurrent use of quinidine and disopyramide: evaluation of serum concentrations and electrocardiographic effects. Am Heart J. 1983;105:12–15. doi: 10.1016/0002-8703(83)90271-5. [DOI] [PubMed] [Google Scholar]
- Barclay CJ. Modelling diffusive O(2) supply to isolated preparations of mammalian skeletal and cardiac muscle. J Muscle Res Cell Motil. 2005;26:225–235. doi: 10.1007/s10974-005-9013-x. [DOI] [PubMed] [Google Scholar]
- Bayer R, Rodenkirchen R, Kaufmann R, Lee JH, Hennekes R. The effects of nifedipine on contraction and monophasic action potential of isolated cat myocardium. Naunyn Schmiedebergs Arch Pharmacol. 1977;301:29–37. doi: 10.1007/BF00501261. [DOI] [PubMed] [Google Scholar]
- Belpaire FM, De Rick A, De Smet F, Bourda A, Rosseel MT, Bogaert MG. Influence of lidocaine on plasma protein binding and pharmacokinetics of verapamil in dogs. Prog Clin Biol Res. 1989;300:437–440. [PubMed] [Google Scholar]
- Brouillette J, Lupien MAS, Michel C, Fiset C. Characterization of ventricular repolarisation in male and female guinea pigs. J Mol Cell Cardiol. 2007;42:357–366. doi: 10.1016/j.yjmcc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Burke GH, Loukides JE, Berman ND. Comparative electropharmacology of mexiletine, lidocaine and quinidine in a canine Purkinje fiber model. J Pharmacol Exp Ther. 1986;237:232–236. [PubMed] [Google Scholar]
- Bussek A, Wettwer E, Christ T, Lohmann H, Camelliti P, Ravens U. Tissue slices from adult mammalian hearts as a model for pharmacological drug testing. Cell Physiol Biochem. 2009;24:527–536. doi: 10.1159/000257528. [DOI] [PubMed] [Google Scholar]
- Carliner NH, Fisher ML, Crouthamel WG, Narang PK, Plotnick GD. Relation of ventricular premature beat suppression to serum quinidine concentraton determined by a new and specific assay. Am Heart J. 1980;100:483–489. doi: 10.1016/0002-8703(80)90660-2. [DOI] [PubMed] [Google Scholar]
- Charnet P, Quadid H, Richard S, Nargeot J. Electrophysiological analysis of the action of nifedipine and nicardipine on myocardial fibers. Fundam Clin Pharmacol. 1987;1:413–431. doi: 10.1111/j.1472-8206.1987.tb00575.x. [DOI] [PubMed] [Google Scholar]
- Cheng J. Evidences of the gender-related differences in cardiac repolarisation and the underlying mechanisms in different animal species and human. Fundam Clin Pharmacol. 2006;20:1–8. doi: 10.1111/j.1472-8206.2005.00384.x. [DOI] [PubMed] [Google Scholar]
- Colbert CM. Preparation of cortical brain slices for electrophysiological recording. Methods Mol Biol. 2006;337:117–125. doi: 10.1385/1-59745-095-2:117. [DOI] [PubMed] [Google Scholar]
- Conrad GJ, Ober RE. Metabolism of flecainide. Am J Cardiol. 1984;53:41B–51B. doi: 10.1016/0002-9149(84)90501-0. [DOI] [PubMed] [Google Scholar]
- Coppi G, Mosconi P, Springolo V. Pharmacokinetics of a fixed combination of atenolol and indapamide in dogs and humans after oral administration. Curr Ther Res. 1987;42:411–418. [Google Scholar]
- Cordeiro JM, Spitzer KW, Giles WR. Repolarising K+ currents in rabbit heart Purkinje cells. J Physiol. 1998;508:811–823. doi: 10.1111/j.1469-7793.1998.811bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranefield PF, Aronson RS, Wit AL. Effect of verapamil on the normal action potential and on a calcium-dependent slow response of canine cardiac Purkinje fibers. Circ Res. 1974;34:204–213. doi: 10.1161/01.res.34.2.204. [DOI] [PubMed] [Google Scholar]
