

Abstract
The genetic mechanisms that contribute to reduced susceptibility to vancomycin in Staphylococcus aureus are complex and heterogeneous. In addition, debate is emerging as to the true effect of reduced susceptibility to vancomycin on staphylococcal virulence. To investigate this, comparative genomics was performed on a collection of vancomycin-exposed isogenic S. aureus pairs (14 strains in total). Previously described mutations were observed in genes such as vraG, agrA, yvqF, and rpoB; however, a new mechanism was identified involving a serine/threonine phosphatase, Stp1. After constructing an stp1 deletion mutant, we showed that stp1 is important in vancomycin susceptibility and cell wall biosynthesis. Gene expression studies showed that stp1 also regulates virulence genes, including a hemolysin, superantigen-like protein, and phenol-soluble modulin, and that the deletion mutant is attenuated in virulence in vivo. Stp1 provides a new link between vancomycin susceptibility and virulence in S. aureus.
(See the editorial commentary by Cheung and Duclos, on pages 1625–7.)
Staphylococcus aureus is a well-known human pathogen that causes both hospital- and community-acquired infections. The severity of disease caused by S. aureus is vast, ranging from superficial skin infections to severe and often complex infections such as osteomyelitis and endocarditis. With the emergence of methicillin-resistant S. aureus, the reliance on last-line antistaphylococcal antibiotics such as vancomycin has increased dramatically. Fortunately, vancomycin resistance remains rare. However, S. aureus strains with reduced susceptibility to vancomycin, or vancomycin-intermediate S. aureus (VISA), are reported more commonly [1, 2] and have been associated with prolonged bacteremia and vancomycin treatment failures [3–5].
The genetic pathway in the evolution of reduced susceptibility to vancomycin in S. aureus remains heterogeneous, with no consistent genetic marker for resistance identified [6–8]. Although a number of genes have been shown to be involved in reduced vancomycin susceptibility, such as vraSR [9], graSR [6, 10], and agr [11], it appears that VISA arises due to the accumulation of mutations in multiple genetic pathways [7]. For example, Mwangi et al used whole genome sequencing of a series of clinical strains derived from the same patient who was treated with vancomycin and showed that it was a cumulative mutation process that correlated with a stepwise decrease in vancomycin susceptibility [7]. Despite the diversity of genes involved, common themes exist, including genes that regulate cell wall biosynthesis and cellular stress response [12]. A deepening of our understanding of the array of genes involved in the evolution of VISA will provide greater insights into possible preventative or therapeutic strategies to fight this problematic organism.
Debate exists over the pathogenic consequences of reduced susceptibility to vancomycin in S. aureus. Initial clinical studies reported a high complication rate and mortality in those infected with VISA strains [3, 13]. However, more contemporary studies do not support these findings and rather highlight the importance of adjusting for other factors that contribute to poor outcomes such as severity of illness and comorbidities [14]. In fact, laboratory studies point toward reduced virulence in VISA strains, which could be explained by mutations in virulence regulator genes such as agr and changes to the bacterial cell surface involving capsule and protein A [15]. Furthermore, we have previously shown in an invertebrate model that S. aureus strains with reduced susceptibility to vancomycin were attenuated in virulence, independent of growth [16].
In this study, we explored not only the genetic mechanism behind VISA formation but also the potential impact of these mutations on S. aureus virulence. Whole genome sequencing was performed on 14 strains making up 5 pairs or series, whereby the first isolate was vancomycin-susceptible and the subsequent isolates were isogenic vancomycin-nonsusceptible daughter strains that arose as a result of vancomycin therapy. We identified mutations in a number of genes previously defined in VISA, including vraG, agrA, dltA, rpoB, and yvqF. Most importantly, we describe a new gene that is involved in reduced susceptibility to vancomycin, stp1. This gene encodes for a serine/threonine phosphatase, and deletion of the stp1 gene resulted in an increase in the minimum inhibitory concentration (MIC) of vancomycin and a cellular phenotype that resembles VISA strains. We also demonstrate the role of Stp1 in S. aureus virulence through gene expression studies and a murine bacteremia model.
METHODS
Bacterial Strains and Susceptibility Testing
Bacterial strains used in this study are shown in Table 1. Four clinical strain pairs or series collected from patients in North America who had failed vancomycin therapy were included in the analysis [11, 16]. All strains within a pair or series were determined as isogenic using pulsed-field gel electrophoresis as described [11]. To assess for differences between in vivo and in vitro evolution of resistance, a laboratory-derived series, which was generated previously [17], was included for genetic analysis (Table 1). MICs for vancomycin were determined by broth dilution, according to the Clinical and Laboratory Standards Institute, and by Etest (bioMérieux), according to the manufacturer's specifications. To assess for vancomycin heteroresistance, population analysis profiles (PAPs) were performed as described previously [18]. In brief, serial dilutions of overnight cultures were plated onto heart infusion agar (Oxoid) at vancomycin concentrations ranging from 0 to 8 μg/mL. Colony-forming units (CFUs) were counted after 48 hours of incubation in aerobic conditions at 37°C.
Table 1.
