

Abstract
Triple-negative breast cancer (TNBC) is a subgroup of breast cancer that is negative for estrogen and progesterone receptor and ERBB2 protein expression. It is characterized by its aggressive behavior and by the lack of targeted therapies. To identify new therapeutic targets in TNBC, we used real-time quantitative RT-PCR to analyze 63 TNBC samples in terms of their mRNA expression of 26 genes coding for the major proteins currently targeted by drugs used to treat other cancers or undergoing clinical trials in breast cancer. Six of the 26 genes tested (VEGFA, SRC, PARP1, PTK2, RAF1, and FGFR3) were significantly upregulated in 13% to 46% of the TNBCs. None of the 6 genes was specifically upregulated in the TNBCs compared with 3 other classical breast tumor subtypes. No association was observed between overexpression of these 6 genes (except for FGFR3) and PIK3CA mutation status. These results confirm the interest of targeting VEGFA and PARP1 in ongoing clinical trials in TNBC patients and also identify new target genes (SRC, PTK2, RAF1, and FGFR3). Clinical trials could be initiated easily with existing drugs. Our results also suggest that these target genes might serve as predictive biomarkers of the TNBC treatment response.
Keywords: breast cancer, triple negative, therapeutic target
Introduction
Breast cancer is the most common female malignancy in industrialized countries, affecting more than a million women per year worldwide. Breast cancer exhibits heterogeneous behavior, clinical outcomes, and treatment responses. Better understanding of breast cancer biology has led to significant improvements in patient survival. The discovery of steroid hormone dependence resulted in the development of estrogen receptor antagonists and aromatase inhibitors, which are currently the gold standard treatments for patients with hormone-receptor–positive breast tumors.1 Similarly, the detection of ERBB2 overexpression/amplification led to ERBB2 targeting with trastuzumab, and lapatinib was recently shown to significantly improve the survival of patients with ERBB2-overexpressing tumors. Hormone (estrogen and progesterone) receptor status, ERBB2 overexpression/amplification, and Ki67 expression are now used to predict the prognosis of breast cancers and to guide treatment.2 However, chemotherapy is the only available systemic therapy for women with so-called triple-negative breast cancer (TNBC), which lacks estrogen receptor (ER) and progesterone receptor (PR) expression and ERBB2 overexpression/gene amplification.
TNBC represents about 15% to 20% of breast cancers. It is characterized by an aggressive clinical course and poor prognosis, owing partly to the lack of targeted therapies.3-5 Most TNBCs have the “basal-like” molecular profile in gene expression arrays, but recent studies have suggested that TNBC is a heterogenous type of cancer; indeed, less common subtypes including “Claudin-low,” “HER2-enriched but without HER2 gene amplification,” and “molecular apocrine” have also been described in TNBC.6-8 Histologically and transcriptionally, TNBCs have many similarities to BRCA1-associated breast cancers, and most BRCA1-associated breast tumors are triple-negative and basal-like.9 BRCA1 is rarely mutated in sporadic breast cancer, but it has been suggested that BRCA1 (or associated pathways) is inactivated in triple-negative tumors via other molecular mechanisms. Toyama et al.10 showed that BRCA1 mRNA expression was significantly decreased in TNBCs compared with luminal subtype breast cancers.
The overlap between BRCA1-mutated breast cancers and triple-negative tumors suggests that some triple-negative tumors might respond to therapeutics targeting BRCA1-deficient cells, such as PARP inhibitors.11
The addition of iniparib, a PARP inhibitor, to chemotherapy improved the clinical benefit and survival of patients with metastatic TNBC without significantly increased toxic effects.12 On the basis of these results, a phase 3 trial evaluated overall survival and progression-free survival for women with metastatic TNBC but was negative. Given the structural and mechanistic differences between iniparib and other PARP inhibitors, these negative results do not necessarily imply a class effect, and further study of TNBCs with other PARP inhibitors should be encouraged,13 unless the drugs work in some molecular subtypes of TNBC but not others.
There is increasing evidence that the DNA-repair defects characteristic of BRCA1-related cancers, and especially defective homologous recombination, confer tumor sensitivity to certain systemic agents. Indeed, patients with TNBC have higher pCR (partial Complete Remission) rates than patients with non-TNBC, and neoadjuvant trials have shown higher relapse-free survival in TNBC patients who achieve pCR than in patients with residual disease.14 Despite this relative chemosensitivity, local and systemic TNBC relapse rates remain higher than in other breast cancer subtypes.15,16
Molecularly directed therapy targets tumor cells and the tumor microenvironment by blocking the effects of tumor-specific molecular changes. Targeted treatments are directed at a specific molecular target that is not present in normal breast cells and that is important for tumor growth and progression.
