

Abstract
A major issue in the control of malaria is the evolution of drug resistance. Ecological theory has demonstrated that pathogen superinfection and the resulting within-host competition influences the evolution of specific traits. Individuals infected with Plasmodium falciparum are consistently infected by multiple parasites; however, while this probably alters the dynamics of resistance evolution, there are few robust mathematical models examining this issue. We developed a general theory for modelling the evolution of resistance with host superinfection and examine: (i) the effect of transmission intensity on the rate of resistance evolution; (ii) the importance of different biological costs of resistance; and (iii) the best measure of the frequency of resistance. We find that within-host competition retards the ability and slows the rate at which drug-resistant parasites invade, particularly as the transmission rate increases. We also find that biological costs of resistance that reduce transmission are less important than reductions in the duration of drug-resistant infections. Lastly, we find that random sampling of the population for resistant parasites is likely to significantly underestimate the frequency of resistance. Considering superinfection in mathematical models of antimalarial drug resistance may thus be important for generating accurate predictions of interventions to contain resistance.
Keywords: Plasmodium falciparum, malaria, superinfection, drug resistance, evolution
1. Introduction
More than a century ago, the Nobel prize-winning German scientist Paul Ehrlich predicted that pathogens subjected to drugs would evolve resistance [1]. The conjecture has been proven true so often and in so many contexts that it can be regarded as Ehrlich's Law. There is, nevertheless, uncertainty about virtually every other question of academic or public health interest—in particular, the role of superinfection, defined as the simultaneous infection with multiple strains of the same pathogen, and its influence on the evolution of drug resistance. Even though superinfection has been shown to alter the evolution of specific traits, particularly virulence [2–4], a robust theory of the effect of superinfection on the evolution of drug resistance is still lacking. This is important in the study of malaria, because superinfection, with the concomitant increase in within-host competition, is likely to change the dynamics of resistance evolution and have ramifications for control efforts. Here, we extend some of the classic models in malaria to consider the consequences of within-host competition among drug-sensitive and drug-resistant Plasmodium falciparum malaria parasites, the species of malaria associated with the highest levels of morbidity and mortality.
Early studies of P. falciparum malaria suggested that high rates of exposure to malaria could result in simultaneous infection with multiple parasites, termed superinfection [5]. This was not conclusively confirmed until the 1970s [6], but it is now known that P. falciparum malaria infections are typically composed of several genetically distinct lineages, called the multiplicity of infection (MOI), even in areas of low transmission [7]. Because of the complexity of the parasite's life cycle, in which parasite meiosis and recombination occurs in the mosquito, genetic and phenotypic variation between clonal P. falciparum populations can be significant, which can impact their competitive ability.
Antimalarial drug resistance is encoded by mutations in, or changes to, the copy number of genes relating to the drug's target or influx–efflux pumps that affect intraparasitic concentrations of the drug [8]. These changes, which are beneficial in the presence of drugs, also have a biological fitness cost to the parasite, lowering its competitiveness [9,10]. In the presence of therapy, the drug-resistant parasites have an advantage. Removal of the drug exposes the parasites to increased competition and can lead to a decline in the frequency of resistance-conferring mutations, as occurred in Malawi after the country stopped using chloroquine (CQ) [11]. However, in areas with lower transmission (and lower MOI and lower rates of competition), the frequency of resistance has declined at a slower rate [12]. Thus, understanding how superinfection affected historical examples of resistance to the former first-line antimalarial drugs CQ and sulphadoxine-pyrimethamine (SP) is of great interest for understanding the factors that are likely to be important for the evolution of resistance to artemisinin-class drugs and to the artemisinin combination therapies that are now the first-line treatment for falciparum malaria in much of the world.
The existing mathematical theory describing the evolution of resistance is poorly developed with respect to superinfection and within-host competition. Models of superinfection in other organisms [2,3] either have assumed that a dominant strain could displace another pathogen and take over the host, but not vice versa, or have not allowed for transmission from coinfected states [13,14]. Neither of these approaches is biologically relevant in malaria, where superinfection is the rule rather than the exception, and evidence from the field [15,16] and murine models [17–19] has demonstrated the impact of within-host competition on the survival and transmissibility of genetically distinct malaria clones.