Characteristics of Vancomycin-Exposed Strains of Staphylococcus aureus
| MIC (µg/mL) |
||||
|---|---|---|---|---|
| Isolate | Clinical Syndrome | MLST | Vn | Dp |
| Pair 1 | ||||
| A5937 | Bacteremia, endocarditis | 5 | 1.5 | 0.12 |
| A5940 | 4 | 0.25 | ||
| Pair 2 | ||||
| A6224 | Bacteremia | 5 | 2 | 0.25 |
| A6226 | 3 | 2 | ||
| Pair 3 | ||||
| A6300 | Bacteremia, prosthetic joint infection | 5 | 2 | 0.25 |
| A6298 | 4 | 1 | ||
| Series 1 | ||||
| A9635 | Bacteremia, vertebral osteomyelitis | 1892 | 1 | 0.5 |
| A9636 | 1 | 0.5 | ||
| A9637 | 2 | 1 | ||
| A9638 | 3 | 2 | ||
| A9639 | 4 | 4 | ||
| Series 2a | ||||
| A8117 | Laboratory-derived series | 5 | 1 | … |
| A8118 | 4 | … | ||
| A8392 | 8 | … | ||
Genome Sequencing
Whole genome sequencing of the vancomycin-susceptible parent genomes was performed using 454 FLX pyrosequencing (Roche) with an average 24-fold coverage for each genome (range, 14- to 39-fold). Vancomycin-exposed daughter genomes (hVISA/VISA) were sequenced to higher coverage (>100-fold) using the Illumina sequencing platform. Genomes were assembled in Newbler (Roche) and open reading frames identified and annotated using both ab initio (Genemark, Glimmer3, Metagene, and ZCURVEb models) and evidence-based approaches (BLASTX against nonredundant S. aureus protein database). Single-nucleotide polymorphisms (SNPs) were determined between vancomycin-exposed clinical pairs using the variant ascertainment algorithm polymorphism discovery algorithm (Broad Institute) [19]. Select mutations were independently confirmed by polymerase chain reaction (PCR) and sequencing. Multilocus sequence type (MLST) typing was performed for all strains (http://saureus.mlst.net/). Sequences of parent strains were deposited in the National Center for Biotechnology Information GenBank under the accession numbers ACKE00000000 (A6224), ACKC00000000 (A5937), ACKF00000000 (A6300), ACYO00000000 (A8117), and ACKI00000000 (A9635). Genome sequences and SNP data for all isolates can be viewed at the Broad Institute S. aureus Drug Resistance Project group database (http://broadinstitute.org/annotation/genome/staphylococcus_aureus_drug_resistance).
Genetic Manipulation
An stp1 (SA1062) in-frame deletion mutant was generated using the Escherichia coli/S. aureus shuttle vector pKOR1 [20]. In brief, approximately 1 kb of DNA flanking up- and downstream of stp1 was PCR-amplified from clinical S. aureus isolate A5937 using primers AP1/AP2 and AP3/AP4, respectively (see Supplementary Table 1). The products were ligated and subcloned into pKOR1 using BP Clonase (Invitrogen) per the manufacturer's instructions. The final construct was electroporated into vancomycin-susceptible clinical isolate A5937, and deletion of the stp1 gene was confirmed using DNA sequencing, Southern blot analysis, and reverse-transcription (RT) PCR. Complementation was performed by subcloning the intact stp1 gene into pALC2073 using primers AP153/AP154 (see Supplementary Table 1) and transforming the stp1 deletion mutant using chloramphenicol (10 μg/mL) selection [21]. Tetracycline at a concentration of 200 ng/mL was used to induce expression of the stp1 gene, and complementation was confirmed using RT-PCR (Supplementary Figure 1).
Microarray Analysis
Total RNA was extracted during the exponential growth phase (Optical Density600 of 0.5). Cells were first incubated for 15 minutes in TE buffer supplemented with lysostaphin (Sigma-Aldrich) to a final concentration of 400 μg/mL. RNA was prepared using the RNeasy Mini Kit (Qiagen). Microarray transcriptional analysis was performed using The Institute of Genome Research (TIGR) version 9.0 S. aureus slides as described previously [22]. RNA extraction and microarray hybridizations were performed in triplicate, including 1 dye swap analysis. Slides were imaged using a Genetic Microsystems 418 scanner and analyzed with Imagene 5.2 software using the Bioarray Software Environment (BASE) [23], with normalization being performed within and between arrays using Linear Models for Microarray (LIMMA) and Statistics for Microarray (SMA) (Bioconductor) [24]. P values, adjusted for multiple comparisons, and fold ratios were determined using moderated t tests [25]. Fold changes of >1.5 with an adjusted P value of <.05 were considered significant.
Quantitative RT-PCR (qRT-PCR) of select genes was performed to validate the microarray data. Five micrograms of RNA from 2 biological replicates was reverse transcribed using Superscript II (Invitrogen) per the manufacturer's instructions, except 1 μg of random primers was used. Primers used for qRT-PCR are listed in Supplementary Table 1. The qRT-PCR reactions were performed in triplicate, as described [26], using a Mastercycler ep realplex real-time PCR machine (Eppendorf). Normalization using 16S ribosomal RNA and fold-change calculations were performed as described previously [27].
Electron Microscopy
Electron microscopy was performed as described previously [28]. Cells were sectioned using an Ultracut T microtome (Leica), then stained with lead citrate and 2% uranyl acetate. Cells were visualized using an H-7500 transmission electron microscope (Hitachi). Four separate points on 25 equatorially cut cells were measured for each strain in a blinded fashion, with significance determined by Student t test at a significance level of P ≤ .05.
Murine Infection Model
Wild-type 6-week-old female C57BL/6 mice were injected via the tail vein with approximately 1.0 × 108 CFU of the desired bacterial strain in 0.1 mL of sterile saline. A subset of mice was euthanized 120 hours after infection, and histopathology was performed on the liver at these time points. Slides were analyzed using an Olympus B×51 microscope and imaged using an Olympus DP70 camera. The remaining mice were monitored at least 3 times daily, with those showing signs of stress euthanized by CO2 inhalation. Kaplan-Meier curves and log-rank tests were performed using STATA 6 with a significance level of P ≤ .05. All animal experiments were performed in accordance with the Animal Research Platform Ethics Committee, Monash University, Australia.
RESULTS AND DISCUSSION
Characteristics of Vancomycin-Exposed S. aureus Isolates
Characteristics of the clinical isolates used in this study are described in Table 1. The strains were isolated from patients with complicated S. aureus bacteremia including endocarditis, osteomyelitis, and foreign-body infections [11, 16, 29]. All patients failed vancomycin therapy, and this was associated with reduced susceptibility to vancomycin (VISA). Despite no exposure to daptomycin, all vancomycin-nonsusceptible isolates had elevated MICs to daptomycin, an observation that has previously been reported [30]. Four of the 5 parent strains were MLST type 5, which is one of the more common hospital-associated S. aureus sequence types in North America [31].
Vancomycin Exposure Leads to a Diverse Range of Mutations in S. aureus
To characterize the genetic mechanisms behind VISA formation and its relationship with virulence, we performed whole genome sequencing and comparative genomics using our vancomycin-exposed strains. In total, 14 S. aureus genomes were sequenced, including 3 clinical pairs, 1 clinical series (consisting of 5 isolates), and 1 laboratory-derived series (consisting of 3 isolates). The average genome size was 2.8 Mb (range, 2.72–2.87 Mb) consisting of an average of 2653 predicted protein-encoding genes (range, 2594–2755). An average of 7 coding region mutations (range, 5–13) were identified in each of the vancomycin-exposed pairs or series (Table 2). The majority were SNPs (mean, 6 [range, 3–7]), with an average of 2 deletions and 1 insertion in each of the pairs. All of the point mutations, excluding 4, resulted in a change in amino acid sequence.