- Davidenko JM, Cohen L, Goodrow R, Antzelevitch C. Quinidine-induced action potential prolongation, early afterdepolarisations, and triggered activity in canine Purkinje fibers. Effects of stimulation rate, potassium, and magnesium. Circulation. 1989;79:674–686. doi: 10.1161/01.cir.79.3.674. [DOI] [PubMed] [Google Scholar]
- Davie C, Valentin JP, Pollard C, Standen N, Mitcheson J, Alexander P, et al. Comparative pharmacology of guinea pig cardiac myocyte and cloned hERG (I-Kr) channel. J Cardiovasc Electrophysiol. 2004;15:1302–1309. doi: 10.1046/j.1540-8167.2004.04099.x. [DOI] [PubMed] [Google Scholar]
- Davies MK, McAinsh J. Tissue atenolol levels following chronic beta-adrenoceptor blockade using oral atenolol in dogs. J Pharm Pharmacol. 1986;38:316–319. doi: 10.1111/j.2042-7158.1986.tb04577.x. [DOI] [PubMed] [Google Scholar]
- Demolis JL, Martel C, Funck-Brentano C, Sachse A, Weimann HJ, Jaillon P. Effects of tedisamil, atenolol and their combination on heart and rate-dependent QT interval in healthy volunteers. Br J Clin Pharmacol. 1997;44:403–409. doi: 10.1046/j.1365-2125.1997.t01-1-00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducroq J, Printemps R, Guilbot S, Gardette J, Salvetat C, Le Grand M. Action potential experiments complete hERG assay and QT-interval measurements in cardiac preclinical studies. J Pharmacol Toxicol Methods. 2007;56:159–170. doi: 10.1016/j.vascn.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Duff HJ, Roden DM, Maffucci RJ, Vesper BS, Conard GJ, Higgins SB, et al. Suppression of resistant ventricular arrhythmias by twice daily dosing with flecainide. Am J Cardiol. 1981;48:1133–1140. doi: 10.1016/0002-9149(81)90331-3. [DOI] [PubMed] [Google Scholar]
- Duff HJ, Wyse DG, Manyari D, Mitchell LB. Intravenous quinidine: relations among concentration, tachyarrhythmia suppression and electrophysiologic actions with inducible sustained ventricular tachycardia. Am J Cardiol. 1985;55:92–97. doi: 10.1016/0002-9149(85)90306-6. [DOI] [PubMed] [Google Scholar]
- Dumotier BM, Adamantidis MM, Puisieux FL, Bastide MM, Dupuis BA. Repercussions of pharmacologic reduction in ionic currents on action potential configuration in rabbit Purkinje fibers: are they indicative of proarrhythmic potential? Drug Dev Res. 1999;47:63–76. [Google Scholar]
- Echizen H, Manz M, Eichelbaum M. Electrophysiologic effects of dextro- and levo-verapamil on sinus node and AV node function in humans. J Cardiovasc Pharmacol. 1988;12:543–546. doi: 10.1097/00005344-198811000-00007. [DOI] [PubMed] [Google Scholar]
- Edvardsson N, Varnauskas E. Clinical course, serum concentrations and elimination rate in a case of massive sotalol intoxication. Eur Heart J. 1987;8:544–548. doi: 10.1093/oxfordjournals.eurheartj.a062316. [DOI] [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflugers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- Elonen E, Neuvonen PJ, Tarssanen L, Kala R. Sotalol intoxication with prolonged Q-T interval and severe tachyarrhythmias. Br Med J. 1979;1:1184. doi: 10.1136/bmj.1.6172.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes NA, Garan H, Ruskin JN. Electrophysiologic properties of flecainide acetate. Am J Cardiol. 1984;53:26B–29. doi: 10.1016/0002-9149(84)90498-3. [DOI] [PubMed] [Google Scholar]
- Fossa AA, Depasquale MJ, Raunig DL, Avery MJ, Leishman DJ. The relationship of clinical QT prolongation to outcome in the conscious dog using a beat-to-beat QT-RR interval assessment. J Pharmacol Exp Ther. 2002;302:828–833. doi: 10.1124/jpet.102.035220. [DOI] [PubMed] [Google Scholar]
