

Abstract
Shewanella yellow enzyme (SYE-4), a novel recombinant enoate reductase, was screened against a variety of different substrates bearing an activated double bond, such as unsaturated cyclic ketones, diesters, and substituted imides. Dimethyl- and ethyl esters of 2-methylmaleic acid were selectively reduced to (R)-configured succinic acid derivatives and various N-substituted maleimides furnished the desired (R)-products in up to >99% enantiomeric excess. Naturally occurring (+)-carvone was selectively reduced to (−)-cis-dihydrocarvone and (−)-carvone was converted to the diastereomeric product, respectively. Overall SYE-4 proved to be a useful biocatalyst for the selective reduction of activated C C double bonds and complements the pool of synthetic valuable enoate reductases.
Keywords: Enoate reductase, Shewanella yellow enzyme, Bioreduction, Biocatalysis
Graphical abstract

1. Introduction
Biocatalytic conversion of alkenes to chiral alkanes has received significant attention in recent years as a complementary strategy to metal-assisted hydrogenation reactions. This biotransformation is merely a nucleophilic attack by hydride originating from NAD(P)H across an activated C C double bond via a Michael-type reaction.1, 2, 3, 4 Enzymes involved are referred to as enoate reductases (EREDs).5 These biocatalysts are able to reduce a wide variety of substrates, such as conjugated enals, enones, α,β-unsaturated carboxylic acids, imides, nitroalkenes, and ynones (Fig. 1).1, 6, 7, 8
Fig. 1.

Schematic overview of enoate reductase mediated reduction of activated C C double bonds.
Recently, old yellow flavoprotein (OYE) from Saccharomyces carlsbergensis was re-discovered for the chemo- and stereoselective conversion of enones and nitro-olefins into saturated compounds.9, 10 Ligand binding properties of related enzymes and mechanistic aspects of the bioreduction were investigated extensively.11, 12 The catalytic cycle was found to proceed via a ping–pong bi–bi mechanism. The catalytic potential (reducing ‘activated’ alkenes in the presence of NADPH) and possible synthetic applications of EREDs were partly discovered in the last decade.13 α,β-Unsaturated cyclic ketones represent the most common substrates for the majority of the old yellow enzyme (OYE) family, producing optically active saturated ketones.14, 15
Much less is known about the asymmetric bioreduction of carboxylic acid derivatives. Whereas simple alkyl esters seem to be accepted to a certain degree, ester hydrolysis represents a major side reaction in whole-cell bioreductions.16, 17 Another interesting substrate class is N-protected α-methylmaleimides that are stereoselectively reduced by plant cell cultures (tobacco, liverwort) or whole cells of the cyanobacterium Synechococcus sp. to yield (R)-2-methylsuccinimide.18
Although the remarkable synthetic potential of enoate reductases has been recognized long ago,13, 19 preparative scale applications were severely impeded by two major problems: application of simple to use whole-cell systems (most prominent baker's yeast, but also fungi and yeasts, such as Geotrichum candidum, Rhodotorula rubra, Beauveria bassiana, Aspergillus niger, etc.) is largely compromised by undesired side reactions, such as carbonyl reduction (catalyzed by competing alcohol dehydrogenases/carbonyl reductases),20, 21, 22, 23 or ester hydrolysis (mediated by carboxyl ester hydrolases).24 On the other hand, the first generation of isolated (cloned) enoate reductases was obtained from (strict or facultative) anaerobe microbes, which were inapplicable to preparative-scale transformations owing to their sensitivity toward traces of molecular oxygen. Recently, this bottleneck was resolved by providing oxygen-stable OYEs from yeasts.7, 9, 25, 26
Homologs of old yellow enzyme from Shewanella oneidensis (SYEs) were shown to be catalytically active when recombinantly expressed as GST fusion proteins. S. oneidensis is an important model organism in bioremediation studies because it is characterized by unique respiratory capabilities, such as the possibility to reduce heavy metals.27 Recently, the biochemical properties of different SYE originating enzymes were compared to those of other OYE family members28 and also the structure of SYE-4 was described.29
We investigated the substrate scope of the novel enoate reductase from Shewanella sp. in details by using a diverse range of activating groups, such as ketones, imides, carboxylic ester moieties, as well as by varying cyclic and acyclic structural scaffolds.
