

Abstract
BACKGROUND AND PURPOSE
Airway smooth muscle (ASM) phenotype plasticity, characterized by reversible switching between contractile and proliferative phenotypes, is considered to contribute to increased ASM mass and airway hyper-responsiveness in asthma. Further, increased expression of collagen I has been observed within the ASM bundle of asthmatics. Previously, we showed that exposure of intact bovine tracheal smooth muscle (BTSM) to collagen I induces a switch from a contractile to a hypocontractile, proliferative phenotype. However, the functional relevance of this finding for intact human ASM has not been established.
EXPERIMENTAL APPROACH
We investigated the effects of exposure of human tracheal smooth muscle (HTSM) strips to monomeric collagen I and PDGF on contractile responses to methacholine and KCl. Expression of contractile proteins sm-α-actin and sm-MHC was assessed by Western blot analysis. The proliferation of HTSM cells was assessed by cell counting, measuring mitochondrial activity (Alamarblue conversion) and [3H]-thymidine incorporation. Proliferation of intact tissue slices was assessed by [3H]-thymidine incorporation.
KEY RESULTS
Culturing HTSM strips in the presence of collagen I or PDGF for 4 days reduced maximal contractile responses to methacholine or KCl and the expression of contractile proteins. Conversely, collagen I and PDGF increased proliferation of HTSM cells and proliferative responses in tissue slices. PDGF additively increased the proliferation of HTSM cells cultured on collagen I; this additive effect was not observed on contractility, contractile protein expression or proliferation of intact tissue.
CONCLUSION AND IMPLICATIONS
These findings indicate that collagen I and PDGF induce a functionally hypocontractile, proliferative phenotype of human ASM, which may contribute to airway remodelling in asthma.
Keywords: collagen type I, platelet-derived growth factor, airway smooth muscle contractility, airway smooth muscle proliferation, phenotype modulation
Introduction
Structural changes in the airway wall, including increased airway smooth muscle (ASM) mass, are characteristic features of airway remodelling, which may contribute to airway hyper-responsiveness (AHR) and decline in lung function in asthma (Bousquet et al., 2000; Meurs et al., 2008). Increased ASM mass is at least partially caused by hyperplasia, and in vitro ASM proliferation is increased by serum and various growth factors that may synergize with neurotransmitters and inflammatory mediators (Ebina et al., 1993; Woodruff et al., 2004; Gosens et al., 2008). Exposure of cultured ASM cells to mitogenic stimuli causes switching from a contractile, hypoproliferative to a proliferative, hypocontractile phenotype, as indicated by changes in molecular phenotypic markers (Hirst et al., 2000; Halayko et al., 2008). The potential effect of these molecular changes on ASM contractile function has recently been demonstrated in intact bovine tracheal smooth muscle (BTSM). Thus, prolonged exposure of BTSM strips to serum or peptide growth factors like platelet-derived growth factor (PDGF) and insulin-like growth factor-1 decreased maximal methacholine- and KCl-induced contractions, which was inversely correlated with the proliferative responses of BTSM cells to these mitogenic stimuli (Gosens et al., 2002; Dekkers et al., 2007). ASM phenotype switching is reversible, as indicated by the observation that removal of mitogenic stimuli, for example by serum deprivation, results in the re-establishment of a (hyper)contractile ASM phenotype associated with increased expression of contractile marker proteins like sm-α-actin, sm-MHC and calponin, which is further enhanced in the presence of insulin or TGF-β (Ma et al., 1998; Schaafsma et al., 2007; Hirota et al., 2009; Dekkers et al., 2009b).
Increased deposition of extracellular matrix (ECM) proteins within the airway wall is another hallmark of airway remodelling in asthma (Fernandes et al., 2006; Dekkers et al., 2009a). Expression of various ECM proteins, including collagen I, is increased in the subepithelial basement membrane (Roche et al., 1989; Fernandes et al., 2006). Within the ASM bundle, increased deposition of collagen I has also been observed (Roberts and Burke, 1998; Bai et al., 2000; Araujo et al., 2008), which may be due to increased production of this matrix protein by asthmatic ASM cells (Johnson et al., 2004). In addition, increased proliferative responses of asthmatic ASM cells have also been shown, which depend on the ECM produced by these cells (Johnson et al., 2004), indicating that collagen I may affect the phenotype of these cells. Indeed, results from in vitro studies on the effects of collagen I on human ASM cell function have indicated that cells grown on this ECM protein show increased growth factor-induced proliferation, increased synthetic capabilities, decreased apoptosis and reduced expression of contractile proteins (Hirst et al., 2000; Freyer et al., 2001; Nguyen et al., 2005; Peng et al., 2005). Using BTSM, we have recently shown that exposure of intact strip preparations to monomeric (denatured) collagen I induced a hypocontractile state, characterized by decreased contractile responses to methacholine and KCl and reduced expression of contractile marker proteins, which was associated with increased proliferation of cultured BTSM cells (Dekkers et al., 2007). In addition, PDGF augmented collagen I-induced proliferation in an additive fashion, without an additional effect on contractility or contractile protein expression (Dekkers et al., 2007). The functional significance of ECM- and growth factor-induced phenotype switching on intact human ASM is currently unknown. Therefore, in the present study, we investigated the effects of monomeric collagen I and PDGF on the contractility of human tracheal smooth muscle (HTSM) strips and on contractile protein expression as well as on proliferation of HTSM cells and tissue slices. The results demonstrate that phenotype modulation by collagen I and PDGF is of relevance to human ASM contractile function and may contribute to airway remodelling in asthma.