Targeted treatments tend to have fewer adverse effects, but their use must be guided by biomarker assays. For example, immunohistochemical assays are used to identify the therapeutic target in the breast tumor before prescribing hormone therapy or trastuzumab, and detection of EGFR activating mutations is an obligatory prerequisite to EGFR inhibitor prescription in lung cancer.
To identify new therapeutic targets in TNBC, we applied real-time quantitative RT-PCR assays to 63 triple-negative tumor samples. We quantified the mRNA expression of a panel of 26 genes coding for the major proteins that are currently targeted by drugs used to treat other cancer types or that are undergoing clinical trials in breast cancer.
Results
mRNA expression of the 26 target genes in the 63 triple-negative breast tumors
We used real-time quantitative RT-PCR to analyze mRNA expression of the 26 target genes in a series of 63 TNBCs and 12 normal breast tissues. The mRNA levels of all 26 target genes were high in both the normal and tumorous breast tissues and were thus reliably quantifiable by real-time quantitative RT-PCR based on fluorescence SYBR Green method (Cycle Threshold, Ct < 32). Target gene mRNA levels in the 63 TNBCs were expressed relative to the median mRNA levels observed in the 12 normal breast tissues. For each gene, normalized mRNA values of 3 or more were considered to represent gene overexpression in tumor samples, and values 0.33 or less represented gene underexpression. Medians and ranges of mRNA levels for the 26 target genes are shown in Table 1, along with the percentages of overexpression or underexpression.
Table 1.
mRNA Expression of MKI67 and the 26 Target Genes in TNBC Tissues Relative to Normal Breast Tissues and Percentages of Overexpression and Underexpression
| Genes | Normal (n = 12) | TNBC (n = 63) | P a | Overexpression, % | Underexpression, % |
|---|---|---|---|---|---|
| BRAF | 1.0 (0.74-1.19)b | 0.63 (0.15-2.03)b | 0.00016 | 0.0 | 12.7 |
| CSF1R | 1.0 (0.48-2.75) | 0.80 (0.16-9.04) | NS | 4.8 | 12.7 |
| EGFR | 1.0 (0.64-1.81) | 0.52 (0.04-115.94) | 0.00021 | 3.2 | 27.0 |
| FGFR1 | 1.0 (0.67-1.27) | 0.29 (0.05-10.20) | 0.0000098 | 3.2 | 54.0 |
| FGFR2 | 1.0 (0.56-1.42) | 0.56 (0.01-25.30) | NS | 7.9 | 38.1 |
| FGFR3 | 1.0 (0.08-2.92) | 2.60 (0.00-60.83) | 0.049 | 41.3 | 11.1 |
| FLT3 | 1.0 (0.24-1.88) | 0.47 (0.00-4.30) | 0.046 | 1.6 | 42.9 |
| HGF | 1.0 (0.39-1.75) | 0.23 (0.00-1.04) | 0.00000036 | 0.0 | 76.2 |
| IGF1R | 1.0 (0.07-1.77) | 0.64 (0.05-27.04) | NS | 6.3 | 34.9 |
| JAK2 | 1.0 (0.70-1.59) | 1.17 (0.23-5.52) | NS | 7.9 | 7.9 |
| KIT | 1.0 (0.13-2.64) | 0.11 (0.00-3.78) | 0.0000088 | 1.6 | 79.4 |
| KITLG | 1.0 (0.20-1.99) | 0.55 (0.06-5.85 ) | 0.0073 | 4.8 | 23.8 |
| MET | 1.0 (0.31-1.74) | 0.61 (0.00-7.55) | NS | 7.9 | 31.7 |
| PARP1 | 1.0 (0.84-1.45) | 2.26 (0.68-29.75) | 0.000064 | 38.1 | 0.0 |
| PDGFRA | 1.0 (0.67-1.89) | 0.33 (0.04-2.14) | 0.0000017 | 0.0 | 49.2 |
| PDGFRB | 1.0 (0.63-1.45) | 0.38 (0.06-5.85) | 0.000034 | 1.6 | 44.4 |
| PTGS2 | 1.0 (0.19-2.56) | 0.27 (0.00-32.69) | 0.0041 | 9.5 | 52.4 |
| PTK2 | 1.0 (0.69-1.27) | 1.78 (0.28-45.81) | 0.0044 | 15.9 | 1.6 |
| RAF1 | 1.0 (0.73-1.45) | 1.37 (0.09-11.66) | 0.025 | 12.7 | 1.6 |
| RET | 1.0 (0.00-2.17) | 0.68 (0.00-30.04) | NS | 11.1 | 39.7 |
| SRC | 1.0 (0.77-1.48) | 2.07 (0.33-26.61) | 0.000039 | 20.6 | 1.6 |
| STAT3 | 1.0 (0.74-1.49) | 0.89 (0.30-4.25) | NS | 1.6 | 4.8 |
| VEGFA | 1.0 (0.79-1.93) | 2.65 (0.56-39.12) | 0.0000091 | 46.0 | 0.0 |
| VEGFR1 | 1.0 (0.67-1.90) | 0.80 (0.11-3.54) | NS | 1.6 | 4.8 |
| VEGFR2 | 1.0 (0.55-2.10) | 0.38 (0.12-2.09) | 0.0000061 | 0.0 | 36.5 |
| VEGFR3 | 1.0 (0.27-2.89) | 0.23 (0.00-4.23) | 0.0000065 | 1.6 | 73.0 |
| MKI67 | 1.0 (0.18-3.70) | 24.71 (6.98-113.94) | <0.0000001 | 100.0 | 0.0 |
Note: NS = not significant; TNBC = triple-negative breast cancer.