2. Results and discussion
The results from all biotransformations of various substrates with SYE-4 protein are summarized in Table 1. All biotransformations were performed with the crude cell extract of a SYE-4 over expressing Escherichia coli strain and in the presence of a NADP+/G6P/G6PDH cofactor recycling system. The bioreduction of 2-methylcyclopent-2-enone (1a) gave 92% of the desired product with a moderate enantiomeric purity of 52% after 6 h reaction time. The time screening for substrate 1a showed a gradual decrease in enantiomeric excess caused by either the acidity of α-protons or a competing enoate reductase type enzyme of the host (data not shown). Interestingly, neither 3-methylcyclopenten- (1b) nor cyclohexenone (1c) yielded the desired bioreduction products, even not after 6 h biotransformation time, which is in contrast to the well-known EREDs from Saccharomyces cerevisiae (OYE1-3) and Zymomonas mobilis (NCR).30 Since substrate acceptance for 3-substituted cyclic ketones was rather narrow and 2-substituted ketones gave reasonable results, we expanded the substrate scope toward more sterically demanding 2-substituted cyclopenten- and hexenones (entries 4 & 5).31
Table 1.
Bioreduction of different substrates with SYE-4 proteins
| Entry | Substrate | Time | Conv. (%)a | ee (%) | Abs. config. | Product | |
|---|---|---|---|---|---|---|---|
| Ketones | |||||||
| 1 | 1a (R=2-Me) | 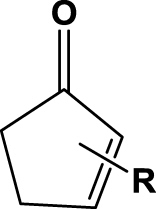 |
6 h | 92 | 52 | nd | 2a |
| 2 | 1b (R=3-Me) | 6 h | nc | na | na | 2b | |
| 3 | 1c | 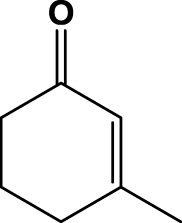 |
6 h | nc | na | na | 2c |
| 4 | 1d (n=1) | 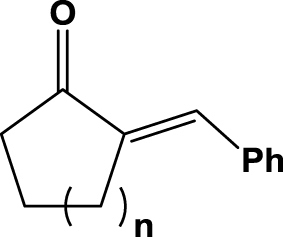 |
6 h | 4 | rac | na | 2d |
| 5 | 1e (n=2) | 6 h | 92 | 51 | nd | 2e | |
| 6 | 1f (n=1) | 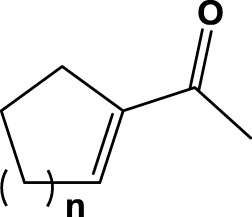 |
6 h | 30 | na | na | 2f |
| 7 | 1g (n=2) | 6 h | 71 | na | na | 2g | |
| 8 | 1h (+)-Carvone | 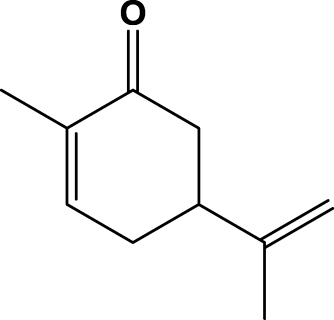 |
1 h | 100 | 97b | 2R,5R | 2h |
| 9 | 1i (−)-Carvone | 1 h | 100 | 95c | 2R,5S | 2i | |
| Diesters | |||||||
| 10 | 1j (R=Me) | 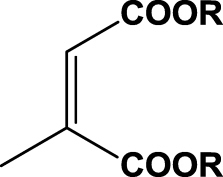 |
6 h | 99 | 98 | R | 2j |
| 11 | 1k (R=Et) | 6 h | 99 | 99 | R | 2k | |
| 12 | 1l (R=Me) | 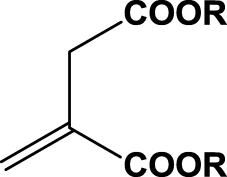 |
6 h | 19 | 90 | R | 2j |
| 13 | 1m (R=Et) | 6 h | nc | na | na | 2k | |
| Imides | |||||||
| 14 | 1n (R1=Me; R2=Me) | 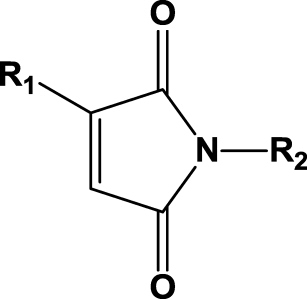 |
1 h | 99 | 99 | R | 2n |
| 15 | 1o (R1=Me; R2=Et) | 3 h | 99 | 99 | R | 2o | |
| 16 | 1p (R1=Me; R2=Propyl) | 3 h | >99 | 99 | R | 2p | |
| 17 | 1q (R1=Me; R2=Butyl) | 3 h | 99 | 99 | R | 2q | |
| 18 | 1r (R1=Me; R2=Allyl) | 3 h | 99 | 99 | R | 2r | |
| 19 | 1s (R1=Me; R2=Bn) | 3 h | 99 | 99 | R | 2s | |
| 20 | 1t (R1=H; R2=Allyl) | 1 h | 99 | na | na | 2t | |
| 21 | 1u (R1=H; R2=Bn) | 1 h | 98 | na | na | 2u | |
nd=not determined; na=not applicable.