In malaria, the mathematical theory of superinfection was developed by Walton, Macdonald, Irwin, Dietz and Bailey [5,20–24]. Their models allow for superinfection, meaning that multiple genetically distinct clones coinfect a host, and were developed to aid in matching assumptions of the transmission rate with clearance rates. Although the models generally fit the data on prevalence better than the assumption that additional infections have no effect on the clearance rate [20], epidemiological models of this type have not been applied to problems of within-host competition and drug resistance, where it is often assumed that individuals are infected by either drug-resistant or drug-sensitive parasites only [25–27].
In their model of antimalarial resistance, Koella & Antia [28] assume that individuals can be superinfected by both drug-resistant and drug-sensitive infections (heterotypic) but not by two drug-resistant or drug-sensitive types (homotypic). They assume that individuals with heterotypic infections transmit both parasites at the same rate regardless of the composition of drug-resistant and drug-sensitive parasites in the population. This significantly biases transmission, particularly when resistance is first emerging, and drug-resistant parasites are rare. As has been shown in species coexistence models [29,30], the only way that one species can exclude another is if there is significant competition in superinfected hosts, and in malaria, there is strong evidence that drug-resistant and drug-sensitive parasites compete in a host, and removing one will increase the fitness advantage of the other [31]. Because the full costs of competition are not embedded in the model (i.e. individuals cannot be multiply infected by the same type of parasite), when the fitness cost of resistance is low (but not zero), the model predicts coexistence even when there is no treatment in the population [28], a result that is not biologically plausible (see electronic supplementary material, appendix SI).
In this paper, using a general formulation of malaria superinfection, we present a general theory for the evolution of resistance when superinfection occurs. Building on population genetics models for malaria [32–34] as well as past epidemiological models [25], the approach presented here allows for an examination of some of the basic questions involved in how competition affects the spread of drug resistance in different environments. Our model provides a new way of thinking about modelling antimalarial drug resistance, incorporating many of the concepts that are in common use in malaria epidemiology today [35], and suggests ways to better understand the important control points and identify new directions for future research.
2. Methods
(a). Superinfection model
To model the evolution of resistance with superinfection, we modified a Markov chain model for superinfection and clearance to include clinical malaria and antimalarial drugs; the original model was developed for malaria [22]. MOI (the number of pathogens per host individual) increases as new infections occur, but decreases as they clear. The state variables in the model represent the fraction of the population that has a given MOI: Xi denotes the fraction of hosts with an MOI of i. We have extended the model to track the MOI of sensitive and resistant types (figure 1). Let Xi,j denote the fraction of the population with i sensitive and j resistant strains. The equations are formulated so that 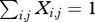 , and hence the sum of the time derivatives
, and hence the sum of the time derivatives 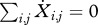 . Thus, the values of the state variables describe the joint distribution of resistant and sensitive phenotypes in a population.
. Thus, the values of the state variables describe the joint distribution of resistant and sensitive phenotypes in a population.
Figure 1.
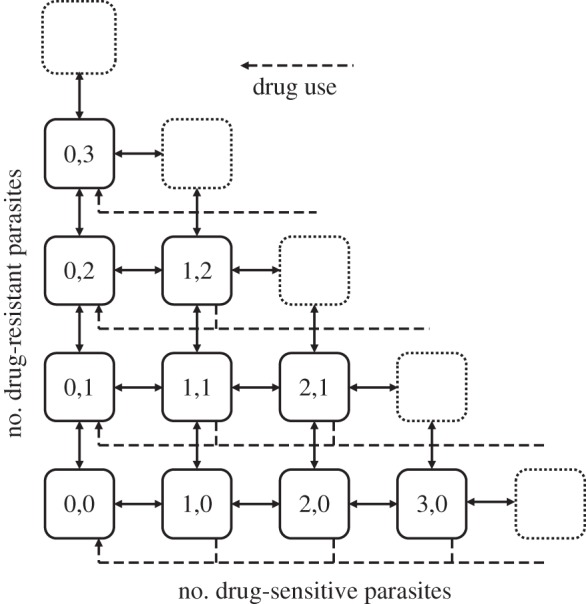
The MOI model with sensitive and resistant types. The state variables describe the proportion of the population with a given MOI for both drug-sensitive and drug-resistant parasites, and the solid lines show changes that are the result of transmission and clearance. Antibiotic use (dashed lines) reduces the MOI of drug-sensitive parasites, but not drug-resistant ones. Biological costs of resistance are introduced as an increased propensity to clear or a reduced propensity to transmit drug-resistant types.
