

Abstract
HG-7-85-01(22) and HG-7-86-01(26) are thiazolo[5,4-b]pyridine containing type II tyrosine kinase inhibitors with potent cellular activity against both wild-type and ‘gatekeeper’ mutant T315I- Bcr-Abl. Here we report on the ‘hybrid design’ concept and subsequent structure activity guided optimization efforts that resulted in the development of these inhibitors.
Keywords: T315I Bcr-Abl, Kinase inhibitor, Gatekeeper mutant, Type II inhibitor, Thiazolo[5, 4-b]pyridine
1. Introduction
Despite the tremendous success of the first generation tyrosine kinase inhibitor imatinib for the treatment of chronic myelogenous leukemia (CML), a portion of the patient population eventually develops resistance1,2. Three ‘second generation’ inhibitors: nilotinib,3 dasatinib4 and bosutinib,5 have been approved to treat patients with imatinib resistance however these drugs remain ineffective against T315I ‘gatekeeper’ mutant form of Bcr-Abl.6 A ‘third generation’ of type II inhibitors that effectively inhibit the T315I Bcr-Abl mutant has recently been developed. These inhibitors include AP24163,7 AP24534,7 DCC-2036,8 BGG463,9 GNF-7,10 DSA series compounds,11 and a series of alkynyl inhibitors,12 of which only AP24534 and DCC-2036 are reported to be undergoing clinical evaluation. Recently, we described the biological characterization of two Type II tyrosine kinase inhibitors: HG-7-85-01(22)13 and HG-7-86-01(26)14 which can potently inhibit the proliferation of cells expressing the major imatinib-resistant gatekeeper mutants T315I-BCR-ABL, T670I-Kit, T674M/I-PDGFRα and T341M/I-Src and which potently and selectively target mutant FLT3. Herein, we describe the hybrid design strategy and medicinal chemistry effort leading to the development of these ATP-competitive type II inhibitors.
2. Results & Discussion
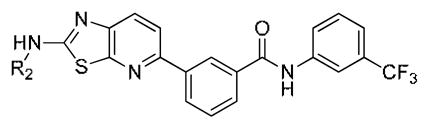
We have previously reported on the use of a rational “hybrid-design” strategy to convert well known Type I scaffolds into corresponding Type II inhibitors. This approach consists of appending the moiety from a Type II inhibitor that occupies the region adjacent to the ATP-binding site created by the flip of the ‘DFG-motif’ to the portion of a Type I inhibitor that makes contacts with the hinge region of the kinase.15 We designed the thiazolopyridine tyrosine kinase inhibitors reported here by hybridizing the hinge interacting thiazole functionality of the Type I inhibitor dasatinib with the 3-trifluoromethylbenzamide pharmacophore present in Type II inhibitors such as imatinib, nilotinib and sorafenib.
The first hybrid compounds that we designed are exemplified by HG-7-85-01 (22) and HG-7-86-01 (26) and contain a thiazolopyridine core, a 3-trifluoromethylbenzamide Type II ‘tail’ and various groups appended to the thiazolopyridine (Figure 1). The synthesis of the thiazolo[5,4-b]pyridine core commenced with a Suzuki coupling between commercially available 2-chloro-5-nitropyridine and various phenylboronic acids (Scheme 1). The nitro group was reduced using 5% Pd/C and the resulting product was readily brominated using N-bromosuccinimide at low temperature. One-pot 2-(methylthio)thiazole formation was accomplished using potassium ethyl xanthogenate and iodomethane to yield compound 5. The sulfide group was oxidized with oxone to sulfone compound 6 which could be easily displaced using ammonia in isopropanol. Saponification of ester compound 8 followed by amide coupling using HATU and DIEA provided the target compound 9a.
Figure 1.

Design rational for the thiazolopyridine scaffold
Scheme 1. Reagent and conditions.