Kruskal-Wallis H test.
Median (range) of mRNA levels relative to normal.
Eighteen (69.2%) of the 26 genes were significantly dysregulated in the TNBCs. Six (23.1%) genes were mainly upregulated (FGFR3, PARP1, PTK2, RAF1, SRC and VEGFA), and 12 (46.1%) were down-regulated.
In the same set of 63 TNBC samples, we also examined the expression of MKI67, which encodes the proliferation-related antigen Ki-67. As expected, MKI67 was upregulated in all tumor samples.
Table 1 shows the mRNA expression levels of the 26 target genes in breast tumors relative to the TBP endogenous control. The same results were obtained when other endogenous RNA controls (RPLP0 or PPIA) were used.
Comparison of mRNA levels of the 6 upregulated genes according to the tumor subtype
We then examined whether the 6 genes upregulated in TNBCs were specific to this tumor subtype by analyzing their mRNA expression in the other 3 major breast tumor subtypes: NNP (ERα-negative, PR-negative, ERBB2-positive tumors), PPN (ERα-positive, PR-positive, ERBB2-negative tumors), and PPP (ERα-positive, PR-positive, ERBB2-positive tumors). The results are shown in Figure 1.
Figure 1.
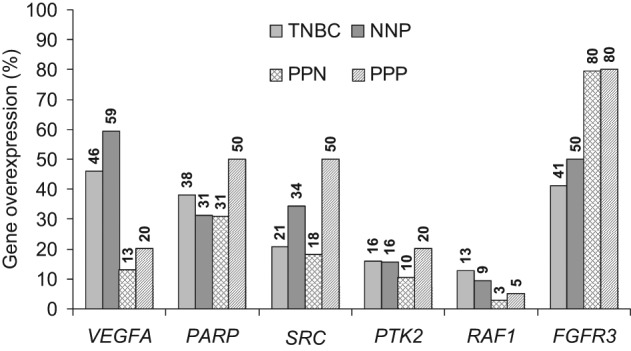
Distribution and frequency of the overexpression of the 6 upregulated genes (VEGFA, PARP1, SRC, PTK2, RAF1, and FGFR3) in TNBCs relative to the 3 other main breast cancer subgroups, NNP (ERα-negative, PR-negative, ERBB2-positive tumors), PPN (ERα-positive, PR-positive, ERBB2-negative tumors), and PPP (ERα-positive, PR-positive, ERBB2-positive tumors).
None of the 6 genes were specifically upregulated in the TNBCs compared with the other 3 tumor subtypes. Nevertheless, it is noteworthy that VEGFA was predominantly upregulated in ERα-negative tumors (TNBC and NNP subtypes) (P = 0.00001 vs. the PPN and PPP subtypes). FGFR3 was significantly more strongly overexpressed in the ERα-positive tumors (PPN and PPP) than in the ERα-negative tumors (TNBC and NNP) (P = 0.00002). SRC overexpression was significantly stronger in the ERBB2-positive tumors (NNP and PPP) than in the ERBB2-negative tumors (PPN and TNBC) (P = 0.006).