(b). Entomology
The dynamics of infection in the model follow a notation similar to that of single-infection models by Macdonald [36], as modified by Smith & McKenzie [37]. Vectorial capacity (V), the number of infectious bites by a mosquito over its lifetime, is given by the formula V = ma2e−gn/g, where m denotes the number of mosquitoes per human and a is the number of bites on humans per mosquito per day. The instantaneous death rate is g (e−g is the probability of a mosquito surviving 1 day) and n is the number of days required for sporogony.
The daily entomological inoculation rate (EIR), the number of infectious bites per person per day, is calculated as the product of vectorial capacity and the fraction of mosquitoes that are infectious (P/(1 + aP/g)), where P is the proportion of the bites on the infected human population that infect mosquitoes, assuming transmission efficiency c from humans to mosquitoes (P = cXi,j, where i ≠ 0 or j ≠ 0). The force of infection, or happenings rate (h), is bEIR, where b, the infectivity rate, is the fraction of bites on humans that produce a patent infection.
(c). Competition and a biological cost of resistance
The model ignores fluctuations in the abundance of parasites within a host, but competition is naturally incorporated into the model as a transmission bottleneck at the mosquito. In the absence of any biases, the probability of a mosquito transmitting either a resistant or a sensitive parasite is a function of the proportion of each type ingested (assuming there is no preference for selfing). We assume that resistant genotypes in each gamete in the mosquito are lower than the proportion of the resistant genotypes in the human infection; thus, the parameter λ weighs all the factors that could produce a biological cost of resistance in the dynamics of parasites from hepatocyte to gametocyte.
The number of genotypes that are transmitted by each infectious bite is limited both by the number of ookinetes that have contributed sporozoites, and by the number of liver-stage schizonts that arise from each infectious bite. The proportion of sporozoites in a mosquito's salivary glands injected during a feeding event is typically small [38–40], though the number of genetically distinct sporozoites injected is unclear [41]. Because the bottlenecks inherent within the system are likely to make the infection of an individual with a large number of genetically diverse sporozoites an uncommon event [42], we assume that each mosquito transmits the offspring of only one gamete, regardless of the MOI of the host that infected it.
Because competition for transmission occurs only when a mosquito bites an individual with a mixed infection, the overall probability that an individual sporozoite in the next generation is sensitive (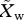 ) or resistant (
) or resistant (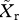 ) can be represented as
) can be represented as
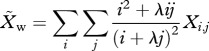 |
2.1a |
and
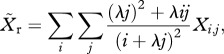 |
2.1b |
where λ < 1 (see electronic supplementary material, appendix SII for derivation). Thus, the frequency of resistance in new infections is 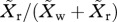 , and the force of infection for sensitive (hw) and resistant (hr) parasites is defined as
, and the force of infection for sensitive (hw) and resistant (hr) parasites is defined as
| 2.2a |
and
| 2.2b |
We also assume that resistant parasites face an additional cost in clearance. This is denoted by q, where qi,j = [(i + j)/j]α and α > 0. Thus, α describes the bias in the proportion of cleared parasites that are drug-resistant, relative to the frequency of drug-resistant types among all types in a host. While numerous reports have presented evidence of fitness costs of resistance in malaria [10–12], there is no clear understanding of how these costs are paid [10]. However, as the resistance alleles are seemingly lost at a much higher rate in high-transmission areas relative to low-transmission areas [11,12], we assume that within-host competition is the most important driver, and thus drug-resistant parasites are assumed to have a biased clearance rate only when in competition with drug-sensitive parasites.
