

Abstract
In eukaryotic cells, protein phosphorylation is an important and widespread mechanism used to regulate protein function. Yet, of the thousands of phosphosites identified to date, only a few hundred at best have a characterized function. It was recently shown that these functional sites are significantly more conserved than phosphosites of unknown function, stressing the importance of considering evolutionary conservation in assessing the global functional landscape of phosphosites. This leads us to review studies that examined the impact of phosphorylation on evolutionary conservation. While all these studies have shown that conservation is greater among phosphorylated sites compared with non-phosphorylated ones, the magnitude of this difference varies greatly. Further, not all studies have considered key factors that may influence the rate of phosphosite evolution. Such key factors are their localization in ordered or disordered regions, their stoichiometry or the abundance of their corresponding protein. Here we take into account all of these factors simultaneously, which reveals remarkable evolutionary patterns. First, while it is well established that protein conservation increases with abundance, we show that phosphosites partly follow an opposite trend. More precisely, Saccharomyces cerevisiae phosphosites present among abundant proteins are 1.5 times more likely to diverge in the closely related species Saccharomyces bayanus when compared with phosphosites present in the 5 per cent least abundant proteins. Second, we show that conservation is coupled to stoichiometry, whereby sites frequently phosphorylated are more conserved than those rarely phosphorylated. Finally, we provide a model of functional and noisy or ‘accidental’ phosphorylation that explains these observations.
Keywords: phosphosite evolution, promiscuous phosphorylation, protein abundance, phosphosite stoichiometry, protein disorder
1. Introduction
(a). Phosphorylation as a widespread and important mechanism to regulate protein function
Proteins are the workhorses of the cell and their activity is regulated at multiple levels [1]. Among the best-described modes of protein regulation are the post-translational modifications of specific residues that affect their stability, localization, activity and ability to interact. The most studied and arguably one of the most abundant types of post-translational modifications is protein phosphorylation, as reflected by the large number of genes that encode kinases and phosphatases. For example, in humans, about 700 proteins play a direct role in phosphorylating and dephosphorylating other proteins [2,3].
Because of the importance of protein phosphorylation in regulating protein function, significant efforts have recently been dedicated to probe, map and catalogue phosphoproteomes [4–24]. These studies have revealed several important characteristics about phosphoproteomes, including that (i) protein phosphorylation is common: at least 30 per cent of proteins have been found to be phosphorylated on at least one residue in organisms such as the budding yeast (2000 out of 6000 proteins [25]); (ii) some proteins are heavily phosphorylated, with more than 30 sites for some proteins [26]; (iii) phosphorylation is dynamic across growth conditions [4,10,17–20]. This deepening knowledge of the importance of protein phosphorylation has sparked a profound interest regarding the role of this type of regulation in evolution and whether changes in protein phosphorylation could play key roles during evolution of cellular and organismal phenotypes [27].
(b). A consensus view of the evolution of phosphorylation sites
Large-scale mass-spectrometry studies of protein phosphorylation in several model organisms have also enabled the study of the rate at which this post-translational modification evolves. Despite the fact that many studies have used different species as the basis of their comparisons and have used different metrics to estimate the rate at which phosphorylated residues evolve, a consensus emerges from these investigations (table 1). First, most studies (e.g. [15,30,32] including Landry et al. [25]), show that phosphorylation sites (pS/pT) are on average more conserved than non-phosphorylated equivalent residues (S/T). Second, not all pS/pT show signs of purifying selection and the data are consistent with a significant proportion of pS/pT evolving at a rate that is indistinguishable from that of equivalent non-phosphorylated S/T [25,32,34,35]. Finally, sites that are known to be functional (associated with a phenotype when mutated) are significantly more conserved than sites with no characterized function [25,28,31,33]. Landry et al. [25] and others [28,36] have concluded from these results that a significant fraction of sites identified in large-scale studies have little to no functional role and could result from the random and neutral birth and death of sites that kinases can phosphorylate by accident (off-target activity).
Table 1.
Studies aiming at measuring the relative rate of evolution of phosphorylation sites (comparison of phosphorylated and non-phosphorylated sites).
| study | dataset | comparative method | conclusion |
|---|---|---|---|
| Gnad et al. [15] | large-scale phosphoproteomic experiments on human HeLa cells | examine whether sites are conserved across organisms (70 organisms from E. coli to mouse) | (i) phosphosites are more conserved than non-phosphorylated sites: 88% versus 81% for pS and S when compared with mouse and considering loop regions only |
| Malik et al. [28] | phosphoproteomics data derived from the human mitotic spindle apparatus | derived a normalized, relative score from the number of vertebrate species with a conserved residue (among six total species) | (i) phosphosites are more conserved than non-phosphorylated sites: 92% of pS/pT/pY conserved in mouse compared with 84% for S/T/Y (ii) phosphorylation sites with experimentally verified biological functions are significantly more conserved. Median conservation score: S/T/Y: −0.0893; pS/pT/pY: 0.1144; functional pS/pT/pY: 0.183 |
| Boekhorst et al. [29] | comparisons of phosphosites for six eukaryote species (human, mouse, fly, yeast, plant, zebrafish) | all-against-all Smith–Waterman searches to measure overlap | (i) overlap of orthologous phosphosites among the datasets greater than expected by chance alone(ii) conserved phosphosites are slightly more likely to be found in globular domains (iii) three-way comparisons show conservation from 1.6 to 7.4 fold over the expectation |
| Landry et al. [25] | comparisons of rate of evolution of human phosphosites among vertebrates and yeast phosphosites among yeasts | measure relative rate of evolution across the proteome using concatenated multiple species alignment | (i) phosphorylation sites are significantly older that non-phosphorylated residues in both the yeast and human lineages (ii) phosphorylation sites with experimentally verified biological functions are significantly more conserved |
| Tan et al. [30] | human, fly, worm and yeast phosphorylation sites from large-scale and small scale experiments. | compare the conservation of phosphorylation in three model organisms, fly, worm and yeast and conservation of residues in other species. | (i) phosphorylation sites are more conserved than non-phosphorylated ones |
| Ba & Moses [31] | analysis of high-confidence annotated sites in budding yeast | compare rates of substitution at phosphosites and flanking sites by comparing with four closely related species | (i) overall, significant reduction in amino acid substitution at phosphosites compared with flanking regions (0.07 versus 0.12) (ii) sites grouped by protein kinases show that some sites evolve slower, though not all |
| Chen et al. [32] | human and mouse experimentally verified phosphosites. | measure the rate of evolution of phosphosites by inferring the ancestral phosphoproteome of human and mouse. Ancestral phosphorylation states are predicted | (i) phosphorylated S/T evolve slower than non-phosphorylated sites in both ordered and disordered regions (overall S: 0.097 versus 0.127; T: 0.121 versus 0.139) (ii) phosphorylated Tyr evolve at similar rate as non-phosphorylated Tyr |
| Boulais et al. [23] | compare the phosphorylation of Mouse, Drosophila and Dictyostelium phagosome proteins | compare the number of orthologous sites phosphorylated in two species with what is expected by chance | (i) phosphosites are more likely to occur at homologous positions between species |
| Wang et al. [33] | human phosphosites from large-scale and small-scale (annotated) studies | compare human phosphosites to orthologous sites in a second species | (i) phosphosites evolve slower than their flanking sequences for some functional categories and not others (ii) phosphosites of known function evolve slower than those of unknown function |
| Gray et al. [34] | human phosphosites, including annotated ones | estimate the absolute rates of evolution by mapping sequence differences among 44 species and dividing by the total time; P-sites of protein are compared with all equivalent sites of the same protein | (i) evidence for higher purifying selection in about 70% of phosphorylated sites; 30% of phosphorylation sites evolve at the same rate or faster than equivalent residues on the same proteins |
| Park et al. [35] | analysis of mouse phosphorylation sites in nine mouse tissues (36 000 sites) | measure conservation by comparison with rat, human and chicken separately | (i) found no significant difference in the rate of evolution of phosphorylated and non-phosphorylated S, T and Y |
From a practical perspective, among the thousands of phosphosites commonly observed in phosphoproteomic studies, biologists need to make educated predictions about which sites are most likely to be functionally important. As described already, phosphosite conservation is a good indicator of functional importance. However, we know that factors such as protein disorder and protein abundance are tightly coupled to each other [1] as well as to varying degrees of protein conservation [37,38]. Therefore, we anticipate that the successful identification of phosphosites most likely to be functional requires taking into account a more global cellular context for each site. This leads us to discuss and investigate the existence of phosphosites with little functional relevance and their relationship to global cellular parameters, namely protein disorder, protein abundance and phosphosite stoichiometry.