Conversion based on GC.
Diastereomeric excess (trans).
Diastereomeric excess (cis).
Surprisingly, compound 1d was hardly converted at all, whereas the six-membered analog 1e gave almost full conversion (92%) after 6 h reaction time with a moderate enantiomeric excess (51% ee). Compounds 1a, 1d and 1e possess structural similarities, so they behave in a similar fashion upon enzyme mediated bioreduction; these findings are also in accordance with previous observations by Faber et al. testing the same substrates on different enoate reductases.6, 30 Furthermore, we investigated cyclic ketones 1f and 1g, which were reduced to saturated ketones 2f and 2g by the SYE-4 protein in reasonable to good yields (Table 1, entries 6 & 7).
We continued our investigations by examining enantiopure carvone analogues, which were selectively reduced to dihydrocarvones. (R)-Carvone (1h) was converted to (+)-(2R,5R)-trans-dihydrocarvone (2h) with excellent stereoselectivity (59% isolated yield; 97% ee; +14.2 (c 0.8, CHCl3) and (S)-carvone (1i) gave (−)-(2R,5S)-cis-dihydrocarvone (2i) quantitatively without compromising the optical purity of the precursor (53% isolated yield; −16.9 (c 1.2, CHCl3) Table 1, (entries 8 & 9). According to recently published data on a similar set of substrates by the group of Stewart et al. the SYE-4 enzyme shows a better performance with respect to conversion and selectivity.32
Furthermore, we studied the substrate conversion of SYE-4 enzyme on maleic acid derivatives and homologs bearing diester functionalities. Maleic acid analogs 1j and 1k were fully converted and gave optically pure succinic acid derivatives 2j and 2k (entries 10 & 11).20 Dimethyl 2-methylmaleate (1j) was reduced to (2R)-dimethyl 2-methylsuccinate (2j) in very good yield (85%) and optical purity (98%ee) ( +7.4 (c 2.9, CHCl3)). Accordingly, (2R)-diethyl 2-methylsuccinate (2k) was isolated from the bioreduction of diethyl 2-methylmaleate (1k) in 83% yield and with 99% enantiomeric excess ( +3.1 (c 0.9, CHCl3)). Absolute configuration was determined by comparing the sign of optical rotation of literature known reference compounds to the isolated bio-products. The absolute configuration of dimethyl 2-methylsuccinate (2j) and diethyl 2-methylsuccinate (2k) was assigned to (R) based on a comparison of specific rotation.
In contrast to maleic acid derivatives, exo-methylene analogs 1l and 1m were found to be rather poor substrates for SYE-4. In the case of 1l, low conversion (19%) and of 1m, no conversion at all was observed (entries 12 & 13). Although α,β-unsaturated esters were suspected to be good substrates for enoate reductases,6 a long reaction time and overall slow enzyme kinetics were observed. Whole-cell mediated bioreduction was dominated by ester hydrolysis, leading to the more reactive maleic acid derivatives, which undergo subsequent reduction to the corresponding succinates. This drawback was successfully circumvented by using crude cell lysate, entirely eliminating undesired ester hydrolysis.
Finally, compounds with an imide functionality (1n–u, Table 1, entries 14–21) were investigated to extend the substrate profile of SYE-4 protein. According to the literature, imides are good substrates for several enoate reductases, such as OPR1, OPR3, YqjM and other OYEs,26, 33 but the investigated substrate scope was limited to derivatives substituted at C-3 (Table 1, entries 14–21; compounds bearing an R1 group). A more comprehensive examination of the influence of different nitrogen protecting substituents (R2) on substrate acceptance and stereospecificity was performed. These α-methylmaleimides turned out to be excellent substrates for SYE-4 and gave almost 100% conversions with a high stereopreference in a reasonable timeframe (Table 1, entries 14–19). Sterically more demanding substrates were accepted, as well as less demanding structures, and no significant substitution effect on the stereopreference was observed. All products showed the (R)-configuration; this assignment is based on a preparative scale experiment of 1s by SYE-4 to 2s (76% isolated yield and an optical rotation of +12.7 (c 2.4, CHCl3)). The absolute configuration of imides was determined by comparing the signs of optical rotation of isolated products with reference compounds from literature.34, 35 All other imides were assigned accordingly, due to similar properties and chromatographic behavior.