a) Boronic acids[(3-(Ethoxycarbonyl)phenylboronic Acid(R1 = H), 5-Methoxycarbonyl-2-methylphenylboronic acid (R1 = Me), 5-Ethoxycarbonyl-2-fluorophenylboronic acid (R1 = F)], Pd(PPh3)2Cl2, tert-Butyl XPhos, 2N Na2CO3(aq), dioxane, 90 °C, 10 h, b) 5% Pd/C, EtOH, 16 h, c) NBS, DMF, 0 °C, 5-10 min, d) Potassium ethyl xanthogenate, AcOH, NMP, 150 °C, 16 h and MeI, 50 °C, 30 min, e) Oxone, MeOH, THF, Water, rt, 16 h, f) 2.0 N NH3 in IPA, 90 °C, 24 h, g) LiOH·H2O, THF, MeOH, H2O, rt, 16 h, h) HATU, DIEA, 3-(trifluoromethyl)aniline, DMF, rt, 16 h.
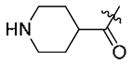
The synthesis of compounds 10–13 was accomplished by acylation or amidation of the NH2 moiety in 9a. Urea formation to form 14 was accomplished via acylation of 9a with 4-nitrophenyl chloroformate followed by displacement of the 4-nitrophenyl group with 4-amino-1-Boc-piperidine. The aryl/heteroaryl substituted compounds 15–16 were achieved by palladium catalyzed coupling reactions between 9a and the appropriate aryl halide and protecting group manipulations.
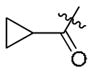
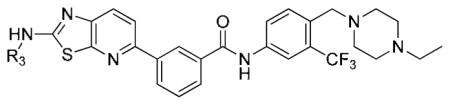
To evaluate the ability of the compounds to inhibit Bcr-Abl in a cellular context we used the murine pre-B cell line Ba/F3 transformed with the Bcr-Abl oncogene. Wild-type Ba/F3 cells proliferate only in the presence of interleukin-3 (IL-3) while Ba/F3 cells transformed with oncogenic kinases such as Bcr-Abl become capable of growing in the absence of IL-3. This provides a robust and commonly used assay for selective kinase inhibition.16 The smallest compound 9a did not display antiproliferative activity against wild-type Bcr-Abl or T315I-Bcr-Abl and the larger compounds 12–16 possessed only weak anti-proliferative activity effect (Table 1). Interestingly, only one compound containing a small cyclopropyl amide displayed submicromolar antiproliferative activity against Bcr-Abl and T315I-Bcr-Abl. Compound 11 exhibited EC50’s of 420 nM on wild-type Bcr-Abl and 960 nM onT315I-Bcr-Abl; however 11 also showed a similar antiproliferative effect against parental Ba/F3 cells suggesting that targets other than Bcr-Abl may contribute to the antiproliferative activity of this compound. To explore this chemical series further we next prepared compounds 20–23 which possess an extended Type II tail containing an (4-ethyl-piperazin-1-yl)methyl moiety appended to the 4-position of the trifluoromethylbenzamide (Table 2). This chemical maneuver resulted in an approximate 6-fold improvement in cellular antiproliferative activity relative to 11 against T315I-Bcr-Abl and a 3-fold decrease in cytotoxicity against parental Ba/F3 cells (Table 2, compound 22).
Table 1.
Cellular antiproliferative activity of compounds 9a-16.
| ||||
|---|---|---|---|---|
| Compds | R2 | Cellular antiproliferative activity (EC50, μM)
|
||
| Bcr-Abl (wt)a | Bcr-Abl (T315I)b | Ba/F3c | ||
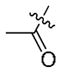
| 9a | H | >10 | >10 | >10 |
| 10 |
|
5.0 | 3.77 | >10 |
| 11 |
|
0.42 | 0.96 | 0.91 |
| 12 |
|
>10 | >10 | >10 |
| 13 |
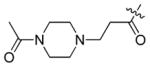
|
5.21 | >10 | >10 |
| 14 |
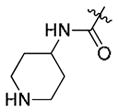
|
7.6 | 7.18 | >10 |
| 15 |
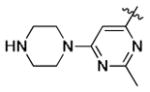
|
1.90 | 2.41 | 6.74 |
| 16 |
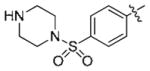
|
6.21 | >10 | >10 |
Cellular antiproliferative activity (EC50, μM) on wt Bcr-Abl-Ba/F3.
Cellular antiproliferative activity (EC50, μM) on mutant Bcr-Abl-T315I-Ba/F3.
Cytotoxicity (EC50, μM) on wt-Ba/F3.
Table 2.