(c). Organism fitness and non-functional phosphorylation are compatible with each other
From an evolutionary standpoint, it may be argued that off-target phosphorylation should be eliminated. However, such off-target activity would be eliminated by natural selection only to the extent that it is deleterious for the cell and that other important conditions that affect the efficiency of natural selection be met [39]. In fact, off-target or ‘promiscuous’ phosphorylation might be functionally neutral but in the long run might contribute to the evolvability of signalling networks [40]. These non-adaptive phosphorylations may represent a burden for investigators in terms of identifying key functional phosphosites in the cell but may nevertheless contribute to the evolution of biological network architecture and therefore deserve to be considered if we are to fully understand the evolution of signalling and protein interaction networks [39,41–44].
(d). Linking cellular crowding and random encounters to non-functional phosphorylation
When considering that random encounters may result in non-functional phosphorylation of S/T, one key factor to consider is protein abundance. In cells, protein abundances span several orders of magnitude. In yeast for example, while some proteins are present in only a few dozen copies, others are synthesized in over a million copies [45]. Therefore, competition is severe between sites phosphorylated in the context of a specific function and other sites for which phosphorylation has little or no functional consequence [39,46–49]. For example, let us consider protein X, whose average abundance is approximately 1000 copies per cell, and whose function requires a phosphorylation at a particular position. Knowing that a yeast cell contains an estimated total of 50 000 000 protein molecules [50], how likely is it for a kinase to be able to specifically phosphorylate all the copies of ‘protein X’ without any off-target activity among the 49 990 000 other proteins present in the cell?
Answering this question requires considering two opposing forces: specificity mechanisms and the randomness of cellular encounters. On the one hand, protein X might be phosphorylated without any off-target activity thanks to mechanisms that increase the specificity of kinase–protein interactions and phosphorylation [51]. This is for instance achieved by subcellular organization of kinase and phosphatase complexes with their substrates via scaffold and adaptor proteins [52,53]. On the other hand, the kinase(s) responsible for the phosphorylation of X may also randomly encounter other proteins within its subcellular compartment and, further, many kinases are active in more than one compartment [36]. In the latter scenario, the probability of random encounters is expected to be proportional to the abundance of proteins present in the same cellular compartment. Considering chance alone, it is indeed significantly more likely to encounter a protein present in a million copies than the rare protein X. In addition, the random nature of these encounters and low probability that they result in phosphorylation imply that only a small fraction of a protein population should be phosphorylated by chance. These concepts are summarized in figure 1.
Figure 1.

Illustration of functional and noisy phosphorylation events expected in cells. Two concepts investigated in this paper are described. First, that kinases are more likely to encounter abundant proteins than rare proteins by chance. Second, that a kinase is likely to phosphorylate only a small fraction of a protein population by chance.
In sum, if protein phosphorylation is influenced by the randomness of cellular encounters, we expect two factors to deserve attention: abundance and stoichiometry, where abundance refers to the total copy number per cell of a given protein, and stoichiometry to the fraction of the copies that are phosphorylated. Considering these two factors, we formulate the following predictions regarding non-functional phosphorylation sites:
-
(1)
If protein abundance increases the probability of random encounters with protein kinases and therefore of off-target phosphorylation (figure 1), we predict that highly abundant proteins are enriched in phosphorylation sites.
-
(2)
Since off-target phosphorylations exhibit no specific function, conservation of phosphosites on highly abundant proteins should be equivalent to that of non-phosphorylated sites. We thus predict that the ratio C(pS|pT)/C(S|T) decreases as protein abundance increases (where C denotes conservation, pS|pT phosphorylated serines and threonines and S|T non-phosphorylated equivalent residues).
-
(3)
Off-target phosphorylation should only affect a small fraction of a protein population, i.e. the stoichiometry at these sites should be low owing to the random nature of the encounters and to the low probability that phosphorylation occurs. We thus predict that highly abundant proteins are enriched in phosphorylation sites of low stoichiometry.
-
(4)
Following prediction 3, we predict that the ratio C(pS|pT)/C(S|T) increases with stoichiometry.
We investigate these predictions in the upcoming sections.
2. Results and discussion
(a). Highly abundant proteins are enriched in phosphorylation sites
We first ask whether abundant proteins are more frequently subject to phosphorylation than those with lower abundances (figure 1). For this, we used the dataset of 6092 phosphosites assembled in Landry et al. [25] (see §4) and divide proteins into 10 classes of increasing abundance (0–10%, 10–20%, etc. upto 90–100%; i.e. dataset percentiles). In the least abundant class, 22 per cent of proteins are detected as being phosphorylated on at least one position in a predicted disordered region—we denote this Plow+diso = 0.22. This proportion increases among higher abundance proteins, although it does not exceed 40 per cent: Phigh+diso ∼ 0.35–0.4). Therefore, the ratio Phigh+diso/Plow+diso increases with abundance but remains below two. When considering ordered regions, this ratio increases even more, as less than 10 per cent of low-abundance proteins contain at least one known phosphorylation site in an ordered region (Plow+ord < 0.1), while almost 50 per cent of proteins in the most abundant class do so (Phigh+ord ∼ 0.5)—therefore the ratio Phigh+ord/Plow+ord is close to five. The weaker contrast observed in disordered regions is due to a combination of the following opposing trends: (i) most phosphosites are in disordered regions, but (ii) abundant proteins contain few disordered regions. We thus control for these effects in figure 2b as well as in figure 3, as described next.
