

Abstract
Background. Quinolone-resistant Neisseria gonorrhoeae (QRNG) arise from mutations in gyrA (intermediate resistance) or gyrA and parC (resistance). Here we tested the consequence of commonly isolated gyrA91/95 and parC86 mutations on gonococcal fitness.
Methods. Mutant gyrA91/95 and parC86 alleles were introduced into wild-type gonococci or an isogenic mutant that is resistant to macrolides due to an mtrR−79 mutation. Wild-type and mutant bacteria were compared for growth in vitro and in competitive murine infection.
Results. In vitro growth was reduced with increasing numbers of mutations. Interestingly, the gyrA91/95 mutation conferred an in vivo fitness benefit to wild-type and mtrR−79 mutant gonococci. The gyrA91/95, parC86 mutant, in contrast, showed a slight fitness defect in vivo, and the gyrA91/95, parC86, mtrR−79 mutant was markedly less fit relative to the parent strains. A ciprofloxacin-resistant (CipR) mutant was selected during infection with the gyrA91/95, parC86, mtrR−79 mutant in which the mtrR−79 mutation was repaired and the gyrA91 mutation was altered. This in vivo–selected mutant grew as well as the wild-type strain in vitro.
Conclusions. gyrA91/95 mutations may contribute to the spread of QRNG. Further acquisition of a parC86 mutation abrogates this fitness advantage; however, compensatory mutations can occur that restore in vivo fitness and maintain CipR.
(See the editorial commentary by Dillon and Parti, on pages 1775–7.)
Neisseria gonorrhoeae is a Gram-negative diplococcus that plays a major role in urogenital tract and perinatal infections [1]. Gonorrhea is the second most frequently reported bacterial sexually transmitted infection (STI) in the United States, with an estimated 700 000 new cases each year. The rate of gonorrhea is highest among females aged <25 years old, especially those that engage in high-risk sexual behaviors [2]. The high prevalence of gonorrhea is particularly concerning because it is a major cause of pelvic inflammatory disease and can lead to serious outcomes such as ectopic pregnancy, infertility, and chronic pelvic pain. Gonorrhea is also a cofactor in human immunodeficiency virus transmission [3].
Antibiotic resistance emerges rapidly in N. gonorrhoeae, which challenges treatment options and threatens current control measures. Currently, only a single class of antibiotics, the third-generation cephalosporins, is recommended for routine treatment of gonorrhea in the Centers for Disease Control and Prevention (CDC) sexually transmitted disease guidelines [2]. The status of N. gonorrhoeae as a “superbug” continues to increase with the recent isolation of a ceftriaxone-resistant strain [4]. Fluoroquinolones were removed from the CDC list of recommended first-line antibiotics for treatment of gonorrhea in 2007 [5]. First used clinically in the mid-1980s [6], over 80% of gonococcal isolates in the western Pacific region were ciprofloxacin resistant (CipR) by 2002 [7, 8]. A steady increase in quinolone-resistant N. gonorrhoeae (QRNG) followed in the United States, with 891 of 6009 (14.8%) clinical isolates identified as QRNG in 2007 [9]. Resistance to this relatively inexpensive class of antibiotics, which target topoisomerase II (DNA gyrase) and topoisomerase IV [6], in N. gonorrhoeae is due to amino acid substitutions in the quinolone resistance–determining regions (QRDRs) of the A subunits of DNA gyrase (GyrA) and ParC, the A subunit of DNA topoisomerase IV [10–13].
Traditionally, antibiotic resistance is accompanied by a fitness loss, particularly when it occurs via mutation of genes that are important for basic cellular functions [14]. Some resistance mutations, however, provide a growth benefit in vitro or increase fitness when tested in pathogenesis models [15–19]. This phenomenon can be due to compensatory mutations that balance the detrimental effects of resistance mutations [20]. Increased microbial fitness during infection can also be a direct consequence of the resistance mutation, as shown for mutations that increase the efflux of antimicrobial substances [15, 18]. For example, in N. gonorrhoeae, multitransferable resistance (mtr) mutations, defined here as mutations in the mtrR repressor gene or its promoter region, increase resistance to macrolide antibiotics and high levels of penicillin G through overexpression of the MtrC-MtrD-MtrE active efflux pump. The mtr mutations also confer a fitness advantage during experimental infection of female mice [18], which is most likely due to increased resistance to host innate effectors that are also substrates of the MtrC-MtrD-MtrE active efflux pump [21–23]. Whether fluoroquinolone resistance mutations also promote increased in vivo fitness of N. gonorrhoeae is not known. This question warrants testing based on the wide prevalence of QRNG strains and reports that gyrA and gyrA, parC mutations in some pathogens confer a fitness advantage in animal models [16, 17].
To further explore the rapid spread of QRNG worldwide, here we introduced commonly isolated gyrA and parC mutations into a wild-type N. gonorrhoeae strain and tested the consequence on gonococcal fitness in vitro and in the mouse infection model. We also examined the effect of these mutations in an mtr mutant of the same strain background to determine if the fitness benefit conferred by overproduction of the MtrC-MtrD-MtrE active efflux pump is lost or further increased in QRNG.
