

Abstract
The switch between the Krebs cycle and the glyoxylate bypass is controlled by isocitrate dehydrogenase kinase/phosphatase (AceK). AceK, a bifunctional enzyme, phosphorylates and dephosphorylates isocitrate dehydrogenase (IDH) with its unique active site that harbours both the kinase and ATP/ADP-dependent phosphatase activities. AceK was the first example of prokaryotic phosphorylation identified, and the recent characterization of the structures of AceK and its complex with its protein substrate, IDH, now offers a new understanding of both previous and future endeavours. AceK is structurally similar to the eukaryotic protein kinase superfamily, sharing many of the familiar catalytic and regulatory motifs, demonstrating a close evolutionary relationship. Although the active site is shared by both the kinase and phosphatase functions, the catalytic residues needed for phosphatase function are readily seen when compared with the DXDX(T/V) family of phosphatases, despite the fact that the phosphatase function of AceK is strictly ATP/ADP-dependent. Structural analysis has also allowed a detailed look at regulation and its stringent requirements for interacting with IDH.
Keywords: kinase, phosphatase, AceK, isocitrate dehydrogenase, phosphorylation
1. Introduction
Isocitrate dehydrogenase kinase/phosphatase (AceK) is a rare example of reversible protein phosphorylation in prokaryotes, regulating the action of isocitrate dehydrogenase (IDH) as the metabolic switch between the Krebs cycle and the glyoxylate bypass (figure 1a) in response to nutrient availability. As a key metabolic enzyme at the branch point between the glyoxylate bypass and Krebs cycle, IDH represents a strategic position for regulation. AceK, a bifunctional enzyme, uniquely contains both kinase and phosphatase activities and is unusual with its two opposing activities at the same highly adaptable active site [1]. Although the discovery of a protein with both kinase and phosphatase activities involved in the regulation of the Krebs cycle, known as AceK, occurred nearly 30 years ago and became the founding member of prokaryotic protein phosphorylation [2], a full understanding of AceK remained elusive until structural information became available. The recent characterization of the structures of AceK and its complex with its substrate IDH [3] has provided the first structural glimpse and offered a renewed look at this enzyme. The structures revealed a striking likeness to the eukaryotic kinase superfamily and a new class of phosphatases, and insight into its regulation and association with its protein substrate.
Figure 1.

The Krebs cycle and the glyoxylate bypass mediated by AceK. (a) In the complete Krebs cycle, isocitrate is converted to α-ketoglutarate and carbon dioxide by the enzyme isocitrate dehydrogenase (IDH). When acetate is the sole carbon source, AceK phosphorylates IDH causing its inactivation. The inactivation of IDH increases the conversion of isocitrate to glyoxylate by isocitrate lyase (ICL). Glyoxylate is then combined with acetyl-CoA to form malate, a downstream product of the Krebs cycle, thus completing the glyoxylate bypass. (b) The transcription of the aceBAK and icIR operons. The aceBAK operon encodes three proteins: malate synthase (aceB), ICL (aceA) and isocitrate dehydrogenase kinase/phosphatase (aceK). These three enzymes are involved in the glyoxylate bypass.
The structure of AceK is composed of two distinct domains (figure 2a). The C-terminal domain or kinase domain (KD), which binds adenosine triphosphate (ATP) and is the site of kinase, phosphatase and ATPase function, resembles both the eukaryotic serine/threonine protein kinases and the tyrosine protein KD of signal transduction receptors such as interleukin-1 receptor [4], fibroblast growth factor receptor 2 (FGFR2; figure 2b) [5] and the proto-oncogene tyrosine protein kinase receptor [6], suggesting a distant evolutionary relationship between AceK and the eukaryotic kinases [3]. The N-terminal domain or regulatory domain (RD) of AceK represents a unique protein fold with no structural homologues (figure 2a). The RD contains binding pockets for small molecules that can regulate the function of the catalytic KD of AceK [3].
Figure 2.
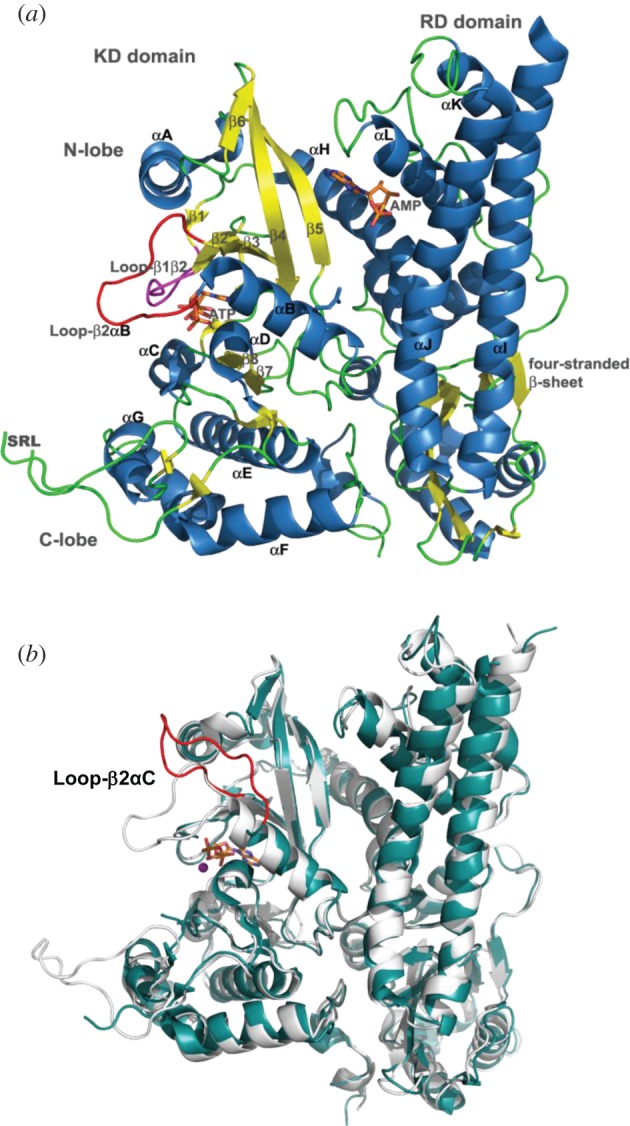
(a) The overall structure of AceK. The active site of AceK includes a buried ATP molecule. The structure also includes a bound AMP at the interface of the two domains. The kinase domain (KD) on the left resembles eukaryotic protein kinases. The regulatory domain (RD) on the right does not have any structural homologues. Loop-β2αC and P-loop are coloured red and pink, respectively. (b) Structural comparison of AceK in AMP-free mode (open, light blue) and AMP-binding mode (closed, light grey).
