

Abstract
Uropathogenic Escherichia coli (UPEC) are the chief cause of urinary tract infections. Although neutrophilic inflammation is a hallmark of disease, previous data indicate that UPEC promotes local dampening of host innate immune responses. Here, we show that UPEC attenuates innate responses to epithelial infection by inducing expression of indoleamine 2,3-dioxygenase (IDO), a host enzyme with previously defined roles in adaptive immune regulation. UPEC induced IDO expression in human uroepithelial cells and polymorphonuclear leukocytes (PMN) in vitro and in bladder tissue during murine cystitis via a noncanonical, interferon-independent pathway. In the bladders of UPEC-infected IDO-deficient mice, we observed augmented expression of proinflammatory cytokines and local inflammation, correlated with reduced survival of extracellular bacteria. Pharmacologic inhibition of IDO also increased human PMN transepithelial migration. Stimulation of IDO expression therefore represents a pathogen strategy to create local immune privilege at epithelial surfaces, attenuating innate responses to promote colonization and the establishment of infection.
A significant component of mammalian evolution has been the development of means to detect and respond to microbial intrusion. Host–pathogen interactions are characterized by competition for essential nutrients as well as a balance between pro- and anti-inflammatory responses. Thus, ancient metabolic enzymes have often gained novel immunity functions beyond nutrient acquisition. The innate immune system is the first line of microbial defense, and polymorphonuclear leukocytes (PMN; neutrophils) are considered first responders. Beyond killing invading microbes, PMN mediate inflammatory signaling by coordinating cues from the site of infection with the recruitment of additional immune cells. Illuminating the balance between pro- and anti-inflammatory signaling is critical to understanding the host–pathogen interaction, and manipulation of this equilibrium by pathogens is essential for productive infection.
The pathogenic cascade of events in Escherichia coli urinary tract infection (UTI), characterized in a murine cystitis model and corroborated by evidence from human samples [1, 2], represents a well-defined paradigm for bacterial epithelial infection. Following attachment to and invasion of superficial bladder epithelial cells [3, 4], bacteria replicate in the host cell cytosol to form intracellular bacterial communities [1, 5]. Bacteria emerge from these cells to infect naive superficial cells, or they can establish quiescent intracellular reservoirs in underlying cells, thought to seed recurrent infections [4, 6]. Despite the observed neutrophil influx and proinflammatory cytokine response during the course of uropathogenic Escherichia coli (UPEC) infection [7, 8], uropathogens appear to resist neutrophil killing and phagocytosis in vivo [1, 9]. Furthermore, recent studies have demonstrated that UPEC actively suppress early inflammatory responses [10–12] and neutrophil function [13]. Insight into the mechanism of differential host responses to pathogenic versus commensal E. coli was gained by transcriptional profiling. A number of potentially anti-inflammatory molecules were relatively induced in PMN and epithelial cells after exposure to UPEC, including IDO1, encoding indoleamine 2,3-dioxygenase (IDO) [13].
IDO catalyzes the initial, rate-limiting step in tryptophan degradation along the kynurenine pathway. Via local depletion of tryptophan and the production of bioactive kynurenines, IDO induction has varied and complex biological roles and is considered an important regulator of adaptive immunity, with functions in pregnancy, autoimmunity, transplantation, and neoplasia [14]. Although widely expressed at basal levels in a variety of tissues and immune cells, IDO is considered an inducible enzyme, responding primarily to interferon α (IFN-α), interferon β (IFN-β), and interferon γ (IFN-γ) as part of a proinflammatory host response [15]. By altering lymphocyte proliferation and survival, IDO induction limits the pathologic consequences of excessive inflammatory and cell-mediated reactions [14]. In the context of infectious diseases, IDO was initially recognized for its role in tryptophan starvation of intracellular pathogens such as Toxoplasma and Chlamydia [16, 17]. Recent data suggest the existence of other circumstances in which IDO may influence host defense against bacteria, viruses, fungi, and parasites, although no unifying hypothesis explains the mechanism underlying these observations [18]. Even less well understood is the ability of some pathogens to exploit the immunomodulatory effects of IDO for pathogenic benefit. Induction of IDO in T cells and dendritic cells by Mycobacterium leprae [19], Leishmania major [20], and human immunodeficiency virus [21] has been shown to benefit these pathogens. Investigation of in vitro Candida–host interactions implicated a complex role for IDO in control of fungal morphology and adaptive antifungal responses involving both PMN and dendritic cells [22]. These examples demonstrate an underappreciated role for IDO activation in microbial pathogenesis; given our previous data, we therefore hypothesized that UPEC upregulates IDO during infection to create local immune privilege in the urinary tract. Consistent with this hypothesis, we show that UPEC infection induces local IDO expression that inhibits proinflammatory humoral and cellular innate responses and promotes bacterial colonization of epithelial tissues. With this new role in innate defense, we propose that IDO represents a critical regulator of early host–pathogen crosstalk at the epithelial interface.
METHODS
Animals
Breeding pairs of B6.129-Ido1tm1Alm/J (Ido1−/−), B6.129S7-Ifngtm1Ts/J (Ifnγ−/−), and C57BL/6J (B6) mice were obtained from Jackson Laboratory. B6 mice with a null mutation in the Ifnar1 gene (IfnαβR−/−) [23] were a gift from Dr Anthony French (Washington University, St. Louis, MO). Results from experiments in which age-matched control mice were purchased directly from the commercial vendor were similar to those in which control mice were bred in the same Washington University facility under the same conditions as mutant mice and are included in aggregate data.