Comparison of mRNA levels of the 6 upregulated genes according to PIK3CA status
In the TKR–RAS-PI3K-AKT signaling pathway, PIK3CA is the oncogene that shows the highest frequency of gain-of-function mutations in breast cancer. Indeed, EGFR, BRAF, and KRAS, although frequently mutated in other cancers (colon, lung, etc.), are rarely mutated in breast cancer. PIK3CA status may play an important role in the response to therapies targeting tyrosine kinase receptors in breast cancer.17,18 We therefore examined the overall series of 154 tumor samples (4 subgroups) for PIK3CA mutations in exons 9 and 20. PIK3CA mutations were detected in 6 (9.5%) of 63 TNBCs, 7 (21.8%) of 32 NNP tumors, 16 (41.0%) of 39 PPN tumors, and 7 (35.0%) of 20 PPP tumors. PIK3CA mutations were thus detected in 13 (13.7%) of the 95 ERα-negative tumors and in 23 (39.0%) of the 59 ERα-positive tumors, in agreement with the literature.19 We then looked for an association between the 6 overexpressed genes and PIK3CA mutation status. A positive association, at the limit of statistical significance (P = 0.045), was only observed between FGFR3 overexpression and PIK3CA mutation status.
Discussion
We used real-time RT-PCR to analyze the expression of 26 selected genes in a large sample of breast tumors. We found that 6 genes (VEGFA, FGFR3, PARP1, SRC, PTK2/FAK, and RAF1) were frequently upregulated in the TNBC subgroup relative to normal breast tissue. However, none of these 6 genes was specifically upregulated in TNBCs compared with the other 3 main breast tumor subgroups (NNP, PPN, and PPP), further highlighting the heterogeneity of TNBC subgroup and the difficulty of finding a specific target.6-8
Several genes were also down-regulated in TNBCs. It is more difficult to restore tumor-suppressor protein expression than to inhibit oncoprotein overexpression. Moreover, the observed down-regulations could be partly explained by differences in proportion of epithelial cells and stromal cells (fibroblasts, adipocytes, endothelial cells, and circulating cells) between normal control breast tissue and tumor samples. Likewise, underexpression of stroma cell–specific genes could be explained by a lower abundance of a particular cell type (fibroblasts, adipocytes, endothelial cells, or circulating cells) in TNBCs relative to normal breast tissue.
By using high-resolution oligonucleotide comparative genomic hybridization arrays, Andre et al.20 demonstrated that 3 genes including VEGFA were specifically gained in TNBC. Moreover, those investigators established that the occurrence of a DNA gain leads to an unregulated overexpression of mRNA and concluded that such dysregulated genes may represent novel therapeutic targets.
It is noteworthy that PARP inhibitors are effective on tumors that carry a DNA-repair defect, such as BRCA1-deficient tumors.11,21 The most appropriate biomarkers for anti-PARP sensitivity will thus be markers of DNA-repair defects, particularly homologous recombination DNA-repair defects, in non–BRCA1- and non–BRCA2-associated tumors. However, PARP upregulation, which was demonstrated at the protein level by Ossovskaya et al.,22 could be an additional biomarker of PARP inhibitor sensitivity in triple-negative breast tumors. These results and ours for VEGFA and PARP1 support the validity of current clinical trials testing bevacizumab and PARP inhibitors such as BSI-2014 and AZD2281/Olaparib21 in TNBC patients. Concerning the other 4 genes identified (RAF1, SRC, FGFR3, and FAK), clinical trials in breast cancer are less advanced.
Recently it has been shown that accumulation of recurrent RAF1 gene amplification is due to EZH2 expression–mediated downregulation of DNA damage repair in breast tumor initiating cells (BTICs), which activates p-ERK-β-catenin signaling to promote BTIC expansion.23 Overexpression of Polycomb protein EZH2, essential in stem cell self-renewal, has been linked to breast cancer progression. This amplification of RAF1 may be compatible with the RAF1 overexpression that we detected in TBNCs. RAF1 is targeted by sorafenib, a drug currently used in the treatment of kidney cancer and hepatocellular carcinoma. Several clinical trials of chemotherapy with or without sorafenib will start shortly in patients with advanced breast cancer.24
Several inhibitors of SRC, such as dasatinib and bosutinib, are currently being evaluated in breast cancer patients.25 It has been suggested that SRC could be a potential target for the treatment of TNBC because SRC expression was more intense in cytoplasm and on the membrane of triple-negative samples than in non–triple-negative samples.26 However, our results suggest that it is mainly patients with ERBB2-overexpressing tumors who are likely to benefit from SRC inhibitors (Figure 1). Recently, Zhang et al.27 showed that increased SRC activation conferred considerable trastuzumab resistance to breast cancer cells and correlated with clinical trastuzumab resistance. Thus, SRC targeting might overcome trastuzumab resistance.27
According to our results at the transcriptional level, FGFR3 expression in invasive breast cancer was not significantly associated with specific clinicopathological/molecular parameters.28 Yom et al.29 demonstrated that FAK overexpression by immunohistochemistry and amplification by FISH were significantly associated with the triple-negative subgroup, whereas our transcript results did not associate FAK overexpression with a particular subgroup. No drugs targeting FGFR3 or FAK are currently being tested in breast cancer, although results cited here support such trials. Dovitinib (a specific FGFR3 inhibitor), PD173074 (a highly potent selective pan-FGFR inhibitor), and BIBF-1120 (a multityrosine kinase inhibitor), which target FGFR3 as well as PF-00562271 and PND-1186 (two FAK inhibitors), are currently in phase 1 trials.30-34
It is noteworthy that Turner et al. 35 identified FGFR2 gene amplification (associated with strong overexpression) in 4% (6/165) of TNBCs but not in other subtypes (0/214). Our results show FGFR2 overexpression in 8% of 63 TNBCs but also in 8% of 39 PPNs and 5% of 20 PPPs. None of 32 NNPs overexpressed FGFR2 (data not shown). These results confirm this rare genetic alteration as a potential therapeutic target in breast cancer but not specifically for TNBCs.