2. Acek and eukaryotic kinases
Eukaryotic protein kinases (ePKs) are one of the largest gene families and are important in regulating a multitude of cellular processes. This superfamily of proteins has retained its basic function and uses a similar mechanism for phosphotransfer. Yet these proteins have evolved to phosphorylate a wide range of substrates, interact with a variety of proteins and respond to different regulatory mechanisms [7]. The catalytic cores in all kinases share a similar fold composed of two lobes. The N-terminal, smaller, subdomain (N-lobe) is predominantly β-sheet and harbours many of the ATP-interacting residues. The C-terminal, larger, subdomain (C-lobe) is primarily α-helical and contains the catalytic loop and residues needed for protein substrate recognition. The active site is located in the cleft formed between these two lobes [8]. The residues involved in ATP binding and those required to transfer the γ-phosphate moiety from ATP to the targeted serine, threonine or tyrosine residue are ubiquitously conserved, even in atypical kinases, making up a functional cassette essential to this activity. The C-terminal residues 307–574 in AceK show many structural similarities to ePKs; hence it is termed the KD, despite it (largely) lacking sequence homology. As with ePKs, the organization of AceK KD is further divided into two lobes with ATP bound at the interface between them (figure 2a). The N- and C-lobes are composed of helices and strands, respectively, with some structural alterations when compared with ePKs; however, the key residues that form the ATP binding site are conserved. These similarities and differences are best illustrated when the KD of AceK is compared with the structure of FGFR2 (PDB 3CLY) [5] (figure 3a,b). These proteins are structurally similar with a root mean square deviation of 3.6 Å despite having only 14.9 per cent sequence identity. The conserved structural elements, particularly in the ATP binding region, are in a good alignment. However, several regions in the N-lobe display significant variations, particularly in helices αA, αB, αH, and β4, β5 and β6. In the C-lobes, the α-helices of these proteins do not align well, except for the region near the ATP binding site [3]. There is a restricted sequence homology between residues 315–340 of AceK and the corresponding ATP-binding residues in ePKs [3] (figure 3d). This includes a glycine-rich motif (P-loop) in the loop between β1 and β2 (315APGIRG320), a multifunctional element that interacts with the γ-phosphate and helps stabilize the ATP binding site [9]. Also conserved is a lysine residue (Lys336) found in the ATP binding site that interacts with and stabilizes the α-phosphate of bound ATP [10]. Several other classical kinase motifs, including the APE, HRD and DFG sequences, appear to be absent or modified in AceK.
Figure 3.

(a) An overall structural comparison between FGFR2 kinase (PDB accession code: 3CLY; light green) and the AceK kinase domain (KD, PDB accession code: 3EPS; dark grey). (b) Overlays of ATP binding sites between AMP-bound AceK (dark grey) and FGFR2 (light green). In FGFR2, the AMP-PCP molecule is shown in cyan and the two Mg2+ ions are shown as yellow spheres. In AceK, ATP is shown in purple. The residue numbering of FGFR2 is in brackets. (c) (left) AceK R- and C-spines flank the αF-helix and span the AceK KD core. The AceK KD structure is shown in AMP-binding mode. The hydrophobic part of the R-spine is shown as a red molecular surface. The C-spine is coloured yellow. The adenine ring of ATP completes the C-spine. Tyr414, proposed to serve an important role in transmitting the allosteric effects from the regulatory domain, is coloured green. (Right) Five hydrophobic residues that constitute the R-spine are from three different AceK conformational modes (AMP-binding, cyan; AMP-free, green; AceK–IDH complex, red). (d) Structured-based sequence alignment of AceK KD with PKA (http://www.bioinformatics.org/strap/#LINKING). Important segments are highlighted in boxes with green and blue boxes. The residues of the R- and C-spine in AceK are indicated by red and yellow boxes, respectively. The conserved hydrophobic (black), acidic (red) and basic (blue) residues are shown.
(a). Catalytic ‘triad’
A ‘catalytic triad’ is identified in ePKs and consists of Asp166 (catalytic base), and Asn171 and Asp184 (cAMP-dependent protein kinase (PKA) numbering), which chelate one of the bound Mg2+ ions [8]. In the absence of structural information, sequence analysis and mutagenesis studies were used to predict this triad in AceK. The catalytic base was believed to be Asp317, along with Asn377 and Asp403 completing the triad [11]. When the structure of AceK was solved, it was clear that this previously proposed ‘catalytic triad’ was incorrect. The triad still exists in AceK; however, the residues involved are Asp457, Asn462 and Asp475, placing these residues in the same spatial positioning as those in ePKs (figure 3d). In accordance with their critical role in ATP binding and catalysis, mutations in this signature motif (e.g. D475A) completely abolished kinase activity [3]. In addition to this catalytic element, a slightly modified phosphotransferase motif is also evident in AceK. This motif in phosphotransferases, which include kinases, is HXDhXXXNhhh, where h is [FLIMVWY] in addition to a downstream aspartate residue (part of the DFG motif) [12]. The corresponding sequence in AceK is 455PGDMLFKNFGV465 with the downstream aspartate at position 475. In ePKs, the histidine residue (or sometimes tyrosine) is part of the HRD motif and is responsible for orienting two catalytic aspartates residues to coordinate the ATP molecule and Mg2+ ion for catalysis. AceK lacks this histidine residue, yet a flexible glycine residue neighbours the catalytic Asp457, allowing for additional freedom of movement for it to shift in and out of the ATP pocket. The absence of histidine may also reflect the observation that only one Mg2+ is found in AceK, whereas two are present in ePKs [3].
(b). Comparison with atypical kinases
Atypical kinases have a similar catalytic core to ePKs and possess kinase activity despite not conserving many of the kinase motifs. For example, in haspin (haploid germ-cell-specific nuclear protein kinase, encoded by germ-cell-specific gene 2: Gsg2), the DFG motif involved in ATP and Mg2+ binding is replaced with DYT and completely lacks the APE motif at the C-terminus of the activation segment. There is only weak sequence homology shared with ePKs; its KD is highly divergent and contains several unique inserts, and variations exist in its activation segment. In fact, its activation segment is highly structured, which may indicate that it is constitutively active and ready for substrate binding. The haspin structure contains several structural features conserved among haspin-like kinases but not seen previously in kinases [13,14]. Although not structurally similar to haspin, AceK shares the idea that a protein can perform the phosphorylation reaction even when divergence in the KD exists.
(c). Hydrophobic ‘spines’
Recently, the conformations of active and inactive kinases were compared, and additional conserved non-contiguous motifs termed hydrophobic spines were proposed that connect the N- and C-lobes [15]. The regulatory spine (R-spine) consists of four hydrophobic residues (Leu95, Leu106, Phe185 and Tyr164; PKA numbering) and is assembled in active kinases yet is disordered in inactive kinases [16]. A catalytic spine (C-spine) also exists that spans the kinase and requires the adenine ring of ATP [17]. Analysis of the KD of AceK shows that both these kinase spines may exist in this bifunctional protein (figure 3c,d). The four hydrophobic residues are proposed to be Val360, Thr372, Phe454 and Tyr476. The AceK R-spine, however, does not anchor to the αF-helix through the commonly observed hydrogen bond as seen in ePKs but may continue the hydrophobicity to Trp545, allowing more flexibility to modulate between its two functions. The C-spine is not as apparent in the AceK structure. The upper portion of the spine is in reasonable agreement, Val325, Val334, ATP, Phe463, Gly464 and Val465; however, the hydrophobicity path to the αF-helix is not as well defined. The residues that may correlate are Leu422, Leu522 and Ile528 with different or additional residues assisting in completing the network (Ile438, Tyr441 and Phe532). A residue such as Tyr474 may be required to substitute for the lack of side chain of Gly464 to interact with the adenine of ATP. Variations in AceK are not surprising as it is a more distant, atypical kinase [18]. Another important feature of the spine model is the role of the ‘gatekeeper’, which is located between the R- and C-spines and may modulate these spines [15,19]. In this position in AceK is Tyr414, a residue already proposed to serve an important role in transmitting the allosteric effects from the RD (discussed later); it may also help switch the kinase on and off by affecting the spines.