SAR of non-bonded sulfur and oxygen(nitrogen) interaction derivatives.
| ||||
|---|---|---|---|---|
| Compds | R3 | Cellular antiproliferative activity (EC50, μM)
|
||
| Bcr-Abl (wt) | Bcr-Abl (T315I) | BaF3 | ||
| 20 | H | >10 | >10 | >10 |
| 21 | acetyl | 0.139 | 0.193 | 4.16 |
| 22 | cyclopropanecarbonyl | 0.098 | 0.156 | 3.085 |
| 23 | 2-pyridinyl | 0.146 | 0.276 | 3.532 |
We hypothesized that an intramolecular interaction between the thiazole sulfur and the oxygen atom of the adjacent amide in compounds 21–22 contributes to their conformation and biological activity.17 This is supported by the similar cellular potencies of 21–22 with the 2-pyridine derivative 23. Compound 23 is predicted to exhibit a similar bioactive conformation to 21–22 as a result of a lone pair of electrons from the pyridine N interacting with the unfilled orbitals of the thiazole sulfur as shown by previous computational analysis.18 Finally we established the bioactive conformation of this series of compounds by elucidation of a co-crystal structure between HG-7-85-01(22) and Src (Figure 2, PDB code: 4AGW). The structure revealed that the distance between the sulfur and oxygen is 2.7 Å which is shorter than the sum of the normal oxygen and sulfur van der Waals radii of 3.3 Å. Consistent with the ‘hybrid-design’ strategy, compound 22 was confirmed to bind to the DFG-out ‘inactive’ conformation of Src.11
Figure 2.

Compound 22 (turquois sticks) forms five hydrogen-bonding interactions with Src; A. Two hydrogen bonds are made with the hinge region: between the thiazole N and backbone NH of M341 and between the cyclopropyl amide NH and the backbone carbonyl of M341. B. Hydrogen bonding interactions exist between the benzamide carbonyl of 22 and the backbone NH of D404 of the DFG-motif, between the benzamide NH and side-chain carboxylate of E310 from the C-helix, and from the presumably protonated distal piperazine nitrogen and the backbone carbonyls of V383 and H384.
We next further explored substitution on the 3-trifluorophenyl moiety which occupies the pocket created by the ‘DFG-out’ flip of the activation loop (Table 3, 24–26). Compound 24, which eliminates the methylene spacer between the aryl group and piperazine in 22, lost approximately 10-fold cellular activity relative to 22. Compound 25 lacking the CF3 group was completely inactive up to the highest concentration tested (10μM). Compound 26, which possesses the ‘tail’ moiety of nilotinib exhibited slightly diminished potency relative to compound 22 but substantially improved the overall kinase selectivity as demonstrated below. We next explored the effects of introducing a flag-methyl or 2-fluoro onto the central phenyl ring (Table 3, 27–30). The compounds exhibited enhanced activity on Bcr-Abl but not on T315I-Bcr-Abl. For example, compound 27 is an exceptionally potent as inhibitor of wild-type Bcr-Abl dependent Ba/F3 cell proliferation. We also investigated the effect of reversing the amide between the central phenyl and CF3-substituted ring and replacing the amide with a urea moiety. The synthesis of these compounds is depicted in Scheme 2B. Reversing the amide bond orientation, compound 33 compared to 22, resulted in approximately a 4-fold reduction in cellular activity against T315I-Bcr-Abl. Interestingly, introduction of a urea linkage (32) resulted in 5–10 fold improved activity on both wild-type and T315I Bcr-Abl, but increased cytotoxicity towards parental Ba/F3 cells by approximately 5-fold.
Table 3.
Cellular antiproliferation activity of analogs 24–33.
| Compds | Structure | Cellular antiproliferative activity (EC50, μM)
|
||
|---|---|---|---|---|
| Bcr-Abl (wt) | Bcr-Abl (T315I) | Ba/F3 | ||
| 24 |
|
2.31 | 2.30 | 2.30 |
| 25 |
|
>10 | >10 | >10 |
| 26 |
|
0.38 | 0.19 | 1.99 |
| 27 |
|
0.001 | 0.06 | 1.23 |
| 28 |
|
0.03 | 0.56 | 4.67 |
| 29 |
|
0.02 | 0.30 | >10 |
| 30 |
|
0.02 | 0.59 | >10 |
| 31 |
|
0.2 | 0.78 | >10 |
| 32 |
|
0.018 | 0.013 | 0.69 |
| 33 |
|
1.24 | 0.55 | >10 |
Scheme 2. Reagent and conditions.