Table 2.
Mutations Observed Between Isogenic Staphylococcus aureus Strain Pairs or Series Exposed to Vancomycin
| Mutationsc |
|||||||
|---|---|---|---|---|---|---|---|
| ORF IDa | Gene | Product | Function | Protein Sizeb | Type | Change | Truncated Sizec |
| Clinically derived pairs | |||||||
| 1. A5937/A5940 | |||||||
| SA1062 | stp1 | Serine/threonine phosphatase | Signal transduction | 247 | Deletion | Frameshift | 113 |
| SA1844 | agrA | Accessory gene regulator A | Transcription | 238 | Insertion | Glu162Stop | 161 |
| SA0500 | rpoB | RNA polymerase subunit β | Transcription | 1183 | SNP | His481Tyr | |
| SA1505 | lysP | Lysine-specific permease | Amino acid metabolism/transport | 315 | SNP | Leu290Phe | |
| SA2093 | ssaA2 | Staphylococcal secretory antigen | Immune interaction | 267 | Insertion | Frameshift | 190 |
| SA0022 | 5′-nucleotidase | Nucleic acid metabolism/transport | 773 | SNP | Asp537Asp | ||
| 2. A6224/A6226 | |||||||
| SA0793 | dltA | d-alanine-poly(phosphoribitol) ligase, subunit 1 | Cell wall/outer membrane metabolism | 485 | SNP | Ser38Arg | |
| SA0020 | yycI | Regulatory protein, WalKR operon | Cell wall/outer membrane metabolism | 262 | Deletion | Frameshift | 51 |
| SA0905 | atl | Bifunctional autolysin | Cell wall/outer membrane metabolism | 1255 | SNP | Ser752Ser | |
| SA1246 | arlS | Histidine-kinase | Signal transduction | 451 | Insertion | Frameshift | 38 |
| SA1022 | mraW | S-adenosyl-methyltransferase | Amino acid metabolism/transport | 311 | Deletion | Frameshift | 143 |
| SA1721 | pcrA | ATP-dependent helicase | DNA replication and repair | 730 | SNP | Glu455Glu | |
| SA0467 | tilS | tRNA(ile)-lysidine synthetase | Nucleic acid metabolism/transport | 431 | SNP | Met128Ile | |
| SA0587 | PsaA adhesin homologue | Inorganic ion transport/metabolism | 309 | Deletion | Frameshift | 225 | |
| SA1528 | Universal stress protein | Stress response | 137 | SNP | Leu105Leu | ||
| SA0837 | 2-isopropylmalate synthase | Amino acid metabolism/transport | 381 | Insertion | Frameshift | 296 | |
| SA1778 | Hypothetical protein | Unknown | 114 | Deletion | Frameshift | 76 | |
| SA0349 | Hypothetical protein | Unknown | 495 | SNP | Pro474Ser | ||
| SA0668 | Hypothetical protein | Unknown | 157 | SNP | Val41Ile | ||
| 3. A6300/A6298 | |||||||
| SA2024 | rpsK | 30S ribosomal protein S11 | Translation | 129 | SNP | Arg127Gly | |
| SA1404 | rpsU | 30S ribosomal protein S21 | Translation | 67 | Deletion | Frameshift | 51 |
| SA0926 | purD | Phosphorybosylamine-glycine ligase | Nucleotide metabolism/transport | 415 | SNP | Val389Ala | |
| SA1491 | hemL | Glutamate-1-semialdehyde-2,1-aminomutase | Secondary metabolism biosynthesis | 425 | SNP | Gly48Asp | |
| SA2480 | drp35 | Lactonase | Carbohydrate metabolism and transport | 324 | SNP | Asn83Ser | |
| SA1398 | Diacylglycerol kinase, putative | Lipid metabolism | 114 | SNP | Glu66Lys | ||
| SA0949 | Hypothetical protein | Unknown | 179 | Deletion | No start codon | ||
| SA0556 | Hypothetical protein | Unknown | 210 | SNP | Phe147Phe | ||
| Clinically derived series | |||||||
| A9635/A9636 | |||||||
| SA1438 | greA | Transcription elongation factor | Transcription | 158 | 1. SNP 2. Insertion | Arg29Pro, 30Ser, 31Cys | 160 |
| A9637 | |||||||
| SA1404 | rpsU | 30S ribosomal protein S21 | Translation | 67 | Deletion | Frameshift | 53 |
| SA1478 | Hypothetical protein | Unknown | 94 | SNP | Glu24Lys | ||
| A9638 | |||||||
| SA0617 | vraG | ABC transporter permease | ABC transporter, defense | 633 | SNP | Ala580Val | |
| SA1702 | yvqF | Protein of the VraSR operon | Cell wall/outer membrane metabolism | 233 | SNP | Asn74Asp | |
| SA2047 | rplC | 50S ribosomal protein L3 | Translation | 220 | SNP | Gly7Val | |
| SA1404 | rpsU | 30S ribosomal protein S21 | Translation | 67 | Deletion | Frameshift | 53 |
| SA1861 | ilvC | Ketol-acid reductoisomerase | Amino acid metabolism and transport | 334 | Deletion | 30Gln/31Gly | 332 |
| A9639 | |||||||
| SA1067 | vraG | ABC transporter permease | ABC transporter, defense | 633 | SNP | Ala580Val | |
| SA1870 | rsbW | Ser-protein kinase, anti-σ β factor | Transcriptional regulation | 159 | Deletion | 88Ser/89Phe | 157 |
| SA1404 | rpsU | Ribosomal protein S21 | Translation | 67 | Deletion | Frameshift | 53 |
| SA1861 | ilvC | Ketol-acid reductoisomerase | Amino acid metabolism/transport | 334 | Deletion | 30Gln/31Gly | 332 |
| Laboratory-derived series | |||||||
| A8117/A8118 | |||||||
| SA0018 | walK | Sensor protein kinase | Cell wall/outer membrane metabolism | 608 | 1. SNP, 2. SNP | Arg263Cys, Ser273Asn | |
| SA2147 | tcaR | Teichoplanin-resistance associated HTH-type transcriptional regulator | Transcription | 151 | SNP-1, SNP-2 | Ile69Ser, Lys95Asn | |
| SA1936 | luxS | S-ribosylhomocysteinase | Amino acid metabolism/transport, quorum sensing | 156 | SNP | Lys15Glu | |
| A8392 | |||||||
| SA0018 | walK | Sensor protein kinase | Cell wall/outer membrane metabolism | 608 | Deletion | 371Gln | 607 |
| SA1702 | yvqF | Protein of the VraSR operon | Cell wall/outer membrane metabolism | 233 | SNP | Leu57Phe | |
| SA0500 | rpoB | RNA polymerase subunit β | Transcription | 1183 | SNP | Ser1052Leu | |
| SA2147 | tcaR | Teicoplanin resistance associated HTH-type transcriptional regulator | Transcription | 151 | 1. SNP, 2. SNP | Ile69Ser, Lys95Asn | |
| SA0375 | guaB | Inosine-5-monophosphate dehydrogenase | Nucleotide metabolism/transport | 488 | SNP | Glu417Glu | |
Abbreviations: ABC, ATP binding casette; ATP, adenosine triphosphate; ID, identification; ORF, open reading frame; SNP, single-nucleotide polymorphism.