- Gwilt M, Arrowsmith JE, Blackburn KJ, Burges RA, Cross PE, Dalrymple HW, et al. UK-68,798: a novel, potent and highly selective class III antiarrhythmic agent which blocks potassium channels in cardiac cells. J Pharmacol Exp Ther. 1991;256:318–324. [PubMed] [Google Scholar]
- Halkin H, Vered Z, Millman P, Rabinowitz B, Neufeld HN. Steady-state serum quinidine concentration: role in prophylactic therapy following acute myocardial infarction. Isr J Med Sci. 1979;15:583–587. [PubMed] [Google Scholar]
- Hancox JC, Convery MK. Inhibition of L-type calcium current by flecainide in isolated single rabbit atrioventricular nodal myocytes. Exp Clin Cardiol. 1997;2:163–170. [Google Scholar]
- Hayashi S, Kii Y, Tabo M, Fukuda H, Itoh T, Shimosato T, et al. QT PRODACT: a multi-site study of in vitro action potential assays on 21 compounds in isolated guinea pig papillary muscles. J Pharmacol Sci. 2005;99:423–437. doi: 10.1254/jphs.qt-a1. [DOI] [PubMed] [Google Scholar]
- Henning R, Nyberg G. Serum quinidine levels after administration of three different quinidine preparations. Eur J Clin Pharmacol. 1973;6:239–244. doi: 10.1007/BF00644739. [DOI] [PubMed] [Google Scholar]
- Himmel HM. Suitability of commonly used excipients for electrophysiological in vitro safety pharmacology assessment of effects on hERG potassium current and on rabbit Purkinje fiber action potential. J Pharmacol Toxicol Methods. 2007;56:145–158. doi: 10.1016/j.vascn.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Himmel HM, Hoffmann M. QTc shortening with a new investigational cancer drug: a brief case study. J Pharmacol Toxicol Methods. 2010;62:72–81. doi: 10.1016/j.vascn.2010.05.012. [DOI] [PubMed] [Google Scholar]
- Hodges M, Haugland JM, Granrud G, Conard GJ, Asinger RW, Mikell FL, et al. Suppression of ventricular ectopic depolarisations by flecainide acetate, a new antiarrhythmic agent. Circulation. 1982;65:879–885. doi: 10.1161/01.cir.65.5.879. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- Hreiche R, Morisette P, Zakrzewski-Jakubiak H, Turgeon J. Gender-related differences in drug-induced prolongation of cardiac repolarisation in prepubertal guinea pigs. J Cardiovasc Pharmacol Ther. 2009;14:28–37. doi: 10.1177/1074248408331018. [DOI] [PubMed] [Google Scholar]
- Jurevicious J, Muckus K, Macianskiene R, Chmel-Dunaj GN. Influence of action potential duration and resting potential on effects of quinidine, lidocaine and ethmozine on Vmax in guinea pig papillary muscle. J Mol Cell Cardiol. 1991;23(Suppl. 1):103–114. doi: 10.1016/0022-2828(91)90029-l. [DOI] [PubMed] [Google Scholar]
- Kawakami K, Nagatomo T, Abe H. Comparison of HERG channel blocking effects of various beta-blockers – implication for clinical strategy. Br J Pharmacol. 2006;147:642–652. doi: 10.1038/sj.bjp.0706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara Y, Inoko M, Hatakeyama N, Momose Y, Sasayama S. Mechanisms of negative inotropic effects of class 1c antiarrhythmic agents: comparative study of the effects of flecainide and pilsicainide on intracellular calcium handling in dog ventricular myocardium. J Cardiovasc Pharmacol. 1996;27:42–51. doi: 10.1097/00005344-199601000-00008. [DOI] [PubMed] [Google Scholar]