(d). Regulatory loop
One of the most striking differences between AceK and the ePK family is the presence of Loopβ3αC in AceK (figure 2b). This regulatory loop shields and exposes ATP in the adenosine monophosphate (AMP)-bound and AMP-free AceK structures, respectively, thereby acting as a regulator of catalytic function as it controls the accessibility to the ATP binding site. Upon dissociation of AMP, Loopβ3αC shifts upwards by 16 Å towards the N-lobe and opens the ATP site, initiating the process of kinase activation. Although this structural element is absent in ePKs, its function is somewhat analogous to the activation loops in these kinases. The activation loop is positioned between the β9 strand and P + 1 loop and regulates kinase activity. The activation loop region of ePKs is completely absent in the KD of AceK, instead the substrate recognition loop (SRL) partially occupies this region. Many ePKs are themselves phosphoproteins and the activation loop contains the phosphorylation site(s), which activates the enzyme. Phosphorylation in the activation loop not only stabilizes the active conformation but also destabilizes the inactive conformation. The addition of the phosphate moiety introduces new hydrogen bonds that orientate the active site and substrate-binding interface, whereas this loop in inactive kinases is disordered and the key catalytic residues are out of position. Phosphorylation also stabilizes the loop in an open and extended conformation, allowing the protein substrate to bind [20]. The specifics of this loop among ePKs are structurally diverse [21]; some kinases lack the phosphorylation site(s) completely and are constitutively active, and those kinases that do not require phosphorylation for activation have acquired alternative strategies to mimic the phosphate moiety [22]. Thus, utilization of a flexible surface loop as a means of regulating activity in kinases has been conserved from bacteria to eukaryotes, despite some significant differences in the sequence, spatial location and factors influencing the conformational change.
(e). Accessory domain
The overall structural design of AceK consisting of both a kinase (catalytic) domain and a RD is analogous to many kinases (figure 2a). ePKs diversify themselves by their non-KDs. Like AceK, these non-KDs are often involved in regulation but may also serve other roles [23]. Unlike the conserved kinase catalytic core, the RD of AceK has a novel fold, thereby its only similarity with the various RDs of ePKs is its function to control the enzyme. Inactive cyclic AMP (cAMP)-dependent PKA is composed of two catalytic kinase subunits and a regulatory (inhibitory) subunit dimer in the absence of cAMP. When cAMP binds the regulatory subunit the catalytic subunits dissociate and become active [24]. Similarly, cGMP-dependent protein kinase has three functional domains: an N-terminal domain, a RD and the KD. The N-terminal domain inhibits kinase function in the absence of cyclic guanosine monophosphate (cGMP). When the two cGMP-binding sites are occupied in the RD, a large conformational change is induced, which relieves the inhibitory control of the N-terminal domain, hence activating the kinase [25]. As in the case of AceK, regulation can also be mediated by allosteric effects, in particular by AMP (see later), such as AMP-activated protein kinase (AMPK). AMPK is a heterotrimeric complex consisting of a catalytic subunit (α) and two regulatory subunits (β and γ) and is activated allosterically by AMP [26]. In contrast, the kinase function of AceK is inhibited when AMP is bound.
It is clear that, despite many differences between AceK and ePKs, the common structural and functional features are present in these kinases, albeit in a varied form and style. If a critical element seems to be missing, a different protein segment would be present and compensate for the ‘loss’ of function. AceK represents a more extreme case of kinase diversity, most likely due to the fact that it also possesses phosphatase function, the complete opposite of kinase activity.
3. Acek atp/adp-dependent phosphatase function
The close structural similarity of the KD of AceK to ePKs was unexpected, but provided a good basis for understanding the kinase function of AceK. In contrast, the mechanism of its unique ATP/adenosine diphosphate (ADP)-dependent phosphatase activity remains unclear despite this structural information. Like kinase activity, phosphatase function is a universal theme in regulation, and so clues may lie in a comparison of AceK with the three major families of phosphatases. The phosphatase families are defined by their amino acid sequences in their active sites. Similarities are seen only when AceK is compared with the new class of phosphatases with the active site sequence DXDX(T/V). This newly emerging family of phosphatases are members of the haloacid dehalogenase (HAD) superfamily; hence we herein term it HADP. HADP acts through a pentacovalent intermediate by an associative (or SN2) mechanism [27,28]. The strictly conserved first aspartate serves as the nucleophile and forms the phosphoenzyme intermediate. The second aspartate acts as a general acid to donate its proton to the leaving hydroxyl group. Conservation of the second aspartate varies from low retention of phosphatase activity to full loss of function. Therefore, the carboxylic side chain of the first aspartate is more position sensitive for its function than the second aspartate. This group is defined by the DXDX(T/V) sequence, but other features include the signature motif being preceded by four hydrophobic residues and a strictly conserved lysine residue with a role in transition state stabilization [29,30].
(a). AceK and haloacid dehalogenase phosphatases
In AceK, the sequence DXDXI is found preceded by four hydrophobic residues and a conserved Lys461 is apparent (figure 4a). Comparison of AceK with a representative of HADP shows good alignment of all important features (figure 4b), further supporting the notion that AceK may be a distant relative of this phosphatase family. One difference is that an isoleucine is present rather than threonine or valine at position 5 in the motif, although it is a relatively conservative substitution and may relate to the multiple functions of this enzyme (phosphatase, kinase and ATPase activities). Regardless of this discrepancy, this motif contains Asp475 and Asp477, which through both structural and mutational studies have been shown to be critical to enzyme function. The D477N and D477A mutant proteins retain kinase activity yet have complete loss of phosphatase activity [3,32]. From the AceK structure, Asp477 and Asp475 interact with the γ-phosphate of ATP, and Asp475 coordinates the ATP-liganded magnesium ion [3]. The D475A mutant, which likely disrupts the binding of a metal cofactor in the ATP binding site, results in the complete loss of ATPase activity [3].
Figure 4.