Methods
Tissue preparation and organ culture
Human tracheal sections of anonymized lung transplantation donors, taken from just above the bifurcation, were obtained from the Department of Cardiothoracic Surgery, University Medical Centre Groningen and transported to the laboratory in ice-cold Krebs–Henseleit (KH) buffer (composition in mM: NaCl 117.5, KCl 5.60, MgSO4 1.18, CaCl2 2.50, NaH2PO4 1.28, NaHCO3 25.00 and glucose 5.50), pre-gassed with 5% CO2 and 95% O2 (pH 7.4). HTSM strips were prepared as described for BTSM (Dekkers et al., 2007; 2009b). After dissection of the smooth muscle layer and careful removal of connective tissue and mucosa, we prepared strips of identical width and length (1 cm × 2 mm, 4–8 strips per donor) by cutting and cultured them for 4 days in Medium Zero [sterile Dulbecco's Modified Eagle's medium (DMEM; Gibco BRL Life Technologies Paisley, UK), supplemented with sodium pyruvate (1 mM; Gibco), non-essential amino acid mixture (1:100; Gibco), gentamicin (45 µg·mL−1; Gibco), penicillin (100 U·mL−1; Gibco), streptomycin (100 µg·mL−1; Gibco), amphotericin B (1.5 µg·mL−1; Gibco), apo-transferrin (5 µg·mL−1, human; Sigma, St. Louis, MO) and ascorbic acid (0.1 mM)]. When used, monomeric denatured collagen I (50 µg·mL−1, calf skin; Fluka, Buchs, Switzerland) and/or PDGF-AB (10 ng·mL−1, human; Bachem, Weil am Rhein, Germany) were present during the entire incubation period. Occasionally, strips were used for isometric tension measurements directly after preparation.
Isometric tension measurements
Isometric contraction experiments were performed as described previously (Dekkers et al., 2007; 2009b). Briefly, HTSM strips were mounted for isometric recording in organ baths, containing KH buffer at 37°C. During a 90 min equilibration period with washouts every 30 min, resting tension was adjusted to 0.5 g, followed by pre-contractions with 20 and 40 mM KCl. Following washout, maximal relaxation was established by the addition of (–)-isoprenaline (0.1 µM; Sigma). Tension was re-adjusted to 0.5 g immediately followed by two changes with KH buffer. After another equilibration period of 30 min, cumulative concentration–response curves were constructed to KCl (5.6–50 mM) or methacholine (1 nM–0.1 mM; ICN Biomedicals, Costa Mesa, CA). When maximal tension was reached, the strips were washed several times, and maximal relaxation was established by using (–)-isoprenaline (10 µM). Contractions were expressed as the percentage of maximal contraction induced by KCl or methacholine in vehicle-treated strips. After the experiment, strips were snap-frozen for Western blot analysis.
Western blot analysis
HTSM strip homogenization and Western blot analysis of sm-α-actin and sm-MHC expression were performed as described previously (Dekkers et al., 2007; 2009b). In short, homogenates were prepared by pulverizing the strips under liquid nitrogen, followed by sonification in homogenization buffer (composition in mM: Tris–HCl 50 mM, NaCl 150.0, EDTA 1.0, PMSF 1.0, Na3VO4 1.0, NaF 1.0, pH 7.4, supplemented with leupeptin 10 µg·mL−1, aprotinin 10 µg·mL−1, pepstatin 10 µg·mL−1, Na-deoxycholate 0.25% and Igepal 1%; all from Sigma). Equal amounts of protein were subjected to SDS-PAGE and transferred onto nitrocellulose membranes, followed by standard immunoblotting techniques. Antibodies [anti-sm-α-actin (Sigma) and anti-sm-myosin heavy chain (anti-sm-MHC; Neomarkers, Fremont, CA)] were visualized on film using enhanced chemiluminescence reagents (Pierce, Breda, NL) and analysed by densitometry (Totallab™, Nonlinear dynamics, Newcastle, UK). Bands were normalized to β-actin expression.
HTSM cell culture
HTSM, prepared free of mucosa and connective tissue, was chopped using a McIlwain tissue chopper, three times at a setting of 500 µm and three times at 100 µm. Tissue slices were washed once with Medium Plus [DMEM, supplemented with sodium pyruvate (1 mM), non-essential amino acid mixture (1:100), gentamicin (45 µg·mL−1), penicillin (100 U·mL−1), streptomycin (100 µg·mL−1), amphotericin B (1.5 µg·mL−1) and fetal bovine serum (FBS, 10%; Gibco)], placed in culture flasks and allowed to adhere. Medium was refreshed every 48–72 h. Upon reaching confluence, cells were passaged by trypsinization. Cells from passages 1–5 were used for the present study.
Coating of culture plates with collagen I
Collagen I was reconstituted in hydrochloric acid (10 mM) at 5 mg·mL−1 and diluted in PBS to a final concentration of 50 µg·mL−1. Diluted collagen I (0.5 mL) was adsorbed to culture plates overnight and air-dried at room temperature. Unoccupied protein-binding sites were blocked by 30 min incubation with 0.1% BSA solution. Subsequently, plates were washed twice with unsupplemented DMEM and dried before further use.