METHODS
Bacterial Strains and Culture Conditions
Bacterial strains are described in Table 1. Streptomycin-resistant (SmR) derivatives of wild-type N. gonorrhoeae strain FA19 and mutant KH15 were used in this study [22]. Mutant KH15 overexpresses the mtrCDE operon due to a single base pair (bp) deletion in a 13-bp inverted repeat in the mtrR promoter that maps 79 bps upstream of the mtrR start codon (mtrR−79) [18, 21, 24]. Neisseria gonorrhoeae strain C29 is a CipR isolate from an Israeli outbreak [25] that carries mutated gyrA (Ser91Phe and Asp95Asn) and parC (Asp86Asn) alleles. DNA from strain C29 (provided by Jonathan Zenilman, Johns Hopkins University) was used to polymerase chain reaction (PCR) amplify a 1.3-kb PCR fragment that carries the desired gyrA91/95 mutations using primers gyrA1-for and gyrA1-rev. The PCR product was cloned into pCR-Blunt and transformed into Escherichia coli Top10 (Invitrogen). Transformants were selected on Luria agar with 50 µg/mL of kanamycin. The same mutated gyrA sequence was amplified from a positive clone and transformed into FA19SmR and KH15 bacteria [26]. Transformants were selected on GC agar with 0.125 µg/mL Cip to obtain mutants AK1 (gyrA91/95) and AK11 (gyrA91/95, mtrR−79). The gyrA91/95, parC86 double mutants were constructed similarly using primers parC1-for and parC1-rev to obtain a 1.6-kb PCR fragment containing the parC86 mutation from strain C29 and mutants AK1 and AK2 as the recipients. Mutants AK2 (gyrA91/95, parC86) and AK12 (gyrA91/95, parC86, mtrR−79) were selected on GC agar with 2.0 µg/mL of Cip. All plasmid clones and mutations were confirmed by nucleotide sequence analysis. Primers gyrA1-for, gyrA1-rev, and gyrA2-for and primers parC1-for, parC1-rev, and parC2-for anneal within the gyrA and parC structural genes, respectively (Table 2). The mtr locus of mutant AK13 was sequenced using primers 5′mtrR and 3′mtrR [27]. Stocks of nonpiliated wild-type and mutant bacteria, as assessed by colony morphology, were used for in vitro cocultures to minimize clumping and in competitive infection experiments to reduce DNA transfer between the strains being compared. Neisseria gonorrhoeae was cultured on GC agar with Kellogg's supplements as described [28].
Table 1.
Bacterial Strains
| Strain | Relevant Characteristics | Reference |
|---|---|---|
| FA19SmR | Spontaneous SmR mutant of wild-type FA19 | [22] |
| KH15 | mtrR−79 mutant; parent strain FA19SmR | [27] |
| AK1 | gyrA91/95; parent strain FA19SmR | Present study |
| AK2 | gyrA91/95, parC86; parent strain AK1 | Present study |
| AK11 | gyrA91/95, mtrR−79; parent strain KH15 | Present study |
| AK12 | gyrA91/95, parC86, mtrR−79; parent strain AK11 | Present study |
| AK13 | in vivo-selected mutant; gyrASer91Leu/95, parC86, wild-type mtr | Present study |
Table 2.
Oligonucleotide Primers Used in This Study
| Primer | Sequence (5′ to 3′) |
|---|---|
| gyrA1-for | GACTTCCTCATGCAGCAAATG |
| gyrA1-rev | CAACCATATTGATGCCGAAACTG |
| gyrA2-for | ACGAAACATTGAAACCATGAC |
| parC1-for | CATAGCGACGGTCTTTGTGTG |
| parC1-rev | GTTGATGAAGGTATCGGTATCGATG |
| parC2-for | ACGCTTCCCATACCGATTC |
Minimal Inhibitory Concentration
The minimum inhibitory concentrations (MICs) of Cip and erythromycin (Em) against wild-type and mutant bacteria were determined by a standard agar dilution assay. Ciprofloxacin MICs of 0.125–0.5 µg/mL were reported as intermediate resistance (CipI); Cip MIC values ≥1 µg/mL were reported as resistance (CipR), as per CDC guidelines [29, 30].
Growth Kinetics and In Vitro Competition Experiments
Wild-type and mutant bacteria were harvested from GC agar plates after 18–20 hours incubation and inoculated into 30 mL of supplemented GC broth with 5 mM NaHCO3 to a starting absorbance value at 600 nm (A600) = A600 = 0.07–0.075. Cultures were incubated in a shaking air incubator at 37°C as described [31]. The average time required to reach an A600 = 1.0 was determined from 3 independent experiments for each strain and compared by a 1-way analysis of variance. For in vitro competition experiments, similar numbers of the strains being compared were inoculated into GC broth, and aliquots were quantitatively cultured on GC agar with 100 µg/mL Sm [total number of colony forming units (CFUs)] and 100 µg/mL Sm plus 0.125 μg/mL Cip [total number of mutant CFUs] over time. The number of mutant CFUs was subtracted from the total number to estimate the number of parent bacteria. The ratio of mutant to parent CFUs at each time point was divided by the ratio of mutant to parent CFUs at time 0 to obtain the competitive index (Ic).
Competitive Experimental Murine Infection
Female BALB/c mice (approximately 6–8 weeks old) were treated with water-soluble 17-β-estradiol [32] and antibiotics (2.4 mg Sm and 0.4 mg vancomycin intraperitoneally twice daily and 0.04 g trimethoprim sulfate per 100 mL drinking water) to increase susceptibility to N. gonorrhoeae and reduce the overgrowth of commensal flora. None of the antibiotics used is a substrate of the MtrC-MtrD-MtrE efflux pump system or target GyrA or ParC. Bacteria were cultured on GC agar plates (18–20 hours), and isolated colonies were suspended in saline and passed through a 1.2-µm filter to remove aggregates. Suspensions with similar numbers of the bacteria to be compared were combined, and 20 µL of the mixed suspensions were inoculated vaginally into mice (total dose, approximately 106 CFUs) (n = 5–8 mice per test group). Vaginal mucus was quantitatively cultured on GC agar with Sm (total CFUs) and GC agar with Sm plus 0.125 µg/mL Cip (to isolate AK1 and AK11) or 2 µg/mL Cip (to isolate AK2 and AK12) on days 1, 3, 5, and 7 postinoculation, as described [31]. The number of parent CFUs was calculated by subtracting the number of mutant CFUs from the total number of CFUs. Data were expressed as an Ic [(mutant CFUs/parent CFUs)output/(mutant CFUs/parent CFUs)input]. The limit of detection (4 CFUs/100 µL vaginal swab suspension) was used for cultures from which no gonococci were isolated. All animal infection experiments were conducted at the Uniformed Services University of the Health Sciences (USUHS), which is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, under a protocol approved by the university's Institutional Animal Care and Use Committee.