Figure 2.

Proteins do not all have the same probability to be phosphorylated. Percentage of proteins (a,c) or S/T residues (b,d) detected as being phosphorylated out of the total population. (There are 4670 proteins with disordered regions and a known abundance, and these contain 141 007 S/T residues and 4670 phosphorylation sites; and there are 4777 proteins with ordered regions and a known abundance, containing 218 974 S/T residues and 856 phosphorylation sites). Note that a protein is considered phosphorylated if it carries at least one phosphosite in the region of interest. We consider separately disordered (a,b) and ordered regions (c,d). In all cases, the probability for a protein or a residue to be phosphorylated is dependent on its cellular abundance, as obtained from the Pax-db database [54]. Abundance classes represent percentiles of the entire Pax-db data. The heterogeneity observed on (a) is due to the low disorder content of highly expressed proteins, i.e. many are unlikely to be detected as being phosphorylated in a disordered region if they contain only a few disordered residues. This effect is however normalized when taking a ‘residue’ viewpoint (b,d).
Figure 3.
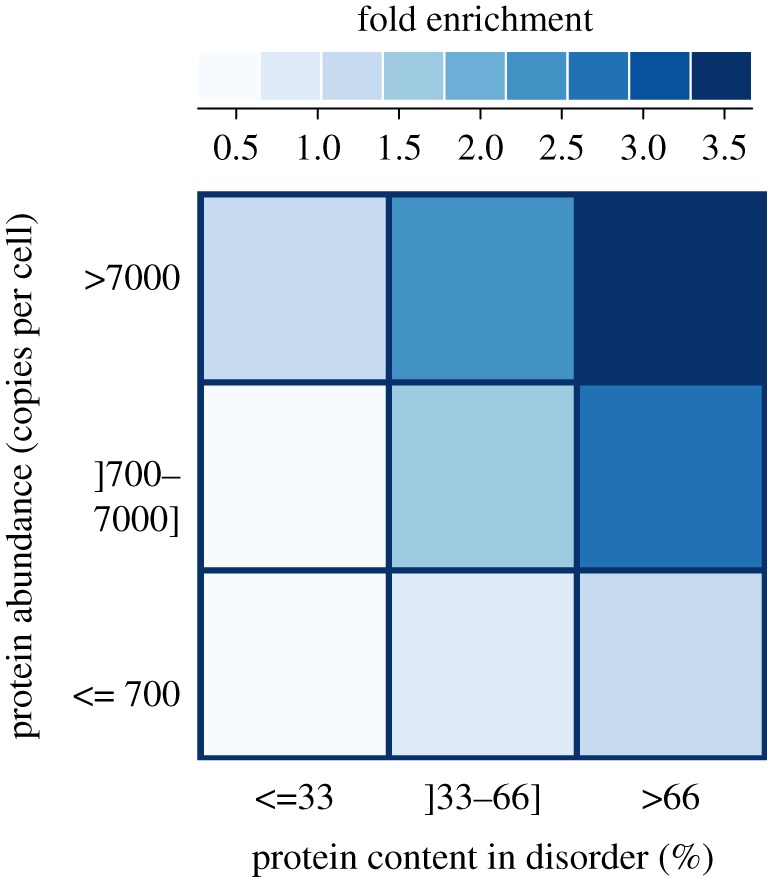
Protein abundance and disorder content affect the probability of an S/T to be phosphorylated. Highly abundant, disordered proteins are over three times more likely to be phosphorylated than expected. The fold enrichment corresponds to the ratio of observed phosphosite that falls in each category over the expected ones. Each protein in the dataset was classified according to its abundance [54] and the proportion of residues that belonged to disordered regions of the proteins. Then, we determined how many phosphorylation sites fell in each category. The expected number of sites for each category was determined by its proportion in S/T multiplied by the total number of phosphosites.
First, we move from a protein perspective to an amino acid perspective and calculate the percentage of all S/T that are phosphorylated among proteins sampled in a given abundance class. We find that the amino acid perspective significantly increases the contrast between abundance classes. While approximately 1 per cent of S/T residues in disordered regions are phosphorylated among low-abundance proteins, this fraction increases to over approximately 7 per cent among highly abundant proteins. Similarly, in ordered regions, approximately 0.2 per cent of S/T residues are known to be phosphorylated among low-abundance proteins versus 2.25 per cent among highly abundant proteins. Second, we create nine categories of proteins according to their abundance and proportion of disordered residues (figure 3), and calculate the ratio of observed to expected phosphorylation sites in each category. This reveals that highly abundant disordered proteins contain more than three times the number of phosphosites expected by chance. In contrast, proteins of low abundance and with little disorder contain less than three times the number of sites expected by chance. A combination of disorder and high expression therefore results in an increased probability of being phosphorylated.
Together, these results show that serines and threonines do not all have the same probability of being phosphorylated. It is known that ordered and disordered regions are associated with different probabilities of S/T sites being phosphorylated [25,55,56]. Here we see that the impact of protein abundance on these probabilities is also strong, and in fact it acts in synergy with disorder (figure 3).
Although this observation is consistent with our first prediction, the trends observed can also be explained by detection methods (i.e. frequent peptides might more likely be detected than rare peptides, despite the fact that phosphopeptide enrichment strategies are employed in these studies). Until methods that can circumvent this limitation are developed, it will be difficult to confirm this observation. This prompts us to probe an additional aspect of phosphorylation sites: their evolutionary conservation.
(b). Conservation of phosphosites on highly abundant proteins is similar to that of non-phosphorylated equivalent sites
To perform the evolutionary analyses described later, we compared aligned protein sequences from Saccharomyces cerevisiae and Saccharomyces bayanus (see §4). These two species have diverged for about 20 Myr and provide a good comparison because they have the same gene content, their orthology calls are unambiguous and yet their level of nucleotide divergence is comparable to that of human and mouse (62 versus 66% nucleotide identity in aligned positions [57]). This limited divergence also reduces alignment problems, which can be problematic in species that have diverged for longer periods of time, especially in disordered regions of proteins where the majority of phosphosites are found.
It was shown over a decade ago [58] and confirmed in several studies since then [38,59] that highly abundant proteins evolve slower than those of lower abundance. The comparison of S. cerevisiae and S. bayanus phosphorylatable residues (S/T) provides the same result (figure 4). In the least abundant half of the proteome, and considering disordered regions first, less than 70 per cent of S and T are conserved on average in S. bayanus (with an S or a T). This fraction increases to 90 per cent when considering the 5 per cent most abundant proteins. However, we observe an opposite trend for pS and pT sites: the conservation in the [0–5%] abundance class is equal to 86 per cent, and decreases to 77 per cent in the [60–80%] abundance class. Therefore, the ratio C(pS|pT)/C(S|T) is highest among low-abundance proteins, decreasing progressively until C(pS|pT)/C(S|T) approaches approximately one among highly expressed proteins (figure 4b). In ordered regions, though it is much weaker, the global trend is identical. pS/pT sites are thus significantly more conserved than equivalent non-phosphorylated S/T in low-abundance proteins, but not in highly expressed proteins.