[3H]-thymidine incorporation
[3H]-thymidine incorporation in HTSM cells was performed as described previously (Dekkers et al., 2007; 2009b). HTSM cells were plated on uncoated or collagen I-coated 24-well culture plates at a density of 30 000 cells per well and allowed to attach overnight in Medium Plus. Cells were washed with PBS and made quiescent by incubation in Medium Zero, supplemented with 1% ITS (insulin, transferrin and selenium; Gibco) for 72 h. Subsequently, cells were washed and incubated in the absence or presence of PDGF (10 ng·mL−1) in Medium Zero for 28 h, the last 24 h in the presence of [methyl-3H]-thymidine (0.25 µCi·mL−1). After incubation, the cells were washed with PBS at room temperature. Subsequently, the cells were treated with ice-cold 5% trichloroacetic acid (TCA) on ice for 30 min, and the acid-insoluble fraction was dissolved in NaOH (1 M). Incorporated [3H]-thymidine was quantified by liquid scintillation counting using a Beckman LS1701 β-counter.
For the determination of thymidine incorporation in intact HTSM tissue, tissue slices were used as described previously (Gosens et al., 2004). After the dissection of the smooth muscle layer, the muscle was chopped three times at a setting of 500 µm and three times at 100 µm (McIlwain tissue chopper). Subsequently, the tissue slices were washed twice with Medium Zero and maintained in Medium Zero overnight. The next day, tissue slices were washed twice with PBS and transferred into 24-well plates at 50 mg wet weight ml−1 in Medium Zero. Slices were stimulated with PDGF (10 ng·mL−1), collagen I (50 µg·mL−1) or the combination of both in the presence of [methyl-3H]-thymidine (0.25 µCi·mL−1) for 48 h. After the incubation, slices were washed twice with PBS at room temperature. Subsequently, slices were treated with ice-cold 5% TCA on ice for 1 h. The acid-insoluble fraction was dissolved in NaOH (1 M). Incorporated [3H]-thymidine was quantified as described above.
Cell number determination
HTSM cells were plated on uncoated or collagen I-coated six-well culture plates at a density of 75 000 cells per well, allowed to attach overnight in medium Plus and made quiescent by incubation in medium Zero, supplemented with 1% ITS for 72 h. Subsequently, cells were washed and incubated in the absence and presence of PDGF (10 ng·mL−1) in Medium Zero for 4 days. Thereafter, cells were incubated with Hanks' balanced salt solution containing 5% Alamar blue solution for 30 min (BioSource, Camarillo, CA). Mitochondrial activity, as a measure of cell number, was assessed by conversion of Alamar blue into its reduced form, as indicated by the manufacturer. In addition, cells were counted in duplicate, using a haemocytometer.
Immunofluorescence
HTSM strips were cultured in the absence or presence of PDGF (10 ng·mL−1) and/or collagen I (50 µg·mL−1) as described above. After culture, tissue strips were frozen at −80°C in isopentane and stored at −80°C. Cryostat sections of HTSM tissue (4 µm) were fixed in methanol at −20°C for 10 min. After fixation, sections were permeabilized with 0.025% Triton X-100 in PBS, followed by incubation in blocking buffer (1% BSA in PBS) for 30 min. Subsequently, sections were stained with primary antibodies [rabbit anti-Ki67 (Neomarkers) and mouse anti-sm-MHC] in blocking buffer for 1 h at room temperature. Bound antibodies were visualized by incubation with FITC- and Cy3-conjugated secondary antibodies (donkey anti-rabbit and donkey anti-mouse, respectively) in blocking buffer for 45 min. Nuclei were labelled with Hoechst 33342 (1 µg·mL−1; Invitrogen, Bleiswijk, the Netherlands) for 5 min. After being stained, sections were mounted using ProLong Gold anti-fade reagent (Invitrogen) and analysed by using an Olympus AX70 microscope equipped with digital image capture system (ColorView Soft System with Olympus UCMAD2 lens, Olympus Corporation, Tokyo, Japan).
Data analysis
Data represent means ± SEM from n separate experiments. Statistical significance of differences was evaluated by Student's paired t-test. Statistical significance was reached at P < 0.05.
Results
Collagen I and PDGF decrease the contractility of HTSM strips
To assess whether exposure to collagen I or PDGF induces a functionally hypocontractile ASM phenotype, HTSM strips were cultured in the absence and presence of monomeric collagen I (50 µg·mL−1) and/or PDGF (10 ng·mL−1) for 4 days. Culturing HTSM strips in the presence of collagen I or PDGF for 4 days significantly (P < 0.05) reduced the maximal contractile force (Emax) induced by methacholine or KCl compared with vehicle-treated control strips (Figure 1, Table 1). No additive effects were observed after combined treatment. The sensitivity to both contractile stimuli was unaffected by all treatments. No significant change in methacholine-induced contractility was observed after 4 days of culturing in vehicle compared with freshly isolated strips (Emax= 2.89 ± 0.68 g and 2.50 ± 0.32 g, –logEC50= 5.95 ± 0.10 and 5.77 ± 0.14, respectively, n= 4–7, P > 0.05 Student's t-test for unpaired observations). In agreement with the reduced contractility, collagen I and PDGF also decreased the protein expression of sm-α-actin and sm-myosin heavy chain (sm-MHC, P < 0.05 all; Figure 2). No additive effects were observed after combined treatment.
Figure 1.