RESULTS
Characterization of gyrA91/95 and gyrA91/95, parC86 Mutants
Fluoroquinolone resistance in N. gonorrhoeae is a 2-step process in which mutations in gyrA lead to a CipI phenotype and an additional mutation in parC confers a CipR phenotype [10]. To test the consequence of intermediate-level Cip resistance on gonococcal fitness, we introduced 2 commonly isolated gyrA mutations that encode Ser91Phe and Asp95Asn substitutions into the CipS N. gonorrhoeae strain FA19SmR. We also introduced these mutations into strain KH15, which is an isogenic mutant of FA19SmR that carries a commonly isolated mtr promoter mutation [27]. As expected, mutants AK1 (gyrA91/95) and AK11 (gyrA91/95, mtrR−79) were CipI (Table 3). Introduction of a parC86 mutation that encodes an Asp86Asn substitution into mutants AK1 and AK11 resulted in mutants AK2 (gyrA91/95, parC86) and AK12 (gyrA91/95, parC86, mtrR−79), respectively, and increased the Cip MIC to a clinically relevant level (6 µg/mL). The mtrR−79 mutation did not alter the Cip MIC, but as expected, Em MICs against parent strain KH15 (mtrR−79) and mutants AK11 and AK12 were 2.5-fold higher than against FA19SmR, AK1, and AK2 (Table 3).
Table 3.
Antibiotic Sensitivity and In Vitro Growth of Wild-Type and Mutant Strains
| MIC (µg/mL) |
Time to Reach A600 = 1.0a |
|||
|---|---|---|---|---|
| Strain | Cip | Em | Minutes (±SE)a | P Value |
| FA19SmR | <0.0625 | 0.5 | 287 (±13) | … |
| AK1 | 0.5 | 0.5 | 325 (±6) | .08b |
| AK2 | 6 | 0.5 | 376 (±17) | <.01c |
| KH15 | <0.0625 | 4 | 340 (±7) | .01d |
| AK11 | 1 | 4 | 360 (±8) | .08e |
| AK12 | 6 | 4 | 410 (±16) | <.01f |
| AK13 | 6 | 0.5 | 294 (±6) | <.05g |
Abbreviations: Cip, ciprofloxacin; Em, erythromycin; MIC, minimum inhibitory concentration; SE, standard error.
a All broth cultures were started at an absorbance reading at 600 nm (A600) an A600 = 0.075–0.085 with the starting A600 for each strain within 0.005 units within each experiment. The A600 was measured at hourly time points and plotted vs time. The number of minutes required to reach an A600 = 1.0 was calculated from the linear part of the curve. Results were compared by a 1-way analysis of variance.
b AK1 compared with parent strain FA19SmR.
c AK2 compared with FA19SmR; P value < .05 for AK2 vs AK1.
d KH15 compared with parent strain FA19SmR.
e AK11 compared with parent strain KH15.
f AK12 mutant compared with FA19SmR, KH15, AK1, or AK11.
g AK13 mutant compared with KH15; P value < .01 for AK13 vs AK2, AK11, or AK12.
Mutations in the QRDR of gyrA and/or parC alter growth rates in other bacterial species [33–38]. When cultured separately in GC broth under standard conditions, growth of strains AK1 and AK11, which carry the gyrA91/95 alleles, appeared slower than the respective parent strains (Figure 1), although there was no significant difference in the time it took the mutant and parent strains to reach mid to late logarithmic phase (A600 = 1.0) (Table 3). In contrast, mutants that carry both gyrA91/95 and parC86 mutations (AK2 and AK12) grew more slowly than the respective parent bacteria and required a significantly longer time to reach an A600 = 1.0, with AK12 the most attenuated (Figure 1; Table 3). We conclude that Ser91Phe and Asp95Asn substitutions in GyrA do not detectably affect growth under these conditions. However, mutants that express both an altered GyrA and ParC protein grow significantly more slowly and are further attenuated by mtrR−79.
Figure 1.