Figure 4.

Phosphosites are proportionally more conserved in proteins with low abundance. Abundance classes represent percentiles of the entire Pax-db data [54], and copy numbers are obtained by linear scaling to fit abundances from [45] (see §4 for details). Considering disordered regions (top), the conservation of non-phosphorylated S/T increases with abundance. Conservation of pS/pT, however, decreases to a minimum, followed by an increase at higher protein abundances. As a result, the ratio of pS/pT (orange lines) conservation over that of S/T (grey lines) decreases with higher abundance (top of b). This suggests that the proportion of pS/pT with important functions decreases as abundance increases. Functional phosphosites indeed tend to be significantly more conserved than structurally equivalent S/T residues, as shown by the high conservation of the manually curated dataset from [31] (green circle). In ordered regions, the signal follows a similar albeit much weaker trend.
These observations are compatible with prediction 2, where a significant proportion of phosphorylation events detected among highly abundant proteins results from off-target kinase activity, while events detected among low-abundance proteins are enriched for important functions. The presence of non-functional phosphorylation sites in abundant proteins is further supported by the comparison with the proportion of conserved sites among a set of curated and arguably functional phosphosites obtained from [31] (green circle in figure 4a: ndiso = 159; nordered = 33). Conservation for these sites is indeed much higher than what is seen in large-scale phosphoproteomic studies. It has also been hypothesized [25] and shown [17] that an overall weak conservation of phosphosites can be explained by compensatory mechanisms. Cases are indeed known where the position of a phosphosite changes [60–62] although the functional mechanisms associated with it remain conserved. Importantly, we do not expect the earlier-mentioned observation to be affected by this evolutionary scenario, because such compensation (and associated lower conservation) is expected to increase among low-abundance proteins (because these proteins evolve faster and are richer in disordered regions, when compared with highly abundant proteins), and we observe the opposite trend.
(c). Highly abundant proteins are enriched in phosphorylation sites of low stoichiometry
In the scenario where highly abundant proteins are off-targets for phosphorylation, we do not expect the corresponding phosphosites to exhibit high stoichiometries. In other words, off-target phosphorylation should affect only a small fraction of an entire protein population. Estimates of phosphosite stoichiometry were recently published [63] and allow us to investigate this hypothesis.
As previously described, we split proteins into classes of increasing abundance, but now measure the mean stoichiometry of their corresponding phosphosites (figure 5a). This analysis reveals a clear relationship between these two variables, with phosphosites on low-abundance proteins having an average stoichiometry close to or more than 50 per cent (for both ordered and disordered regions), while for those on highly abundant proteins the average stoichiometry is down to 15 per cent (disordered regions) and 8 per cent (ordered regions). If we now look at the opposite picture and measure abundance as a function of stoichiometry (figure 5b), we observe a similar trend, where sites of low stoichiometry are more frequently observed on abundant proteins than sites with high stoichiometry. The figure also highlights that this effect is concentrated on sites of very low stoichiometry (]0–10%]), and remains comparatively moderate otherwise (green area). This confirms prediction 3, i.e. that highly abundant proteins are enriched in phosphorylation sites of low stoichiometry, as would be expected from off-target phosphorylation.
Figure 5.

Phosphosites with low stoichiometry are frequently found in highly abundant proteins. (a) We measure phosphosite stoichiometry across classes of increasing protein abundance; or (b) we measure abundance associated with classes of increasing phosphosite stoichiometry. The area of each data point is proportional to the number of sites used to obtain it. We observe a picture similar among ordered and disordered regions, where highly abundant proteins frequently carry phosphosites of low stoichiometry. Note that the impact of stoichiometry on abundance is stronger when considering ordered regions: the average abundance of proteins with a low-stoichiometry site in an ordered region is over four times greater (approx. 200 000 copies per cell) than the abundance of proteins carrying a low-stoichiometry site in a disordered region (less than 50 000 copies per cell).
(i). Phosphosites with high stoichiometry are more conserved than those with low stoichiometry
We have previously observed that phosphosites with high stoichiometry are more conserved than other phosphosites with lower stoichiometry detected on the same peptide [25]. Here, the data from Wu et al. [63] provide relative stoichiometry information at the proteome scale, which allows us to compare stoichiometry values across different peptides. We thus integrate stoichiometry and evolutionary information to ask whether higher stoichiometry of phosphorylation is associated with higher site conservation. Importantly however, answering this question is difficult, as it involves two confounding trends:
— low stoichiometry is strongly associated with high abundance,
— protein conservation is also strongly coupled to abundance, with high abundance equating with high global conservation.
Therefore, in order to investigate the relationship between stoichiometry and conservation, we focus on the range highlighted in green in figure 5, where the coupling between stoichiometry and abundance remains moderate.
We plot the average phosphosite conservation for three stoichiometry classes: ]10–30%], ]30–80%] and ]80–100%] (figure 6). This is shown with orange diamonds, and grey diamonds indicate the conservation of equivalent but non-phosphorylated sites. The black line, shows the ratio C(pS|pT)/C(S|T) as in figure 4b. For both ordered and disordered regions, the ratio increases from the low to high-stoichiometry classes, which is consistent with prediction 4, i.e. that low-stoichiometry sites are enriched in non-functional protein phosphorylation.
Figure 6.
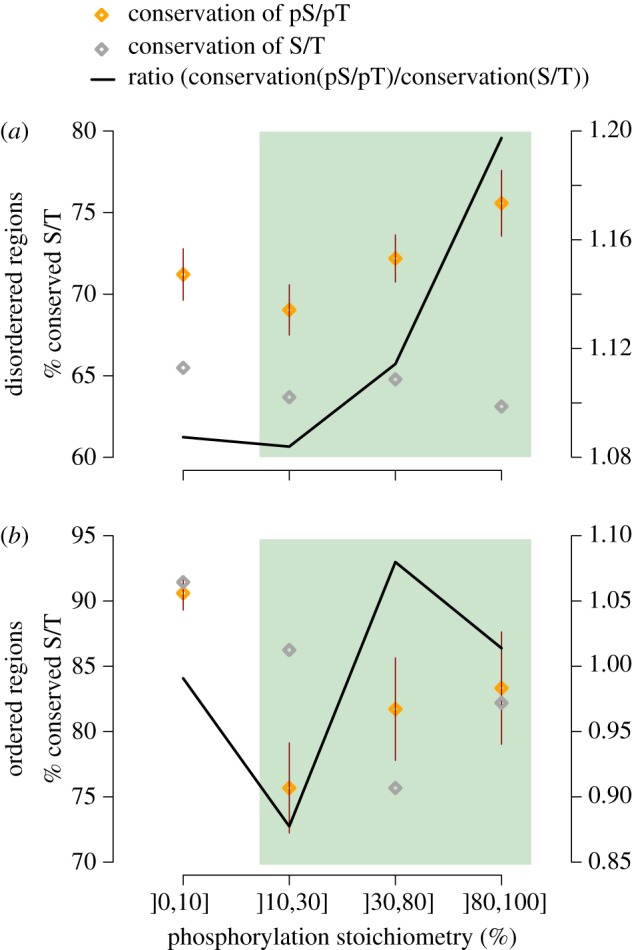
Phosphosites with high stoichiometry are more conserved than those with low stoichiometry. We measure the conservation of pS/pT sites as a function of their stoichiometry. We exclude the lowest stoichiometry class (]0,10]%) from the comparison because we saw above that it is strongly biased towards highly abundant proteins, particularly if considering sites present in ordered regions. Among the three other classes highlighted by the green area, we observe that the ratio C(pS|pT)/C(S|T) increases with site stoichiometry. This is consistent with low-stoichiometry sites being more frequently a by-product of off-target phosphorylation than high-stoichiometry sites.