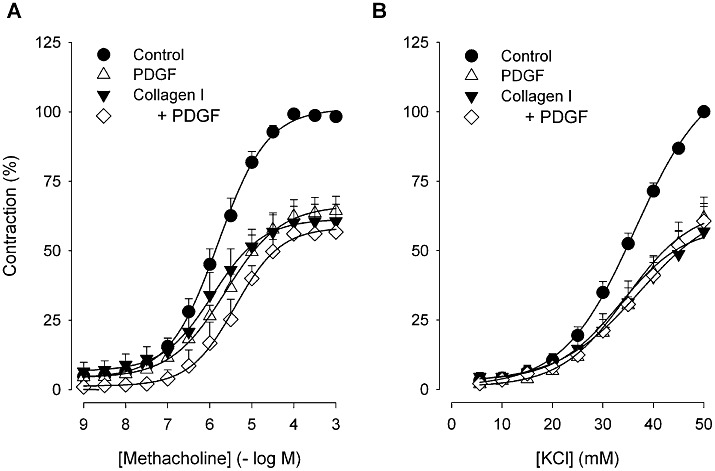
Concentration–response curves of (A) methacholine- and (B) KCl-induced contractions of HTSM strips, pretreated with vehicle (control) or collagen I (50 µg·mL−1) in the absence or presence of PDGF (10 ng·mL−1) for 4 days. Data represent means ± SEM of three to seven experiments performed in duplicate.
Table 1.
Contractile responses of HTSM strips to methacholine or KCl after 4 days of culturing in the absence or presence of collagen I (50 µg·mL−1) and/or PDGF (10 ng·mL−1)
| Methacholine | KCl | |||
|---|---|---|---|---|
| Emax (%) | pEC50 (-log M) | Emax (%) | EC50 (mM) | |
| Control | 100 ± 0 | 5.77 ± 0.14 | 100 ± 0 | 34.6 ± 0.8 |
| +PDGF | 64.8 ± 5.3* | 5.66 ± 0.14 | 61.8 ± 5.2* | 34.7 ± 1.1 |
| Collagen I | 61.0 ± 6.0* | 5.99 ± 0.18 | 56.9 ± 9.0* | 35.2 ± 1.4 |
| +PDGF | 56.7 ± 4.8* | 5.69 ± 0.29 | 60.6 ± 8.6* | 35.6 ± 1.2 |
Data represent means ± SEM of three to seven experiments, performed in duplicate.
P < 0.05, **P < 0.01, ***P < 0.001 compared with control.
Figure 2.
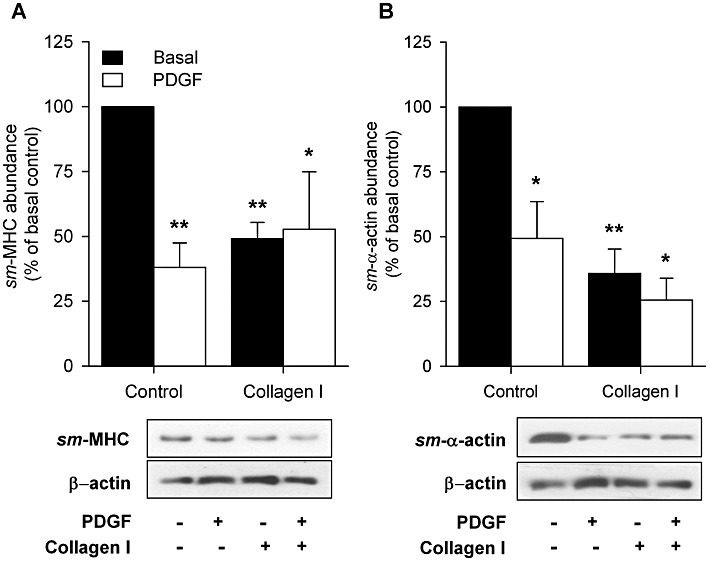
Western blot analysis of (A) sm-MHC and (B) sm-α-actin expression in HTSM strips treated with vehicle (control) or collagen I (50 µg·mL−1) in the absence (basal) or presence of PDGF (10 ng·mL−1) for 4 days. Means ± SEM of three to five experiments are shown. *P < 0.05, **P < 0.01 compared with basal control. Representative immunoblots of sm-MHC, sm-α-actin and β-actin are shown.
Collagen I and PDGF increase proliferation of HTSM cells
To assess whether the decreases in Emax induced by collagen I and PDGF were associated with proliferative changes, [3H]-thymidine incorporation and cell number were assessed using primary HTSM cell cultures. DNA synthesis of these cells was increased by PDGF (P < 0.05) as well as by culturing on a collagen I matrix (P < 0.05, Figure 3A). PDGF-induced DNA synthesis was significantly enhanced when the cells were cultured on collagen I-coated instead of uncoated matrices (P < 0.05). Similarly, PDGF and collagen I also enhanced the number of HTSM cells (P < 0.05, both conditions), which was further enhanced when both stimuli were combined (P < 0.05, Figures 3B and C).
Figure 3.
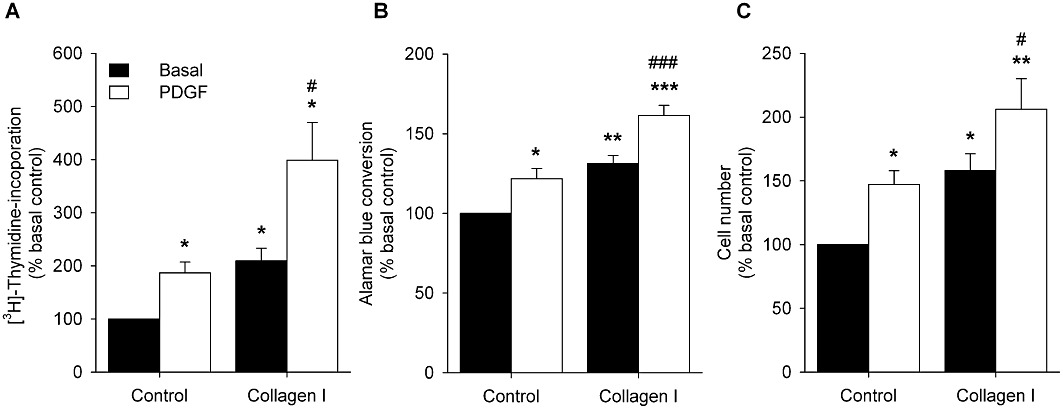
Basal and PDGF (10 ng·mL−1)-stimulated (A) [3H]-thymidine incorporation (a measure of DNA synthesis), (B) Alamar blue conversion and (C) changes in cell count of HTSM cells cultured on uncoated plastic (control) or collagen I (50 µg·mL−1)-coated matrices. Data represent means ± SEM of four to five experiments performed in triplicate (DNA synthesis) or duplicate (Alamar blue, cell count). *P < 0.05, **P < 0.01, ***P < 0.001 compared with basal control. #P < 0.05, ###P < 0.001 compared with PDGF control.