Bacterial Strains and Culture
UPEC strain UTI89 was obtained from a patient with cystitis [24]. MG1655 is a type 1 piliated K-12 laboratory strain [25]. Previously characterized [13] fecal isolates of E. coli from healthy children (gift from P. Tarr; denoted FI-10, FI-11, and FI-12) were also used for comparison. A derivative of UTI89 with the tryptophan biosynthetic operon trpABCDE replaced by a chloramphenicol-resistance cassette (Trp–) was constructed [26] using pKD46 and primers JLP221 and JLP223 (Integrated DNA Technologies) listed in Table 1. Bacteria were grown overnight in standing Luria-Bertani (LB) broth at 37°C, with chloramphenicol (20 µg/mL) when appropriate.
Table 1.
Primers Used in This Study
| Name | Sequence | Description |
|---|---|---|
| JLP121 | GCTCCTCCTGTTCGACAGTCA | Real-time PCR for (HUMAN) GAPDH |
| JLP122 | ACCAGGCGCCCAATACG | |
| JLP209 | GCCCTTCAAGTGTTTCACCAA | Real-time PCR for (HUMAN) IDO1 |
| JLP210 | GACAAATATATGCGAAGAACACTGAAAA | |
| JLP344 | CTCGTCCCGTAGACAAAATGG | Real-time PCR for (MOUSE) Gapdh |
| JLP345 | TGACCAGGCGCCCAATA | |
| JLP329 | AAGGGCTTCTTCCTCGTCTC | Real-time PCR for (MOUSE) Ido1 |
| JLP330 | AAAAACGTGTCTGGGTCCAC | |
| JLP221 | CAAACACAAAAACCGACTCTCGAACTGCT AACCTGCAAAGGTGTAGGCTGGAGCTGCTTC | Construction of UTI89 trpABCDE::cmR |
| JLP223 | ACTGCGCGTCGCCGCTTTCATCGGTTGTACAAAA GCTTTCCATATGAATATCCTCCTTAG | |
| JLP13 | CCAGGGCTACACACGTGCTA | Real-time PCR for rrsA |
| JLP14 | TCTCGCGAGGTCGCTTCT | |
| JLP211 | GCCGCGTTGCGTCATAAT | Real-time PCR for trpA |
| JLP212 | GCAAATCATCGTCGGCATT | |
| JLP213 | GCATGTACGTGCCACAAATCC | Real-time PCR for trpB |
| JLP214 | CGCGCTGACAAAAGCTTCTT | |
| JLP215 | AAAGGCGTGATCCGTGATG | Real-time PCR for trpC |
| JLP216 | GCCGAAGCATAATTTCTGTAAACG | |
| JLP217 | CGTCGCGGCGAATGTC | Real-time PCR for trpD |
| JLP218 | GCATTGGCTTGCAGATCTTCA |
Abbreviation: PCR, polymerase chain reaction.
Murine Cystitis Model
The murine cystitis model has been described in detail elsewhere [4, 27]. All animal studies were performed with approval of the Animal Studies Committee at Washington University School of Medicine. Differences in tissue colonization of wild-type and Ido1−/− mice were examined for significance using the Mann–Whitney U test.
RNA Isolation and Gene Expression Analysis
RNA isolation from encounters of bacteria with human PMN or 5637 bladder epithelial cells has been described elsewhere [13]. Whole bladders from mock- or UPEC-infected mice were homogenized with an FP120 FastPrep reciprocal shaking device in tubes containing lysis buffer (RLT, Qiagen) and a commercially available extraction reagent, lysing matrix B (MP Biomedicals). Total RNA was isolated from this lysate as described elsewhere [13]. First-strand complementary DNA (cDNA) synthesis and real-time polymerase chain reaction (RT-PCR) were performed as described elsewhere [13]. Transcript abundance was normalized to that of an endogenous control gene, Gapdh. Relative target expression was calculated according to the Δ(ΔCt) method as described elsewhere [28], where fold-change in expression is equal to 2−Δ(ΔCt). Data are presented as fold-change in transcript abundance in the experimental condition relative to the mock-infected calibrator condition and represent the mean and standard deviation of triplicate assays, each performed with RNA prepared from at least 5 mice. For analysis of bacterial gene expression, calibrator RNA was isolated from the inoculum or media-only condition and cDNA synthesized as described above. The endogenous control gene was rrsA. Primer sequences are listed in Table 1 and were designed using Primer Express software (Applied Biosystems). Differences in gene expression were examined for significance using an unpaired t test.
Tissue Cytokine Analysis
Cytokine concentrations in whole bladders from mice infected with UTI89 or mock-infected animals were analyzed by a multiplex bead array platform (BioPlex, Bio-Rad, Hercules, CA) as described elsewhere [8]. Data represent the mean and standard deviation of bladders from 4 individual mice per time point, each analyzed in duplicate. Differences between samples from Ido1−/− and wild-type mice were examined for significance using the Mann–Whitney U test.
Myeloperoxidase Activity and Quantification of Urine Neutrophils
Wild-type and Ido1−/− mice were inoculated transurethrally, sacrificed at the indicated time points, and bladders were homogenized. Fifty-microliter aliquots of undiluted homogenates were assayed for myeloperoxidase (MPO) activity (Cell Technology) by fluorescent detection of an MPO substrate as described elsewhere [13]. Samples from at least 3 independent experiments with groups of at least 4 mice were analyzed in triplicate.
To quantify urine neutrophils, wild-type and Ido1−/− mice were inoculated as described above, and urines were collected into sterile microcentrifuge tubes at 2, 6, and 24 hours post infection. Leukocytes (>95% neutrophils by staining) in urine were quantified using a hemacytometer. Samples from 3 independent experiments with groups of 3 mock-infected and at least 5 UTI89-infected mice were analyzed. Differences between mouse strains were examined for significance using the Mann–Whitney U test.
Microscopic Analysis of Intracellular Bacterial Community Formation
Bladders of wild-type and Ido1−/− mice were examined at 16 hours post infection with green fluorescent protein (GFP)–expressing UTI89 as described elsewhere [29]. Intracellular bacterial communities (IBCs) were enumerated by X-gal staining of bladders from wild-type and Ido1−/− mice harvested at 6 hours post infection with UTI89 as described elsewhere [29]. Numbers of IBCs formed were compared using the Mann–Whitney U test.