a ORF ID derived from N315 genome annotation.
b Sizes of the gene or protein based on the susceptible strain of S. aureus.
c Size of the truncated/extended protein in the nonsusceptible strain of S. aureus.
The mutational profile of each of the vancomycin-exposed pairs varied (Table 2), highlighting the heterogeneity of genetic changes associated with vancomycin exposure and VISA formation. Despite the absence of a consistent gene mutation to explain the phenotype, mutations in genes of similar functional categories were observed, including those involved in cell wall metabolism, regulation, and protein synthesis (Table 2). We observed a number of mutations that have been associated with reduced susceptibility to vancomycin, including the ABC transporter permease vraG [32]; yvqF (SA1702) [7], which is a member of the vraSR operon; and the DNA-dependent RNA polymerase subunit β, rpoB, which has recently been shown to be important in the development of reduced susceptibility to both vancomycin and daptomycin [7, 33]. However, the point mutations and subsequent amino acid changes identified in this study were different from those described previously (Table 2), further highlighting the diversity of mutations that can result due to vancomycin exposure. Compared with that described by Mwangi et al [7], we did not observe a clear stepwise accumulation in mutations in our vancomycin-exposed series (Table 2). We observed mutations in genes in early strains that were not conserved throughout the series. For example, an SNP in yvqF emerged in an early vancomycin-exposed isolate (A9638) but was absent from a later isolate (A9639; Table 2), which suggests that the emergence of VISA may be due to the selection of resistant subpopulations within a strain, as opposed to the stepwise accumulation of mutations that are the result of prolonged vancomycin exposure.
Given our previous findings of attenuated virulence of S. aureus strains with reduced susceptibility to vancomycin [16], we were particularly interested in gene mutations that not only contribute to reduced vancomycin susceptibility but also alter staphylococcal virulence. An ideal example of this is a mutation within the agr operon, which is known to be an important virulence regulator in S. aureus as well as being associated with VISA development in the presence of vancomycin exposure [11]. We identified a single nucleotide insertion in the response regulator gene agrA within the clinical VISA isolate A5940, which resulted in gene truncation (Table 2). It has been shown that this strain has agr dysfunction due to a lack of δ-hemolysin activity [11]. We also identified point mutations in walK (previously yycG), which is a sensor kinase of an essential 2-component regulatory system that is emerging as a likely contributor to VISA formation [34, 35]. None of our clinical pairs or series had a walKR mutation, but the laboratory-derived series (A8117, A8118, and A8392) had 2 nonsynonymous point mutations as well as an amino acid deletion in walK (Table 2). The deletion (Gln371) and its role in vancomycin susceptibility have been described recently [35]. It has also been shown that WalKR has a role in S. aureus virulence [36, 37]. The operon directly regulates up to 5 virulence determinants in S. aureus that are important for bacterial adhesion to host surfaces [36]. Furthermore, a temperature-sensitive walR mutant has been shown to be attenuated in virulence in vivo [38].
In Bacillus spp, the walKR operon is negatively regulated by both YycH and YycI [39]. Mutation within yycH has been described previously in a VISA clinical series [7], but its direct effect on vancomycin susceptibility remains to be established. Here, we describe a truncation (∼10% of wild-type) of yycI in 1 of the clinical VISA isolates (A6226; Table 2). We propose that this mutation may lead to increased WalKR activity, which may be contributing to the VISA phenotype in this strain. We also identified mutations in tcaR, a teicoplanin resistance–associated transcriptional regulator, and dltA, which is integral to the d-alanation of teichoic acids within the cell wall. Both these genes have been described previously to be associated with reduced susceptibility to glycopepetides as well as S. aureus virulence [40–42].
Serine/Threonine Phosphatase Stp1 Influences Vancomycin Susceptibility in S. aureus
Of great interest was a mutation in a serine/threonine phosphatase gene known as stp1 that, along with its cognate kinase, encoded by pknB, forms a eukaryotic-like signal transduction system. The identified mutation was a single nucleotide deletion in 1 of the clinical pairs (A5937/A5940) that resulted in a frame shift truncation (∼45% of wild-type). To further characterize the role of the stp1 gene in VISA formation independent of other mutations, we created an stp1 deletion mutant from the clinical vancomycin-susceptible parent strain (A5937) using allelic exchange methodology. An in-frame deletion of stp1 resulted in a rise in MIC for vancomycin from 1.5 μg/mL to 3 μg/mL (Figure 1A), which is within the nonsusceptible range. Importantly, complementation of stp1 returned the MIC for vancomycin to the level of the vancomycin-susceptible parent strain (Figure 1A), confirming the role of stp1 in reduced vancomycin susceptibility in S. aureus. However, the MIC for vancomycin of the stp1 mutant was not as high as the clinical VISA daughter isolate (A5940; MIC, 4 μg/mL), which suggests that other mutations apart from stp1 also contribute to the reduced vancomycin susceptibility. In fact, an additional 5 genetic mutations exist between the pair, including mutations in rpoB, lysP, and agrA (Table 2), which have all been described as being associated with VISA. These data were supported by the findings of the PAP analysis, which showed that the stp1 mutant had a heterogeneous population of cells with MICs of vancomycin into the nonsusceptible range (Figure 1B). This change, however, was not as pronounced as the change for the clinical VISA daughter strain (Figure 1B). Taken together, these data confirm the importance of stp1 in reduced vancomycin susceptibility in S. aureus. Furthermore, they correlate with a recent study by Renzoni et al, which assessed the impact of stp1 mutation on reduced susceptibility to a different glycopeptide antibiotic, teicoplanin [43]. Using a laboratory-derived strain that was exposed to teicoplanin, they showed that stp1 mutation resulted in a reduction in teicoplanin susceptibility [43]. Although they were unable to show clear differences in MIC for vancomycin using their stp1 mutant strain, they did show subtle growth differences in the presence of vancomycin using a highly sensitive spot population analysis method [43]. Their data also supported recent work that has shown that stp1 is an important regulator of cell wall biosynthesis and that deletion of the stp1 gene leads to an increase in cell wall thickness [28, 43], which is a consistent phenotype of VISA isolates.