Collagen I and PDGF increase proliferation in intact HTSM tissue
To determine whether collagen I- and PDGF-induced proliferation was also of relevance in intact HTSM tissue, [3H]-thymidine incorporation was assessed in HTSM tissue slices as described previously for BTSM (Gosens et al., 2004). PDGF significantly (P < 0.01) increased DNA synthesis in intact HTSM tissue slices by ∼1.4-fold (Figure 4) and collagen I similarly increased DNA synthesis in these slices (P < 0.01). No additive effects were observed after combined treatment with PDGF and collagen I.
Figure 4.
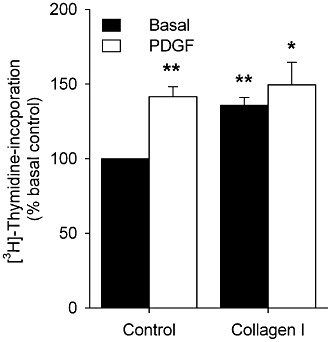
DNA synthesis of intact HTSM tissue in the presence of medium (basal control), PDGF (10 ng·mL−1), collagen I (50 µg·mL−1) or the combination of both. Data represent means ± SEM of five experiments performed in triplicate. *P < 0.05, **P < 0.01 compared with basal control.
Immunohistochemistry for the proliferative marker Ki67 and contractile marker sm-MHC was performed in sections of HTSM strips cultured for 4 days as described above, to identify the cells contributing to the enhanced proliferative response. Staining for the contractile marker revealed that the vast majority of the cells within the tissue strip were positive for sm-MHC (Figure 5). In the strips cultured in the presence of collagen I and/or PDGF, a small fraction of the nuclei stained positive for the proliferative marker Ki67 as well. In addition, the majority of the Ki67-positive nuclei co-localized with the staining for sm-MHC in these sections. No positive staining for Ki67 was observed in the vehicle-treated strips.
Figure 5.
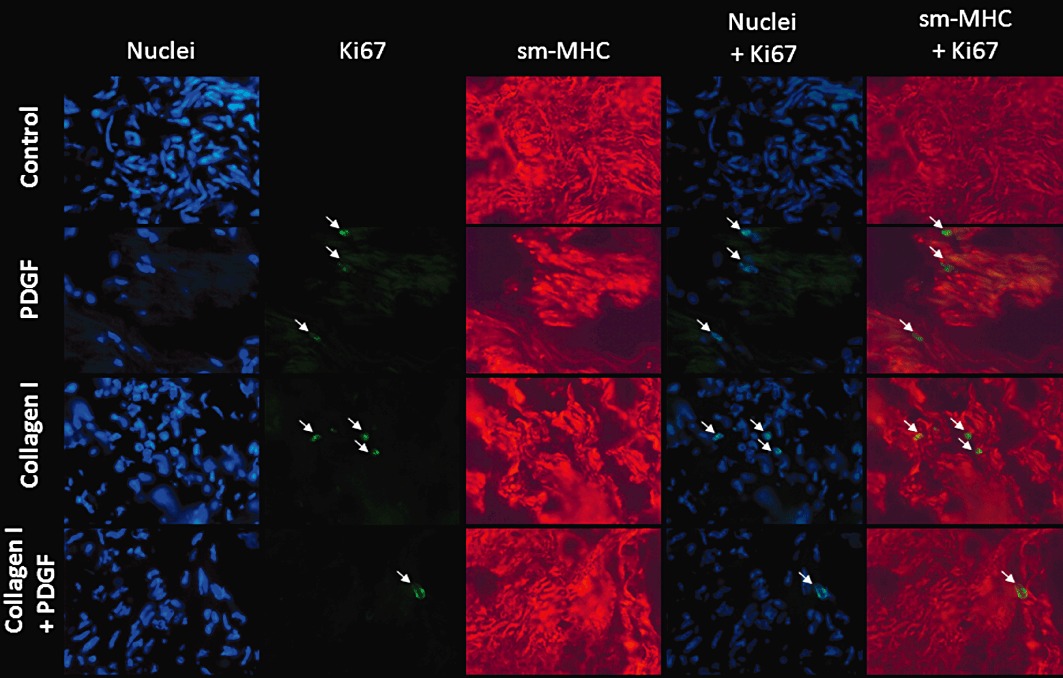
Immunofluorescent stainings of nuclei (Hoechst 33342), Ki67 (green), sm-MHC (red) in cryostat sections of HTSM strips treated without (control) or with collagen I (50 µg·mL−1) in the absence or presence of PDGF (10 ng·mL−1) for 4 days. Pictures were taken at a 400× magnification. Arrows indicate Ki67-positive nuclei.
Collectively, our results indicate that exposure of intact HTSM tissue to collagen I and PDGF induces a functionally hypocontractile ASM phenotype, which is associated with increased proliferative responses in HTSM cells and intact tissue.