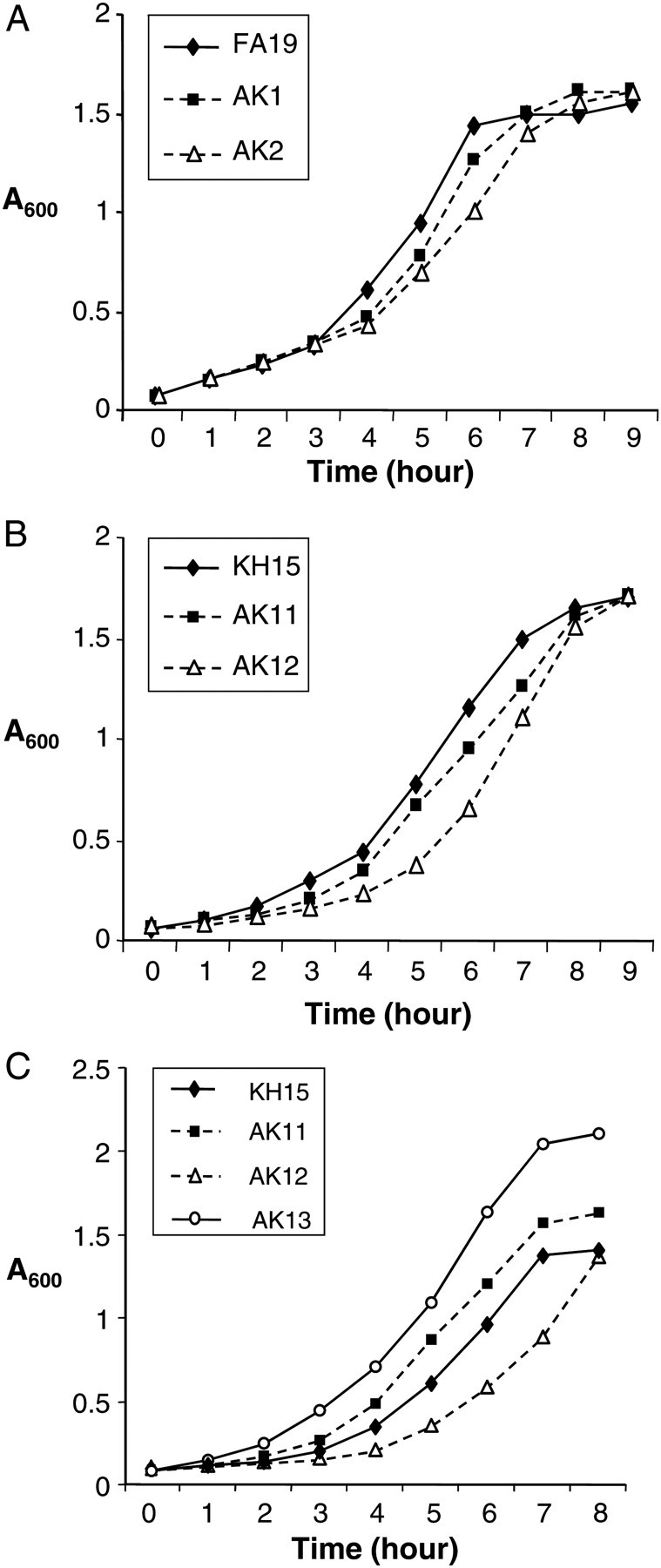
In vitro growth kinetics of wild-type and mutant strains. Representative growth curves for FA19SmR, AK1 (gyrA91/95), and AK2 (gyrA91/95, parC86) (A); KH15 (mtr−79), AK11 (mtr−79, gyrASer91Phe/95), and AK12 (mtr−79, gyrASer91Phe/95, parC86) (B); and KH15, AK11, AK12, and AK13 (gyrALeu91Phe/95, parC86) (C) when cultured separately in GC broth. Mutants AK2 and AK12, but not AK1 and AK11 grew more slowly than the respective parent strains. The in vivo–selected mutant AK13 grew significantly faster than mutants KH15, AK11, and AK12. Mutants were cultured separately in GC broth at a starting absorbance reading at 600 nm (A600) A600 = 0.070–0.075 and the A600 reading at each hour plotted. Experiments were performed 3 times per strain set.
Effect of gyrA91/95 and parC86 Mutations on In Vivo Fitness
We next examined whether the gyrA91/95 or gyrA91/95, parC86 mutants exhibited altered fitness in vivo using a well-characterized female mouse model of lower genital tract infection [39]. Mice were inoculated with mixed suspensions of FA19SmR and AK1 or AK2 bacteria. The relative number of each strain among vaginal isolates was determined over time and normalized to the ratio of mutant and parent gonococci in the inocula. A significantly higher proportion of AK1 bacteria was isolated as infection progressed, with a >10-fold increase in the mean Ic observed on days 5 and 7 pos-inoculation (Figure 2B). In contrast, mutant AK1 showed a slight growth disadvantage when cocultured with FA19SmR in broth (Ic, approximately 3-fold decreased at 3 and 5 hours; 10-fold decreased at 7 hours [stationary phase]) (Figure 2A, closed symbols).
Figure 2.

A gyrA91/95 mutant is more fit than the parent wild-type strain in vivo. Strains FA19SmR, AK1 and AK2 were cocultured in vitro and coinoculated into mice. The competitive index (Ic) for in vitro and in vivo competition experiments was calculated as described in the Methods. Ic values = 1.0 indicate no fitness difference; <1 = reduced fitness; and >1 = increased fitness. (A) Ic values for FA19SmR vs AK1 or AK2 when cocultured in GC broth. B–D show results from competitive murine infection experiments with FA19SmR vs AK1 (B), FA19SmR vs AK2 (C), and AK1 vs AK2 (D). Symbols represent the Ic for individual mice at each time point. AK1 but not AK2 bacteria showed a marked in vivo fitness advantage relative to FA19SmR, and consistent with this result, AK2 was highly attenuated compared with AK1 (D). In B and C, open circles correspond to mice from which only AK1 or AK2 bacteria were recovered. In D, cross hatches correspond to mice from which only AK1 was recovered. The open circle in D corresponds to a mouse from which only ciprofloxacin-resistant colonies were recovered. Results are combined data from 2 independent experiments.
Unlike the CipI gyrA91/95 mutant, the CipR mutant AK2 (gyrA91/95, parC86) exhibited an approximately 2-fold reduced recovery from infected mice relative to FA19SmR on days 1 and 3 postinoculation. A slight increase in the mean Ic occurred over time, with similar numbers of mice exhibiting increased or reduced fitness on days 5 and 7 (Figure 2C). Mutant AK2 also showed reduced fitness when cocultured with strain FA19SmR in vitro (Ic, approximately 5- and 10-fold decreased at 5 and 7 hours, respectively) (Figure 2A, open symbols). Competitive infection experiments with mutants AK1 and AK2 confirmed the in vivo fitness differences of these mutants. Only AK1 bacteria were recovered from a majority of mice on days 3 and 5 postinoculation (Figure 2D, cross hatches). We conclude that gyrA91/95 mutant bacteria have a growth or survival advantage within the murine genital tract. This fitness benefit does not occur during broth culture in vitro. Introduction of the parC86 allele abrogates the in vivo fitness benefit associated with gyrA91/95 alone and significantly slows growth in vitro.