3. Conclusions
In eukaryotic cells, a large fraction of proteins are post-translationally modified, and each of these modifications has the potential to affect their function. The ever-increasing throughput, sensitivity and accuracy of mass spectrometers together with the development of new proteomics strategies [6–8] means that these post-translational modifications can be identified on a large scale in an unbiased fashion with respect to the function (or absence thereof) of these modifications. Further, protein and phosphopeptide enrichment strategies are ever-improving the overall sensitivity of mass-spectrometry-based phosphoproteomics. For instance, a recent study on the phosphoproteome of purified yeast centromeres revealed many phosphosites previously not observed in whole-cell studies [64]. Such subcellular analysis of compartments and protein complexes should reveal even more sites, among both low- and highly abundant proteins having residues that are only phosphorylated within specific complexes or under specific circumstances. Probing cellular functions and responses on a large-scale may ultimately lead to our understanding of cells as a system. However, this task has proved difficult. Crucial to our success is the need to consider the contribution of non-adaptive forces (mutation and genetic drift) and biophysical constraints in shaping cellular networks [42]. Such forces are indeed likely to give birth to non-functional elements [39,41]. Because genomic elements of prime importance are expected to be conserved over longer evolutionary times than non-functional elements, we can use evolutionary comparisons to focus on the most conserved and thus most likely functional phosphosites. In the context of protein phosphorylation, it has indeed been shown that phosphorylation sites of known function are significantly more conserved than those of unknown function.
Above and beyond the identification of functional phosphosites, it is of paramount importance to understand what might influence the occurrence of non-functional phosphorylation events in the cell. Here we proposed a simple model that explains global patterns of non-functional phosphorylation (figure 1). Our model proposes that an important factor to consider is protein abundance. Kinases are indeed more likely to randomly encounter abundant proteins rather than rare proteins, as reflected in the fact that abundant proteins are enriched in phosphorylation sites (hypothesis 1, figures 2, 3 and 7a). Accordingly, evolutionary data showed that abundant proteins are enriched in non-functional phosphosites, and that low-abundance proteins are enriched in functional ones (hypothesis 2, figure 4). Consistently also, abundant proteins are enriched in low-stoichiometry sites (hypothesis 3, figure 5), and when abundance is taken into account, phosphosites with higher stoichiometry are more conserved (and arguably more functional) than those with lower stoichiometry (hypothesis 4, figures 6 and 7b). Importantly, it is possible that current technical limitations contribute to the signals observed with respect to hypotheses 1 and 3, i.e. it might be more likely to detect peptides from highly abundant proteins than peptides from low-abundance proteins, which would result in an increased probability of highly abundant proteins to appear phosphorylated. This could also result in an apparent increased probability for highly abundant proteins to contain low-stoichiometry sites. Yet, we do not expect our results to be a simple consequence of such technical limitations because hypotheses 2 and 4 are based on evolutionary data, which is independent from such a potential bias, and which nevertheless corroborate hypotheses 1 and 3. Future technological developments will be needed to unambiguously address these questions.
Figure 7.

Schematic depiction of the results obtained. (a) Two opposing trends are at play when considering the conservation of phosphorylation sites: on the one hand off-target phosphorylation occurs predominantly on abundant proteins. For these sites, conservation may be explained by abundance rather than function (orange). On the other hand, phosphorylation sites on low-abundance proteins are less likely to result from off-target activity. The conservation for these sites is therefore more likely due to function (green). (b) Two opposing trends are linked to high conservation when considering phosphosites stoichiometry. On the one hand, off-target phosphorylation results in low-stoichiometry sites that are predominantly on abundant proteins. For these low-stoichiometry sites (orange), conservation is more likely due to abundance than function. On the other hand, sites with high stoichiometry are also associated with higher conservation and this is more likely due to function (green).
Many mechanisms that contribute to the specificity of protein kinases have been described [51], and our results do not question the importance of these mechanisms for cellular functions. In fact, without these mechanisms, phosphorylation would certainly be much less efficient in regulating specific protein functions. However, it is likely that these mechanisms could not be optimized to a point where they can compensate for the three to four orders of magnitude differences we see in protein abundances and other effects not investigated here such as cellular crowding in specific compartments. One could argue that many more complex mechanisms could evolve to optimize these specificities further but they may actually represent a higher cost than the cost imposed by non-functional phosphorylation. There may also be a tradeoff between the pace at which protein kinases can phosphorylate their substrates (reactivity) and their specificity. Optimization of one parameter would inadvertently lead to reduction in the other and thus to a cost that counterbalances the benefits. Ultimately, measuring the cost of non-functional phosphorylation may be key to our full understanding of why it may be so frequent in the cell.
4. Methods
(a). Proteins: alignments, conservation, disorder and abundance
Protein sequences and alignments were identical to those used in Landry et al. [25], and were originally obtained from Wapinski et al. [65]. However, we extracted the couple S. cerevisiae–S. bayanus from these alignments. A serine or threonine was considered conserved in S. cerevisiae (value = 1) if the corresponding amino acid in S. bayanus was also a serine or a threonine, and considered non-conserved (value = 0) if it was a non-phosphorylatable amino acid, a tyrosine or an indel. Disorder was obtained with DISOPRED [37], as described in Landry et al. [25]. Protein abundances were obtained from the Pax-db database [54]. This database provides abundances in arbitrary units. In order to relate these to protein copy numbers per cell, we use the copy numbers estimated in Ghaemmaghami et al. [45]. Linear regression between the data from Pax-db and those from Ghaemmaghami et al. [45] results in the following relationship: log10(Pax) = −1.848 × log10(Ghaemmaghami). We thus multiply all values from Pax-db by 101.848 = 70.46. This is not intended to provide an accurate estimate and we only use it to obtain an approximation. Importantly, our analyses are based on relative abundances and thus do not depend on these absolute copy numbers. For figures 2, 4 and 5a, we indeed bin proteins into abundance classes, which represent percentiles of the entire dataset of proteins present in Pax-db.