Discussion
In the present study, we demonstrated that prolonged exposure of intact HTSM tissue to the ECM protein collagen I and to the peptide growth factor PDGF induces a functionally hypocontractile, proliferative phenotype. Thus, pretreatment of HTSM strips with collagen I and with PDGF decreased maximal contractions in response to the receptor-dependent agonist methacholine and the receptor-independent stimulus KCl, which was associated with decreased contractile protein expression, whereas both stimuli increased proliferation of cultured HTSM cells and proliferation in intact HTSM tissue. Culturing the HTSM cells on collagen I additively increased the proliferative response to PDGF, whereas no additional effects were observed on contractility, contractile protein expression or proliferation of intact tissue, suggesting differential regulation of these processes between cells and intact tissue. Our findings are well in line with previous observations that exposure of human bronchiole ring segments to serum–containing pro-proliferative factors reduced the contractions induced by carbachol, histamine and KCl and decreased the expression of the contractile protein calponin (Moir et al., 2003).
ASM cells display phenotype plasticity, characterized by reversible and dynamic changes in the expression of contractile and proliferative markers that may be governed by a variety of growth factors and ECM proteins present in the asthmatic airway wall, including collagen I and PDGF (Gosens et al., 2008; Halayko et al., 2008). The peptide growth factor PDGF is a well-characterized proliferative stimulus for ASM, which may be released from inflammatory and structural cells within the airway, including eosinophils, macrophages and fibroblasts (Fabisiak et al., 1992; Taylor et al., 1994; Ohno et al., 1995). Exposure of human ASM cells to PDGF increases their proliferation and the expression of the proliferative marker Ki67, whereas the expression of the contractile markers sm-α-actin and sm-MHC is reduced (Hirst et al., 2000). Previous studies by our laboratory have indicated that exposure of intact BTSM to peptide growth factors, including PDGF, induces a functionally hypocontractile phenotype, which was inversely correlated with DNA synthesis of isolated BTSM cells (Gosens et al., 2002). In addition, DNA synthesis of intact BTSM tissue slices was shown to be increased by stimulation with PDGF, indicating that growth factor-induced changes in contractility are associated with increased proliferation in intact tissue (Gosens et al., 2004). In the present study, we demonstrated the functional relevance of these findings in intact HTSM tissue in which cell-to-cell contacts and endogenous ECM components are preserved, the results being highly reminiscent of previous findings in BTSM (Gosens et al., 2002; 2004; Dekkers et al., 2007).
Collagens are widespread throughout the body, provide structural support and fulfil a variety of biological functions (Prockop and Kivirikko, 1995). In vitro, it has been found that culturing of human ASM cells on monomeric collagen I augments growth factor-induced proliferation and enhances the reduction in contractile marker expression by PDGF (Hirst et al., 2000). In the airways of asthmatics, deposition of collagen I is increased in the extracellular microenvironment of the ASM cells (Bai et al., 2000; Fernandes et al., 2006). In addition, ASM cells from asthmatic patients produce more collagen I than those obtained from healthy subjects, which, via an autocrine mechanism, could contribute to the increased proliferation of asthmatic ASM cells (Johnson et al., 2004). Interestingly, culturing of ASM cells on fibrillar collagen I instead of monomeric collagen I did not promote growth factor-induced ASM proliferation (Nguyen et al., 2005), whereas recently, fibrillar collagen I was even shown to inhibit both basal and growth factor-induced proliferation (Schuliga et al., 2010). In addition, inhibition of collagen degradation by the MMP inhibitor ilomastat further enhanced the growth-attenuating effects of fibrillar collagen I, indicating that degradation of collagen to its monomeric isoform may enhance ASM proliferation (Schuliga et al., 2010). In BTSM cells, monomeric collagen I has been shown to increase basal as well as PDGF-induced proliferation (Bonacci et al., 2003; Dekkers et al., 2007), which was tightly correlated with decreased contractility of intact BTSM strips (Dekkers et al., 2007). We now show that these findings can be translated to HTSM. Interestingly, these observations also suggest that BTSM is a representative experimental model for human ASM phenotype plasticity.
Collectively, the findings described above indicate that changes in the extracellular environment surrounding the ASM may contribute to ASM accumulation in asthma. Indeed, using a guinea pig model of allergic asthma, we have recently shown that ASM remodelling induced by repeated allergen challenges is inhibited by the integrin-blocking peptide Arg-Gly-Asp-Ser (RGDS), containing the RGD binding motif, which also inhibits human ASM cell proliferation induced by monomeric collagen I (Dekkers et al., 2010).
Increased ASM mass is considered to be a major factor contributing to airway hyper-responsiveness and decline in lung function in asthmatics (Lambert et al., 1993; Oliver et al., 2007). Our present findings demonstrating that exposure of intact HTSM preparations to collagen I and PDGF has an effect on contractile function provides more insight into the functional consequences of phenotype switching. Next to increased mass, however, asthmatic ASM also shows increased expression of contractile proteins (Leguillette et al., 2008), suggesting an increased rather than decreased contractile function. Phenotypic plasticity, however, is a dynamic and reversible process and in patients, episodes of increased levels of growth factors, Gq-coupled neurotransmitters such as ACh and inflammatory mediators – which may (synergistically) promote a proliferative, hypocontractile phenotype (Gosens et al., 2002; Halayko et al., 2008) – alternate with episodes of reduced levels. In vitro, the latter is mimicked by serum deprivation of cultured ASM cells, particularly in the presence of insulin or TGF-β, which redirects the hypocontractile phenotype to a (hyper)contractile state (Schaafsma et al., 2007; Hirota et al., 2009; Dekkers et al., 2009b). Similar processes could contribute to ASM hypercontractility and AHR in asthma.