(d). Clinical infections and drug use
Drug use is assumed to be associated with clinical symptoms (primarily fever), which develop at rate ψ. The rate at which clinical symptoms arise is independent of MOI. A fraction, ρ, of symptomatic patients are assumed to use drugs and successfully clear all sensitive parasites. The drug usage rate is assumed to be constant over each simulation, but is varied among simulations as noted. Treatment of resistant parasites is assumed to be ineffective.
(e). Equations
The dynamics of the state variable, Xi,j, which denotes the fraction of the population with i sensitive and j resistant strains, are thus described by the following set of coupled ordinary differential equations.
The equation describing the change in the proportion of uninfected hosts (i = 0, j = 0) is
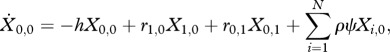 |
2.3a |
where r is the recovery rate of an infection consisting of i sensitive and j resistant strains, h is the force of infection, ψ is the rate that clinical symptoms develop, and ρ is the fraction of those patients that are treated and clear the infection successfully.
For individuals infected only with drug-sensitive clones (i > 0, j = 0),
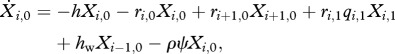 |
2.3b |
where q is the reduction in the duration of infection owing to the biological cost of resistance, and hw is the force of infection for drug-sensitive parasites.
For individuals infected only with drug-resistant clones (i = 0, j > 0),
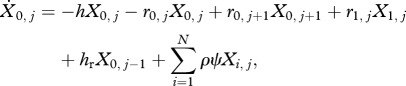 |
2.3c |
where hr is the force of infection for drug-resistant parasites.
For individuals infected with both drug-sensitive and drug-resistant clones (i > 0, j > 0),
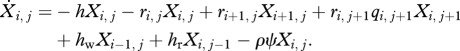 |
2.3d |
The equations are constrained such that the model has a ‘triangular’ formulation (as in figure 1).
(f). Triangularity and the neutrality condition
The model presupposes coexistence in the parasite population of a large number of distinct genotypes, but it considers competition solely between drug-sensitive and drug-resistant parasites, which differ both genetically and phenotypically. To describe the effect of competition on the evolution of resistance, it was necessary to establish that the model satisfied general principles of ecological and population genetic neutrality as described by Lipsitch et al. [43]. These principles note that the models of competition between genotypically different but phenotypically similar strains should meet two criteria: (i) the relative fraction of infected and uninfected hosts should not depend on the frequency of either strain; and (ii) the relative frequency of both strains should remain stable for all time greater than zero. In other words, the structure of the model should not, in and of itself, generate coexistence of indistinguishable strains, but mechanisms that could induce coexistence should be introduced explicitly. These principles suggest that the prior superinfection model published by Koella & Antia [28] needs modification because, as formulated, the model structure itself promotes coexistence. In the electronic supplementary material, appendix SI, we describe a model with similar properties to the Koella & Antia superinfection model, in which homotypic superinfection was not allowed, and rigorously demonstrate that coexistence is always an outcome of the model, even when the resistant strain has a fitness cost.
To ensure that coexistence in our model of superinfection, described earlier, is not an artefact of the model formulation, we analysed a special case of the general form of the model, in which the maximum MOI was two. In this case, the model allows for both heterotypic superinfection and homotypic superinfection. Numerical simulations confirm that in the absence of drug treatment, drug-resistant parasites can neither invade nor persist when they have a cost of resistance. We also found that in the absence of either drug pressure or a biological cost of resistance, the model is neutrally stable, provided the transmission rate and the recovery rate of each type in a heterotypic superinfection are exactly equal to each type in a homotypic superinfection. In other words, the rate that individuals become doubly infected with either the same type or a different type must be equal, and the transmission rate of heterotypic and homotypic infections must be the same regardless of strain composition, with an equal rate of transmission of each type from heterotypic infections. Thus, the model in this form does not predict coexistence when there is no specific mechanism promoting its spread.