(b). Phosphorylation sites
Phosphorylation sites were identical to those used in Landry et al. [25] and were originally compiled from the following studies: Ficarro et al. [9], Gruhler et al. [66], Reinders et al. [67], Chi et al. [68], Li et al. [69], Smolka et al. [70] and Albuquerque et al. [71]. Note that we did not consider tyrosine phosphorylation in this study. The aggregation of these datasets yields 6092 phosphosites for which the conservation can be calculated. This dataset is used for figures 2–4.
(c). Phosphorylation stoichiometry
Phosphorylation site stoichiometry was obtained from Wu et al. [63], providing 4754 sites for which we have both stoichiometry information and conservation information. Note that we did not consider sites for which the stoichiometry information was ambiguous—that is, we considered sites annotated only by Wu et al. as being on ‘singly phosphorylated’ peptides. This yields a dataset of 3803 sites for which we have both stoichiometry information as well as conservation information. This dataset is used for figures 5 and 6.
(d). Comparing phosphorylated sites with equivalent non-phosphorylated sites
In order to compare the evolutionary conservation between phosphorylated and non-phosphorylated sites, it is important to consider ‘equivalent sites’, which we define as:
— present on the same proteins as those considered for phosphorylated ones,
— found within 10 amino acids of a known phosphorylation site, and
— found in the same structural region (ordered and disordered regions were never compared with each other).
(e). Confidence intervals
Evolutionary conservation is given in the form of zeros and ones. Given a set of sites, we calculate the mean conservation by the mean of the values. A 95% CI for the mean is approximated by the following formula derived from the central limit theorem: CI = 1.96 × ((p1 × (1 – p1))/(N/p1))0.5, where p1 is the estimated mean and N the number of sites used for its calculation.
Acknowledgements
E.D.L. acknowledges the Human Frontier Science Program for support through a long-term postdoctoral fellowship. C.R.L. is a CIHR new investigator and this work is supported by a CIHR grant (GMX-191597). This work was supported in part by a CIHR grant (GMX-192838) to S.W.M. We thank Alan Moses, Pedro Beltrao and Tony Hunter for reviews of the manuscript, and Steven Gygi for helpful advice regarding the data on phosphosite stoichiometry.
References
- 1.Gsponer J., Futschik M. E., Teichmann S. A., Babu M. M. 2008. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science 322, 1365–1368 10.1126/science.1163581 (doi:10.1126/science.1163581) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. 2002. The protein kinase complement of the human genome. Science 298, 1912–1934 10.1126/science.1075762 (doi:10.1126/science.1075762) [DOI] [PubMed] [Google Scholar]
- 3.Alonso A., Sasin J., Bottini N., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., Mustelin T. 2004. Protein tyrosine phosphatases in the human genome. Cell 117, 699–711 10.1016/j.cell.2004.05.018 (doi:10.1016/j.cell.2004.05.018) [DOI] [PubMed] [Google Scholar]
- 4.Yu Y., et al. 2011. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332, 1322–1326 10.1126/science.1199484 (doi:10.1126/science.1199484) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boekhorst J., Boersema P. J., Tops B. B., van Breukelen B., Heck A. J., Snel B. 2011. Evaluating experimental bias and completeness in comparative phosphoproteomics analysis. PLoS ONE 6, e23276 10.1371/journal.pone.0023276 (doi:10.1371/journal.pone.0023276) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aebersold R., Mann M. 2003. Mass spectrometry-based proteomics. Nature 422, 198–207 10.1038/nature01511 (doi:10.1038/nature01511) [DOI] [PubMed] [Google Scholar]
- 7.Choudhary C., Mann M. 2010. Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 11, 427–439 10.1038/nrm2900 (doi:10.1038/nrm2900) [DOI] [PubMed] [Google Scholar]
- 8.Ahrens C. H., Brunner E., Qeli E., Basler K., Aebersold R. 2010. Generating and navigating proteome maps using mass spectrometry. Rev. Mol. Cell Biol. 11, 789–801 10.1038/nrm2973 (doi:10.1038/nrm2973) [DOI] [PubMed] [Google Scholar]
- 9.Ficarro S. B., McCleland M. L., Stukenberg P. T., Burke D. J., Ross M. M., Shabanowitz J., Hunt D. F., White F. M. 2002. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20, 301–305 10.1038/nbt0302-301 (doi:10.1038/nbt0302-301) [DOI] [PubMed] [Google Scholar]
- 10.Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 10.1016/j.cell.2006.09.026 (doi:10.1016/j.cell.2006.09.026) [DOI] [PubMed] [Google Scholar]
- 11.Tan C. S. 2011. Sequence, structure, and network evolution of protein phosphorylation. Sci. Signal 4, mr6 10.1126/scisignal.2002093 (doi:10.1126/scisignal.2002093) [DOI] [PubMed] [Google Scholar]
- 12.Ren J., Gao X., Liu Z., Cao J., Ma Q., Xue Y. 2011. Computational analysis of phosphoproteomics: progresses and perspectives. Curr. Protein Pept. Sci. 12, 591–601 [DOI] [PubMed] [Google Scholar]
- 13.Keshava Prasad T. S, et al. 2009. Human protein reference database: 2009 update. Nucleic Acids Res. 37, D767–D772 10.1093/nar/gkn892 (doi:10.1093/nar/gkn892) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dinkel H., Chica C., Via A., Gould C. M., Jensen L. J., Gibson T. J., Diella F. 2011. Phospho.ELM: a database of phosphorylation sites: update 2011. Nucleic Acids Res. 39, D261–267 10.1093/nar/gkq1104 (doi:10.1093/nar/gkq1104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gnad F., Ren S., Cox J., Olsen J. V., Macek B., Oroshi M., Mann M. 2007. PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8, R250 10.1186/gb-2007-8-11-r250 (doi:10.1186/gb-2007-8-11-r250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kratchmarova I., Blagoev B., Haack-Sorensen M., Kassem M., Mann M. 2005. Mechanism of divergent growth factor effects in mesenchymal stem cell differentiation. Science 308, 1472–1477 10.1126/science.1107627 (doi:10.1126/science.1107627) [DOI] [PubMed] [Google Scholar]
- 17.Holt L. J., Tuch B. B., Villen J., Johnson A. D., Gygi S. P., Morgan D. O. 2009. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 10.1126/science.1172867 (doi:10.1126/science.1172867) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodenmiller B., et al. 2010. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal. 3, rs4 10.1126/scisignal.2001182 (doi:10.1126/scisignal.2001182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bensimon A., Schmidt A., Ziv Y., Elkon R., Wang S. Y., Chen D. J., Aebersold R., Shiloh Y. 2010. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 3, rs3 10.1126/scisignal.2001034 (doi:10.1126/scisignal.2001034) [DOI] [PubMed] [Google Scholar]