In conclusion, our findings indicate that collagen I and PDGF induce a shift of human ASM phenotype to a hypocontractile, proliferative state, which has a functional effect on the muscle and may contribute to airway remodelling in asthma.
Acknowledgments
This study was financially supported by the Netherlands Asthma Foundation (NAF grant 3.2.03.36). We are grateful to the Department of Cardiothoracic Surgery of the University Medical Centre Groningen for providing the human tracheal sections. The authors wish to thank Carolina Elzinga for expert technical assistance.
Glossary
- ASM
airway smooth muscle
- BTSM:
bovine tracheal smooth muscle
- DMEM
Dulbecco's modified Eagle's medium
- ECM
extracellular matrix
- Emax
maximal contraction
- FBS
fetal bovine serum
- HTSM
human tracheal smooth muscle
- ITS
insulin, transferrin and selenium
- KH
Krebs–Henseleit
- PDGF
platelet-derived growth factor
- RGDS
arginine–glycine–aspartic acid–serine
Conflicts of interest
The authors declare no conflicts of interest.
References
- Araujo BB, Dolhnikoff M, Silva LF, Elliot J, Lindeman JH, Ferreira DS, et al. Extracellular matrix components and regulators in the airway smooth muscle in asthma. Eur Respir J. 2008;32:61–69. doi: 10.1183/09031936.00147807. [DOI] [PubMed] [Google Scholar]
- Bai TR, Cooper J, Koelmeyer T, Pare PD, Weir TD. The effect of age and duration of disease on airway structure in fatal asthma. Am J Respir Crit Care Med. 2000;162:663–669. doi: 10.1164/ajrccm.162.2.9907151. [DOI] [PubMed] [Google Scholar]
- Bonacci JV, Harris T, Stewart AG. Impact of extracellular matrix and strain on proliferation of bovine airway smooth muscle. Clin Exp Pharmacol Physiol. 2003;30:324–328. doi: 10.1046/j.1440-1681.2003.03838.x. [DOI] [PubMed] [Google Scholar]
- Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000;161:1720–1745. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- Dekkers BG, Schaafsma D, Nelemans SA, Zaagsma J, Meurs H. Extracellular matrix proteins differentially regulate airway smooth muscle phenotype and function. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1405–L1413. doi: 10.1152/ajplung.00331.2006. [DOI] [PubMed] [Google Scholar]
- Dekkers BG, Maarsingh H, Meurs H, Gosens R. Airway structural components drive airway smooth muscle remodeling in asthma. Proc Am Thorac Soc. 2009a;6:683–692. doi: 10.1513/pats.200907-056DP. [DOI] [PubMed] [Google Scholar]
- Dekkers BG, Schaafsma D, Tran T, Zaagsma J, Meurs H. Insulin-induced laminin expression promotes a hypercontractile airway smooth muscle phenotype. Am J Respir Cell Mol Biol. 2009b;41:494–504. doi: 10.1165/rcmb.2008-0251OC. [DOI] [PubMed] [Google Scholar]
- Dekkers BG, Bos IS, Gosens R, Halayko AJ, Zaagsma J, Meurs H. The integrin-blocking peptide RGDS inhibits airway smooth muscle remodeling in a guinea pig model of allergic asthma. Am J Respir Crit Care Med. 2010;181:556–565. doi: 10.1164/rccm.200907-1065OC. [DOI] [PubMed] [Google Scholar]
- Ebina M, Takahashi T, Chiba T, Motomiya M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma. A 3-D morphometric study. Am Rev Respir Dis. 1993;148:720–726. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- Fabisiak JP, Absher M, Evans JN, Kelley J. Spontaneous production of PDGF A-chain homodimer by rat lung fibroblasts in vitro. Am J Physiol. 1992;263:L185–L193. doi: 10.1152/ajplung.1992.263.2.L185. [DOI] [PubMed] [Google Scholar]
- Fernandes DJ, Bonacci JV, Stewart AG. Extracellular matrix, integrins, and mesenchymal cell function in the airways. Curr Drug Targets. 2006;7:567–577. doi: 10.2174/138945006776818700. [DOI] [PubMed] [Google Scholar]
- Freyer AM, Johnson SR, Hall IP. Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2001;25:569–576. doi: 10.1165/ajrcmb.25.5.4605. [DOI] [PubMed] [Google Scholar]
- Gosens R, Meurs H, Bromhaar MM, McKay S, Nelemans SA, Zaagsma J. Functional characterization of serum- and growth factor-induced phenotypic changes in intact bovine tracheal smooth muscle. Br J Pharmacol. 2002;137:459–466. doi: 10.1038/sj.bjp.0704889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosens R, Bromhaar MM, Tonkes A, Schaafsma D, Zaagsma J, Nelemans SA, et al. Muscarinic M(3) receptor-dependent regulation of airway smooth muscle contractile phenotype. Br J Pharmacol. 2004;141:943–950. doi: 10.1038/sj.bjp.0705709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosens R, Roscioni SS, Dekkers BG, Pera T, Schmidt M, Schaafsma D, et al. Pharmacology of airway smooth muscle proliferation. Eur J Pharmacol. 2008;585:385–397. doi: 10.1016/j.ejphar.2008.01.055. [DOI] [PubMed] [Google Scholar]