(a) Sequence alignment of AceK from several species. The DXDX(T/V) motif is highlighted in red. The preceding four hydrophobic residues are underlined, and the conserved lysine involved in transition state stabilization is shown in blue. (b) Structural alignment of AceK 475DXDX(I)479 motif (pink) with that of d,d-heptose 1,7-bisphosphate phosphatase [31] (PDB accession code: 3L8H; light blue). The yellow rectangle contains the four hydrophobic residues, and the strictly conserved lysine coordinated with Mg2+ ion. (c) Proposed phosphatase mechanism for AceK. (step 1) Asp475 acts as the nucleophile attacking the phosphate moeity on Ser113 of IDH through an associative mechanism to form a pentacovalent phosphate intermediate. (step 2) Asp477 acts as a general acid to donate its proton to Ser113 of IDH. The phosphoenzyme intermediate at Asp475 is formed. (step 3) The aspartatyl-phosphate undergoes nucleophilic attack by the ‘catalytic residue’ ADP to produce ATP. The breaking of the high-energy apartatyl-phosphate bond provides the energy for this reaction. (step 4) Water donates its proton to Asp477 and then attacks the γ-phosphate of ATP (ATP hydrolysis reaction). (step 5) The completed reaction with the enzyme available to perform its next round of reaction.
(b). Proposed AceK phosphatase mechanism
If AceK is a member of HADP, it is likely that phosphorylated IDH binds to AceK and positions its phosphoryl group into the active site of AceK near the catalytic Asp475. Asp475, as the nucleophile, attacks the phosphate on IDH to form a phosphoenzyme intermediate. Asp477 donates a proton to the hydroxyl side chain of Ser113 on IDH as it leaves. The phosphoryl-aspartate transfers the phosphate moiety to the bound ADP molecule to form ATP, whereby ADP plays a catalytic role with the high energy of the acyl bond on aspartate driving this phosphatase reaction. It is known that the phosphatase activity of AceK is ATP/ADP-dependent, although the specifics are still unclear with evidence that it is a kinase back reaction [33]. Perhaps it is simply related to the regeneration of the ‘catalytic’ ADP and hence the ATPase function of AceK is just one step in the dephosphorylation reaction (figure 4c). ATP hydrolysis activity is surprisingly strong, with a 10-fold increased activity compared with both kinase and phosphatase function [33], and therefore must be stringently controlled. In the default phosphatase state of AceK, water must be excluded to prevent the wasteful hydrolysis of ATP. The allosteric regulator AMP stimulates phosphatase activity (or inhibits kinase activity). With AMP bound, Loopβ3αC is in the closed conformation (figure 2b) owing to a salt bridge between Lys346 and Glu478 [3]. In fact, the AceK structures with the loop closed show the ATP molecule bound in the active site, whereas in the AMP-free AceK structure, Loopβ3αC is in the open conformation and ADP is found [3]. In the latter case, water was accessible to the active site and ATP hydrolysis occurred. This raises the question, if ATPase activity is one step in the phosphatase reaction, how does water enter the active site when it is in the locked position? In the AceK/IDH complex structure, the salt bridge between Lys346 and Glu478 is broken, and the γ-phosphate of ATP moves out from inside of the active site pocket although Loopβ3αC is still not open. This conformational change is caused by the binding of IDH, suggesting that with binding of phosphorylated IDH there may be an even larger local conformational change, which might allow water to enter, or the phosphate group to leave. However, details regarding the ATPase mechanism remain a point to be investigated.
4. Regulation
IDH represents a rate-limiting step in the Krebs cycle and includes the steps where carbon is lost to carbon dioxide, therefore forming a critical point of regulation. Phosphorylation of IDH is a means of regulating the branch point between the Krebs cycle and glyoxylate bypass (figure 1a). When a rich carbon source is available, the energy-generating Krebs cycle continues to flow as IDH is dephosphorylated; however, if a less-preferred carbon source, such as acetate or fatty acids is available, the cell responds by phosphorylating IDH, activating the glyoxylate bypass [1]. Isocitrate is the substrate for both IDH and isocitrate lyase (ICL); however, IDH has a higher affinity for isocitrate. By inactivating IDH, through the action of AceK, the increased accumulation of isocitrate allows ICL to operate. Regulation as a consequence of varying affinities is called the branch point effect, so that the glyoxylate bypass is highly sensitive to the phosphorylation state of IDH [1,34].
(a). Gene regulation
AceK is tightly controlled from the transcriptional to the post-translational level, demonstrating both global and specific regulatory systems and how highly sensitive the glyoxylate pathway responds to the metabolic needs of the cell. The aceBAK operon (figure 1b) encodes the enzymes of the glyoxylate bypass. In addition to aceK, aceB encodes malate synthase, and aceA encodes ICL. These genes are all controlled by the same promoter, yet are expressed at differing levels, with AceK expressed the least. Codons for aceK are less favourable compared with aceB and aceA, allowing control at the elongation phase of translation. Also preceding the aceK gene is a palidromic sequence that forms a stable stem and loop structure, in consequence decreasing its expression. The aceK gene is controlled at the transcriptional level with Ic1R and FadR acting as repressors and FruR and IHF as activators (figure 1b) [35].
The IclR repressor represses the expression of the aceBAK operon through binding at the promoter region competing with RNA polymerase for the same binding site (figure 1b). When cultures are grown in acetate, IclR is released from the promoter region, allowing the expression of the aceBAK operon. Glyoxylate and pyruvate bind the same site on the IclR protein but have opposing effects. Glyoxylate prevents the formation of the IclR–operator complex, favouring the inactive dimeric state of IclR activating aceBAK gene expression; however, pyruvate promotes binding of IclR to the aceBAK promoter and stabilizes the active tetrameric form of IclR repressing the operon [36]. This is in agreement with the previous observation that when pyruvate is added to a culture grown in acetate, IDH is quickly dephosphorylated and activated [37]. Surprisingly, neither the aceBAK operon nor the AceK protein is activated by acetate or acetyl-CoA, at least not directly. However, phosphoenolpyruvate (PEP), acting as an inducer, will prevent the binding of IclR to the promoter [38]. In vitro, fructose-1-phosphate or fructose-1,6-bisphosphate can remove and prevent FruR from binding the operon, thereby repressing aceBAK expression [35].