- Knilans TK, Lathrop DA, Nanasi PP, Schwartz A, Varro A. Rate and concentration-dependent effects of UK-68,798, a potent new class III antiarrhythmic, on canine Purkinje fibre action potential duration and Vmax. Br J Pharmacol. 1991;103:1568–1572. doi: 10.1111/j.1476-5381.1991.tb09828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konzen G, Reichardt B, Hauswirth O. Fast and slow blockade of sodium channels by flecainide in rabbit cardiac Purkinje fibres. Naunyn Schmiedebergs Arch Pharmacol. 1990;341:565–576. doi: 10.1007/BF00171738. [DOI] [PubMed] [Google Scholar]
- Kvetina J, Svoboda Z, Nobilis M, Pastera J, Anzenbacher P. Experimental goettingen minipig and beagle dog as two species used in bioequivalence studies for clinical pharmacology (5-aminosalicylic acid and atenolol as model drugs) Gen Physiol Biophys. 1999;18:80–85. [PubMed] [Google Scholar]
- Lathrop DA. Electromechanical characterization of the effects of racemic sotalol and its optical isomers on isolated canine ventricular trabecular muscles and Purkinje strands. Can J Physiol Pharmacol. 1985;63:1506–1512. doi: 10.1139/y85-248. [DOI] [PubMed] [Google Scholar]
- Lee HC, Cai JJ, Arnar DO, Shibata EF, Martins JB. Mechanism of alpha-2 adrenergic modulation of canine Purkinje fiber action potential. J Pharmacol Exp Ther. 1996;278:597–606. [PubMed] [Google Scholar]
- Lehmann MH, Hardy S, Archibald D, MacNeil DJ. JTc prolongation with d,l-sotalol in women versus men. Am J Cardiol. 1999;83:354–359. doi: 10.1016/s0002-9149(98)00868-6. [DOI] [PubMed] [Google Scholar]
- Liu YK, Katchman A, Drici MD, Ebert SN, Ducic I, Morad M, et al. Gender difference in the cycle length-dependent QT and potassium currents in rabbits. J Pharmacol Exp Ther. 1998;285:672–679. [PubMed] [Google Scholar]
- Lu HR, Marien R, Saels A, De Clerck F. Are there sex-specific differences in ventricular repolarisation or in drug-induced early afterdepolarisations in isolated rabbit Purkinje fibers? J Cardiovasc Pharmacol. 2000;36:132–139. doi: 10.1097/00005344-200007000-00018. [DOI] [PubMed] [Google Scholar]
- Lu HR, Marien R, De Clerck F. Species plays an important role in drug-induced prolongation of action potential duration and early afterdepolarisations in isolated Purkinje fibers. J Cardiovasc Electrophysiol. 2001;12:93–102. doi: 10.1046/j.1540-8167.2001.00093.x. [DOI] [PubMed] [Google Scholar]
- Lu HR, Vlaminckx E, Van Ammel K, De Clerck F. Drug-induced long QT in isolated rabbit Purkinje fibers: importance of action potential duration, triangulation and early afterdepolarisations. Eur J Pharmacol. 2002;452:183–192. doi: 10.1016/s0014-2999(02)02246-x. [DOI] [PubMed] [Google Scholar]
- Lui HK, Lee G, Dietrich P, Low RI, Mason DT. Flecainide-induced QT prolongation and ventricular tachycardia. Am Heart J. 1982;103:567–569. doi: 10.1016/0002-8703(82)90346-5. [DOI] [PubMed] [Google Scholar]
- MacCannell KA, Bazzazi H, Chilton L, Shibukawa Y, Clark RB, Giles WR. A mathematical model of electrotonic interactions between ventricular myocytes and fibroblasts. Biophys J. 2007;92:4121–4132. doi: 10.1529/biophysj.106.101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley BS, Alexopoulos D, Robinson GJ, Cobbe SM. Subsidiary class III effects of beta blockers? A comparison of atenolol, metoprolol, nadolol, oxprenolol and sotalol. Cardiovasc Res. 1986;20:705–709. doi: 10.1093/cvr/20.10.705. [DOI] [PubMed] [Google Scholar]
- McAinsh J, Holmes BF. Pharmacokinetic studies with atenolol in the dog. Biopharm Drug Dispos. 1983;4:249–261. doi: 10.1002/bdd.2510040306. [DOI] [PubMed] [Google Scholar]