Additional substrates 1r and 1t bearing two unsaturated C C moieties were tested to investigate functional group tolerance. As expected, activated double bonds were attacked in a ‘Michael-type’ addition whereas non-activated C C bonds remained unaffected.
3. Conclusions
Within this study we have demonstrated the synthetic potential of SYE-4, an enoate reductase from Shewanella sp. We investigated the anti bioreduction of different activated alkenes, such as α,β-unsaturated ketones, α,β-unsaturated esters and α-methylmaleimides. In general, the catalytic performance was very good and most of the desired products were obtained in good yields and optical purities. Remarkable results were obtained for the optically pure terpenes (+) and (−) carvone, which were selectively reduced to the cis and trans diastereoisomers. Furthermore, we could demonstrate that the active site of SYE-4 does accept sterically more demanding residues. Investigations of N-substituted maleimides clearly showed that neither the stereoselectivity nor the substrate acceptance was affected by the N-residue. Overall SYE-4 is a versatile and synthetically useful biocatalyst for the reduction of various activated C C double bonds and will complement the biocatalytic toolbox of already known enoate reductases.
4. Experimental
4.1. General
Chemicals and microbial growth media were purchased from commercial suppliers and used without further purification. All solvents were distilled prior to use. Screening experiments were performed in sterile multi-well plates. Flash column chromatography was performed on silica gel 60 from Merck (40–63 μm). NMR spectra were recorded on a Bruker AC 200 (200 MHz) spectrometer; chemical shifts are reported in parts per million using TMS as internal standard. Enantiomeric purity was determined by chiral phase GC using a BGB 175 column (30 m×0.25 mm ID, 0.25 μm film) on a Thermo Finnigan Trace or Focus chromatograph and compared to reference material obtained by chemical conversion, if applicable. GC–MS analyses were carried out on a GC–MS Thermo Scientific DSQ II using a standard capillary column BGB5 (30 m×0.25 mm ID, 0.25 μm film). Specific rotation was determined using a Perkin Elmer Polarimeter 241.
4.2. Typical screening procedure for bioreductions
Initially, SYE-4 was overexpressed in a BL21(DE3) E. coli strain carrying the pGEX-4 T-2 plasmid. The protein was obtained from recombinant cell cultures according to previous literature protocols.28
An Erlenmeyer flask containing sterile TB medium (200 mL) supplemented with chloroamphenicol (34 μg/ml) (200 μL) was incubated with a 2% vol of an overnight culture grown on LB medium (chloroamphenicol) up to an OD600=1 within 3 h at 28 °C. Enzyme synthesis was induced using 160 μL IPTG (0.5 mM). The flask was shaken (120 rpm) at 28 °C for 24 h. The cell pellet was collected by centrifugation (6000 rpm for 15 min), washed with 10 mM PBS buffer (30 mL), centrifuged (6000 rpm for 4 min) again, and finally for further use resuspended in PBS (5 mL) for further use. Protease inhibitor (phenyl methane sulfonyl fluoride) was added to a final conc. of 0.2 mM (0.2 M stock in EtOH). Cells were ruptured by sonication (6 cycles, pulse for 10 s, 60 s cooling). The cell debris was centrifuged (10,000 rpm for 15 min) to obtain the crude cell lysate, which was stored at −20 °C until further use. A Bradford protein assay was conducted to calculate the protein concentration in the cell lysate. Biotransformations were performed with SYE-4 protein (25 mg/mL) in sterile multi-well plates (500 μL each well) in the presence of Tris–HCl (50 mM, pH 8), NADP+ (200 μM), glucose-6-phosphate (4 mM), glucose-6-phosphate dehydrogenease (1 unit), and substrate 2 mM, 0.8 μL (stock solution in EtOH/H2O (2:1)) at 28 °C for 6 h. Samples were collected after 1 h, 3 h and 6 h. The reaction mixture was extracted with ethyl acetate containing the internal standard methyl benzoate and samples were analyzed by chiral GC and GC–MS (Table 1).