Ex Vivo Gentamicin Protection Assay
Quantification of extracellular and intracellular bacteria in murine bladder was performed by a modified ex vivo gentamicin protection assay as described elsewhere [30]. Aggregate data from 3 independent experiments with groups of 4 mice are shown, and statistically significant differences were determined by the Mann–Whitney U test.
Inhibition of Indoleamine 2,3-dioxygenase in Human Neutrophils
In accordance with a protocol approved by the Washington University Human Research Protection Office, PMN were isolated from venous blood of healthy volunteers and PMN transepithelial migration assayed as described elsewhere [13]. The influence of IDO in this model was assessed by quantifying transepithelial migration in the presence or absence of 10 µM 1-methyl-d/l-tryptophan (1-MT; Sigma-Aldrich), a specific inhibitor [31]. The migration assay included treatment groups where PMN, or 5637 cells, or both were pretreated for 60 minutes with 1-MT. For experiments where only PMN or 5637 cells were treated with 1-MT, inhibitor was not added to media in the lower or upper chamber during experimental incubation, respectively. The results represent triplicate assays from at least 3 independent experiments with different PMN donors, and statistically significant differences were assessed by an unpaired t test.
Statistical Analysis
Statistical tests for significance were performed using GraphPad Prism software version 4.00 for Windows (San Diego, CA).
RESULTS
UPEC Induces Host IDO Expression by an Interferon-Independent Mechanism
We previously reported pathogen-specific induction of IDO in human neutrophils as demonstrated by global transcriptional profiling [13] and first aimed to extend these data to include another UTI-relevant cell type, human bladder epithelial cells. We queried IDO1 expression in human PMN or 5637 bladder epithelial cells exposed to UTI89, a model UPEC strain; MG1655, a nonpathogenic E. coli strain; or several fecal E. coli isolates, relative to a media-only control by RT-PCR. Consistent with our published data, IDO1 was induced 3-fold in PMN exposed to UTI89. Induction in infected uroepithelial cells was even more profound, with a 35-fold increase relative to mock-treated cells (Figure 1A). In contrast, exposure to commensal E. coli resulted in reduced IDO1 expression, yielding a relative upregulation in UPEC-exposed PMN and 5637 cells of approximately 8-fold and 100-fold, respectively (see Figure 1A: compare UTI89 to MG1655, FI-10, FI-11, FI-12).
Figure 1.
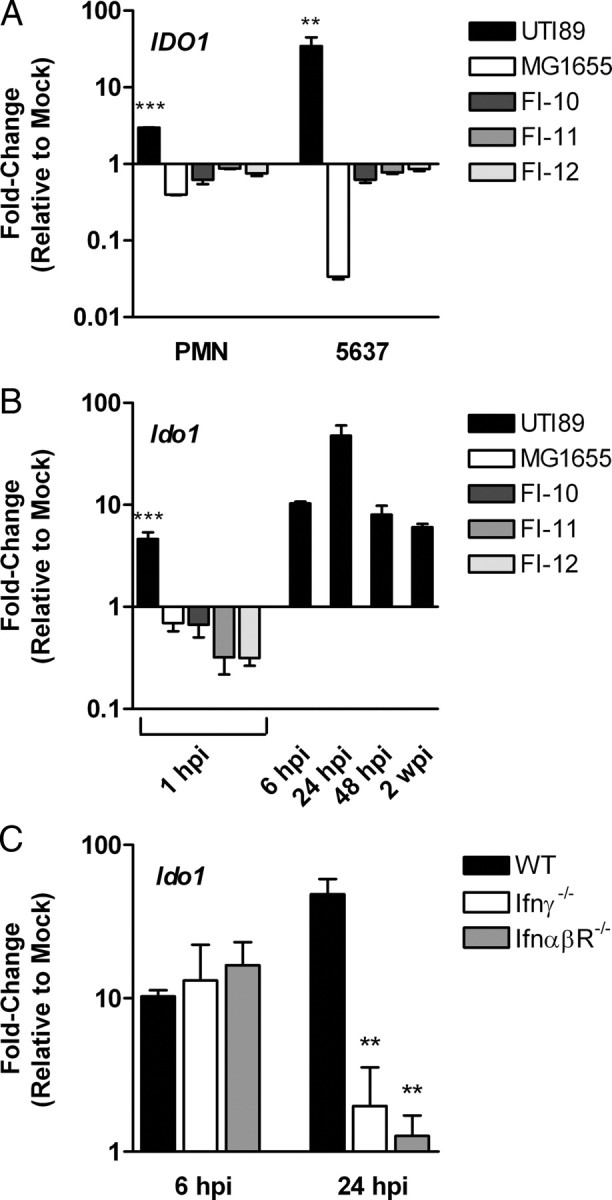
Uropathogenic Escherichia coli (UPEC) induces host expression of indoleamine 2,3-dioxygenase (IDO) by an interferon-independent mechanism. Expression of genes encoding IDO was determined by quantitative real-time polymerase chain reaction. A, Gene expression in 5637 human bladder epithelial cells or human PMN following exposure to UPEC strain UTI89 or nonpathogenic E. coli strains MG1655, FI-10, FI-11, and FI-12, relative to mock-infected cells. Bars represent the mean and standard deviation of at least 3 independent experiments with samples analyzed in triplicate. A significant difference in UTI89-infected PMN (***P < .001) and 5637 cells (**P < .01) compared to the nonpathogenic strains is indicated. B, Gene expression in whole bladders from wild-type mice infected with UTI89 or nonpathogenic strains at the indicated time points (hpi, hours post infection; wpi, weeks post infection), relative to mock-infected mice. Gene expression in UTI89-infected mice was significantly different from mock-infected mice at each time point (P < .01), and a significant difference in response to UTI89 compared to the nonpathogenic strains at 1 hpi is indicated (***P < .001). C, Gene expression in whole bladders from wild-type (WT) or interferon signaling–deficient mice infected with UTI89 at the indicated time points relative to mock-infected mice of the same strain. A significant difference in expression compared to infected wild-type mice at the same time point is indicated (**P < .01). For in vivo gene expression (panels B and C), bars represent the mean and standard deviation of groups of at least 5 mice from at least 2 independent experiments analyzed in triplicate.