- Mikus G, Eichelbaum M, Fischer C, Gumulka S, Klotz U, Kroemer HK. Interaction of verapamil and cimetidine: stereochemical aspects of drug metabolism, drug disposition and drug action. J Pharmacol Exp Ther. 1990;253:1042–1048. [PubMed] [Google Scholar]
- Morganroth J, Horowitz LN. Flecainide: its proarrhythmic effect and expected changes on the surface electrocardiogram. Am J Cardiol. 1984;53:89B–94B. doi: 10.1016/0002-9149(84)90509-5. [DOI] [PubMed] [Google Scholar]
- Mortensen E, Tande PM, Klow NE, Refsum H. Plasma concentrations and hemodynamic effects of d-sotalol after beta-blockade in dogs. Pharmacol Toxicol. 1991;68:420–425. doi: 10.1111/j.1600-0773.1990.tb00856.x. [DOI] [PubMed] [Google Scholar]
- Nemeth M, Varro A, Virag L, Hala O, Thormaehlen D, Papp JG. Frequency-dependent cardiac electrophysiologic effects of tedisamil: comparison with quinidine and sotalol. J Cardiovasc Pharmacol Ther. 1997;2:273–284. doi: 10.1177/107424849700200405. [DOI] [PubMed] [Google Scholar]
- Neuvonen PJ, Elonen E, Vuorenmaa T, Laakso M. Prolonged Q-T interval and severe tachyarrhythmias, common features of sotalol intoxication. Eur J Clin Pharmacol. 1981;20:85–89. doi: 10.1007/BF00607142. [DOI] [PubMed] [Google Scholar]
- Nitta J, Sunami A, Marumo F, Hiraoka M. States and sites of actions of flecainide on guinea pig cardiac sodium channels. Eur J Pharmacol. 1992;214:191–197. doi: 10.1016/0014-2999(92)90118-n. [DOI] [PubMed] [Google Scholar]
- Numaguchi H, Mullins FM, Johnson JP. Probing the interaction between inactivation gating and d-sotalol block of HERG. Circ Res. 2000;87:1012–1018. doi: 10.1161/01.res.87.11.1012. [DOI] [PubMed] [Google Scholar]
- Parrish AR, Gandolfi AJ, Brendel K. Precision-cut tissue slices: applications in pharmacology and toxicology. Life Sci. 1995;57:1887–1901. doi: 10.1016/0024-3205(95)02176-j. [DOI] [PubMed] [Google Scholar]
- Paul AA, Witchel HJ, Hancox JC. Inhibition of the current of heterologously expressed HERG potassium channels by flecainide and comparison with quinidine, propafenone and lignocaine. Br J Pharmacol. 2002;136:717–729. doi: 10.1038/sj.bjp.0704784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Rakhit A, Holford NH, Effeney DJ, Riegelman S. Induction of quinidine metabolism and plasma protein binding by phenobarbital in dogs. J Pharmacokinet Biopharm. 1984;12:495–515. doi: 10.1007/BF01060128. [DOI] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- Roden DM, Hoffman BF. Action potential prolongation and induction of abnormal automaticity by low quinidine concentrations in canine Purkinje fibers. Relationship to potassium and cycle length. Circ Res. 1985;56:857–867. doi: 10.1161/01.res.56.6.857. [DOI] [PubMed] [Google Scholar]
- Rosen MR, Wit AL. Electropharmacology of antiarrhythmic drugs. Am Heart J. 1983;106:829–839. doi: 10.1016/0002-8703(83)90005-4. [DOI] [PubMed] [Google Scholar]
- Rosen MR, Ilvento JP, Gelband H, Merker C. Effects of verapamil on electrophysiologic properties of canine cardiac Purkinje fibers. J Pharmacol Exp Ther. 1974;189:414–422. [PubMed] [Google Scholar]
- Salata JJ, Wasserstrom JA. Effects of quinidine on action potentials and ionic currents in isolated canine ventricular myocytes. Circ Res. 1988;62:324–337. doi: 10.1161/01.res.62.2.324. [DOI] [PubMed] [Google Scholar]
- Scamps F, Undrovinas A, Vassort G. Inhibition of ICa in single frog cardiac cells by quinidine, flecainide, ethmozin and ethazin. Am J Physiol. 1989;256:C549–C559. doi: 10.1152/ajpcell.1989.256.3.C549. [DOI] [PubMed] [Google Scholar]