Both conditions remain true as MOI increases, provided that the model structure maintains a triangular formulation in which every possible combination of the maximum MOI is included (i.e. in a model with a maximum MOI of three, all possible states where the MOI is equal to three must be possible: X3,0 and X2,1 and X1,2 and X0,3). Otherwise, the model structurally creates a niche that allows for coexistence. All our numerical simulations were done using a general form of the model with the triangularity condition in place and in which the maximum MOI was 30. Parameters used in simulations were consistent with malaria epidemiological literature [35,37,44], and are listed in the electronic supplementary material, table S1.
(g). Frequency of resistance
We employ two measures of the frequency of resistance in complex infections: (i) the fraction of the population that is infected by at least one resistant strain, 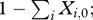 and (ii) the fraction of the parasite load that is resistant,
and (ii) the fraction of the parasite load that is resistant, 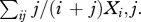 The former is important because it tracks nearly exactly with the frequency that a treated clinical infection is resistant, which is the measure by which resistance is often tracked in a population [45,46]. On the other hand, the latter is particularly useful when discussing the role that asymptomatic infections play in the spread of resistance.
The former is important because it tracks nearly exactly with the frequency that a treated clinical infection is resistant, which is the measure by which resistance is often tracked in a population [45,46]. On the other hand, the latter is particularly useful when discussing the role that asymptomatic infections play in the spread of resistance.
3. Results
The model is based on the ideas of malaria superinfection as first laid out by Walton [24] and Macdonald [5] and written down in equation form by Bailey [22]. However, unlike previous models of drug resistance in malaria [25,28], we explicitly model the effect of within-host competition between drug-resistant and drug-sensitive parasites. This allows for an examination of the importance of different types of fitness costs as well as how competition affects the rate at which resistance spreads across differential transmission rates.
To evaluate the model, we ran the system to equilibrium without resistance and then introduced resistance at a low frequency (10−7). At low transmission rates (an annual EIR of approximately one or less), the model predicts that drug-resistant parasites will invade and spread, and eventually dominate the drug-sensitive parasites. However, as the transmission rate increases, the ability of drug-resistant parasites to competitively exclude drug-sensitive parasites decreases (see electronic supplementary material, figure S1). This is because the mean MOI increases with the intensity of transmission, measured in terms of either vectorial capacity or EIR, and so competition increases. Thus, at low transmission rates, most individuals are infected with approximately one infection only, and the influence of within-host competition is limited. As the mean MOI increases, it becomes more difficult for resistant parasites to invade because competition within the host increases, overcoming the ability of resistant parasites to spread when they are rare. This is consistent with historical suggestions that resistance to the former first-line malaria drugs, CQ and SP, both emerged from areas of low or unstable transmission [47,48]. Changing the drug treatment rate for any particular transmission level increases the competitive ability of resistant parasites, allowing them to invade even when within-host competition increases at higher transmission levels.
The model also predicts that superinfection modulates coexistence. This result depends on the biological fitness costs as well as the treatment rate. At high treatment rates (80% of clinical infections treated), the drug-resistant parasite competitively excludes the drug-sensitive parasite at all fitness costs at low transmission. As the transmission rate increases (annual EIR approx. 7), coexistence can occur over a large range of fitness costs—even where the fitness cost of clearance is zero (figure 2). At higher transmission rates (annual EIR approx. 25), the range over which coexistence is possible contracts. Lowering the treatment rate (20% of clinical cases treated) shifts the results and allows for coexistence at lower transmission rates, and shrinks the parameter range over which coexistence can occur at higher transmission rates (see electronic supplementary material, figure S2).
Figure 2.