Impact of gyrA and gyrA, parC Mutations on mtr Mutant Gonococci
Quinolone-resistant N. gonorrhoeae frequently carry other resistance mutations [40–42]. We were particularly interested in the consequence of gyrA and parC mutations on the fitness of mtr mutant gonococci because we previously showed that mtr mutations increased gonococcal fitness in the mouse model [18]. We therefore performed competitive infections between strain KH15 (mtrR−79) and either AK11 or AK12, which carry the gyrA91/95 or the gyrA91/95, parC86 mutations, respectively, in the KH15 background. Strains AK11 and KH15 showed no fitness difference when cocultured in vitro (Figure 3A, open symbols). However, when inoculated into mice, significantly more AK11 (gyrA91/95, mtrR−79) bacteria were recovered relative to KH15 within 1 day postinoculation (mean Ic, approximately 40-fold increased on days 1 and 3), and only AK11 bacteria were recovered from several mice over the course of infection (Figure 3B, open circles).
Figure 3.

The mutant gyrA91/95 allele increases the in vivo fitness of an mtr mutant. In vitro and in vivo competition experiments were performed between strains KH15, AK11, and AK12 and competitive index (Ic) values were calculated as described in the legend for Figure 2. A, The Ic values for KH15 vs AK11 or AK12 when cocultured in GC broth. B–D, results from competitive murine infections with KH15 vs AK11(B), KH15 vs AK12 (C), and AK11 vs AK12 (D). AK11 bacteria showed a marked in vivo fitness advantage relative to the mtr mutant parent strain KH15 early in infection. In contrast, AK12 was attenuated in vivo. This result is consistent with the increased fitness of AK11 over AK12 shown in D. In B, open circles correspond to mice from which only AK11 bacteria were recovered. In C, cross hatches refer to mice from which only KH15 bacteria were recovered. In D, cross hatches correspond to mice from which only AK11 colony-forming units were recovered. The open circle on day 5 in D corresponds to the mouse from which equal numbers of bacteria were recovered on GC media with streptomycin (Sm) and GC media with Sm and 2.0 ug/mL ciprofloxacin, as occurs with strain AK12. A ciprofloxacin-resistant isolate from this mouse was colony purified and later characterized as mutant AK13. All results are combined data from 2 independent experiments.
In contrast, in vivo competition of strains KH15 and AK12 demonstrated a clear fitness disadvantage to having mtrR−79, gyrA91/95 and parC86 relative to mtrR−79 alone (Ic, approximately 50-fold decreased on days 3 and 5) (Figure 3C). When cocultured in vitro, AK12 bacteria were approximately 5-fold less fit than the parent strain (Figure 3A, closed symbols). As predicted, strain AK11 out-competed AK12 during mouse infections (Figure 3D). We conclude that the mutant gyrA91/95 allele confers an in vivo fitness advantage to gonococci that already benefit from a mutation that derepresses the mtrCDE operon, but that additional acquisition of parC86 confers a fitness cost to the gonococcus.
In Vivo Selection of a High-Level CipR Mutant With Increased Fitness
In the experiments described above, CipI mutants demonstrated an in vivo fitness advantage, regardless of the mtr genotype, but CipR mutants were compromised. We noted, however, that occasionally only CipR colonies were recovered from mice that were inoculated with CipR (AK2 or AK12) gonococci mixed with either wild-type or CipI strains (Figure 2C [day 5 and 7], 2D [day 3], and 3D, [day 5], open circles). To further investigate this finding, we analyzed CipR bacteria isolated on day 5 from a mouse inoculated with strains AK11 and AK12. Significantly more AK11 (3.3 × 104 CFUs) than AK12 (3.3 × 102 CFUs) were isolated from this mouse on day 1 (Ic, 0.018), followed by isolation of only 8 CipI CFUs on day 3, and high numbers (3.3 × 106 CFUs) of only CipR bacteria on day 5 (Figure 3D, open circle on day 5). One colony from day 5 was propagated further and frozen as mutant AK13. Because both AK11 and AK12 were constructed in the KH15 background, we expected AK13 bacteria to be EmR. However, surprisingly, Em MICs against FA19SmR, AK13, and several other isolates from this mouse were similar (Table 3). Consistent with this finding, the nucleotide sequence of the mtr locus of mutant AK13 was identical to the wild-type sequence found in the parent strain FA19SmR in that the single bp deletion in the mtrR promoter was repaired. The parC gene of mutant AK13 carried the same parC86 mutation found in mutant AK12, but interestingly, the gyrA gene of AK13 was predicted to encode Ser91Leu instead of Ser91Phe due to a single bp change. The in vivo–selected mutant AK13 grew more rapidly than KH15, AK11, and AK12 bacteria in vitro (Figure 1C; Table 3). We conclude that >1 compensatory mutations occurred that allowed CipR gonococci to out-compete CipI bacteria in the murine genital tract.
DISCUSSION
Selection for antibiotic resistance mutations in the face of increasing antibiotic pressure is a commonly observed phenomenon. Resistance mutations often reduce the fitness of bacterial pathogens in the absence of antibiotics [37, 43–46]. Resistance mutations can also confer a fitness benefits. Examples of mutations that increase in vivo fitness include repressor gene mutations that result in overproduction of multidrug active efflux pumps that expel both antibiotics and antimicrobial substances of the host innate defense [15, 18, 27] and topoisomerase mutations in Campylobacter jejuni [16] and E. coli [17], which increased bacterial colonization in chicken and mouse infection models, respectively.