- Schneider J, Hauser R, Andreas JO, Linz K, Jahnel U. Differential effects of human ether-a-go-go-related gene (HERG) blocking agents on QT duration variability in conscious dogs. Eur J Pharmacol. 2005;512:53–60. doi: 10.1016/j.ejphar.2005.01.042. [DOI] [PubMed] [Google Scholar]
- Schnelle K, Garrett ER. Pharmacokinetics of the beta-adrenergic blocker sotalol in dogs. J Pharmacol Sci. 1973;62:362–375. doi: 10.1002/jps.2600620303. [DOI] [PubMed] [Google Scholar]
- Sellin LC, McArdle JJ. Multiple effects of 2,3-butanedione monoxime. Pharmacol Toxicol. 1994;74:305–313. doi: 10.1111/j.1600-0773.1994.tb01365.x. [DOI] [PubMed] [Google Scholar]
- Shen JB, Jiang B, Pappano AJ. Comparison of L-type calcium channel blockade by nifedipine and/or cadmium in guinea pig ventricular myocytes. J Pharmacol Exp Ther. 2000;294:562–570. [PubMed] [Google Scholar]
- Shiina H, Sugiyama A, Takahara A, Satoh Y, Hashimoto K. Comparison of the electropharmacological effects of verapamil and propranolol in the halothane-anesthetized in vivo canine model under monophasic action potential monitoring. Jpn Circ J. 2000;64:777–782. doi: 10.1253/jcj.64.777. [DOI] [PubMed] [Google Scholar]
- Sims C, Reisenweber S, Viswanathan PC, Choi BR, Walker WH, Salama G. Sex, age, and regional differences in L-type calcium current are important determinants of arrhythmia phenotype in rabbit hearts with drug-induced long QT type 2. Circ Res. 2008;102:e86–e100. doi: 10.1161/CIRCRESAHA.108.173740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawsky MT, Castle NA. K+ channel blocking actions of flecainide compared with those of propafenone and quinidine in adult rat ventricular myocytes. J Pharmacol Exp Ther. 1994;269:66–74. [PubMed] [Google Scholar]
- Smith DA, Rasmussen HS, Stopher DA, Walker DK. Pharmacokinetics and metabolism of dofetilide in mouse, rat, dog and man. Xenobiotica. 1992;22:709–719. doi: 10.3109/00498259209053133. [DOI] [PubMed] [Google Scholar]
- Snook CP, Sigvaldason K, Kristinsson J. Severe atenolol and diltiazem overdose. J Toxicol Clin Toxicol. 2000;38:661–665. doi: 10.1081/clt-100102018. [DOI] [PubMed] [Google Scholar]
- Snyders DJ, Hondeghem LM. Effects of quinidine on the sodium current of guinea pig ventricular myocytes. Evidence for a drug-associated rested state with altered kinetics. Circ Res. 1990;66:565–579. doi: 10.1161/01.res.66.2.565. [DOI] [PubMed] [Google Scholar]
- Somberg JC, Preston RA, Ranade V, Cvetanovic I, Molnar J. Gender differences in cardiac repolarisation following intravenous sotalol administration. J Cardiovasc Pharmacol Ther. 2011 doi: 10.1177/1074248411406505. DOI: 10.1177/1074248411406505.? [DOI] [PubMed] [Google Scholar]
- Tande PM, Bjoernstad H, Yang T, Refsum H. Rate-dependent class III antiarrhythmic action, negative chronotropy, and positive inotropy of a novel IK blocking drug, UK-68,798: potent in guinea pig but no effect in rat myocardium. J Cardiovasc Pharmacol. 1990;16:401–410. doi: 10.1097/00005344-199009000-00008. [DOI] [PubMed] [Google Scholar]
- Tashibu H, Miyazaki H, Aoki K, Akie Y, Yamamoto K. QT PRODACT: in vivo QT assay in anesthetized dog for detecting the potential for QT interval prolongation by human pharmaceuticals. J Pharmacol Sci. 2005;99:473–486. doi: 10.1254/jphs.qt-a3. [DOI] [PubMed] [Google Scholar]