- 20.Steen J. A., Steen H., Georgi A., Parker K., Springer M., Kirchner M., Hamprecht F., Kirschner M. W. 2008. Different phosphorylation states of the anaphase promoting complex in response to antimitotic drugs: a quantitative proteomic analysis. Proc. Natl Acad. Sci. USA 105, 6069–6074 10.1073/pnas.0709807104 (doi:10.1073/pnas.0709807104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dephoure N., Zhou C., Villen J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. 2008. A quantitative atlas of mitotic phosphorylation. Proc. Natl Acad. Sci. USA 105, 10 762–10 767 10.1073/pnas.0805139105 (doi:10.1073/pnas.0805139105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weintz G., et al. 2010. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 10.1038/msb.2010.29 (doi:10.1038/msb.2010.29) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boulais J., Trost M., Landry C. R., Dieckmann R., Levy E. D., Soldati T., Michnick S. W., Thibault P., Desjardins M. 2010. Molecular characterization of the evolution of phagosomes. Mol. Syst. Biol. 6, 423 10.1038/msb.2010.80 (doi:10.1038/msb.2010.80) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graumann J., Hubner N. C., Kim J. B., Ko K., Moser M., Kumar C., Cox J., Scholer H., Mann M. 2008. Stable isotope labeling by amino acids in cell culture (SILAC) and proteome quantitation of mouse embryonic stem cells to a depth of 5,111 proteins. Mol. Cell Proteomics 7, 672–683 10.1074/mcp.M700460-MCP200 (doi:10.1074/mcp.M700460-MCP200) [DOI] [PubMed] [Google Scholar]
- 25.Landry C. R., Levy E. D., Michnick S. W. 2009. Weak functional constraints on phosphoproteomes. Trends Genet. 25, 193–197 10.1016/j.tig.2009.03.003 (doi:10.1016/j.tig.2009.03.003) [DOI] [PubMed] [Google Scholar]
- 26.Freschi L., Courcelles M., Thibault P., Michnick S. W., Landry C. R. 2011. Phosphorylation network rewiring by gene duplication. Mol. Syst. Biol. 7, 504 10.1038/msb.2011.43 (doi:10.1038/msb.2011.43) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moses A. M., Landry C. R. 2010. Moving from transcriptional to phospho-evolution: generalizing regulatory evolution?. Trends Genet. 26, 462–467 10.1016/j.tig.2010.08.002 (doi:10.1016/j.tig.2010.08.002) [DOI] [PubMed] [Google Scholar]
- 28.Malik R., Nigg E. A., Korner R. 2008. Comparative conservation analysis of the human mitotic phosphoproteome. Bioinformatics 24, 1426–1432 10.1093/bioinformatics/btn197 (doi:10.1093/bioinformatics/btn197) [DOI] [PubMed] [Google Scholar]
- 29.Boekhorst J., van Breukelen B., Heck A., Jr, Snel B. 2008. Comparative phosphoproteomics reveals evolutionary and functional conservation of phosphorylation across eukaryotes. Genome Biol. 9, R144 10.1186/gb-2008-9-10-r144 (doi:10.1186/gb-2008-9-10-r144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan C. S., et al. 2009. Comparative analysis reveals conserved protein phosphorylation networks implicated in multiple diseases. Sci. Signal. 2, ra39 10.1126/scisignal.2000316 (doi:10.1126/scisignal.2000316) [DOI] [PubMed] [Google Scholar]
- 31.Ba A. N., Moses A. M. 2010. Evolution of characterized phosphorylation sites in budding yeast. Mol. Biol. Evol. 27, 2027–2037 10.1093/molbev/msq090 (doi:10.1093/molbev/msq090) [DOI] [PubMed] [Google Scholar]
- 32.Chen S. C., Chen F. C., Li W. H. 2010. Phosphorylated and nonphosphorylated serine and threonine residues evolve at different rates in mammals. Mol. Biol. Evol. 27, 2548–2554 10.1093/molbev/msq142 (doi:10.1093/molbev/msq142) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z., Ding G., Geistlinger L., Li H., Liu L., Zeng R., Tateno Y., Li Y. 2011. Evolution of protein phosphorylation for distinct functional modules in vertebrate genomes. Mol. Biol. Evol. 28, 1131–1140 10.1093/molbev/msq268 (doi:10.1093/molbev/msq268) [DOI] [PubMed] [Google Scholar]
- 34.Gray V. E., Kumar S. 2011. Rampant purifying selection conserves positions with posttranslational modifications in human proteins. Mol. Biol. Evol. 28, 1565–1568 10.1093/molbev/msr013 (doi:10.1093/molbev/msr013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park C., Zhang J. 2011. Genome-wide evolutionary conservation of N-glycosylation sites. Mol. Biol. Evol. 28, 2351–2357 10.1093/molbev/msr055 (doi:10.1093/molbev/msr055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lienhard G. 2008. Non-functional phosphorylations? Trends Biochem. Sci. 33, 351–352 10.1016/j.tibs.2008.05.004 (doi:10.1016/j.tibs.2008.05.004) [DOI] [PubMed] [Google Scholar]
- 37.Ward J. J., McGuffin L. J., Bryson K., Buxton B. F., Jones D. T. 2004. The DISOPRED server for the prediction of protein disorder. Bioinformatics 20, 2138–2139 10.1093/bioinformatics/bth195 (doi:10.1093/bioinformatics/bth195) [DOI] [PubMed] [Google Scholar]
- 38.Rocha E. P.C. 2006. The quest for the universals of protein evolution. Trends Genet. 22, 412–416 10.1016/j.tig.2006.06.004 (doi:10.1016/j.tig.2006.06.004) [DOI] [PubMed] [Google Scholar]
- 39.Levy E. D., Landry C. R., Michnick S. W. 2009. How perfect can protein interactomes be? Sci. Signal. 2, pe11 10.1126/scisignal.260pe11 (doi:10.1126/scisignal.260pe11) [DOI] [PubMed] [Google Scholar]
- 40.Tawfik D. S. 2006. Messy biology and the origins of evolutionary innovations. Nat. Chem. Biol. 6, 692–696 [DOI] [PubMed] [Google Scholar]
- 41.Lynch M. 2007. The frailty of adaptive hypotheses for the origins of organismal complexity. Proc. Natl Acad. Sci. USA 104(Suppl 1), 8597–8604 10.1073/pnas.0702207104 (doi:10.1073/pnas.0702207104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lynch M. 2007. The evolution of genetic networks by non-adaptive processes. Nat. Rev. Genet. 8, 803–813 10.1038/nrg2192 (doi:10.1038/nrg2192) [DOI] [PubMed] [Google Scholar]
- 43.Levy E. D., Landry C. R., Michnick S. W. 2010. Cell signaling. Signaling through cooperation. Science 328, 983–984 [DOI] [PubMed] [Google Scholar]
- 44.Breitkreutz A., et al. 2010. A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046 10.1126/science.1176495 (doi:10.1126/science.1176495) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. 2003. Global analysis of protein expression in yeast. Nature 425, 737–741 10.1038/nature02046 (doi:10.1038/nature02046) [DOI] [PubMed] [Google Scholar]