4.3. Typical procedure for preparative biotransformations
Biotransformation with SYE-4 protein (35.5 mg/mL) in a sterile baffled Erlenmeyer flask was performed using the same constituents as mentioned above, except that a substrate concentration of 0.5 mM was used. Product was extracted with ethyl acetate and purified via column chromatography. Samples were analyzed by chiral GC, GC–MS and NMR spectroscopy.
4.3.1. (+)-(2R,5R)-2-Methyl-5-(prop-1-en-2-yl)cyclohexanone (2h)36
(−)-Carvone (10 mg in ethanol/water (2:1)) was reduced, applying the general bioreduction protocol to (+)-trans-dihydrocarvone (2h) (6 mg, 59%) ( +14.2 (c 0.8, CHCl3). 1H NMR (200 MHz; CDCl3; Me4Si), δ=1.02 (3H, d, J=6.8 Hz), 1.46–1.84 (4H, m), 1.66 (3H, s), 2.28–2.56 (4H, m), 4.62–4.76 (2H, m). 13C NMR (50 MHz; CDCl3; Me4Si): δ=15.6 (q), 21.5 (q), 26.3 (t), 30.6 (t), 43.9 (d), 44.1 (d), 44.6 (t), 111.5 (t), 146.8 (s), 214.0 (s). GC: MS, m/z=152 (M+, 27), 137 (12), 95 (81), 82 (42), 67 (100).
4.3.2. (−)-(2R,5S)-2-Methyl-5-(prop-1-en-2-yl)cyclohexanone (2i)
(+)-Carvone (10 mg in ethanol/water (2:1)) was converted to (−)-cis-dihydrocarvone (2i) (5.4 mg, 53%) ( −16.9 (c 1.2, CHCl3)). 1H NMR (200 MHz; CDCl3; Me4Si), δ=0.98 (3H, d, J=6.5 Hz), 1.28–1.90 (4H, m), 1.46 (3H, s), 2.01–2.18 (4H, m), 4.45–4.48 (2H, m). 13C NMR (50 MHz; CDCl3; Me4Si): δ=14.3 (q), 20.4 (q), 30.7 (t), 34.8 (t), 44.6 (d), 46.8 (d), 46.9 (t), 109.5 (t), 147.5 (s), 212.4 (s). GC: MS, m/z=152 (M+, 15), 137 (11), 95 (66), 81 (43), 67 (100).
4.3.3. (2R)-Dimethyl 2-methylsuccinate (2j)37
Dimethyl 2-methylmaleate (1j) (20 mg in ethanol/water (2:1)) was reduced to (2R)-dimethyl 2-methylsuccinate (2j) (18 mg, 85%) ( +7.4 (c 2.9, CHCl3)). 1H NMR (200 MHz; CDCl3; Me4Si): δ=1.23 (3H, d, J=7.2 Hz), 2.35–2.47 (1H, m), 2.68–2.94 (2H, m), 3.68 (3H, s), 3.70 (3H, s). 13C NMR (50 MHz; CDCl3; Me4Si): δ=16.8 (q), 35.6 (t), 37.3 (d), 51.5 (q), 51.7 (q), 172.0 (s), 175.5 (s).
4.3.4. (2R)-Diethyl 2-methylsuccinate (2k)38
Diethyl 2-methylmaleate (1k) (20 mg in ethanol/water (2:1)) was reduced to (2R)-diethyl 2-methylsuccinate (2k) (16 mg, 83%) ( +3.1 (c 0.9, CHCl3)). 1H NMR (200 MHz; CDCl3; Me4Si): δ=1.22 (3H, d, J=7 Hz), 1.26 (6H, dt, J=7.2, 1.2 Hz), 2.33–2.44 (1H, m), 2.66–2.92 (2H, m), 4.08–4.20 (4H, dq, J=7.2, 2.4 Hz), 13C NMR (50 MHz; CDCl3; Me4Si): δ=14.0 (q), 16.8 (q), 35.7 (d), 37.6 (t), 60.3 (t), 60.4 (t), 171.6 (s), 175.1 (s).