To test whether UPEC infection induced IDO expression in vivo, we infected B6 mice with UTI89 or commensal E. coli and measured Ido1 transcript in whole bladder homogenates for comparison to samples from mock-infected control mice. At each of the studied time points, Ido1 was upregulated in UPEC-infected mice, with peak induction at 24 hours post infection (see Figure 1B, UTI89). Ido1 transcript was unchanged in the kidneys of infected mice (data not shown). Because commensal strains are rapidly cleared from bladders of normal mice, only a 1-hour-postinfection sample was collected from these inoculations. Although commensal burdens at 1 hour post infection were similar to that observed after UPEC infection (data not shown), nonpathogenic strains did not elicit Ido1 expression, indicating pathogen-specific induction in the bladder consistent with our in vitro analysis (see Figure 1B: MG1655, FI-10, FI-11, FI-12).
Because IDO expression is inducible by Type I and II interferons [15], we hypothesized that signaling through IFN-α, IFN-β, and IFN-γ pathways would be required for UPEC induction of Ido1. We infected B6 mice lacking IFN-α/β receptor or IFN-γ with UTI89 and measured Ido1 transcript in whole bladders harvested 6 and 24 hours post infection relative to samples from mock-infected knock-out mice. Relative Ido1 expression in both knock-out mouse strains was similar to that of wild-type mice at 6 hours post infection; however, at 24 hours post infection, transcript levels in the knock-out mice were sharply reduced (Figure 1C). Taken together, these data indicate that interferon signaling is not essential for initial induction of Ido1 by UPEC but may be required to sustain IDO expression throughout the course of infection.
IDO Attenuates Proinflammatory Cytokine Signaling and Inflammatory Responses During Epithelial Infection
To evaluate the effects of IDO induction on local host immune responses to epithelial infection, we measured cytokines in bladder tissue from UPEC- and mock-infected wild-type and Ido1−/− mice by multiplex bead array analysis. No significant increase in IFN-γ was detected in infected wild-type mice, replicating prior findings [8] and consistent with our above observation that IFN-γ is not required for initial Ido1 induction by UPEC. The IFN-γ requirement for Ido1 expression at 24 hours post infection (Figure 1C) may be fulfilled by basal levels; alternatively, increases in IFN-γ in microenvironments within the infected bladder may not be detected in whole-bladder homogenates. Baseline expression of other cytokines was equivalent in wild-type and Ido1−/− mice, and cytokine levels were also similar in both mouse strains at 1 and 24 hours post infection (Figure 2). In contrast, UPEC infection elicited significantly larger increases in proinflammatory cytokine production in the absence of IDO at 6 hours post infection, including several that are important for PMN recruitment and longevity, such as KC (CXCL1), granulocyte colony-stimulating factor, macrophage inflammatory protein-1, interleukin 17, and interleukin 6 (Figure 2).
Figure 2.

Indoleamine 2,3-dioxygenase (IDO) modulates cytokine signaling during epithelial infection. Wild-type (WT; solid bars) or Ido1−/− (open bars) mice were infected with uropathogenic Escherichia coli (UPEC) strain UTI89 or mock infected with phosphate-buffered saline (PBS). Bladders were homogenized at 1, 6, or 24 hours post infection (hpi) and centrifuged to remove cellular debris before cytokine expression was measured. Bars represent the mean and standard deviation from 4 individual mice per time point with samples assayed in duplicate. Statistically significant differences in samples from Ido1−/− mice compared to those from wild-type mice at the same time point are indicated (*P < .05).
Given the increases in proinflammatory cytokines in samples from infected Ido1−/− mice, we predicted increased local inflammation in the bladders of these animals. We quantified recruitment of PMN to the site of infection by measuring MPO activity in bladder homogenates from wild-type and Ido1−/− mice infected with UTI89 and mock-infected control mice. This enzyme resides specifically in granules of PMN where it oxidizes chloride to generate potent antimicrobial compounds [32]. Although there was no difference in baseline MPO activity, Ido1−/− mice demonstrated significantly more MPO activity than wild-type mice in bladder samples taken at 1, 6, 24, and 48 hours post infection (Figure 3A). Urine PMN counts corroborated these findings; we found significantly more neutrophils in urine samples collected from UTI89-infected mice lacking Ido1 (Figure 3B). Thus, induction of IDO by UPEC acts to reduce bladder inflammation during acute UTIs.
Figure 3.

Indoleamine 2,3-dioxygenase (IDO) reduces bladder inflammation in murine cystitis. Wild-type (WT) or Ido1−/− mice were infected with uropathogenic Escherichia coli (UPEC) strain UTI89 or mock infected with phosphate-buffered saline (PBS). A, Bladders were homogenized at the indicated time points, centrifuged to remove cellular debris, and myeloperoxidase (MPO) activity measured. Data points represent the mean of triplicate measurements from individual bladders, and statistically significant differences from wild-type are indicated (***P < .001 and **P < .01). B, Urine specimens were obtained at the indicated time points and the concentration of white blood cells quantified. Bars represent the mean and standard deviation from groups of at least 5 mice, and statistically significant differences between wild-type and Ido1−/− mice are indicated (*P < .05).