- 46.Zhang J., Maslov S., Shakhnovich E. I. 2008. Constraints imposed by non-functional protein–protein interactions on gene expression and proteome size. Mol. Syst. Biol. 4, 210 10.1038/msb.2008.48 (doi:10.1038/msb.2008.48) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heo M., Maslov S., Shakhnovich E. 2011. Topology of protein interaction network shapes protein abundances and strengths of their functional and nonspecific interactions. Proc. Natl Acad. Sci. USA 108, 4258–4263 10.1073/pnas.1009392108 (doi:10.1073/pnas.1009392108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson M. E., Hummer G. 2011. Nonspecific binding limits the number of proteins in a cell and shapes their interaction networks. Proc. Natl Acad. Sci. USA 108, 603–608 10.1073/pnas.1010954108 (doi:10.1073/pnas.1010954108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janin J. 1996. Quantifying biological specificity: the statistical mechanics of molecular recognition. Proteins 25, 438–445 10.1002/prot.4 (doi:10.1002/prot.4) [DOI] [PubMed] [Google Scholar]
- 50.Futcher B., Latter G. I., Monardo P., McLaughlin C. S., Garrels J. I. 1999. A sampling of the yeast proteome. Mol. Cell Biol. 19, 7357–7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ubersax J. A., Ferrell J. E., Jr 2007. Mechanisms of specificity in protein phosphorylation. Rev. Mol. Cell Biol. 8, 530–541 10.1038/nrm2203 (doi:10.1038/nrm2203) [DOI] [PubMed] [Google Scholar]
- 52.Scott J. D., Pawson T. 2009. Cell signaling in space and time: where proteins come together and when they're apart. Science 326, 1220–1224 10.1126/science.1175668 (doi:10.1126/science.1175668) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhattacharyya R. P., Remenyi A., Yeh B. J., Lim W. A. 2006. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75, 655–680 10.1146/annurev.biochem.75.103004.142710 (doi:10.1146/annurev.biochem.75.103004.142710) [DOI] [PubMed] [Google Scholar]
- 54.Wang M., Weiss M., Simonovic M., Haertinger G., Schrimpf S. P., Hengartner M.O., von Mering C. In press PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell Proteomics. 10.1074/mcp.O111.014704 (doi:10.1074/mcp.O111.014704) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nuhse T. S., Stensballe A., Jensen O. N., Peck S. C. 2004. Phosphoproteomics of the Arabidopsis plasma membrane and a new phosphorylation site database. Plant Cell 16, 2394–2405 10.1105/tpc.104.023150 (doi:10.1105/tpc.104.023150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iakoucheva L. M., Radivojac P., Brown C. J., O'Connor T. R., Sikes J. G., Obradovic Z., Dunker A. K. 2004. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 10.1093/nar/gkh253 (doi:10.1093/nar/gkh253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kellis M., Patterson N., Endrizzi M., Birren B., Lander E. S. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423, 241–254 10.1038/nature01644 (doi:10.1038/nature01644) [DOI] [PubMed] [Google Scholar]
- 58.Pal C., Papp B., Hurst L. D. 2001. Highly expressed genes in yeast evolve slowly. Genetics 158, 927–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drummond D. A., Raval A., Wilke C. O. 2006. A single determinant dominates the rate of yeast protein evolution. Mol. Biol. Evol. 23, 327–337 10.1093/molbev/msj038 (doi:10.1093/molbev/msj038) [DOI] [PubMed] [Google Scholar]
- 60.Serber Z., Ferrell J. E., Jr 2007. Tuning bulk electrostatics to regulate protein function. Cell 128, 441–444 10.1016/j.cell.2007.01.018 (doi:10.1016/j.cell.2007.01.018) [DOI] [PubMed] [Google Scholar]
- 61.Strickfaden S. C., Winters M. J., Ben-Ari G., Lamson R. E., Tyers M., Pryciak P. M. 2007. A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell 128, 519–531 10.1016/j.cell.2006.12.032 (doi:10.1016/j.cell.2006.12.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moses A. M., Liku M. E., Li J. J., Durbin R. 2007. Regulatory evolution in proteins by turnover and lineage-specific changes of cyclin-dependent kinase consensus sites. Proc. Natl Acad. Sci. USA 104, 17 713–17 718 10.1073/pnas.0700997104 (doi:10.1073/pnas.0700997104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu R., Haas W., Dephoure N., Huttlin E. L., Zhai B., Sowa M. E., Gygi S. P. 2011. A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat. Methods 8, 677–683 10.1038/nmeth.1636 (doi:10.1038/nmeth.1636) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keck J. M., et al. 2011. A cell cycle phosphoproteome of the yeast centrosome. Science 332, 1557–1561 10.1126/science.1205193 (doi:10.1126/science.1205193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wapinski I., Pfeffer A., Friedman N., Regev A. 2007. Natural history and evolutionary principles of gene duplication in fungi. Nature 449, 54–61 10.1038/nature06107 (doi:10.1038/nature06107) [DOI] [PubMed] [Google Scholar]
- 66.Gruhler A., Olsen J. V., Mohammed S., Mortensen P., Faergeman N. J., Mann M., Jensen O. N. 2005. Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell Proteomics 4, 310–327 10.1074/mcp.M400219-MCP200 (doi:10.1074/mcp.M400219-MCP200) [DOI] [PubMed] [Google Scholar]
- 67.Reinders J., et al. 2007. Profiling phosphoproteins of yeast mitochondria reveals a role of phosphorylation in assembly of the ATP synthase. Mol. Cell Proteomics 6, 1896–1906 10.1074/mcp.M700098-MCP200 (doi:10.1074/mcp.M700098-MCP200) [DOI] [PubMed] [Google Scholar]
- 68.Chi A., et al. 2007. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl Acad. Sci. USA 104, 2193–2198 10.1073/pnas.0607084104 (doi:10.1073/pnas.0607084104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li X., Gerber S. A., Rudner A. D., Beausoleil S. A., Haas W., Villen J., Elias J. E., Gygi S. P. 2007. Large-scale phosphorylation analysis of alpha-factor-arrested Saccharomyces cerevisiae. J. Proteome Res. 6, 1190–1197 10.1021/pr060559j (doi:10.1021/pr060559j) [DOI] [PubMed] [Google Scholar]
- 70.Smolka M. B., Albuquerque C. P., Chen S. H., Zhou H. 2007. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl Acad. Sci. USA 104, 10 364–10 369 10.1073/pnas.0701622104 (doi:10.1073/pnas.0701622104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Albuquerque C. P., Smolka M. B., Payne S. H., Bafna V., Eng J., Zhou H. 2008. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell Proteomics 7, 1389–1396 10.1074/mcp.M700468-MCP200 (doi:10.1074/mcp.M700468-MCP200) [DOI] [PMC free article] [PubMed] [Google Scholar]