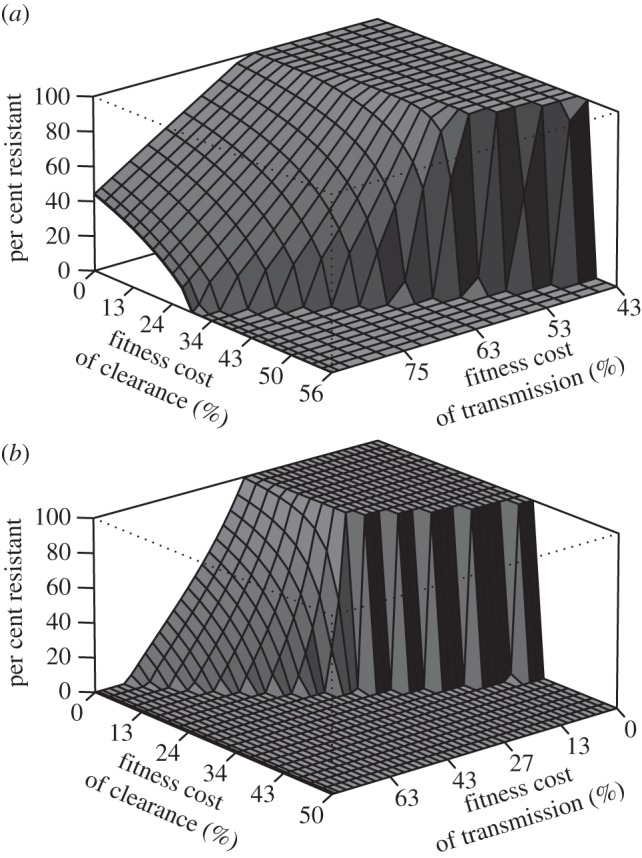
Fitness costs and coexistence. The cost of resistance can occur through either transmission or clearance. The transmission cost is measured as the relative difference in the contribution of resistant and sensitive parasites to transmission when in equal abundance. The fitness cost of clearance is measured as the reduction in the rate of clearance of a resistant parasite relative to a sensitive parasite when in competition. Thus, when there is little or no cost of clearance but a significant cost of transmission, the result is coexistence. As the transmission rate is increased (b), the parameter space over which coexistence can occur is abrogated. In addition, there are significant differences in the range of each parameter over which coexistence can occur. Areas of coexistence are marked by low to moderate fitness costs of clearance and high costs of transmission. In fact, in low-transmission areas, resistant parasites can coexist with sensitive parasites even when there is no fitness cost of clearance. (a) Moderate transmission is defined as an annual EIR of approximately 7, and (b) high transmission is an annual EIR of approximately 25. The treatment rate was assumed to be 80%.
The fitness of drug-resistant parasites is proportional to both the average duration of an infection and the ability to transmit. The former is a function only of within-host competition, whereas the latter is a function of both within-host and among-host competition, because transmission potential is a function of both transmissibility to a susceptible vector (a function of competition between clones within the host) and the propensity for infectious vectors to infect a new host (the relative fraction of each type). Results from the model suggest that resistant parasites can invade even with significant fitness costs of transmission, but not when the fitness cost of clearance increases significantly. This suggests that the ability to remain competitively infectious within hosts is a stronger determinant of the invasion capacity of drug-resistant parasites than their probability of transmission at any single event.
Competition also affects the rate that resistance spreads in a population. At low transmission rates, increasing the transmission intensity increases the rate that resistance spreads in a population because, as noted earlier, most infected individuals have an MOI of one and there is no competition. Thus, the ability of drug-resistant parasites to competitively exclude drug-sensitive parasites increases as transmission increases. However, as the average MOI increases and competition begins to inhibit the ability of the drug-resistant parasite to exclude drug-sensitive parasites, the rate at which resistance spreads in a population decreases—or, stated another way, the waiting time for resistance to reach a certain threshold value grows longer (electronic supplementary material, figure S3). This change is rapid at first, but as the transmission rate continues to increase, the rate of increase slows.