IDO Promotes UPEC Infection and Protects Extracellular Bacteria in the Urinary Tract
We next used the murine model of cystitis to evaluate the consequences of IDO induction on UPEC pathogenesis. The ability of UTI89 to colonize the bladders of Ido1−/− mice was significantly attenuated at 24 and 48 hours post infection compared to wild-type mice (Figure 4A). There was no difference in bacterial burden in the kidneys (data not shown). The observations of increased PMN infiltrate and decreased bacterial colonization in the absence of IDO, at time points when bacteria are known to have increased exposure to PMN [1], suggested that extracellular bacteria may be more susceptible to killing under these circumstances. Consistent with this hypothesis, the number of bacteria present was similar in the intracellular (gentamicin-protected) compartment of bladders in wild-type and Ido1−/− mice at 24 hours post infection but significantly reduced in the extracellular (gentamicin-sensitive) compartment in Ido1−/− mice (Figure 4B). Further evidence that progress along the intracellular pathway of UPEC pathogenesis is not dependent on IDO emerged from microscopic analysis of bladders from wild-type or Ido1−/− mice infected with GFP-expressing UPEC. We observed no difference in the quantity of intracellular bacterial communities formed or in their morphology (Figure 4C and 4D), despite the reduction in total colony-forming units (CFUs) recovered. Taken together, these data suggest that IDO induction promotes survival of extracellular bacteria by reducing early inflammatory responses in the bladder, and that the intracellular niche shelters bacteria from PMN activity.
Figure 4.

Indoleamine 2,3-dioxygenase (IDO) promotes uropathogenic Escherichia coli (UPEC) colonization of bladder in murine cystitis and survival of extracellular bacteria. A, Wild-type (WT) or Ido1−/− mice were infected with UPEC strain UTI89. Bladders were homogenized at the indicated time point, serially diluted, and colony-forming units (CFUs) enumerated. Each point represents 1 mouse; bars are geometric mean. Graph is a composite of 3 or more independent experiments with at least 4 mice in each group. A statistically significant difference between wild-type and Ido1−/− mice is indicated (***P < .001 and **P < .01). B, Bacteria present in the extracellular (luminal) and intracellular compartments of bladders from wild-type or Ido1−/− mice were enumerated at 24 hours post infection (hpi) with UPEC strain UTI89. Aggregate data from 3 independent experiments with 4 mice per group are shown, and bars represent the geometric mean. A statistically significant difference is indicated (**P < .01). C, The morphologies characteristic of UPEC infection (including intracellular bacterial communities and filamentous forms) were observed at 16 hpi of both wild-type and Ido1−/− mice with green fluorescent protein–expressing UTI89. Representative images are shown. D, Intracellular bacterial communities (IBCs) present in bisected bladders from wild-type or Ido1−/− mice infected with UPEC strain UTI89 at 6 hpi were stained with X-gal and enumerated by microscopy. Data from 2 independent experiments with 5 mice per group are shown, and bars represent the geometric mean. There was no statistically significant difference between the 2 mouse strains.
Previous studies demonstrate the capacity of IDO to curtail the growth of some pathogens within the host by limiting tryptophan availability. As with other E. coli, the UPEC genome encodes the machinery for de novo tryptophan synthesis [33]. Transcription of genes in this operon was upregulated during in vitro encounters with human PMN and bladder epithelial cells (Figure 5A) and in infected murine bladders in an Ido1-dependent manner (Figure 5B). Furthermore, a UPEC strain in which the trp locus was replaced with a selectable marker grew poorly in tryptophan-deficient medium, whereas growth was similar to the wild-type parent strain when medium was supplemented with tryptophan (data not shown). Despite this in vitro defect, the UPEC trp mutant was not substantially attenuated in the murine cystitis model (Figure 5C), suggesting that IDO induction by UPEC does not impose significant tryptophan limitation on UPEC in the intracellular or extracellular niches.
Figure 5.
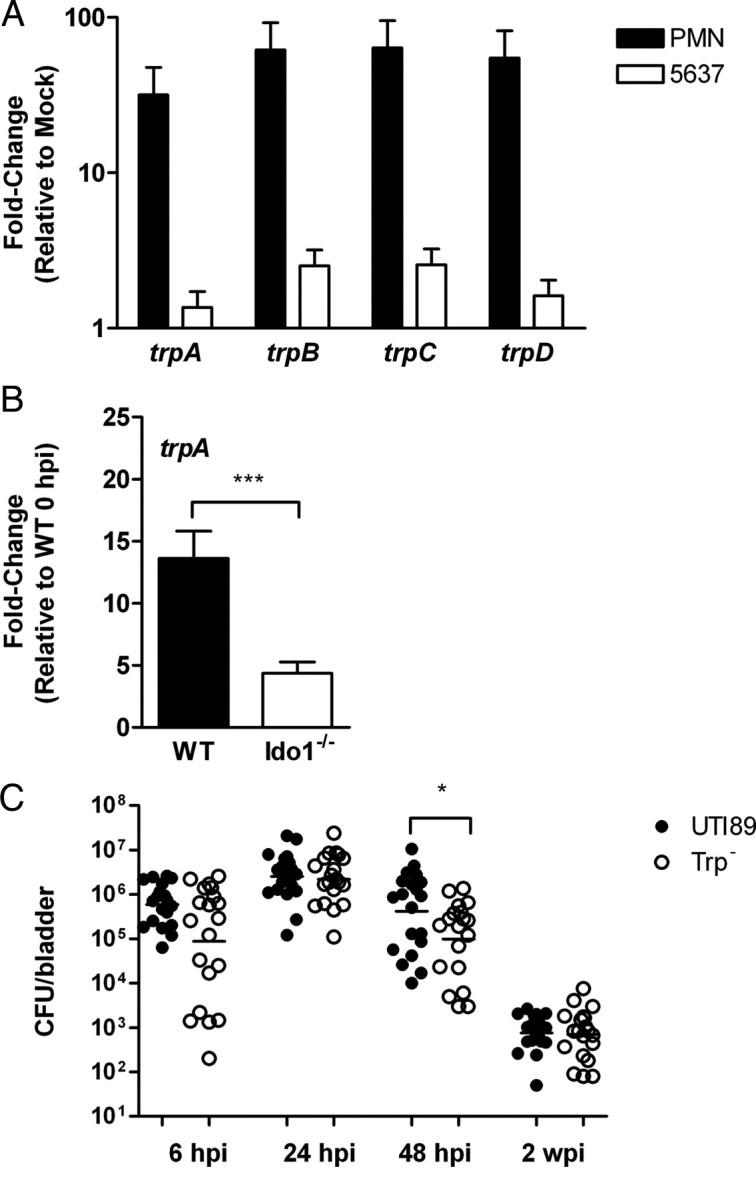