4.3.5. (3R)-1-Benzyl-3-methylpyrrolidine-2,5-dione (2s)39
1-Benzyl-3-methyl-1H-pyrrole-2,5-dione (1s) (30 mg in ethanol/water (2:1)) was bioreduced to (3R)-1-benzyl-3-methylpyrrolidine-2,5-dione (2s) (24 mg, 76%) ( +12.7 (c 2.4, CHCl3)). 1H NMR (200 MHz; CDCl3; Me4Si), δ=1.28–1.32 (3H, d, J=7.2 Hz), 2.14–2.37 (1H, m), 2.80–2.90 (2H, m), 4.62 (2H, s), 7.25–7.34 (5H, m). 13C NMR (50 MHz; CDCl3; Me4Si): δ=16.7 (q), 34.7 (t), 36.4 (d), 42.3 (t), 127.9 (d), 128.6 (d), 128.7 (d), 135.8 (s), 176.1 (s), 180.2 (s). GC: MS, m/z=203 (M+, 100), 174 (23), 160 (70), 104 (61), 91 (58).
Acknowledgements
This project was in part funded by the Austrian Science Fund FWF (P-I723). N.I. acknowledges financial support by the Higher Education Commission, Pakistan, for receiving an international PhD fellowship.
References and notes
- 1.Williams R.E., Bruce N.C. Microbiology. 2002;148:1607. doi: 10.1099/00221287-148-6-1607. [DOI] [PubMed] [Google Scholar]
- 2.Shimoda K., Ito D.I., Izumi S., Hirata T. J. Chem. Soc., Perkin Trans. 1. 1996:355. [Google Scholar]
- 3.Fuganti C., Grasselli P. J. Chem. Soc., Chem. Commun. 1979:995. [Google Scholar]
- 4.Muller A., Hauer B., Rosche B. J. Mol. Catal. B: Enzym. 2006;38:126. [Google Scholar]
- 5.Kohli R.M., Massey V. J. Biol. Chem. 1998;273:32763. doi: 10.1074/jbc.273.49.32763. [DOI] [PubMed] [Google Scholar]
- 6.Stuermer R., Hauer B., Hall M., Faber K. Curr. Opin. Chem. Biol. 2007;11:203. doi: 10.1016/j.cbpa.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 7.Hall M., Stueckler C., Kroutil W., Macheroux P., Faber K. Angew. Chem., Int. Ed. 2007;46:3934. doi: 10.1002/anie.200605168. [DOI] [PubMed] [Google Scholar]
- 8.Muller A., Sturmer R., Hauer B., Rosche B. Angew. Chem., Int. Ed. 2007;46:3316. doi: 10.1002/anie.200605179. [DOI] [PubMed] [Google Scholar]
- 9.Swiderska M.A., Stewart J.D. J. Mol. Catal. B: Enzym. 2006;42:52. [Google Scholar]
- 10.Swiderska M.A., Stewart J.D. Org. Lett. 2006;8:6131. doi: 10.1021/ol062612f. [DOI] [PubMed] [Google Scholar]
- 11.Karplus P.A., Fox K.M., Massey V. FASEB J. 1995;9:1518. doi: 10.1096/fasebj.9.15.8529830. [DOI] [PubMed] [Google Scholar]
- 12.Abramovitz A.S., Massey V. J. Biol. Chem. 1976;251:5327. [PubMed] [Google Scholar]
- 13.Oberdorfer G., Steinkellner G., Stueckler C., Faber K., Gruber K. ChemCatChem. 2011;3:1562. [Google Scholar]
- 14.Vaz A.D.N., Chakraborty S., Massey V. Biochemistry. 1995;34:4246. doi: 10.1021/bi00013a014. [DOI] [PubMed] [Google Scholar]
- 15.Yanto Y., Winkler C.K., Lohr S., Hall M., Faber K., Bommarius A.S. Org. Lett. 2011;13:2540. doi: 10.1021/ol200394p. [DOI] [PubMed] [Google Scholar]
- 16.Hirata T., Shimoda K., Gondai T. Chem. Lett. 2000:850. [Google Scholar]
- 17.Shimoda K., Kubota N., Hamada H. Tetrahedron: Asymmetry. 2004;15:2443. [Google Scholar]
- 18.Leuenberger H.G.W., Boguth W., Barner R., Schmid M., Zell R. Helv. Chim. Acta. 1979;62:455. [Google Scholar]
- 19.Hirata T., Takarada A., Hegazy M.E.F., Sato Y., Matsushima A., Kondo Y., Matsuki A., Hamada H. J. Mol. Catal. B: Enzym. 2005;32:131. [Google Scholar]