The upregulated genes identified here are only upregulated in a fraction (13%-46%) of TNBCs. It will therefore be necessary to test their value as predictive biomarkers of the response to targeted drugs. However, the best such a biomarker is not necessarily the target itself. Indeed, VEGFA (overexpressed in 46% of TNBCs), which codes for a secreted protein measurable in serum, does not seem to predict the response to the anti-VEGF antibody bevacizumab (Avastin®) in some cancers.36
Concerning the mutation status of the PIK3CA gene, which could play a major role in the response to therapies targeting tyrosine kinase receptors in breast cancer,17,18 we found a positive correlation between PIK3CA mutation and FGFR3 overexpression. This correlation seems to be indirect, being related mainly to ERα-positive tumor status. Indeed, FGFR3 is upregulated in ERα-positive tumors, and PIK3CA mutation is also more frequent in our ERα-positive tumor population (39% vs. 13.7% in the ERα-negative population, in agreement with the literature).19 This suggests that PIK3CA mutation does not influence the expression of these 6 genes.
In conclusion, our results confirm the validity of VEGFA and PARP1 targeting in ongoing clinical trials in TNBC patients and also identify new potential target genes (SRC, PTK2, RAF1, and FGFR3). Clinical trials of drugs inhibiting the products of these genes could be initiated rapidly with existing drugs. Our results also warrant studies of these target genes as putative predictive biomarkers for selecting the patients most likely to benefit from these drugs.
Materials and Methods
Patients and samples
We analyzed tumor samples from 63 TNBC patients treated at Institut Curie, René Huguenin Hospital, Saint-Cloud, France. Tumor samples containing more than 70% of tumor cells were considered suitable for analysis. Immediately after surgery, the tumor samples were placed in liquid nitrogen until RNA extraction.
The patients met the following criteria: primary unilateral nonmetastatic breast carcinoma; complete clinical, histological, and biological information available; no radiotherapy or chemotherapy before surgery; and full follow-up at Institut Curie, René Huguenin Hospital. Patients underwent physical examinations every 3 months for 2 years, then annually. Mammograms were done annually. Median follow-up was 7.5 years (range 8 months to 29 years). Twenty-six patients relapsed; the distribution of first relapse events was as follows: 22 metastases alone and 4 with both metastases and local relapse. Standard prognostic factors are shown in Table 2.
Table 2.
Characteristics of the 63 TNBCs
| Number of patients | Number of patients with metastasis (%) | P valuea | |
|---|---|---|---|
| Total | 63 | 26 (41.3) | – |
| Age | |||
| ≤50 y | 24 | 11 (45.8) | NS |
| >50 y | 39 | 15 (38.5) | (0.68) |
| SBR histological gradeb | |||
| I | 0 | 0 | 0.041 |
| II | 8 | 6 (75.0) | |
| III | 50 | 18 (36.0) | |
| Lymph node statusc | |||
| 0 | 27 | 12 (44.4) | NS |
| 1-3 | 28 | 10 (35.7) | (0.40) |
| >3 | 7 | 4 (57.1) | |
| Macroscopic tumor sizec | |||
| ≤25 mm | 24 | 10 (41.7) | NS |
| >25 mm | 38 | 16 (42.1) | (0.71) |
| Histological types | |||
| Lobular | 0 | 0 | NS |
| Ductal | 58 | 25 (43) | (0.74) |
| Other | 5 | 1 (20) | |
| PIK3CA mutation status | |||
| Wild-type | 57 | 23 (40) | NS |
| Mutated | 6 | 3 (50) | (0.52) |
Note: NS = not significant; SRB = Scarff Bloom Richardson classification.