Figure 1.

Deletion of stp1 results in reduced vancomycin susceptibility in Staphylococcus aureus. (A) Vancomycin minimum inhibitory concentrations (MICs) were determined by Etest for A5937, A5937Δstp1, the complemented strain A5937Δstp1-C, and the vancomycin nonsusceptible isolate A5940. Values in parentheses represent MICs based on broth dilution. (B) Determination of vancomycin heteroresistance was performed using population analysis profiling. The stp1 deletion mutant had subpopulations that grew in the nonsusceptible range (>2 µg/mL); this was not seen in the parent strain (A5937) but was more pronounced in the clinical vancomycin-intermediate S. aureus daughter strain (A5940). CFU, colony-forming unit. (C) Transmission electron microscopy was performed to determine cell wall thickness. Three representative cells from each strain are shown, and values represent mean cell wall thickness (95% confidence interval). A5940 had the thickest cell wall, followed by the A5937Δstp1 and finally A5937.
Stp1 Affects Vancomycin Susceptibility by Regulating Cell Wall Metabolism
In order to determine the mechanism by which Stp1 affects vancomycin susceptibility, we assessed the transcriptional profile of the stp1 deletion mutant. Using microarray analysis, we identified upregulation of genes that could potentially contribute to cell wall thickening (Table 3). The first of these genes is uppS, encoding undecaprenyl pyrophosphate synthase, which is involved in the production of undecaprenyl phosphate, an important lipid carrier in the biosynthesis of peptidoglycan and cell wall teichoic acids [44]. The second gene with altered regulation is sceD, encoding a lytic transglycolase, which has been shown to have peptidoglycan hydrolase activity and involvement in cell wall turnover [45]. Given these findings and those in recently published reports [28, 43], we performed TEM analysis on our stp1 mutant (Figure 1C). This confirmed a thickened cell wall in our stp1 deletion mutant compared to its parent strain (P < .001; Figure 1C), and, as seen with the vancomycin susceptibility testing, the cell wall was not as thick as the clinical VISA isolate (A5940), further supporting the importance of multiple mutations in the observed VISA phenotype. It has previously been shown that disorganization of the peptidoglycan and an increase in false vancomycin binding sites prevent the penetration of vancomycin to its active site on the cytoplasmic membrane [8, 46, 47].
Table 3.
Differentially Expressed Genes in the stp1 Deletion Mutanta Compared to Its Parent Strainb
| ORF IDc | Gene | Description | Fold Changed |
|---|---|---|---|
| Downregulated genes | |||
| Virulence | |||
| SACOL1187 | PSM β2 | Phenol-soluble modulin | 1.9 |
| SAS065 | hld | δ-hemolysin | 1.9 |
| SA0387 | set11 | Superantigen-like protein | 1.55 |
| Unknown function | |||
| SAP007 | Hypothetical protein | 2.1 | |
| SAS059 | Hypothetical protein | 1.64 | |
| Upregulated genes | |||
| Cell wall metabolism | |||
| SA1103 | uppS | Undecaprenyl pyrophosphate synthetase | 1.77 |
| SA1898 | sceD | Transglycosylase | 1.86 |
| Other cellular function | |||
| SACOL1053 | yfbB | Acyl-CoA thioester hydrolase | 2.37 |
| SA2186 | nasF | Uroporphyrin-III C-methyl transferase | 2.16 |
| SA1082 | rimM | 16S ribosomal RNA processing protein | 1.69 |
| SA1184 | citB | Aconitate hydratase | 1.62 |
| SA2180 | nreB | Sensor histidine kinase | 1.61 |
| SA0587 | mntC | Manganese ABC transporter permease | 1.53 |
| SA1421 | Putative metal-dependent phosphohydralase | 1.52 | |
| Unknown function | |||
| SACOL1331 | Hypothetical protein | 2.9 | |
| SACOL2365 | Hypothetical protein | 2.15 | |
| SA0287 | Hypothetical protein | 2.09 | |
| SA0289 | Hypothetical protein | 1.96 | |
| SAR0592 | Hypothetical protein | 1.58 | |
| SA0336 | Hypothetical protein | 1.56 | |
Abbreviations: ABC, adenosine triphosphate binding casette; ID, identification; ORF, open reading frame; PSM, phenol-soluble modulin.
a The cotranscribed kinase, pknB, showed no significant change in expression between the stp1 deletion mutant and its parent strain (A5937).
b Differential expression was confirmed using quantitative reverse-transcriptase polymerase chain reaction
normalized to 16S ribosomal RNA expression for hld (downregulated 4.07 [±1.0]-fold), PSM β2 (downregulated 2.31 [±0.7]-fold), and sceD (upregulated 1.51 [±0.2]-fold).
c ORF IDs derived from N315 (SA/SAP/SAS), COL (SACOL), and methicillin-resistant Staphylococcus aureus MRSA252 (SAR) genome annotations.
d Genes with fold change of ≥1.5 with adjusted P value of ≤.05 were included.