Here we report that gyrA mutations that confer a CipI phenotype to N. gonorrhoeae cause a fitness advantage in a genital tract infection model and that this advantage is abrogated by an additional mutation in parC that increases the Cip MIC to a clinically relevant level. This finding is intriguing because it suggests CipI strains may serve as a reservoir for CipR in N. gonorrhoeae because a single-step mutation in parC is all that is then required for resistance. It is important to note that the mutant strains used here may not accurately reflect all the adaptive mutations that are likely to accumulate in antibiotic-resistant clinical isolates in response to the resistance mutations or other pressures; such mutations may indirectly modify fitness costs to the organism, as proposed by Marcusson et al [17] in studies with highly CipR E. coli. Such a process could also occur with N. gonorrhoeae, especially in environments with constant low levels of antibiotic pressure, as seen with indiscriminate use of fluoroquinolones in clinical practice or where self-medication is common.
We do not know why the gyrA mutant studied here displayed a fitness benefit in vivo. The mechanism for increased fitness of mtr mutant gonococci is likely increased resistance to host innate defenses [18, 22, 23]; whether gyrA91/95 mutations further protect against host defenses or alter a different aspect of gonococcal adaptation to the host is not known. Changes in gene expression due to alterations in supercoiling have been proposed to explain the fitness benefit of topoisomerase mutations in other bacterial pathogens [17, 19]. Because secondary compensatory mutations can reverse the detrimental effects of resistance mutations [14, 20], it is possible that compensatory mutations may have occurred in the gyrA mutants that increase survival or growth of the gonococcus in vivo. With regard to the observed in vivo fitness cost of high level CipR, this result may simply reflect the net balance of the in vivo advantage due to the gyrA91/95 mutation and the more severe growth defect construed by mutation in parC86. It is also possible that the parC86 mutation or the combination of the parC86, gyrA91/95 mutations may have a negative impact on the expression of genes important for survival in vivo. Identification of the mechanism by which gyrA mutations enhance the fitness of N. gonorrhoeae during experimental murine infection might help delineate how parC mutations and/or compensatory mutations balance fitness costs or benefits.
An important question is: can compensatory mutations that influence fitness occur during infection? We believe this to be the case based on the isolation of a highly CipR mutant from a mouse in pure culture in which growth in vitro was restored to wild-type levels. The likely parent strain, AK12, exhibited the most pronounced in vitro fitness cost and carried 2 mutated alleles, gyrA, parC, and an mtr mutation. Loss of the mtrR−79 mutation in strain AK13 may have reversed or contributed to reversing this growth defect, although because the mtrR−79 mutation is known to enhance in vivo fitness in this model [18], one might predict that selection against this mutation would be rare. Other compensatory mutations also may be present and whether the altered mutant GyrA subunit produced by mutant AK13 also impacts fitness is not known. Although the Ser91Phe gyrA mutation is most frequently identified among QRNG strains [8, 47], CipR isolates with the Ser91Leu GyrA substitution have been identified [48]. Other factors not yet defined must increase fluoroquinolone resistance beyond that conferred by gyrA and parC mutations. Quinolone-resistant N. gonorrhoea clinical isolates often have a Cip MIC > 16 ug/mL, as was the case for strain C29 used as the source of the gyrA and parC mutations examined in our study. The consequence of high level CipR on microbial fitness is not known and can be studied in the murine model used herein once the responsible mutations are identified.
In summary, the demonstration that gonococci with commonly isolated gyrA mutations exhibit increased fitness in a genital tract infection model suggests that CipI strains may serve as a reservoir for QRNG. Due to the overabundant use of fluoroquinolones for other disease processes, a low level of antibiotic pressure is likely to contribute to the persistence of these strains. Additionally, we have shown that although gyrA, parC mutants are attenuated in vivo, compensatory mutations can occur that restore fitness while maintaining CipR. Continued investigation of the frequency and nature of compensatory mutations is crucial in understanding the spread of QRNG. Additionally, defining the basis of the fitness advantage observed in gyrA mutants may lead to new insights into survival mechanisms utilized by N. gonorrhoeae during infection.
Notes
Disclaimer. The opinions or assertions contained in this article are the private views of the authors and are not to be construed as reflecting the views of the US Army, Air Force, or the Department of Defense.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases at the National Institute of Health (RO1 AI42053 and U19 AI31496 to A. E. J.; R37 AI21150 to W. M. S.), and a Uniformed Services University of the Health Sciences training grant (C073-RT to A. N. K.). W. M. S. was supported in part by Senior Research Career Scientist and VA Merit awards from the VA Medical Research Service.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Hook EW, Handsfield HH. Gonococcal infections in the adult. In: Holmes KK, Mardh PA, Sparling PF, et al., editors. Sexually transmitted diseases. 3rd ed. 1999. pp. 451–66. New York: McGraw-Hill. [Google Scholar]
- 2.Workowski KA, Berman S Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59:1–110. [PubMed] [Google Scholar]