(b). Protein regulation
Regulation of the IDH phosphorylation/dephosphorylation event by AceK also occurs at the protein level. Many of the regulatory effectors are derived from the end products of the glyoxylate bypass, and represent negative feedback inhibition mechanisms, decreasing the amount of isocitrate available to ICL. In general, many of these metabolites will inhibit the kinase activity but activate the phosphatase activity of AceK (3-phosphoglycerate, pyruvate, AMP, ADP, oxaloacetate, α-ketoglutarate and PEP) [1]. Isocitrate, NADP+, citrate, fructose-6-phosphate and glyoxylate inhibit kinase activity only, with no apparent effect on phosphatase function [1,39]. In addition, ATP, a cofactor for kinase activity, activates phosphatase function (AceK phosphatase activity is ATP/ADP-dependent; see §4a), and NADPH blocks both opposing activities. Surprisingly, no regulators that activate AceK kinase activity or inhibit AceK phosphatase activity have been identified, illustrating the stringent control of the glyoxylate bypass, which only needs to be activated when the cell is nutrient deprived. These metabolites provide a direct measure of the cellular needs, and if these effectors accumulate excessively, the phosphatase activity is increased to reactivate IDH and reduce the flux through the glyoxylate bypass. AMP, 3-phosphoglycerate and pyruvate bind directly to AceK and activate phosphatase and inhibit both kinase and intrinsic ATPase activities (in the absence of IDH). Their binding to AceK could cause a conformational change whereby the phosphatase mode is favoured. NADPH and isocitrate, however, bind to IDH rather than AceK, in turn inhibiting kinase activity, but unlike the other effectors, phosphatase activity is not activated and there is no effect on the intrinsic ATPase function. Thus, in the presence of IDH, isocitrate binds to the active site of IDH competing with and reversing the product inhibition, resulting in more dephosphorylation [39]. Isocitrate binds only to dephospho-IDH but not to phospho-IDH. NADPH binds both dephospho- and phospho-IDH, therefore making IDH a poor substrate, which competes with IDH for both kinase and phosphatase activities of AceK. When the binding sites for NADPH or isocitrate are occupied, IDH is unable to interact with AceK, possibly by preventing the important contacts from forming between these two proteins or formation of an unfavourable closed conformation for interaction, thereby preventing access to Ser113, the target residue for (de)phosphorylation. A regulatory model was proposed by Miller et al. [39], whereby AceK exists in either kinase or phosphatase conformation and an equilibrium is controlled by the metabolites that preferably bind to the phosphatase state. IDH is either in the open or in the closed conformation, and AceK will associate and phosphorylate only the open conformation of IDH. NADPH or isocitrate locks IDH into the closed conformation and may interfere with AceK–IDH complex formation. The structures of AceK appear to support this model [3]. When AceK is exhibiting kinase function, the dissociation constant for IDH is 2.4 μM; however, for phosphatase function, a lower binding affinity exists for phospho-IDH (6 μM). With a preference for binding unphosphorylated IDH, the regulatory metabolites help favour the phosphatase state of AceK, perhaps strengthening this interaction with phosphorylated IDH [39].
(c). Regulation by AMP
AMP is a good monitor of the energy needs of the cell. The eukaryotic AMPK regulates cellular metabolism in response to the availability of energy. The AMP/ATP ratio regulates how AMPK acts on its downstream targets [40,41]. Compared with AMPK, AceK appears to have a more complicated regulatory mechanism for sensing changes in cell metabolism. Depletion in AMP levels signals that the cell requires energy and isocitrate will continue through the Krebs cycle with IDH dephosphorylated. The structure of AceK with AMP bound provides the first structural glimpse into how AMP may act as an allosteric regulator. AMP is bound in a pocket between the RD and KD; yet the ATP binding site, where the catalytic action occurs, is nearly 25 Å away [3]. Structural analysis suggests that these sites may communicate through a network of salt bridges and hydrogen bonds, with Tyr414 central to this intricate network between AMP and ATP (figure 5). Tyr414 hydrogen bonds with Glu374, which in turn communicates with AMP through the Glu374, Lys294, AMP, Ser105 and Glu374, Lys294, AMP, Lys291, Asp55 networks. Towards the ATP binding site, Tyr414 further extends the interaction to Tyr357 via π-stacking, where Tyr357, together with Glu416, stabilizes Lys336, a key residue for ATP binding. The network is likely more complex and dynamic than these initial observations show; however, it does provide a plausible explanation of how the allosteric effects of AMP are transmitted through the protein [3]. Fructose 1,6-bisphosphatase is a homotetramer that is inhibited allosterically by AMP. The binding of AMP to its allosteric site, 28 Å away from the closest active site, triggers a conversion from the active R- to the inactive T-state conformation, as the upper subunit pair rotates relative to the bottom subunit pair. In the absence of AMP, the enzyme assumes the R-state with a loop (residues 52–72) engaged at the active site; however, when AMP binds, this loop is stabilized in the disengaged conformation away from the active site [42]. Although the details of the AMP allosteric mechanism probably differ from AceK, similarities exist wherein AMP binding causes movement of a loop in proximity to the active site and this is all triggered by binding of AMP to a distant allosteric site [42,43]. AMP also binds glycogen phosphorylase b at a site distant from the catalytic site, 35 Å away. The allosteric site, like AceK, is situated across the subunit (domain) interface in this dimeric enzyme sandwiched between a loop of one subunit (cap) and a helix for the other subunit. This allosteric site contains partially overlapping binding sites for both AMP and glucose-6-phosphate, with these effectors having opposing roles. AMP is activating (preferentially binding to the active R-state), and glucose-6-phosphate is inhibitory (binding to the inactive T-state) [44–46]. Although no other effectors are known to share the AMP binding site in AceK, the structure of AceK may reveal additional binding sites and regulatory mechanisms. This possibility is supported by the fact that multiple regulators are already known.
Figure 5.

Electrostatic surface potential analysis. The yellow rectangle contains the AMP binding site and the ATP-binding active site. The green rectangle reveals another potential allosteric binding site at the interface between the KD and RD in AceK. The expanded yellow box shows the detailed interactions between AMP- and ATP-binding pockets in AceK and how the two sites communicate.
The previously identified mutations Y414C and Q373R in AceK result in altered sensitivity towards AMP, but also to 3-phosphoglycerate and pyruvate [32,39]. The Y414C mutant eliminates the critical hydrogen bond between Tyr414 and Glu374, and mutation of the neighbouring residue, Gln373, may change the orientation of this critical hydrogen bond in addition to the loss of interactions with Lys294 and Ile414. Thus, Tyr414 is the sensor residue needed to transmit the allosteric effects of not only AMP but also probably 3-phosphoglycerate and pyruvate to maintain AceK in the phosphatase mode.
(d). Regulation by acetylation
AceK is post-translationally modified by reversible lysine acetylation as a means of regulating its action. When AceK is acetylated, its ability to phosphorylate IDH decreases (favours the phosphatase conformation), whereas when AceK is deacetylated its ability to phosphorylate IDH increases (favours the kinase conformation). The acetylated state can be ‘locked’ in by the mimetic triple mutant (K72Q/K83Q/K553Q), whereby mutation neutralizes the positive charge, as would acetylation. Of the three lysines residues in the acetylation mimetic mutant, only Lys83 is strictly conserved and is probably the crucial site of modification. This represents another key point of regulation as the acetylation reaction requires acetyl-CoA and NAD+, both important metabolites [47]. However, the mechanism through which acetylation regulates AceK is not known and represents an interesting and challenging area for structural investigation.
(e). Regulator binding and switch mechanism
An array of ligands bind to and regulate AceK, and we are at the beginning of understanding how these may function and whether they share the same binding site or there are multiple allosteric binding sites. Identification of additional allosteric interaction sites by electrostatic surface potential analysis was performed on the structure of AceK (figure 5). In particular, the interface that bridges the KD and RD was evaluated. In addition to the AMP binding site (described earlier), at least one potential pocket is located at the interface. It has a well-defined pocket size and depth, and is negatively charged. This pocket, along with others yet to be identified, might represent other allosteric binding sites. Regulator binding in these pockets will no doubt interfere with the interactions between the two domains.