Uropathogenic Escherichia coli (UPEC) tryptophan biosynthesis is upregulated during host interaction but not required for bladder colonization in murine cystitis. Expression of genes in the trp operon was determined by quantitative real-time polymerase chain reaction (PCR). A, Gene expression in UTI89 following a 60-minute in vitro encounter with 5637 human bladder epithelial cells or human polymorphonuclear leukocytes (PMN) is shown relative to a media-only control condition. Bars represent the mean and standard deviation of 3 independent experiments with samples analyzed in triplicate. Expression of each gene in the presence of host cells was significantly different from the control condition (PMN; P < .01 and 5637; P < .05). B, Bacterial trpA gene expression in whole bladders from wild-type (WT) or Ido1−/− mice infected with UTI89 at 24 hours post infection (hpi) is shown relative to a sample prepared immediately following inoculation. Bars represent the mean and standard deviation of 5 mice from 2 independent experiments analyzed in triplicate, and a significant difference is indicated (***P < .001). C, Wild-type mice were infected with UPEC strain UTI89 or an isogenic strain mutated in the trpABCDE operon (Trp–). Bladders were homogenized at the indicated time point, serially diluted, and colony-forming units (CFUs) enumerated. Each point represents 1 mouse; bars are geometric mean. Graph is a composite of 3 or more independent experiments with at least 4 mice in each group. A statistically significant difference from UTI89 is indicated (*P < .05).
IDO Activity Suppresses Human Neutrophil Transepithelial Migration
To establish a role for IDO activity in the acute inflammatory response of human cells to UPEC infection, we characterized the effect of IDO inhibition on in vitro transepithelial migration of human PMN across monolayers of 5637 bladder epithelial cells. We previously showed that UPEC actively suppresses PMN migration in this model, impairing host cells’ responses to inflammatory stimuli through an incompletely defined mechanism dependent on bacterial protein synthesis [13]. Treatment of PMN and epithelial cells with a specific inhibitor of IDO activity, 1-MT, significantly enhanced PMN transepithelial migration in response to UPEC (Figure 6: UTI89, +1-MT). Migration of PMN in response to mock infection also increased with IDO inhibitor treatment, implying a role for IDO in maintaining a baseline of epithelial immune quiescence (Figure 6: Mock, +1-MT). Because UPEC induced IDO1 expression in both PMN and 5637 bladder cells, we evaluated the effect of IDO inhibition on each cell type to specify relative contributions to the migration phenotype. Suppression of PMN transepithelial migration by UPEC was similarly abrogated when each cell type was inhibited individually (Figure 6), indicating that stimulation of IDO activity in both PMN and the epithelium may be important for UPEC modulation of innate responses.
Figure 6.

Indoleamine 2,3-dioxygenase (IDO) activity reduces human polymorphonuclear leukocytes (PMN) influx in vitro. The number of PMN recruited to the apical surface of a polarized 5637 bladder epithelial cell monolayer following infection with the indicated E. coli strains or in an uninfected phosphate-buffered saline (PBS) control (mock) in the presence or absence of the IDO inhibitor, 1-methyl-DL-tryptophan (1-MT), is shown. Treatment conditions include both 5637 and PMN (+1-MT), 5637 cells only (+1-MT 5637), and PMN only (+1-MT PMN). Recruitment in the 1-MT treated conditions was significantly different from the untreated samples as indicated (***P < .001), and significant differences between mock and UTI89-infected samples in the same treatment condition are shown (***P < .001, **P < .01, *P < .05). Data represent the mean and standard deviation derived from triplicate assays in at least 3 independent experiments with neutrophils from different donors.
DISCUSSION
In this study, we show that locally induced IDO during UPEC infection facilitates the establishment and course of UTIs by attenuating host proinflammatory innate responses. IDO activity enhanced bacterial burden in the bladder, specifically in the extracellular niche where microbes are more susceptible to neutrophil killing. Inhibiting IDO in human bladder epithelial cells and PMN augmented inflammatory responses, suggesting the importance of this enzyme in maintaining immune balance at the epithelial interface at baseline and in response to proinflammatory stimuli. This is the first report to our knowledge of a role for IDO in epithelial cell and PMN biology in the context of bacterial pathogenesis. Further, we demonstrate a novel influence of IDO activity on the innate response to a bacterial infection, distinct from its previously described roles in adaptive immunity and other T-cell functions. Our findings expand the emerging identity of this catabolic enzyme as a mediator of host–microbe interactions.
Previous studies of IDO in infectious disease progression and persistence have established this enzyme as a host defense factor, whereby local depletion of tryptophan inhibits growth of microbial invaders [16, 17]. Although the availability of this essential amino acid in infected bladders (and specifically within the IBC niche) is unknown, increased expression of UPEC genes encoding the tryptophan biosynthetic machinery indicates that UPEC senses a decrement in nutrient availability resulting from IDO induction. However, persistence of a UPEC tryptophan auxotroph in the urinary tract indicates that tryptophan starvation is unlikely to represent an important challenge for UPEC under these conditions. Therefore, the metabolic activity of IDO fails to control UPEC infection, in contrast to effects previously described for other intracellular pathogens.