- 3.Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex Transm Infect. 1999;75:3–17. doi: 10.1136/sti.75.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohnishi M, Golparian D, Shimuta K, et al. Is Neisseria gonorrhoeae initiating a future era of untreatable gonorrhea? Detailed characterization of the first strain with high-level resistance to ceftriaxone. Antimicrob Agents Chemother. 2011;55:3538–45. doi: 10.1128/AAC.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Workowski KA, Berman SM. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55:1–94. [PubMed] [Google Scholar]
- 6.Chen FJ, Lo HJ. Molecular mechanisms of fluoroquinolone resistance. J Microbiol Immunol Infect. 2003;36:1–9. [PubMed] [Google Scholar]
- 7.Tapsall JW. Antibiotic resistance in Neisseria gonorrhoeae. Clin Infect Dis. 2005;41(Suppl 4):S263–8. doi: 10.1086/430787. [DOI] [PubMed] [Google Scholar]
- 8.Trees DL, Sandul AL, Neal SW, Higa H, Knapp JS. Molecular epidemiology of Neisseria gonorrhoeae exhibiting decreased susceptibility and resistance to ciprofloxacin in Hawaii, 1991–1999. Sex Transm Dis. 2001;28:309–14. doi: 10.1097/00007435-200106000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2009. Sexually transmitted disease surveillance 2007 supplement, Gonoccocal Isolate Surveillance Project (GISP) annual report 2007; pp. 1–110. [Google Scholar]
- 10.Belland RJ, Morrison SG, Ison C, Huang WM. Neisseria gonorrhoeae acquires mutations in analogous regions of gyrA and parC in fluoroquinolone-resistant isolates. Mol Microbiol. 1994;14:371–80. doi: 10.1111/j.1365-2958.1994.tb01297.x. [DOI] [PubMed] [Google Scholar]
- 11.Deguchi T, Saito I, Tanaka M, et al. Fluoroquinolone treatment failure in gonorrhea. Emergence of a Neisseria gonorrhoeae strain with enhanced resistance to fluoroquinolones. Sex Transm Dis. 1997;24:247–50. doi: 10.1097/00007435-199705000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka M, Nakayama H, Huruya K, et al. Analysis of mutations within multiple genes associated with resistance in a clinical isolate of Neisseria gonorrhoeae with reduced ceftriaxone susceptibility that shows a multidrug-resistant phenotype. Int J Antimicrob Agents. 2006;27:20–6. doi: 10.1016/j.ijantimicag.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 13.Trees DL, Sandul AL, Peto-Mesola V, et al. Alterations within the quinolone resistance-determining regions of GyrA and ParC of Neisseria gonorrhoeae isolated in the Far East and the United States. Int J Antimicrob Agents. 1999;12:325–32. doi: 10.1016/s0924-8579(99)00081-3. [DOI] [PubMed] [Google Scholar]
- 14.Schulz zur Wiesch P, Engelstadter J, Bonhoeffer S. Compensation of fitness costs and reversibility of antibiotic resistance mutations. Antimicrob Agents Chemother. 2010;54:2085–95. doi: 10.1128/AAC.01460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding Y, Onodera Y, Lee JC, Hooper DC. NorB, an efflux pump in Staphylococcus aureus strain MW2, contributes to bacterial fitness in abscesses. J Bacteriol. 2008;190:7123–9. doi: 10.1128/JB.00655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo N, Pereira S, Sahin O, et al. Enhanced in vivo fitness of fluoroquinolone-resistant Campylobacter jejuni in the absence of antibiotic selection pressure. Proc Natl Acad Sci USA. 2005;102:541–6. doi: 10.1073/pnas.0408966102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcusson LL, Frimodt-Moller N, Hughes D. Interplay in the selection of fluoroquinolone resistance and bacterial fitness. PLoS Pathog. 2009;5:e1000541. doi: 10.1371/journal.ppat.1000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warner DM, Shafer WM, Jerse AE. Clinically relevant mutations that cause derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol Microbiol. 2008;70:462–78. doi: 10.1111/j.1365-2958.2008.06424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Q, Sahin O, McDermott PF, Payot S. Fitness of antimicrobial-resistant Campylobacter and Salmonella. Microbes Infect. 2006;8:1972–8. doi: 10.1016/j.micinf.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 20.Andersson DI. The biological cost of mutational antibiotic resistance: any practical conclusions? Curr Opin Microbiol. 2006;9:461–5. doi: 10.1016/j.mib.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Hagman KE, Pan W, Spratt BG, Balthazar JT, Judd RC, Shafer WM. Resistance of Neisseria gonorrhoeae to antimicrobial hydrophobic agents is modulated by the mtrRCDE efflux system. Microbiology. 1995;141(Pt 3):611–22. doi: 10.1099/13500872-141-3-611. [DOI] [PubMed] [Google Scholar]
- 22.Jerse AE, Sharma ND, Simms AN, Crow ET, Snyder LA, Shafer WM. A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect Immun. 2003;71:5576–82. doi: 10.1128/IAI.71.10.5576-5582.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shafer WM, Qu X, Waring AJ, Lehrer RI. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc Natl Acad Sci USA. 1998;95:1829–33. doi: 10.1073/pnas.95.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagman KE, Shafer WM. Transcriptional control of the mtr efflux system of Neisseria gonorrhoeae. J Bacteriol. 1995;177:4162–5. doi: 10.1128/jb.177.14.4162-4165.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giles JA, Falconio J, Yuenger JD, Zenilman JM, Dan M, Bash MC. Quinolone resistance–determining region mutations and por type of Neisseria gonorrhoeae isolates: resistance surveillance and typing by molecular methodologies. J Infect Dis. 2004;189:2085–93. doi: 10.1086/386312. [DOI] [PubMed] [Google Scholar]
- 26.Gunn JS, Stein DC. Use of a non-selective transformation technique to construct a multiply restriction/modification-deficient mutant of Neisseria gonorrhoeae. Mol Gen Genet. 1996;251:509–17. doi: 10.1007/BF02173639. [DOI] [PubMed] [Google Scholar]