- Toyoshima S, Kanno A, Kitayama T, Sekiya K, Nakai K, Haruna M, et al. QT PRODACT: in vivo QT assay in the conscious dog for assessing the potential for QT interval prolongation by human pharmaceuticals. J Pharmacol Sci. 2005;99:459–471. doi: 10.1254/jphs.qt-a2. [DOI] [PubMed] [Google Scholar]
- Uehara A, Hume JR. Interactions of organic calcium channel antagonists with calcium channels in single frog atrial cells. J Gen Physiol. 1985;85:621–647. doi: 10.1085/jgp.85.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Water A, Verheyen J, Xhonneux R, Reneman RS. An improved method to correct the QT interval of the electrocardiogram for changes in heart rate. J Pharmacol Methods. 1989;22:207–217. doi: 10.1016/0160-5402(89)90015-6. [DOI] [PubMed] [Google Scholar]
- Vickers AE, Fisher RL. Organ slices for the evaluation of human drug toxicity. Chem Biol Interact. 2004;150:87–96. doi: 10.1016/j.cbi.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Vormberge T, Hoffmann M, Himmel HM. Safety pharmacology assessment of drug-induced QT-prolongation in dogs with reduced repolarisation reserve. J Pharmacol Toxicol Methods. 2006;54:130–140. doi: 10.1016/j.vascn.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Wang T, Bergstrand RH, Thompson KA, Siddoway LA, Duff HJ, Woosley RL, et al. Concentration-dependent pharmacologic properties of sotalol. Am J Cardiol. 1986;57:1160–1165. doi: 10.1016/0002-9149(86)90692-2. [DOI] [PubMed] [Google Scholar]
- Way BP, Forfar JC, Cobbe SM. Comparison of the effects of chronic oral therapy with atenolol and sotalol on ventricular monophasic action potential duration and effective refractory period. Am Heart J. 1988;116:740–746. doi: 10.1016/0002-8703(88)90332-8. [DOI] [PubMed] [Google Scholar]
- Wyse KR, Ye V, Campbell TJ. Action potential prolongation exhibits simple dose-dependence for sotalol, but reverse dose-dependence for quinidine and disopyramide – Implications for proarrhythmia due to triggered activity. J Cardiovasc Pharmacol. 1993;21:316–322. doi: 10.1097/00005344-199302000-00019. [DOI] [PubMed] [Google Scholar]
- Xiao L, Zhang L, Han W, Wang Z, Nattel S. Sex-based transmural differences in cardiac repolarisation and ionic-current properties in canine left ventricles. Am J Physiol. 2006;291:H570–H580. doi: 10.1152/ajpheart.01288.2005. [DOI] [PubMed] [Google Scholar]
- Zhabyeyev P, Missan S, Jones SE, McDonald TF. Low-affinity block of cardiac K(+) currents by nifedipine. Eur J Pharmacol. 2000;401:137–143. doi: 10.1016/s0014-2999(00)00413-1. [DOI] [PubMed] [Google Scholar]
- Zhang S, Sawanobori T, Hirano Y, Hiraoka M. Multiple modulations of action potential duration by different calcium channel blocking agents in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 1997;30:489–496. doi: 10.1097/00005344-199710000-00013. [DOI] [PubMed] [Google Scholar]
- Zhang ST, Zhou ZF, Gong QM, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to hERG potassium channels. Circ Res. 1999;84:989–998. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]
- Zhou ZF, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, et al. Properties of hERG channels stably expressed in HEK293 cells studied at physiological temperature. Biophys J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan R, Padrini R, Bernini V, Piovan D, Ferrari M. Influence of age and gender on the in vitro serum protein binding of flecainide. Pharmacol Res. 1993;28:259–264. doi: 10.1006/phrs.1993.1129. [DOI] [PubMed] [Google Scholar]