Log-rank test.
Data available for 58 patients.
Data available for 62 patients.
ER and PR were routinely analyzed at the time of diagnosis on frozen tumors by ligand binding assay until 1988, by enzyme immunoassay (ER-EIA Monoclonal, PgR-EIA Monoclonal, Abbott Laboratories, Abbott Park, IL) between 1988 and 2000, and then by immunohistochemistry on paraffin sections. HER2 status was routinely analyzed by immunohistochemistry (with confirmation by FISH of the 2+ cases). For this study, the triple negative status of the tumors was confirmed by RT-qPCR on frozen tumors.37,38
To investigate specific dysregulation of candidate genes in the TNBC/NNN subtype, we analyzed a panel of RNAs from the 3 other major breast tumor subtypes: 32 NNP (ERα-negative, PR-negative, ERBB2-positive tumors), 39 PPN (ERα-positive, PR-positive, ERBB2-negative tumors), and 20 PPP (ERα-positive, PR-positive, ERBB2-positive tumors).
Twelve specimens of adjacent normal breast tissue from breast cancer patients or normal breast tissue from women undergoing cosmetic breast surgery were used as sources of normal RNA.
Real-time RT-PCR
The theoretical and practical aspects of real-time quantitative PCR have been described in detail elsewhere.39
By studying the literature, we selected 26 genes coding for the major proteins currently targeted by drugs used to treat other cancers or for proteins targeted in ongoing breast cancer clinical trials (Suppl. Table S1). The 26 target genes tested in this study are listed in Table 3.
Table 3.
The 26 Selected Genes
| Gene symbols | Alternative symbols | Names of genes | Chromosome location | Genbank accession numbers |
|---|---|---|---|---|
| BRAF | v-raf murine sarcoma viral oncogene homolog B1 | 7q34 | NM_004333 | |
| CSF1R | colony stimulating factor 1 receptor | 5q33-q35 | NM_005211 | |
| EGFR | epidermal growth factor receptor | 7p12 | NM_005228 | |
| FGFR1 | fibroblast growth factor receptor 1 | 8p11.2-p11.1 | NM_015850 | |
| FGFR2 | fibroblast growth factor receptor 2 | 10q26 | NM_000141 | |
| FGFR3 | fibroblast growth factor receptor 3 | 4p16.3 | NM_000142 | |
| FLT3 | fms-related tyrosine kinase 3 | 13q12 | NM_004119 | |
| HGF | hepatocyte growth factor | 7q21.1 | NM_000601 | |
| IGF1R | insulin-like growth factor 1 receptor | 15q26.3 | NM_000875 | |
| JAK2 | janus kinase 2 | 9p24 | NM_004972 | |
| KIT | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene | 4q11-q12 | NM_000222 | |
| KITLG | KIT ligand | 12q22 | NM_000899 | |
| MET | met proto-oncogene (hepatocyte growth factor receptor) | 7q31 | NM_000245 | |
| PARP1 | poly (ADP-ribose) polymerase 1 | 1q41-q42 | NM_001618 | |
| PDGFRA | platelet-derived growth factor receptor A | 4q11-q13 | NM_006206 | |
| PDGFRB | platelet-derived growth factor receptor B | 5q31-q32 | NM_002600 | |
| PTGS2 | COX2 | Prostaglandin-endoperoxide synthase 2 | 1q25.2-q25.3 | NM_000963 |
| PTK2 | FAK | PTK2 protein tyrosine kinase 2 | 8q24-qter | NM_005607 |
| RAF1 | v-raf-1 murine leukemia viral oncogene homolog 1 | 3p25 | NM_002880 | |
| RET | ret proto-oncogene | 10q11.2 | NM_020975 | |
| SRC | v-src sarcoma viral oncogene homolog (avian) | 20q12-q13 | NM_005417 | |
| STAT3 | signal transducer and activator of transcription 3 | 17q21.31 | NM_003150 | |
| VEGFA | vascular endothelial growth factor A | 6p12 | NM_003376 | |
| VEGFR1 | FLT1 | fms-related tyrosine kinase 1 | 13q12 | NM_002019 |
| VEGFR2 | KDR | kinase insert domain receptor | 4q11-q12 | NM_002253 |
| VEGFR3 | FLT4 | fms-related tyrosine kinase 4 | 5q35.3 | NM_002020 |
| MKI67 | antigen identified by monoclonal antibody Ki-67 | 10q25-qter | NM_002417 |
The precise amount of total RNA added to each reaction mix (based on optical density) and its quality (i.e., lack of extensive degradation) are both difficult to assess. We therefore also quantified transcripts of 3 endogenous RNA control genes involved in various cellular metabolic pathways, namely TBP 37 (Genbank accession NM_003194), which encodes the TATA box-binding protein (a component of the DNA-binding protein complex TFIID); RPLP0 40 (also known as 36B4; NM_001002), which encodes human acidic ribosomal phosphoprotein P0; and PPIA,41 which encodes peptidylprolyl isomerase A (also known as cyclophilin A; NM_021130).