- 27.Warner DM, Folster JP, Shafer WM, Jerse AE. Regulation of the MtrC-MtrD-MtrE efflux-pump system modulates the in vivo fitness of Neisseria gonorrhoeae. J Infect Dis. 2007;196:1804–12. doi: 10.1086/522964. [DOI] [PubMed] [Google Scholar]
- 28.Jerse AE. Experimental gonococcal genital tract infection and opacity protein expression in estradiol-treated mice. Infect Immun. 1999;67:5699–708. doi: 10.1128/iai.67.11.5699-5708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Centers for Disease Control and Prevention. Agar dilution antimicrobial susceptibility testing. http://www.cdc.gov/STD/Gonorrhea/lab/agar.htm. Accessed June 1, 2009. [Google Scholar]
- 30.Knapp JS, Hale JA, Neal SW, Wintersheid K, Rice RJ, Whittington WL. Proposed criteria for interpretation of susceptibilities of strains of Neisseria gonorrhoeae to ciprofloxacin, ofloxacin, enoxacin, lomefloxacin, and norfloxacin. Antimicrob Agents Chemother. 1995;39:2442–5. doi: 10.1128/aac.39.11.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H, Jerse AE. Alpha-2,3-sialyltransferase enhances Neisseria gonorrhoeae survival during experimental murine genital tract infection. Infect Immun. 2006;74:4094–103. doi: 10.1128/IAI.00433-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song W, Condron S, Mocca BT, et al. Local and humoral immune responses against primary and repeat Neisseria gonorrhoeae genital tract infections of 17β-estradiol-treated mice. Vaccine. 2008;26:5741–51. doi: 10.1016/j.vaccine.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balsalobre L, Ferrandiz MJ, Linares J, Tubau F, de la Campa AG. Viridans group streptococci are donors in horizontal transfer of topoisomerase IV genes to Streptococcus pneumoniae. Antimicrob Agents Chemother. 2003;47:2072–81. doi: 10.1128/AAC.47.7.2072-2081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komp Lindgren P, Marcusson LL, Sandvang D, Frimodt-Moller N, Hughes D. Biological cost of single and multiple norfloxacin resistance mutations in Escherichia coli implicated in urinary tract infections. Antimicrob Agents Chemother. 2005;49:2343–51. doi: 10.1128/AAC.49.6.2343-2351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Regan E, Quinn T, Frye JG, et al. Fitness costs and stability of a high-level ciprofloxacin resistance phenotype in Salmonella enterica serotype enteritidis: reduced infectivity associated with decreased expression of Salmonella pathogenicity island 1 genes. Antimicrob Agents Chemother. 2010;54:367–74. doi: 10.1128/AAC.00801-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pope CF, Gillespie SH, Pratten JR, McHugh TD. Fluoroquinolone-resistant mutants of Burkholderia cepacia. Antimicrob Agents Chemother. 2008;52:1201–3. doi: 10.1128/AAC.00799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rozen DE, McGee L, Levin BR, Klugman KP. Fitness costs of fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother. 2007;51:412–6. doi: 10.1128/AAC.01161-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vickers AA, O'Neill AJ, Chopra I. Emergence and maintenance of resistance to fluoroquinolones and coumarins in Staphylococcus aureus: predictions from in vitro studies. J Antimicrob Chemother. 2007;60:269–73. doi: 10.1093/jac/dkm191. [DOI] [PubMed] [Google Scholar]
- 39.Jerse AE, Wu H, Packiam M, Vonck RA, Begum AA, Garvin LE. Estradiol-treated female mice as surrogate hosts for Neisseria gonorrhoeae genital tract infections. Front Microbiol. 2011;2:107. doi: 10.3389/fmicb.2011.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen VG, Farrell DJ, Rebbapragada A, et al. Molecular analysis of antimicrobial resistance mechanisms in Neisseria gonorrhoeae isolates from Ontario, Canada. Antimicrob Agents Chemother. 2011;55:703–12. doi: 10.1128/AAC.00788-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dewi BE, Akira S, Hayashi H, Ba-Thein W. High occurrence of simultaneous mutations in target enzymes and MtrRCDE efflux system in quinolone-resistant Neisseria gonorrhoeae. Sex Transm Dis. 2004;31:353–9. doi: 10.1097/00007435-200406000-00007. [DOI] [PubMed] [Google Scholar]
- 42.Vorobieva V, Firsova N, Ababkova T, et al. Antibiotic susceptibility of Neisseria gonorrhoeae in Arkhangelsk, Russia. Sex Transm Infect. 2007;83:133–5. doi: 10.1136/sti.2006.021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bjorkholm B, Sjolund M, Falk PG, Berg OG, Engstrand L, Andersson DI. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc Natl Acad Sci USA. 2001;98:14607–12. doi: 10.1073/pnas.241517298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Macvanin M, Bjorkman J, Eriksson S, Rhen M, Andersson DI, Hughes D. Fusidic acid-resistant mutants of Salmonella enterica serovar Typhimurium with low fitness in vivo are defective in RpoS induction. Antimicrob Agents Chemother. 2003;47:3743–9. doi: 10.1128/AAC.47.12.3743-3749.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagaev I, Bjorkman J, Andersson DI, Hughes D. Biological cost and compensatory evolution in fusidic acid-resistant Staphylococcus aureus. Mol Microbiol. 2001;40:433–9. doi: 10.1046/j.1365-2958.2001.02389.x. [DOI] [PubMed] [Google Scholar]
- 46.Trzcinski K, Thompson CM, Gilbey AM, Dowson CG, Lipsitch M. Incremental increase in fitness cost with increased beta-lactam resistance in pneumococci evaluated by competition in an infant rat nasal colonization model. J Infect Dis. 2006;193:1296–303. doi: 10.1086/501367. [DOI] [PubMed] [Google Scholar]
- 47.Yang Y, Liao M, Gu WM, et al. Antimicrobial susceptibility and molecular determinants of quinolone resistance in Neisseria gonorrhoeae isolates from Shanghai. J Antimicrob Chemother. 2006;58:868–72. doi: 10.1093/jac/dkl301. [DOI] [PubMed] [Google Scholar]
- 48.Ruiz J, Jurado A, Garcia-Mendez E, et al. Frequency of selection of fluoroquinolone-resistant mutants of Neisseria gonorrhoeae exposed to gemifloxacin and four other quinolones. J Antimicrob Chemother. 2001;48:545–8. doi: 10.1093/jac/48.4.545. [DOI] [PubMed] [Google Scholar]