Another aspect of UPEC induction of IDO that distinguishes it from previously characterized signaling mechanisms is that it does not rely on Type I or II interferons. The only regulatory DNA identified for human IDO corresponds to IFN-α/β and IFN-γ responsiveness [34, 35]; however, there is experimental evidence of interferon-independent pathways for induction of IDO expression. Studies of IDO regulation have revealed complex, cell type–dependent observations that control of IDO transcription can be influenced by several other immunomodulatory agents, including tumor necrosis factor α, interleukin 1, lipopolysaccharide, and ligation of CD80/CD86 [36–38]. The signaling networks that connect these molecules to expression of IDO remain to be fully elucidated. The present model of UPEC epithelial infection will be a useful tool for further characterization of these noncanonical pathways, including the relative contributions of different host cell types to the IDO upregulation that we observed.
Studies in our laboratory and others support a model in which early suppression of inflammation affords UPEC the opportunity to establish a protected intracellular niche in bladder tissue, prior to development of the robust inflammatory response characteristic of UTI. In the absence of IDO, despite the consequences for extracellular bacteria, these intracellular reservoirs are still established, allowing the pathogen to persist in the murine cystitis model. These results underscore the importance of intracellular bacterial community formation in UPEC pathogenesis. Additional effects of IDO induction may be revealed by characterizing the effects of IDO inhibition in other host genetic backgrounds where extracellular bacteria represent a major component of a chronic bacterial cystitis phenotype [39]. In addition, the host and bacterial factors affecting the balance of pro- and anti-inflammatory signals during epithelial infection remain an area of active investigation. Of particular interest are identified bacterial effectors that participate in immunomodulation [10, 13, 40–43] and the influence of the early host inflammatory milieu in shaping the outcomes of UPEC infection [39].
The data presented here implicate IDO as a regulator in microbial pathogenesis, which microbes have evolved mechanisms to manipulate in order to alter both innate and adaptive immune responses within the host. Ongoing studies will use the UPEC–host interaction as a model to further elucidate the activities of IDO during epithelial infections.
Notes
Acknowledgments. We thank the neutrophil donors, and we acknowledge A. French for providing the IfnαβR−/− mice, and P. Tarr for providing the fecal isolates. We also thank A. French and M. Dinauer for critical review of the manuscript.
Financial support. This work was supported by the National Institutes of Health (grant R01 DK080752) and the Edward Mallinckrodt Foundation of St. Louis, Missouri. Fecal isolates were collected with support from the US Department of Agriculture National Research Initiative (grant 0202238 to P. T.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Justice SS, Hung C, Theriot JA, et al. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc Natl Acad Sci USA. 2004;101:1333–8. doi: 10.1073/pnas.0308125100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garofalo CK, Hooton TM, Martin SM, et al. Escherichia coli from urine of female patients with urinary tract infections is competent for intracellular bacterial community formation. Infect Immun. 2007;75:52–60. doi: 10.1128/IAI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, Hultgren SJ. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000;19:2803–12. doi: 10.1093/emboj/19.12.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulvey MA, Lopez-Boado YS, Wilson CL, et al. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science. 1998;282:1494–7. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 5.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Intracellular bacterial biofilm-like pods in urinary tract infections. Science. 2003;301:105–7. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 6.Mysorekar IU, Hultgren SJ. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc Natl Acad Sci USA. 2006;103:14170–5. doi: 10.1073/pnas.0602136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schilling JD, Martin SM, Hunstad DA, et al. CD14- and Toll-like receptor-dependent activation of bladder epithelial cells by lipopolysaccharide and type 1 piliated Escherichia coli. Infect Immun. 2003;71:1470–80. doi: 10.1128/IAI.71.3.1470-1480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ingersoll MA, Kline KA, Nielsen HV, Hultgren SJ. G-CSF induction early in uropathogenic Escherichia coli infection of the urinary tract modulates host immunity. Cell Microbiol. 2008;10:2568–78. doi: 10.1111/j.1462-5822.2008.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horvath DJ, Jr, Li B, Casper T, et al. Morphological plasticity promotes resistance to phagocyte killing of uropathogenic Escherichia coli. Microbes Infect. 2011;13:426–37. doi: 10.1016/j.micinf.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunstad DA, Justice SS, Hung CS, Lauer SR, Hultgren SJ. Suppression of bladder epithelial cytokine responses by uropathogenic Escherichia coli. Infect Immun. 2005;73:3999–4006. doi: 10.1128/IAI.73.7.3999-4006.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Billips BK, Forrestal SG, Rycyk MT, Johnson JR, Klumpp DJ, Schaeffer AJ. Modulation of host innate immune response in the bladder by uropathogenic Escherichia coli. Infect Immun. 2007;75:5353–60. doi: 10.1128/IAI.00922-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klumpp DJ, Weiser AC, Sengupta S, Forrestal SG, Batler RA, Schaeffer AJ. Uropathogenic Escherichia coli potentiates type 1 pilus-induced apoptosis by suppressing NF-κB. Infect Immun. 2001;69:6689–95. doi: 10.1128/IAI.69.11.6689-6695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loughman JA, Hunstad DA. Attenuation of human neutrophil migration and function by uropathogenic bacteria. Microbes Infect. 2011;13:555–65. doi: 10.1016/j.micinf.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–74. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 15.Dai W, Gupta SL. Regulation of indoleamine 2,3-dioxygenase gene expression in human fibroblasts by interferon-gamma: upstream control region discriminates between interferon γ and interferon α. J Biol Chem. 1990;265:19871–7. [PubMed] [Google Scholar]