The recent structure of AceK suggests a switch mechanism between the kinase and phosphatase activities of AceK, the two activities that operate at the same active site (figure 6). In the absence of AMP, Loopβ3αC is in the closed conformation, obstructing the ATP-binding pocket (figure 2b). The interactions between residue Gln345 from Loopβ3αC (AceK) and the backbone of Ser113 (IDH) prevent unphosphorylated IDH from approaching the AceK ATP binding site, which inhibits kinase activity and favours phosphatase function. However, phosphorylated-Ser113 IDH could enter the active site and undergo dephosphorylation, as the large, negatively charged phosphate group impairs the interactions between Gln345 and Ser113. Loopβ3αC can be opened when the allosteric effector AMP is released, allowing Ser113 of IDH to be phosphorylated, thus putting the enzyme into kinase mode (figure 6).
Figure 6.

Schematic of the proposed mechanism of AceK activity switch. (1) Closed and open modes of IDH. (2) AMP-bound AceK binds to the open IDH. (3) Upon AMP dissociation, Loop-β2αC opens and AceK in kinase mode phosphorylates IDH. (4) In the absence of AMP, Loop-β2αC remains open and, without substrate, AceK exhibits ATPase activity. (5) AMP-bound AceK, in which Loop-β2αC is closed, binds to phosphorylated IDH. (6) AMP-bound AceK, as a phosphatase, dephosphorylates phospho-IDH.
5. Interaction with isocitrate dehydrogenase as a kinase substrate
The primary sequence surrounding the phosphorylation site (P-site) is often all that is required for a protein kinase to bind and phosphorylate its target substrate. Thus, a short peptide representing this sequence may be used as a substitute substrate. However, AceK strictly forms only an intimate association with the homodimer of IDH [3] and cannot phosphorylate either proteolytic fragments derived from IDH or synthetic peptides that correspond to the sequence surrounding the phosphorylation site [48]. The interface created between AceK and IDH is highly specific [11,48,49]. The SRL (residues 484–510) of AceK deeply extends approximately 32 Å into the active site cleft of IDH with a short α-helix at its tip (figure 7a). An extensive array of interactions is produced between the SRL and both molecules of dimeric IDH, which include hydrophobic packing, salt bridges and hydrogen bonds [3]. Further analysis of the AceK–IDH complex structure reveals two discontinuous regions on IDH that contact AceK: the P-loop from monomer 1 and the twisted antiparallel β-sheet from monomer 2 (ARS; AceK recognition segment) of the homodimeric structure of IDH (figure 7b). If either of these recognition sites on IDH is modified, even slightly, AceK fails to bind and recognize IDH. A structural survey of all IDH proteins revealed that only IDHs from Gram-negative bacteria are a target for AceK. Indeed, Escherichia coli AceK can cross-phosphorylate Burkholderia pseudomallei IDH, demonstrating a close evolutionary relationship among Gram-negative bacterial AceK–IDH systems. It is thus not surprising that all known and putative AceK proteins exist only in Gram-negative bacteria. The IDH protein from other organisms may have evolved structurally to circumvent association with AceK, or the lack of selective pressure eliminated the need for these AceK-binding elements [50].
Figure 7.

(a) The interface between AceK and the IDH active site cleft. IDH dimer is in electrostatic surface (left) and the side chain interactions between AceK SRL and IDH are shown. Residues from IDH are coloured in blue, and residues from AceK are coloured in green (right). (b) The AceK recognition segment (ARS) and P-loop (both dark blue) on IDH (grey) are strictly needed for AceK (orange) to associate. The SRL on AceK is also shown (yellow).
6. Future perspective
Although the AceK field was stalled for some time, the new structural information has sparked new interest into this unusual bifunctional enzyme. With a working hypothesis of the phosphatase mechanism now developed, we now have new avenues to test experimentally. One such goal would be the structure determination of ‘paired’ AceK/IDH complex structures, including AceK–AMP with phosphorylated IDH and AceK with IDH in the absence of AMP, through the use of inactivated mutants or non-hydrolyzable ATP analogues. In essence, this would create a structural movie of the catalytic mechanisms, providing better understanding of the bifunctionality of AceK, particularly phosphatase.
Despite the strides that have been made with the ligand AMP, regulation of AceK remains complex with many potential regulators, each with a possibility of a different communication network running through the AceK protein. Uncovering how all these regulators work together to control AceK function would be highly beneficial. Do these ligands regulate AceK competitively, cooperatively or both? Structural location of these allosteric binding sites may shed some light as we develop a fully integrated model of regulation. Recent work on acetylated AceK has located a potential residue involved, and structural work may suggest how acetylation communicates throughout the protein.
The ultimate goal is to fully understand both the kinase and phosphatase functions of AceK, which may require the design of inhibitors and activators that modulate these functions. Despite this ambitious goal, what remains clear is that we are at the beginning of an exciting journey into uncovering the details of this unusual kinase/phosphatase.
Acknowledgements
This work was supported by Canadian Institutes of Health Research (CIHR) and the National Natural Science Foundation of China (grant no. 21133003). Z.J. is a Canada Research Chair in Structural Biology and a Killam Research Fellow and SPY is a recipient of a CIHR fellowship.