Primers for TBP, RPLP0, ERα, PR, ERBB2, MKI67, and the 26 target genes were chosen with the assistance of the Oligo 5.0 computer program (National Biosciences, Plymouth, MN). We searched the dbEST and nr databases to confirm the total gene specificity of the nucleotide sequences chosen as primers and the absence of single nucleotide polymorphisms. In particular, the primer pairs were selected to be unique relative to the sequences of closely related family member genes or of the corresponding retropseudogenes. The nucleotide sequences of the oligonucleotide primers used to amplify MKI67 and the 26 target genes are shown in Suppl. Table S2.
Each sample was normalized on the basis of its TBP (or RPLPO or PPIA) content. Results, expressed as N-fold differences in target gene expression relative to the TBP (or RPLPO) gene and termed “Ntarget,” were determined as Ntarget = 2ΔCtsample, where the ΔCt value of the sample is determined by subtracting the average Ct value of the target gene from the average Ct value of the TBP (or RPLP0 or PPIA) gene.37-39 The Ntarget values of the samples were subsequently normalized such that the median of the 12 normal breast tissue Ntarget values was 1.
RNA extraction, cDNA synthesis, and PCR conditions were as described elsewhere.39
PIK3CA mutation screening
PIK3CA mutations were screened for in cDNA fragments obtained by RT-PCR amplification of exons 9 and 20 with their flanking exons. Details of the primers and PCR conditions are available on request. The amplified products were sequenced by using the BigDye terminator sequencing kit on an ABI Prism 3130 automatic DNA sequencer (Applied Biosystems, Courtabæuf, France). Sequences were compared with the corresponding cDNA reference sequence (NM_006218).
Statistical analysis
As the mRNA levels did not fit a Gaussian distribution, (a) the mRNA levels in each subgroup of samples were characterized by their median values and ranges rather than their mean values and coefficients of variation, and (b) relationships between the molecular markers and clinical and biological parameters were tested by using the nonparametric Kruskal-Wallis test (links between 1 qualitative parameter and 1 quantitative parameter). Differences between 2 populations were considered significant at confidence levels greater than 95% (P < 0.05).
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Comité départemental des Hauts-de-Seine de la Ligue Nationale Contre le Cancer, Conseil régional d’Ile-de-France, Cancéropôle Ile-de-France and by the Association d’Aide à la Recherche Cancérologique de Saint-Cloud (ARCS).
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
References
- 1. Lumachi F, Luisetto G, Basso SM, Basso U, Brunello A, Camozzi V. Endocrine therapy of breast cancer. Curr Med Chem. 2011;18:513-22 [DOI] [PubMed] [Google Scholar]
- 2. Awada A, Bozovic-Spasojevic I, Chow L. New therapies in HER2-positive breast cancer: a major step towards a cure of the disease? Cancer Treat Rev. Epub 2012. Feb 2. [DOI] [PubMed] [Google Scholar]
- 3. Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: therapeutic options. Lancet Oncol. 2007;8:235-44 [DOI] [PubMed] [Google Scholar]
- 4. Pal SK, Mortimer J. Triple-negative breast cancer: novel therapies and new directions. Maturitas. 2009;63:269-74 [DOI] [PubMed] [Google Scholar]
- 5. Podo F, Buydens LM, Degani H, et al. Triple-negative breast cancer: present challenges and new perspectives. Mol Oncol. 2010;4:209-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma CX, Luo J, Ellis MJ. Molecular profiling of triple negative breast cancer. Breast Dis. 2010;32:73-84 [DOI] [PubMed] [Google Scholar]
- 9. Atchley DP, Albarracin CT, Lopez A, et al. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J Clin Oncol. 2008;26:4282-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toyama T, Yamashita H, Kondo N, et al. Frequently increased epidermal growth factor receptor (EGFR) copy numbers and decreased BRCA1 mRNA expression in Japanese triple-negative breast cancers. BMC Cancer. 2008;8:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235-44 [DOI] [PubMed] [Google Scholar]
- 12. O’Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364:205-14 [DOI] [PubMed] [Google Scholar]
- 13. Javle M, Curtin NJ. The potential for poly (ADP-ribose) polymerase inhibitors in cancer therapy. Ther Adv Med Oncol. 2011;3:257-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26:1275-81 [DOI] [PubMed] [Google Scholar]
- 15. Haffty BG, Yang Q, Reiss M, et al. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol. 2006;24:5652-7 [DOI] [PubMed] [Google Scholar]