Stp1 Affects S. aureus Virulence
As mentioned previously, we have evidence, using a nonmammalian model system, that VISA isolates may be less virulent than their vancomycin-susceptible parent strains [16]. In addition, it has recently been shown that stp1 is important for toxin production and virulence in S. aureus [48]. In support of these findings, our gene expression analysis of the stp1 mutant relative to its parent strain showed downregulation of a number of genes involved in S. aureus virulence, most notably hld, which encodes δ-hemolysin, a toxin that assists in the destruction of leukocytes and a number of other host cell types [49]. Importantly, the hld gene is embedded within RNAIII, which is the effector molecule of agr, suggesting a possible relationship between Stp1 and Agr. In addition to this, we observed downregulation of other genes that are either directly or indirectly regulated by agr, including a superantigen-like protein, encoded by set11, and the phenol-soluble modulin β2 gene (SACOL1187), which encodes a protein that belongs to a family of short peptides that are emerging as important virulence determinants, particularly in community-associated S. aureus [50]. To mimic the bacteremia that these strains caused in our patient, we tested the vancomycin-susceptible parent strain (A5937), its VISA daughter strain (A5940), and the stp1 deletion mutant (A5937Δstp1) in a murine bacteremia model. We found that the stp1 deletion mutant and the VISA daughter strain were significantly attenuated in virulence compared with the parent strain (Figure 2A). Furthermore, hepatic abscess formation was observed in A5937-infected mice at 5 days after infection, whereas this was not present with the stp1 deletion mutant or the VISA daughter strain (Figure 2B). This was supported by histopathological analysis, which showed severe necrosis and abscess formation only in the A5937-infected mice (Figure 2C). These data not only illustrate the importance of the stp1 gene in staphylococcal virulence but also support our previously generated data using the invertebrate Galleria mellonella model, which showed attenuated virulence in clinical VISA isolates when compared with their isogenic vancomycin-susceptible parent strains [16].
Figure 2.
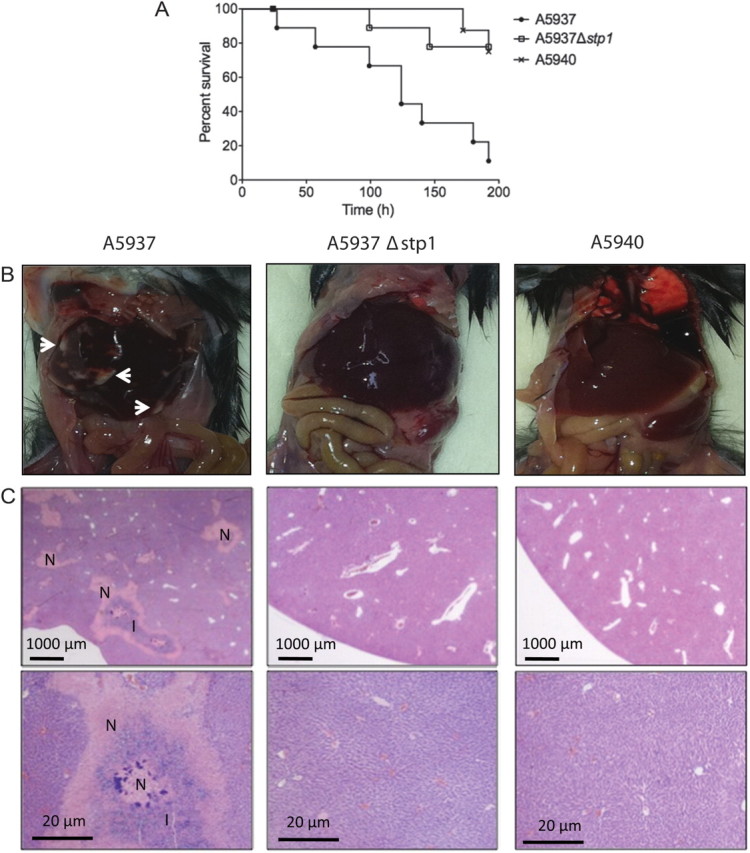
Deletion of stp1 results in attenuated virulence in Staphylococcus aureus. (A) Six-week-old C57BL/6J mice were injected with ∼1.0 × 108 bacterial cells. Kaplan-Meier curves indicate time to euthanasia for each strain. A5937 produced significantly more killing compared with both A5937Δstp1 and A5940 (P = .01; log-rank test). (B) Macroscopic and (C) histopathological analyses showed that the clinical vancomycin-susceptible parent strain (A5937) caused hepatic abscess formation and tissue necrosis, whereas this was not seen with the stp1 deletion mutant (A5937Δstp1) and the clinical vancomycin-intermediate S. aureus daughter strain (A5940). White arrows point to abscess formation in the liver; N represents severe necrosis; and I represents inflammatory infiltrate. Tissues were stained with hematoxylin and eosin.
Conclusion
Comparative genomics of carefully selected strains is a useful tool when trying to understand the genetic mechanisms behind VISA formation. Our data, along with a number of previous studies, have shown that mutations in a range of biological pathways can contribute to reduced vancomycin susceptibility in S. aureus. We have also characterized a new mechanism of reduced vancomycin susceptibility, the serine/threonine phosphatase stp1. Using gene expression analyses, we have shown that stp1 regulates genes that alter cell wall biosynthesis, providing a possible mechanism for its effect on vancomycin susceptibility. We have also shown that this gene has a role in staphylococcal virulence in mammals, which provides support to the hypothesis that virulence may actually be attenuated in S. aureus strains with reduced susceptibility to vancomycin. These data provide important insights into the pathogenic consequences of antibiotic resistance in S. aureus and may assist with the development of novel strategies for the prevention or treatment of infections due to this problematic organism.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://www.oxfordjournals.org/our_journals/jid/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank S. Sykes, T. Hepburn, and S. Young for their assistance with SNP calling. Microarray slides were kindly donated to B. P. H. by the Institute for Genomic Research. We also thank A. Cheung for donating pALC2073.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases; National Institutes of Health, Department of Health and Human Services (HHSN272200900018C, HHSN266200400001C); and in part by the Australian National Health and Medical Research project grant (APP1008973). We also acknowledge the Australian National Health and Medical Research Council Biomedical Fellowship to A. Y. P. (APP606961) and Australian Postgraduate Award to D. C.