- 20.Servi S. Synthesis. 1990:1. [Google Scholar]
- 21.Csuk R., Glanzer B.I. Chem. Rev. 1991;91:49. [Google Scholar]
- 22.Darrigo P., Hogberg H.E., Pedrocchifantoni G., Servi S. Biocatalysis. 1994;9:299. [Google Scholar]
- 23.Ferraboschi P., Grisenti P., Casati R., Fiecchi A., Santaniello E. J. Chem. Soc., Perkin Trans. 1. 1987:1743. [Google Scholar]
- 24.Utaka M., Konishi S., Mizuoka A., Ohkubo T., Sakai T., Tsuboi S., Takeda A. J. Org. Chem. 1989;54:4989. [Google Scholar]
- 25.Muller A., Hauer B., Rosche B. Biotechnol. Bioeng. 2007;98:22. doi: 10.1002/bit.21415. [DOI] [PubMed] [Google Scholar]
- 26.Chaparro-Riggers J.F., Rogers T.A., Vazquez-Figueroa E., Polizzi K.M., Bommarius A.S. Adv. Synth. Catal. 2007;349:1521. [Google Scholar]
- 27.Heidelberg J.F., Paulsen I.T., Nelson K.E., Gaidos E.J., Nelson W.C., Read T.D., Eisen J.A., Seshadri R., Ward N., Methe B., Clayton R.A., Meyer T., Tsapin A., Scott J., Beanan M., Brinkac L., Daugherty S., DeBoy R.T., Dodson R.J., Durkin A.S., Haft D.H., Kolonay J.F., Madupu R., Peterson J.D., Umayam L.A., White O., Wolf A.M., Vamathevan J., Weidman J., Impraim M., Lee K., Berry K., Lee C., Mueller J., Khouri H., Gill J., Utterback T.R., McDonald L.A., Feldblyum T.V., Smith H.O., Venter J.C., Nealson K.H., Fraser C.M. Nature. 2002;20:1118. [Google Scholar]
- 28.Brige A., Van den Hemel D., Carpentier W., De Smet L., Van Beeumen J. Biochem. J. 2006;394:335. doi: 10.1042/BJ20050979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elegheert J., van den Hemel D., Dix I., Stout J., Van Beeumen J., Brige A., Savvides S.N. Acta Crystallogr., Sect. F. 2010;66:85. doi: 10.1107/S1744309109050386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall M., Stueckler C., Hauer B., Stuermer R., Friedrich T., Breuer M., Kroutil W., Faber K. Eur. J. Org. Chem. 2008:1511. [Google Scholar]
- 31.Vatsadze S.Z., Sviridenkova N.V., Manaenkova M.A., Semashko V.S., Zyk N.V. Russ. Chem. Bull. 2005;54:2224. [Google Scholar]
- 32.Padhi S.K., Bougioukou D.J., Stewart J.D. J. Am. Chem. Soc. 2009;131:3271. doi: 10.1021/ja8081389. [DOI] [PubMed] [Google Scholar]
- 33.Satianegara G., Rogers P.L., Rosche B. Biotechnol. Bioeng. 2006;94:1189. doi: 10.1002/bit.20959. [DOI] [PubMed] [Google Scholar]
- 34.Stueckler C., Hall M., Ehammer H., Pointner E., Kroutil W., Macheroux P., Faber K. Org. Lett. 2007;9:5409. doi: 10.1021/ol7019185. [DOI] [PubMed] [Google Scholar]
- 35.Balenovi K., Bregant N. J. Chem. Soc. 1965:5131. [Google Scholar]
- 36.Kato M., Kamat V.P., Tooyama Y., Yoshikoshi A. J. Org. Chem. 1989;54:1536. [Google Scholar]
- 37.Herrera R.P., Monge D., Martin-Zamora E., Fernandez R., Lassaletta J.M. Org. Lett. 2007;9:3303. doi: 10.1021/ol071292c. [DOI] [PubMed] [Google Scholar]
- 38.Morita M., Drouin L., Motoki R., Kimura Y., Fujimori I., Kanai M., Shibasaki M. J. Am. Chem. Soc. 2009;131:3858. doi: 10.1021/ja9005018. [DOI] [PubMed] [Google Scholar]
- 39.Puertas S., Rebolledo F., Gotor V. Tetrahedron. 1995;51:1495. [Google Scholar]