- 16. Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429-34 [DOI] [PubMed] [Google Scholar]
- 17. Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395-402 [DOI] [PubMed] [Google Scholar]
- 18. Eichhorn PJ, Gili M, Scaltriti M, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68:9221-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andre F, Job B, Dessen P, et al. Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res. 2009;15:441-51 [DOI] [PubMed] [Google Scholar]
- 21. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123-34 [DOI] [PubMed] [Google Scholar]
- 22. Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of poly (ADP-Ribose) polymerase-1 (PARP1) in triple-negative breast cancer and other primary human tumor types. Genes Cancer. 2010;1:812-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang CJ, Yang JY, Xia W, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19:86-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moreno-Aspitia A, Morton RF, Hillman DW, et al. Phase II trial of sorafenib in patients with metastatic breast cancer previously exposed to anthracyclines or taxanes: North Central Cancer Treatment Group and Mayo Clinic Trial N0336. J Clin Oncol. 2009;27:11-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mayer EL, Krop IE. Advances in targeting SRC in the treatment of breast cancer and other solid malignancies. Clin Cancer Res. 2010;16:3526-32 [DOI] [PubMed] [Google Scholar]
- 26. Tryfonopoulos D, Walsh S, Collins DM, et al. Src: a potential target for the treatment of triple-negative breast cancer. Ann Oncol. 2011;22:2234-40 [DOI] [PubMed] [Google Scholar]
- 27. Zhang S, Huang WC, Li P, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17:461-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuroso K, Imai Y, Kobayashi M, et al. Immunohistochemical detection of fibroblast growth factor receptor 3 in human breast cancer: correlation with clinicopathological/molecular parameters and prognosis. Pathobiology. 2010;77:231-40 [DOI] [PubMed] [Google Scholar]
- 29. Yom CK, Noh DY, Kim WH, Kim HS. Clinical significance of high focal adhesion kinase gene copy number and overexpression in invasive breast cancer. Breast Cancer Res Treat. 2011;128:647-55 [DOI] [PubMed] [Google Scholar]
- 30. Molckovsky A, Siu LL. First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American Society of Clinical Oncology meeting. J Hematol Oncol. 2008;1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roberts WG, Ung E, Whalen P, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68:1935-44 [DOI] [PubMed] [Google Scholar]
- 32. Mross K, Stefanic M, Gmehling D, et al. Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res. 2010;16:311-9 [DOI] [PubMed] [Google Scholar]
- 33. Huynh H. Molecularly targeted therapy in hepatocellular carcinoma. Biochem Pharmacol. 2010;80:550-60 [DOI] [PubMed] [Google Scholar]
- 34. Walsh C, Tanjoni I, Uryu S, et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Ther. 2010;9:778-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Turner N, Lambros MB, Horlings HM, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29:2013-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dowlati A, Gray R, Sandler AB, Schiller JH, Johnson DH. Cell adhesion molecules, vascular endothelial growth factor, and basic fibroblast growth factor in patients with non-small cell lung cancer treated with chemotherapy with or without bevacizumab—an Eastern Cooperative Oncology Group Study. Clin Cancer Res. 2008;14:1407-12 [DOI] [PubMed] [Google Scholar]
- 37. Bieche I, Onody P, Laurendeau I, et al. Real-time reverse transcription-PCR assay for future management of ERBB2-based clinical applications. Clin Chem. 1999;45:1148-56 [PubMed] [Google Scholar]
- 38. Bieche I, Parfait B, Laurendeau I, Girault I, Vidaud M, Lidereau R. Quantification of estrogen receptor alpha and beta expression in sporadic breast cancer. Oncogene. 2001;20:8109-15 [DOI] [PubMed] [Google Scholar]
- 39. Bieche I, Parfait B, Le Doussal V, et al. Identification of CGA as a novel estrogen receptor-responsive gene in breast cancer: an outstanding candidate marker to predict the response to endocrine therapy. Cancer Res. 2001;61:1652-8 [PubMed] [Google Scholar]
- 40. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817-26 [DOI] [PubMed] [Google Scholar]
- 41. McNeill RE, Miller N, Kerin MJ. Evaluation and validation of candidate endogenous control genes for real-time quantitative PCR studies of breast cancer. BMC Mol Biol. 2007;8:107. [DOI] [PMC free article] [PubMed] [Google Scholar]