A. a) Acyl chloride, pyridine, DCM, 0 °C, 4 h or acid, EDCI, HOBT, DMAP, DMF, rt, 8 h, b) i. 4-nitrophenyl chloroformate, pyridine, DCM, rt, 1 h, ii. tert-butyl 4-aminopiperidine-1-carboxylate, TEA, THF, rt, 4 h, iii. TFA, DCM, 4 h, c) i. tert-butyl 4-(6-chloro-2-methylpyrimidin-4-yl)piperazine-1-carboxylate, Pd2(dba)3, Xanthphos, K2CO3, 2-BuOH, 90 °C, 4 h, ii. TFA, DCM, 4 h. B. a) Cyclopropanecarbonyl chloride, pyridine, DCM, 0 °C, 4 h, b) TFA, DCM, 4 h, c) i. benzoic acid, HATU, DIEA, DMF, 16 h. or tert-butyl 4-aminopiperidine-1-carboxylate, TEA, THF, rt, 4h.
The kinase selectivity of HG-7-85-01(22) and HG-7-86-01(26) was evaluated using the KINOMEscan™ approach (Ambit Biosciences, San Diego, CA) against a panel of 353 kinases at a concentration of 10 μM.19 The kinases which interacted with 22 and 26 with an Ambit score less than 10% of the DMSO control are highlighted with a red circle in a spot tree (Figure 3, supplemental material contains full screening data for 22 and 26). To provide a measure of overall kinase selectivity we calculated S(10) scores, which is defined as the ratio of the number of kinases inhibited below a score of ten versus total number of kinases tested at a specified concentration. This analysis revealed that compound 26 possess an S(10) of 0.36 and compound 22 possesses an S(10) of 0.10 at a concentration of 10 uM.
Figure 3.

Kinase Selectivity of HG-7-85-01(22) and HG-7-86-01(26)
We assessed the pharmacological properties of compound 22 and 26 in rats following oral and intravenous delivery (Table 4) prior to performing the murine tumor models which we have previously reported.13 HG-7-85-01(22) has limited oral bioavailability (%F = 18.8), a moderate half-life (T1/2 = 5.8 hours), a relatively low maximal serum concentration (Cmax = 292 ng/mL at 2 mg/kg ) and a relatively high clearance (Cl = 13 ml/min/kg). HG-7-86-01(26) has poor oral bioavailability (%F = 4.7), however has better exposure as indicated by AUC (111167 min*ng/ml).
Table 4.
Pharmacokinetic results following intravenous 1 mg/kg and oral 2 mg/kg dosing.
| Compds | Route of Administration | t1/2 (hr) | Tmax | Cmax ng/ml | AUC min*ng/ml | AUC (%extrapolated) | Cl ml/min/kg | MRT (hr) | VSS L/kg | %F |
|---|---|---|---|---|---|---|---|---|---|---|
| 22 | PO | 4.7 | 2.7 | 53.1 | 27733 | 35 | 74.4 | 7.7 | 18.8 | |
| IV | 5.8 | 0.08 | 291.7 | 73800 | 35.3 | 13.7 | 8.1 | 6.5 | ||
| 26 | PO | 3.7 | 1.1 | 32.0 | 8160 | 21 | 303.3 | 5.3 | 3.7 | |
| IV | 2.4 | 0.14 | 671.7 | 111167 | 8.7 | 9.5 | 3.1 | 1.7 |
In summary, we have used a rational hybrid-design approach to design a new type II scaffold by combining functionality from the type I inhibitor dasatinib with moieties from the type II inhibitors imatinib, nilotinib and sorafenib. The compounds such as 22 and 26 are capable of potently inhibiting wild-type and T315I-Bcr-Abl activity in biochemical and cellular assays. Further optimization of this scaffold for inhibition of Bcr-Abl and other potential targets identified by the kinase profiling are currently on-going.