Potential conflicts of interest. R. C. M. has served as a consultant to Cubist, Forest, Merck, Novartis, Ortho, Johnson & Johnson, Pfizer, Theravance, and Wyeth. G. M. E. has served on scientific advisory boards for Cubist, Bayer Schering, Johnson & Johnson Pharmaceutical Research and Development, Novartis, Pfizer, Shionogi, and Theravance; has received research training support from Cubist and research contracts from Novexel, Pfizer, and Theravance; has received speaking honoraria from Novartis; and serves on the board of directors of the National Foundation for Infectious Diseases. A. Y. P. has been to 1 advisory board meeting for Abbott Molecular and Ortho-McNeil-Janssen and has received a speaker's honorarium from AstraZeneca and Merck Sharp & Dohme for 1 presentation each. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Hiramatsu K, Aritaka N, Hanaki H, et al. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet. 1997;350:1670–3. doi: 10.1016/S0140-6736(97)07324-8. [DOI] [PubMed] [Google Scholar]
- 2.Chang S, Sievert DM, Hageman JC, et al. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N Engl J Med. 2003;348:1342–7. doi: 10.1056/NEJMoa025025. [DOI] [PubMed] [Google Scholar]
- 3.Charles PG, Ward PB, Johnson PD, Howden BP, Grayson ML. Clinical features associated with bacteremia due to heterogeneous vancomycin-intermediate Staphylococcus aureus. Clin Infect Dis. 2004;38:448–51. doi: 10.1086/381093. [DOI] [PubMed] [Google Scholar]
- 4.Howden BP, Ward PB, Charles PG, et al. Treatment outcomes for serious infections caused by methicillin-resistant Staphylococcus aureus with reduced vancomycin susceptibility. Clin Infect Dis. 2004;38:521–8. doi: 10.1086/381202. [DOI] [PubMed] [Google Scholar]
- 5.Howden BP, Johnson PD, Charles PG, Grayson ML. Failure of vancomycin for treatment of methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis. 2004;39:1544. doi: 10.1086/425129. reply 1544–5. [DOI] [PubMed] [Google Scholar]
- 6.Neoh HM, Cui L, Yuzawa H, Takeuchi F, Matsuo M, Hiramatsu K. Mutated response regulator graR is responsible for phenotypic conversion of Staphylococcus aureus from heterogeneous vancomycin-intermediate resistance to vancomycin-intermediate resistance. Antimicrob Agents Chemother. 2008;52:45–53. doi: 10.1128/AAC.00534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mwangi MM, Wu SW, Zhou Y, et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci U S A. 2007;104:9451–6. doi: 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanaki H, Kuwahara-Arai K, Boyle-Vavra S, Daum RS, Labischinski H, Hiramatsu K. Activated cell-wall synthesis is associated with vancomycin resistance in methicillin-resistant Staphylococcus aureus clinical strains Mu3 and Mu50. J Antimicrob Chemother. 1998;42:199–209. doi: 10.1093/jac/42.2.199. [DOI] [PubMed] [Google Scholar]
- 9.Kuroda M, Kuroda H, Oshima T, Takeuchi F, Mori H, Hiramatsu K. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol Microbiol. 2003;49:807–21. doi: 10.1046/j.1365-2958.2003.03599.x. [DOI] [PubMed] [Google Scholar]
- 10.Howden BP, Stinear TP, Allen DL, Johnson PD, Ward PB, Davies JK. Genomic analysis reveals a point mutation in the two-component sensor gene graS that leads to intermediate vancomycin resistance in clinical Staphylococcus aureus. Antimicrob Agents Chemother. 2008;52:3755–62. doi: 10.1128/AAC.01613-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakoulas G, Eliopoulos GM, Moellering RC, Jr., et al. Accessory gene regulator (agr) locus in geographically diverse Staphylococcus aureus isolates with reduced susceptibility to vancomycin. Antimicrob Agents Chemother. 2002;46:1492–502. doi: 10.1128/AAC.46.5.1492-1502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fridkin SK, Hageman J, McDougal LK, et al. Epidemiological and microbiological characterization of infections caused by Staphylococcus aureus with reduced susceptibility to vancomycin, United States, 1997–2001. Clin Infect Dis. 2003;36:429–39. doi: 10.1086/346207. [DOI] [PubMed] [Google Scholar]
- 14.Musta AC, Riederer K, Shemes S, et al. Vancomycin MIC plus heteroresistance and outcome of methicillin-resistant Staphylococcus aureus bacteremia: trends over 11 years. J Clin Microbiol. 2009;47:1640–4. doi: 10.1128/JCM.02135-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howden BP, Smith DJ, Mansell A, et al. Different bacterial gene expression patterns and attenuated host immune responses are associated with the evolution of low-level vancomycin resistance during persistent methicillin-resistant Staphylococcus aureus bacteraemia. BMC Microbiol. 2008;8:39. doi: 10.1186/1471-2180-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peleg AY, Monga D, Pillai S, Mylonakis E, Moellering RC, Jr., Eliopoulos GM. Reduced susceptibility to vancomycin influences pathogenicity in Staphylococcus aureus infection. J Infect Dis. 2009;199:532–6. doi: 10.1086/596511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakoulas G, Eliopoulos GM, Fowler VG, Jr., et al. Reduced susceptibility of Staphylococcus aureus to vancomycin and platelet microbicidal protein correlates with defective autolysis and loss of accessory gene regulator (agr) function. Antimicrob Agents Chemother. 2005;49:2687–92. doi: 10.1128/AAC.49.7.2687-2692.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wootton M, Howe RA, Hillman R, Walsh TR, Bennett PM, MacGowan AP. A modified population analysis profile (PAP) method to detect hetero-resistance to vancomycin in Staphylococcus aureus in a UK hospital. J Antimicrob Chemother. 2001;47:399–403. doi: 10.1093/jac/47.4.399. [DOI] [PubMed] [Google Scholar]