- 16.Pfefferkorn ER. Interferon γ blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc Natl Acad Sci USA. 1984;81:908–12. doi: 10.1073/pnas.81.3.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrne GI, Lehmann LK, Landry GJ. Induction of tryptophan catabolism is the mechanism for γ-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect Immun. 1986;53:347–51. doi: 10.1128/iai.53.2.347-351.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zelante T, Fallarino F, Bistoni F, Puccetti P, Romani L. Indoleamine 2,3-dioxygenase in infection: the paradox of an evasive strategy that benefits the host. Microbes Infect. 2009;11:133–41. doi: 10.1016/j.micinf.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 19.de Souza Sales J, Lara FA, Amadeu TP, et al. The role of indoleamine 2, 3-dioxygenase in lepromatous leprosy immunosuppression. Clin Exp Immunol. 2011;165:251–63. doi: 10.1111/j.1365-2249.2011.04412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makala LH, Baban B, Lemos H, et al. Leishmania major attenuates host immunity by stimulating local indoleamine 2,3-dioxygenase expression. J Infect Dis. 2011;203:715–25. doi: 10.1093/infdis/jiq095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boasso A. Wounding the immune system with its own blade: HIV-induced tryptophan catabolism and pathogenesis. Curr Med Chem. 2011;18:2247–56. doi: 10.2174/092986711795656126. [DOI] [PubMed] [Google Scholar]
- 22.Bozza S, Fallarino F, Pitzurra L, et al. A crucial role for tryptophan catabolism at the host/Candida albicans interface. J Immunol. 2005;174:2910–8. doi: 10.4049/jimmunol.174.5.2910. [DOI] [PubMed] [Google Scholar]
- 23.Geurs TL, Zhao YM, Hill EB, French AR. Ly49H engagement compensates for the absence of type I interferon signaling in stimulating NK cell proliferation during murine cytomegalovirus infection. J Immunol. 2009;183:5830–6. doi: 10.4049/jimmunol.0901520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hultgren SJ, Schwan WR, Schaeffer AJ, Duncan JL. Regulation of production of type 1 pili among urinary tract isolates of Escherichia coli. Infect Immun. 1986;54:613–20. doi: 10.1128/iai.54.3.613-620.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blattner FR, Plunkett G, 3rd, Bloch CA, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–74. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 26.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hung CS, Dodson KW, Hultgren SJ. A murine model of urinary tract infection. Nat Protoc. 2009;4:1230–43. doi: 10.1038/nprot.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−Δ Δ CT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Nicholson TF, Watts KM, Hunstad DA. OmpA of uropathogenic Escherichia coli promotes postinvasion pathogenesis of cystitis. Infect Immun. 2009;77:5245–51. doi: 10.1128/IAI.00670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Justice SS, Lauer SR, Hultgren SJ, Hunstad DA. Maturation of intracellular Escherichia coli communities requires SurA. Infect Immun. 2006;74:4793–800. doi: 10.1128/IAI.00355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cady SG, Sono M. 1-Methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and β-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch Biochem Biophys. 1991;291:326–33. doi: 10.1016/0003-9861(91)90142-6. [DOI] [PubMed] [Google Scholar]
- 32.Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92:3007–17. [PubMed] [Google Scholar]
- 33.Chen SL, Hung CS, Xu J, et al. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc Natl Acad Sci USA. 2006;103:5977–82. doi: 10.1073/pnas.0600938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai W, Gupta SL. Molecular cloning, sequencing and expression of human interferon γ-inducible indoleamine 2,3-dioxygenase cDNA. Biochem Biophys Res Commun. 1990;168:1–8. doi: 10.1016/0006-291x(90)91666-g. [DOI] [PubMed] [Google Scholar]
- 35.Hassanain HH, Chon SY, Gupta SL. Differential regulation of human indoleamine 2,3-dioxygenase gene expression by interferons γ and α. Analysis of the regulatory region of the gene and identification of an interferon γ–inducible DNA-binding factor. J Biol Chem. 1993;268:5077–84. [PubMed] [Google Scholar]
- 36.Babcock TA, Carlin JM. Transcriptional activation of indoleamine dioxygenase by interleukin 1 and tumor necrosis factor α in interferon-treated epithelial cells. Cytokine. 2000;12:588–94. doi: 10.1006/cyto.1999.0661. [DOI] [PubMed] [Google Scholar]
- 37.Fujigaki S, Saito K, Sekikawa K, et al. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN γ-independent mechanism. Eur J Immunol. 2001;31:2313–8. doi: 10.1002/1521-4141(200108)31:8<2313::aid-immu2313>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 38.Mellor AL, Chandler P, Baban B, et al. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. 2004;16:1391–401. doi: 10.1093/intimm/dxh140. [DOI] [PubMed] [Google Scholar]
- 39.Hannan TJ, Mysorekar IU, Hung CS, Isaacson-Schmid ML, Hultgren SJ. Early severe inflammatory responses to uropathogenic E. coli predispose to chronic and recurrent urinary tract infection. PLoS Pathog. 2010;6:e1001042. doi: 10.1371/journal.ppat.1001042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis JM, Rasmussen SB, O'Brien AD. Cytotoxic necrotizing factor type 1 production by uropathogenic Escherichia coli modulates polymorphonuclear leukocyte function. Infect Immun. 2005;73:5301–10. doi: 10.1128/IAI.73.9.5301-5310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Billips BK, Schaeffer AJ, Klumpp DJ. Molecular basis of uropathogenic Escherichia coli evasion of the innate immune response in the bladder. Infect Immun. 2008;76:3891–900. doi: 10.1128/IAI.00069-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caprioli A, Falbo V, Roda LG, Ruggeri FM, Zona C. Partial purification and characterization of an Escherichia coli toxic factor that induces morphological cell alterations. Infect Immun. 1983;39:1300–6. doi: 10.1128/iai.39.3.1300-1306.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cirl C, Wieser A, Yadav M, et al. Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nat Med. 2008;14:399–406. doi: 10.1038/nm1734. [DOI] [PubMed] [Google Scholar]