Supplementary Material
Acknowledgments
We thank Nam Doo Kim for assistance with molecular modeling and Sara Buhrlage for critical reading of the manuscript. We acknowledge research funding from Novartis and NIH (R01 CA130876-02).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Nat Med. 1996;2(5):561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. N Eng J Med. 2001;344(14):1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 3.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Neuberg D, Wright RD, Gilliland DG, Griffin JD. Cancer Cell. 2005;7(2):129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Science. 2004;305(5682):399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 5.Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, Andreoni F, Scapozza L, Formelli F, Gambacorti-Passerini C. Cancer Res. 2006;66(23):11314–22. doi: 10.1158/0008-5472.CAN-06-1199. [DOI] [PubMed] [Google Scholar]
- 6.O’Hare T, Walters DK, Deininger MW, Druker BJ. Cancer Cell. 2005;7(2):117–9. doi: 10.1016/j.ccr.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 7.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang W-S, Xu Q, Metcalf CA, Iii, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoorthi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sawyer TK, Dalgarno DC, Deininger MWN, Druker BJ, Clackson T. Cancer Cell. 2009;16(5):401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eide CA, Adrian LT, Tyner JW, MacPartlin M, Anderson DJ, Wise SC, Smith BD, Petillo PA, Flynn DL, Deininger MWN, O’Hare T, Druker B. J Cancer Res. 2011;(71):3189–3195. doi: 10.1158/0008-5472.CAN-10-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Program of the Fall Meeting 2008. CHIMIA International Journal for Chemistry. 2008;62(7–8):547–557. [Google Scholar]
- 10.Choi HG, Ren P, Adrian F, Sun F, Lee HS, Wang X, Ding Q, Zhang G, Xie Y, Zhang J, Liu Y, Tuntland T, Warmuth M, Manley PW, Mestan Jr, Gray NS, Sim T. 2010;53(15):5439–5448. doi: 10.1021/jm901808w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seeliger MA, Ranjitkar P, Kasap C, Shan Y, Shaw DE, Shah NP, Kuriyan J, Maly DJ. Cancer Res. 2009;69(6):2384–92. doi: 10.1158/0008-5472.CAN-08-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng X, Lim SM, Zhang J, Gray NS. Bioorg Med Chem Lett. 2010;20(14):4196–4200. doi: 10.1016/j.bmcl.2010.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisberg E, Choi HG, Ray A, Barrett R, Zhang J, Sim T, Zhou W, Seeliger M, Cameron M, Azam M, Fletcher JA, Debiec-Rychter M, Mayeda M, Moreno D, Kung AL, Janne PA, Khosravi-Far R, Melo JV, Manley PW, Adamia S, Wu C, Gray N, Griffin JD. Blood. 2010;115(21):4206–4216. doi: 10.1182/blood-2009-11-251751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weisberg E, Choi HG, Barrett R, Zhou W, Zhang J, Ray A, Nelson EA, Jiang J, Moreno D, Stone R, Galinsky I, Fox E, Adamia S, Kung AL, Gray NS, Griffin JD. Mol Cancer Ther. 2010;9(9):2468–2477. doi: 10.1158/1535-7163.MCT-10-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okram B, Nagle A, Adrian FJ, Lee C, Ren P, Wang X, Sim T, Xie Y, Xia G, Spraggon G, Warmuth M, Liu Y, Gray NS. Chem Biol. 2006;13(7):779–86. doi: 10.1016/j.chembiol.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL, Beigi R, Xia G, Harig RA, Asatryan H, Yan SF, Zhou Y, Gu XJ, Saadat A, Zhou V, King FJ, Shaw CM, Su AI, Downs R, Gray NS, Schultz PG, Warmuth M, Caldwell JS. Proc Natl Acad Sci USA. 2006;103(9):3153–8. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagao Y, Hirata T, Goto S, Sano S, Kakehi A, Iizuka K, Shiro M. J Am Chem Soc. 1998;120(13):3104–3110. [Google Scholar]
- 18.Bilodeau MT, Rodman LD, McGaughey GB, Coll KE, Koester TJ, Hoffman WF, Hungate RW, Kendall RL, McFall RC, Rickert KW, Rutledge RZ, Thomas KA. Bioorg Med Chem Lett. 2004;14(11):2941–2945. doi: 10.1016/j.bmcl.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 19.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Nat Biotechol. 2011;29(11):1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.