- 19.Nusbaum C, Ohsumi TK, Gomez J, et al. Sensitive, specific polymorphism discovery in bacteria using massively parallel sequencing. Nat Methods. 2009;6:67–9. doi: 10.1038/nmeth.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bae T, Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006;55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 21.Bateman BT, Donegan NP, Jarry TM, Palma M, Cheung AL. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect Immun. 2001;69:7851–7. doi: 10.1128/IAI.69.12.7851-7857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howden BP, Johnson PD, Ward PB, Stinear TP, Davies JK. Isolates with low-level vancomycin resistance associated with persistent methicillin-resistant Staphylococcus aureus bacteremia. Antimicrob Agents Chemother. 2006;50:3039–47. doi: 10.1128/AAC.00422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saal LH, Troein C, Vallon-Christersson J, Gruvberger S, Borg A, Peterson C. BioArray Software Environment (BASE): a platform for comprehensive management and analysis of microarray data. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-8-software0003. SOFTWARE0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smyth GK, Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–73. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- 25.Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–75. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- 26.Peleg AY, Adams J, Paterson DL. Tigecycline efflux as a mechanism for nonsusceptibility in Acinetobacter baumannii. Antimicrob Agents Chemother. 2007;51:2065–9. doi: 10.1128/AAC.01198-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 28.Beltramini AM, Mukhopadhyay CD, Pancholi V. Modulation of cell wall structure and antimicrobial susceptibility by a Staphylococcus aureus eukaryote-like serine/threonine kinase and phosphatase. Infect Immun. 2009;77:1406–16. doi: 10.1128/IAI.01499-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pillai SK, Wennersten C, Venkataraman L, Eliopoulos GM, Moellering RC, Karchmer AW. Development of reduced vancomycin susceptibility in methicillin-susceptible Staphylococcus aureus. Clin Infect Dis. 2009;49:1169–74. doi: 10.1086/605636. [DOI] [PubMed] [Google Scholar]
- 30.Cui L, Tominaga E, Neoh HM, Hiramatsu K. Correlation between reduced daptomycin susceptibility and vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50:1079–82. doi: 10.1128/AAC.50.3.1079-1082.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA) Proc Natl Acad Sci U S A. 2002;99:7687–92. doi: 10.1073/pnas.122108599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meehl M, Herbert S, Gotz F, Cheung A. Interaction of the GraRS two-component system with the VraFG ABC transporter to support vancomycin-intermediate resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51:2679–89. doi: 10.1128/AAC.00209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cui L, Isii T, Fukuda M, et al. An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob Agents Chemother. 2010;54:5222–33. doi: 10.1128/AAC.00437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jansen A, Turck M, Szekat C, Nagel M, Clever I, Bierbaum G. Role of insertion elements and yycFG in the development of decreased susceptibility to vancomycin in Staphylococcus aureus. Int J Med Microbiol. 2007;297:205–15. doi: 10.1016/j.ijmm.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 35.Shoji M, Cui L, Iizuka R, et al. walK and clpP mutations confer reduced vancomycin susceptibility in Staphylococcus aureus. Antimicrob Agents Chemother. 2011;55:3870–81. doi: 10.1128/AAC.01563-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dubrac S, Msadek T. Identification of genes controlled by the essential YycG/YycF two-component system of Staphylococcus aureus. J Bacteriol. 2004;186:1175–81. doi: 10.1128/JB.186.4.1175-1181.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner C, Saizieu Ad A, Schonfeld HJ, et al. Genetic analysis and functional characterization of the Streptococcus pneumoniae vic operon. Infect Immun. 2002;70:6121–8. doi: 10.1128/IAI.70.11.6121-6128.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin PK, Li T, Sun D, Biek DP, Schmid MB. Role in cell permeability of an essential two-component system in Staphylococcus aureus. J Bacteriol. 1999;181:3666–73. doi: 10.1128/jb.181.12.3666-3673.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szurmant H, Mohan MA, Imus PM, Hoch JA. YycH and YycI interact to regulate the essential YycFG two-component system in Bacillus subtilis. J Bacteriol. 2007;189:3280–9. doi: 10.1128/JB.01936-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brandenberger M, Tschierske M, Giachino P, Wada A, Berger-Bachi B. Inactivation of a novel three-cistronic operon tcaR-tcaA-tcaB increases teicoplanin resistance in Staphylococcus aureus. Biochim Biophys Acta. 2000;1523:135–9. doi: 10.1016/s0304-4165(00)00133-1. [DOI] [PubMed] [Google Scholar]
- 41.Collins LV, Kristian SA, Weidenmaier C, et al. Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J Infect Dis. 2002;186:214–9. doi: 10.1086/341454. [DOI] [PubMed] [Google Scholar]
- 42.McCallum N, Bischoff M, Maki H, Wada A, Berger-Bachi B. TcaR, a putative MarR-like regulator of sarS expression. J Bacteriol. 2004;186:2966–72. doi: 10.1128/JB.186.10.2966-2972.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Renzoni A, Andrey DO, Jousselin A, et al. Whole genome sequencing and complete genetic analysis reveals novel pathways to glycopeptide resistance in Staphylococcus aureus. PLoS One. 2011;6:e21577. doi: 10.1371/journal.pone.0021577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Apfel CM, Takacs B, Fountoulakis M, Stieger M, Keck W. Use of genomics to identify bacterial undecaprenyl pyrophosphate synthetase: cloning, expression, and characterization of the essential uppS gene. J Bacteriol. 1999;181:483–92. doi: 10.1128/jb.181.2.483-492.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stapleton MR, Horsburgh MJ, Hayhurst EJ, et al. Characterization of IsaA and SceD, two putative lytic transglycosylases of Staphylococcus aureus. J Bacteriol. 2007;189:7316–25. doi: 10.1128/JB.00734-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui L, Murakami H, Kuwahara-Arai K, Hanaki H, Hiramatsu K. Contribution of a thickened cell wall and its glutamine nonamidated component to the vancomycin resistance expressed by Staphylococcus aureus Mu50. Antimicrob Agents Chemother. 2000;44:2276–85. doi: 10.1128/aac.44.9.2276-2285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sieradzki K, Tomasz A. Gradual alterations in cell wall structure and metabolism in vancomycin-resistant mutants of Staphylococcus aureus. J Bacteriol. 1999;181:7566–70. doi: 10.1128/jb.181.24.7566-7570.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burnside K, Lembo A, de Los Reyes M, et al. Regulation of hemolysin expression and virulence of Staphylococcus aureus by a serine/threonine kinase and phosphatase. PLoS One. 2010;5:e11071. doi: 10.1371/journal.pone.0011071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rogolsky M. Nonenteric toxins of Staphylococcus aureus. Microbiol Rev. 1979;43:320–60. doi: 10.1128/mr.43.3.320-360.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang R, Braughton KR, Kretschmer D, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–4. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.