- Halayko AJ, Tran T, Gosens R. Phenotype and functional plasticity of airway smooth muscle: role of caveolae and caveolins. Proc Am Thorac Soc. 2008;5:80–88. doi: 10.1513/pats.200705-057VS. [DOI] [PubMed] [Google Scholar]
- Hirota JA, Nguyen TT, Schaafsma D, Sharma P, Tran T. Airway smooth muscle in asthma: phenotype plasticity and function. Pulm Pharmacol Ther. 2009;22:370–378. doi: 10.1016/j.pupt.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Hirst SJ, Twort CH, Lee TH. Differential effects of extracellular matrix proteins on human airway smooth muscle cell proliferation and phenotype. Am J Respir Cell Mol Biol. 2000;23:335–344. doi: 10.1165/ajrcmb.23.3.3990. [DOI] [PubMed] [Google Scholar]
- Johnson PR, Burgess JK, Underwood PA, Au W, Poniris MH, Tamm M, et al. Extracellular matrix proteins modulate asthmatic airway smooth muscle cell proliferation via an autocrine mechanism. J Allergy Clin Immunol. 2004;113:690–696. doi: 10.1016/j.jaci.2003.12.312. [DOI] [PubMed] [Google Scholar]
- Lambert RK, Wiggs BR, Kuwano K, Hogg JC, Pare PD. Functional significance of increased airway smooth muscle in asthma and COPD. J Appl Physiol. 1993;74:2771–2781. doi: 10.1152/jappl.1993.74.6.2771. [DOI] [PubMed] [Google Scholar]
- Leguillette R, Laviolette M, Bergeron C, Zitouni NB, Kogut P, Solway J, et al. Myosin, transgelin, and myosin light chain kinase: expression and function in asthma. Am J Respir Crit Care Med. 2008;179:194–204. doi: 10.1164/rccm.200609-1367OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Wang Y, Stephens NL. Serum deprivation induces a unique hypercontractile phenotype of cultured smooth muscle cells. Am J Physiol. 1998;274:C1206–C1214. doi: 10.1152/ajpcell.1998.274.5.C1206. [DOI] [PubMed] [Google Scholar]
- Meurs H, Gosens R, Zaagsma J. Airway hyperresponsiveness in asthma: lessons from in vitro model systems and animal models. Eur Respir J. 2008;32:487–502. doi: 10.1183/09031936.00023608. [DOI] [PubMed] [Google Scholar]
- Moir LM, Ward JP, Hirst SJ. Contractility and phenotype of human bronchiole smooth muscle after prolonged fetal bovine serum exposure. Exp Lung Res. 2003;29:339–359. doi: 10.1080/01902140303758. [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Ward JP, Hirst SJ. beta1-Integrins mediate enhancement of airway smooth muscle proliferation by collagen and fibronectin. Am J Respir Crit Care Med. 2005;171:217–223. doi: 10.1164/rccm.200408-1046OC. [DOI] [PubMed] [Google Scholar]
- Ohno I, Nitta Y, Yamauchi K, Hoshi H, Honma M, Woolley K, et al. Eosinophils as a potential source of platelet-derived growth factor B-chain (PDGF-B) in nasal polyposis and bronchial asthma. Am J Respir Cell Mol Biol. 1995;13:639–647. doi: 10.1165/ajrcmb.13.6.7576701. [DOI] [PubMed] [Google Scholar]
- Oliver MN, Fabry B, Marinkovic A, Mijailovich SM, Butler JP, Fredberg JJ. Airway hyperresponsiveness, remodeling, and smooth muscle mass: right answer, wrong reason? Am J Respir Cell Mol Biol. 2007;37:264–272. doi: 10.1165/rcmb.2006-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Lai D, Nguyen TT, Chan V, Matsuda T, Hirst SJ. Multiple beta 1 integrins mediate enhancement of human airway smooth muscle cytokine secretion by fibronectin and type I collagen. J Immunol. 2005;174:2258–2264. doi: 10.4049/jimmunol.174.4.2258. [DOI] [PubMed] [Google Scholar]
- Prockop DJ, Kivirikko KI. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem. 1995;64:403–434. doi: 10.1146/annurev.bi.64.070195.002155. [DOI] [PubMed] [Google Scholar]
- Roberts CR, Burke AK. Remodelling of the extracellular matrix in asthma: proteoglycan synthesis and degradation. Can Respir J. 1998;5:48–50. [PubMed] [Google Scholar]
- Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet. 1989;1:520–524. doi: 10.1016/s0140-6736(89)90067-6. [DOI] [PubMed] [Google Scholar]
- Schaafsma D, McNeill KD, Stelmack GL, Gosens R, Baarsma HA, Dekkers BG, et al. Insulin increases the expression of contractile phenotypic markers in airway smooth muscle. Am J Physiol Cell Physiol. 2007;293:C429–C439. doi: 10.1152/ajpcell.00502.2006. [DOI] [PubMed] [Google Scholar]
- Schuliga M, Ong SC, Soon L, Zal F, Harris T, Stewart AG. Airway smooth muscle remodels pericellular collagen fibrils: implications for proliferation. Am J Physiol Lung Cell Mol Physiol. 2010;298:L584–L592. doi: 10.1152/ajplung.00312.2009. [DOI] [PubMed] [Google Scholar]
- Taylor IK, Sorooshian M, Wangoo A, Haynes AR, Kotecha S, Mitchell DM, et al. Platelet-derived growth factor-beta mRNA in human alveolar macrophages in vivo in asthma. Eur Respir J. 1994;7:1966–1972. [PubMed] [Google Scholar]
- Woodruff PG, Dolganov GM, Ferrando RE, Donnelly S, Hays SR, Solberg OD, et al. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med. 2004;169:1001–1006. doi: 10.1164/rccm.200311-1529OC. [DOI] [PubMed] [Google Scholar]