References
- 1.Cozzone A. J. 1998. Regulation of acetate metabolism by protein phosphorylation in enteric bacteria. Annu. Rev. Microbiol. 52, 127–164 10.1146/annurev.micro.52.1.127 (doi:10.1146/annurev.micro.52.1.127) [DOI] [PubMed] [Google Scholar]
- 2.LaPorte D. C., Koshland D. E., Jr 1982. A protein with kinase and phosphatase activities involved in regulation of tricarboxylic acid cycle. Nature 300, 458–460 10.1038/300458a0 (doi:10.1038/300458a0) [DOI] [PubMed] [Google Scholar]
- 3.Zheng J., Jia Z. 2010. Structure of the bifunctional isocitrate dehydrogenase kinase/phosphatase. Nature 465, 961–965 10.1038/nature09088 (doi:10.1038/nature09088) [DOI] [PubMed] [Google Scholar]
- 4.Wang Z., Liu J., Sudom A., Ayres M., Li S., Wesche H., Powers J. P., Walker N. P. 2006. Crystal structures of IRAK-4 kinase in complex with inhibitors: a serine/threonine kinase with tyrosine as a gatekeeper. Structure. 14, 1835–1844 10.1016/j.str.2006.11.001 (doi:10.1016/j.str.2006.11.001) [DOI] [PubMed] [Google Scholar]
- 5.Chen H., et al. 2008. A crystallographic snapshot of tyrosine trans-phosphorylation in action. Proc. Natl Acad. Sci. USA 105, 19 660–19 665 10.1073/pnas.0807752105 (doi:10.1073/pnas.0807752105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knowles P. P., Murray-Rust J., Kjaer S., Scott R. P., Hanrahan S., Santoro M., Ibanez C. F., McDonald N. Q. 2006. Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem. 281, 33 577–33 587 10.1074/jbc.M605604200 (doi:10.1074/jbc.M605604200) [DOI] [PubMed] [Google Scholar]
- 7.Scheeff E. D., Bourne P. E. 2005. Structural evolution of the protein kinase-like superfamily. PLoS Comput. Biol. 1, e49 10.1371/journal.pcbi.0010049 (doi:10.1371/journal.pcbi.0010049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor S. S., Knighton D. R., Zheng J., Ten Eyck L. F., Sowadski J. M. 1992. Structural framework for the protein kinase family. Annu. Rev. Cell Biol. 8, 429–462 10.1146/annurev.cb.08.110192.002241 (doi:10.1146/annurev.cb.08.110192.002241) [DOI] [PubMed] [Google Scholar]
- 9.Bossemeyer D. 1994. The glycine-rich sequence of protein kinases: a multifunctional element. Trends Biochem. Sci. 19, 201–205 10.1016/0968-0004(94)90022-1 (doi:10.1016/0968-0004(94)90022-1) [DOI] [PubMed] [Google Scholar]
- 10.Taylor S. S., Knighton D. R., Zheng J., Sowadski J. M., Gibbs C. S., Zoller M. J. 1993. A template for the protein kinase family. Trends Biochem. Sci. 18, 84–89 10.1016/0968-0004(93)80001-R (doi:10.1016/0968-0004(93)80001-R) [DOI] [PubMed] [Google Scholar]
- 11.Oudot C., Cortay J. C., Blanchet C., LaPorte D. C., Di P. A., Cozzone A. J., Jault J. M. 2001. The ‘catalytic’ triad of isocitrate dehydrogenase kinase/phosphatase from E. coli and its relationship with that found in eukaryotic protein kinases. Biochemistry 40, 3047–3055 10.1021/bi001713x (doi:10.1021/bi001713x) [DOI] [PubMed] [Google Scholar]
- 12.Brenner S. 1987. Phosphotransferase sequence homology. Nature 329, 21 10.1038/329021a0 (doi:10.1038/329021a0) [DOI] [PubMed] [Google Scholar]
- 13.Eswaran J., Patnaik D., Filippakopoulos P., Wang F., Stein R. L., Murray J. W., Higgins J. M., Knapp S. 2009. Structure and functional characterization of the atypical human kinase haspin. Proc. Natl Acad. Sci. USA 106, 20 198–20 203 10.1073/pnas.0901989106 (doi:10.1073/pnas.0901989106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eswaran J., Knapp S. 2010. Insights into protein kinase regulation and inhibition by large scale structural comparison. Biochim. Biophys. Acta 1804, 429–432 10.1016/j.bbapap.2009.10.013 (doi:10.1016/j.bbapap.2009.10.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kornev A. P., Taylor S. S. 2010. Defining the conserved internal architecture of a protein kinase. Biochim. Biophys. Acta 1804, 440–444 10.1016/j.bbapap.2009.10.017 (doi:10.1016/j.bbapap.2009.10.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kornev A. P., Haste N. M., Taylor S. S., Eyck L. F. 2006. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl Acad. Sci. USA 103, 17 783–17 788 10.1073/pnas.0607656103 (doi:10.1073/pnas.0607656103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kornev A. P., Taylor S. S., Ten Eyck L. F. 2008. A helix scaffold for the assembly of active protein kinases. Proc. Natl Acad. Sci. USA 105, 14 377–14 382 10.1073/pnas.0807988105 (doi:10.1073/pnas.0807988105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burk D. L., Hon W. C., Leung A. K., Berghuis A. M. 2001. Structural analyses of nucleotide binding to an aminoglycoside phosphotransferase. Biochemistry 40, 8756–8764 10.1021/bi010504p (doi:10.1021/bi010504p) [DOI] [PubMed] [Google Scholar]
- 19.Azam M., Seeliger M. A., Gray N. S., Kuriyan J., Daley G. Q. 2008. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol. 15, 1109–1118 10.1038/nsmb.1486 (doi:10.1038/nsmb.1486) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huse M., Kuriyan J. 2002. The conformational plasticity of protein kinases. Cell 109, 275–282 10.1016/S0092-8674(02)00741-9 (doi:10.1016/S0092-8674(02)00741-9) [DOI] [PubMed] [Google Scholar]
- 21.Nolen B., Taylor S., Ghosh G. 2004. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 15, 661–675 10.1016/j.molcel.2004.08.024 (doi:10.1016/j.molcel.2004.08.024) [DOI] [PubMed] [Google Scholar]
- 22.Rabiller M., Getlik M., Kluter S., Richters A., Tuckmantel S., Simard J. R., Rauh D. 2010. Proteus in the world of proteins: conformational changes in protein kinases. Arch. Pharm. (Weinheim) 343, 193–206 10.1002/ardp.201000028 (doi:10.1002/ardp.201000028) [DOI] [PubMed] [Google Scholar]
- 23.Deshmukh K., Anamika K., Srinivasan N. 2010. Evolution of domain combinations in protein kinases and its implications for functional diversity. Prog. Biophys. Mol. Biol. 102, 1–15 10.1016/j.pbiomolbio.2009.12.009 (doi:10.1016/j.pbiomolbio.2009.12.009) [DOI] [PubMed] [Google Scholar]
- 24.Kim C., Xuong N. H., Taylor S. S. 2005. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science 307, 690–696 10.1126/science.1104607 (doi:10.1126/science.1104607) [DOI] [PubMed] [Google Scholar]
- 25.Osborne B. W., et al. 2011. Crystal structure of cGMP-dependent protein kinase reveals novel site of interchain communication. Structure 19, 1317–1327 10.1016/j.str.2011.06.012 (doi:10.1016/j.str.2011.06.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carling D. 2005. AMP-activated protein kinase: balancing the scales. Biochimie 87, 87–91 10.1016/j.biochi.2004.10.017 (doi:10.1016/j.biochi.2004.10.017) [DOI] [PubMed] [Google Scholar]
- 27.Lahiri S. D., Zhang G., Dunaway-Mariano D., Allen K. N. 2003. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science 299, 2067–2071 10.1126/science.1082710 (doi:10.1126/science.1082710) [DOI] [PubMed] [Google Scholar]
- 28.Hausmann S., Erdjument-Bromage H., Shuman S. 2004. Schizosaccharomyces pombe carboxyl-terminal domain (CTD) phosphatase Fcp1: distributive mechanism, minimal CTD substrate, and active site mapping. J. Biol. Chem. 279, 10 892–10 900 10.1074/jbc.M312513200 (doi:10.1074/jbc.M312513200) [DOI] [PubMed] [Google Scholar]
- 29.Collet J. F., Stroobant V., Pirard M., Delpierre G., Van S. E. 1998. A new class of phosphotransferases phosphorylated on an aspartate residue in an amino-terminal DXDX(T/V) motif. J. Biol. Chem. 273, 14 107–14 112 [DOI] [PubMed] [Google Scholar]
- 30.Collet J. F., Stroobant V., Van S. E. 2002. Evidence for phosphotransferases phosphorylated on aspartate residue in N-terminal DXDX(T/V) motif. Methods Enzymol. 354, 177–188 10.1016/S0076-6879(02)54014-3 (doi:10.1016/S0076-6879(02)54014-3) [DOI] [PubMed] [Google Scholar]
- 31.Nguyen H. H., Wang L., Huang H., Peisach E., Dunaway-Mariano D., Allen K. N. 2010. Structural determinants of substrate recognition in the HAD superfamily member D-glycero-D-manno-heptose-1,7-bisphosphate phosphatase (GmhB). Biochemistry 49, 1082–1092 10.1021/bi902019q (doi:10.1021/bi902019q) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ikeda T. P., Houtz E., LaPorte D. C. 1992. Isocitrate dehydrogenase kinase/phosphatase: identification of mutations which selectively inhibit phosphatase activity. J. Bacteriol. 174, 1414–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller S. P., Karschnia E. J., Ikeda T. P., LaPorte D. C. 1996. Isocitrate dehydrogenase kinase/phosphatase. Kinetic characteristics of the wild-type and two mutant proteins. J. Biol. Chem. 271, 19 124–19 128 10.1074/jbc.271.32.19124 (doi:10.1074/jbc.271.32.19124) [DOI] [PubMed] [Google Scholar]
- 34.LaPorte D. C., Walsh K., Koshland D. E., Jr 1984. The branch point effect. Ultrasensitivity and subsensitivity to metabolic control. J. Biol. Chem. 259, 14 068–14 075 [PubMed] [Google Scholar]
- 35.Cozzone A. J., El-Mansi M. 2005. Control of isocitrate dehydrogenase catalytic activity by protein phosphorylation in Escherichia coli. J. Mol. Microbiol. Biotechnol. 9, 132–146 10.1159/000089642 (doi:10.1159/000089642) [DOI] [PubMed] [Google Scholar]
- 36.Lorca G. L., Ezersky A., Lunin V. V., Walker J. R., Altamentova S., Evdokimova E., Vedadi M., Bochkarev A., Savchenko A. 2007. Glyoxylate and pyruvate are antagonistic effectors of the Escherichia coli IclR transcriptional regulator. J. Biol. Chem. 282, 16 476–16 491 10.1074/jbc.M610838200 (doi:10.1074/jbc.M610838200) [DOI] [PubMed] [Google Scholar]
- 37.el-Mansi E. M., Nimmo H. G., Holms W. H. 1986. Pyruvate metabolism and the phosphorylation state of isocitrate dehydrogenase in Escherichia coli. J. Gen. Microbiol. 132, 797–806 [DOI] [PubMed] [Google Scholar]
- 38.Cortay J. C., Negre D., Galinier A., Duclos B., Perriere G., Cozzone A. J. 1991. Regulation of the acetate operon in Escherichia coli: purification and functional characterization of the IclR repressor. EMBO J. 10, 675–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller S. P., Chen R., Karschnia E. J., Romfo C., Dean A., LaPorte D. C. 2000. Locations of the regulatory sites for isocitrate dehydrogenase kinase/phosphatase. J. Biol. Chem. 275, 833–839 10.1074/jbc.275.2.833 (doi:10.1074/jbc.275.2.833) [DOI] [PubMed] [Google Scholar]
- 40.Amodeo G. A., Rudolph M. J., Tong L. 2007. Crystal structure of the heterotrimer core of Saccharomyces cerevisiae AMPK homologue SNF1. Nature 449, 492–495 10.1038/nature06127 (doi:10.1038/nature06127) [DOI] [PubMed] [Google Scholar]
- 41.Xiao B., et al. 2007. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449, 496–500 10.1038/nature06161 (doi:10.1038/nature06161) [DOI] [PubMed] [Google Scholar]
- 42.Iancu C. V., Mukund S., Fromm H. J., Honzatko R. B. 2005. R-state AMP complex reveals initial steps of the quaternary transition of fructose-1,6-bisphosphatase. J. Biol. Chem. 280, 19 737–19 745 10.1074/jbc.M501011200 (doi:10.1074/jbc.M501011200) [DOI] [PubMed] [Google Scholar]
- 43.Choe J. Y., Fromm H. J., Honzatko R. B. 2000. Crystal structures of fructose 1,6-bisphosphatase: mechanism of catalysis and allosteric inhibition revealed in product complexes. Biochemistry 39, 8565–8574 10.1021/bi000574g (doi:10.1021/bi000574g) [DOI] [PubMed] [Google Scholar]
- 44.Johnson L. N., Snape P., Martin J. L., Acharya K. R., Barford D., Oikonomakos N. G. 1993. Crystallographic binding studies on the allosteric inhibitor glucose-6-phosphate to T state glycogen phosphorylase b. J. Mol. Biol. 232, 253–267 10.1006/jmbi.1993.1380 (doi:10.1006/jmbi.1993.1380) [DOI] [PubMed] [Google Scholar]
- 45.Rath V. L., et al. 2000. Activation of human liver glycogen phosphorylase by alteration of the secondary structure and packing of the catalytic core. Mol. Cell 6, 139–148 10.1016/S1097-2765(05)00006-7 (doi:10.1016/S1097-2765(05)00006-7) [DOI] [PubMed] [Google Scholar]
- 46.Barford D., Hu S. H., Johnson L. N. 1991. Structural mechanism for glycogen phosphorylase control by phosphorylation and AMP. J. Mol. Biol. 218, 233–260 10.1016/0022-2836(91)90887-C (doi:10.1016/0022-2836(91)90887-C) [DOI] [PubMed] [Google Scholar]
- 47.Wang Q., et al. 2010. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 10.1126/science.1179687 (doi:10.1126/science.1179687) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKee J. S., Hlodan R., Nimmo H. G. 1989. Studies of the phosphorylation of Escherichia coli isocitrate dehydrogenase. Recognition of the enzyme by isocitrate dehydrogenase kinase/phosphatase and effects of phosphorylation on its structure and properties. Biochimie 71, 1059–1064 10.1016/0300-9084(89)90111-9 (doi:10.1016/0300-9084(89)90111-9) [DOI] [PubMed] [Google Scholar]
- 49.Manning G., Plowman G. D., Hunter T., Sudarsanam S. 2002. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 27, 514–520 10.1016/S0968-0004(02)02179-5 (doi:10.1016/S0968-0004(02)02179-5) [DOI] [PubMed] [Google Scholar]
- 50.Yates S. P., Edwards T. E., Bryan C. M., Stein A. J., Van Voorhis W. C., Myler P. J., Stewart L. J., Zheng J., Jia Z. 2011. Structural basis of the substrate specificity of bifunctional isocitrate dehydrogenase kinase/phosphatase. Biochemistry 50, 8103–8106 10.1021/bi200809p (doi:10.1021/bi200809p) [DOI] [PMC free article] [PubMed] [Google Scholar]