Superinfection also affects how the frequency of resistance is calculated. Although the prevalence of resistance (the fraction of infected people that harbour at least one resistant parasite) increases concomitantly with the fraction of the population that harbours at least one resistant parasite, the resistance load (the proportion of types that are resistant) changes at a different rate (figure 3). As the transmission intensity increases, the resistance load increases at a rate that is similar to the other measures. However, the rate of increase slows after a point and then falls, while the prevalence of resistance continues to increase. At high transmission levels, this spread can be more than five percentage points different when the fraction of clinical infections reaches 10 per cent.
Figure 3.
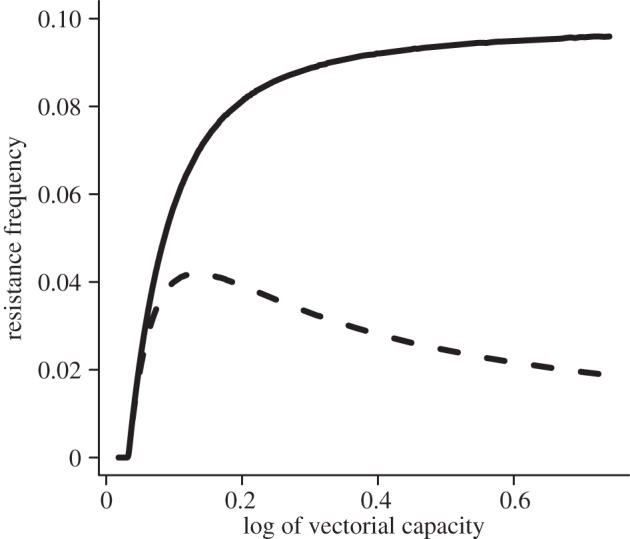
Measuring the frequency of resistance. There are multiple ways to calculate the frequency of resistance in a population: (i) the prevalence of resistance (the fraction of individuals infected with a resistant parasite); (ii) the fraction of clinical infections that fail treatment; and (iii) the resistance load (the proportion of the parasite population that is resistant). Depending on the measurement, the frequency of resistance could be wildly different. To measure these differences, we calculated the resistance load at the point in time when the percentage of clinical infections that fail drug treatment reached 10%. Although both measures increase nearly concurrently at low transmission rates, as transmission increases (and thus so does competition), the fraction of the parasite population (dashed line) that is resistant soon peaks and then begins to fall, even as the proportion of clinical infections harbouring resistant parasites (solid line) increases.
4. Discussion
A prior model of superinfection in malaria with drug-resistant and drug-sensitive parasites assumes that heterotypic superinfection is possible but homotypic superinfection is not [28]. However, this assumption means that coexistence will always occur (see electronic supplementary material, appendix SI). The structure of this prior model [28] is also similar to earlier models of species coexistence [29,30], which always predict coexistence unless there is some type of competition in individuals heterotypically superinfected. Thus, coexistence is an artefact of the mathematical model, not a generic property of the underlying biological process.
In this paper, we replace those assumptions with a model that implements a more robust competition framework that allows for a thorough examination of the effect of competition on the spread of drug-resistant malaria parasites in an epidemiological context. We found general conditions for the evolution of resistance as the outcome of within- and among-host competition between two classes of parasite types with differing fitness; the conditions depend on competition, drug pressure and the biological cost of resistance.
Results from a general model demonstrate that the estimate of disease prevalence as a function of the transmission rate was qualitatively similar to that found in other models [5] and similar to estimates of the same relationship in the field [49–51]. Our results also support earlier studies showing a strong relationship between the fitness cost of resistance and transmission [25,33]. When the fitness cost of resistance is low, drug-resistant parasites are able to invade and spread across all transmission levels. However, as the fitness cost of resistance increases, the ability of drug-resistant parasites to invade and spread is reduced. One possible reason previously suggested for this relationship is that the parasite is exposed to a higher level of drug pressure per infection in low-transmission areas because a higher fraction of infections in these areas result in clinical symptoms [33]. Our results suggest that the low force of infection when recolonization of infected individuals is rare plays a significant role in low-transmission areas as well. After drug use eliminates drug-sensitive parasites, individuals harbouring resistant parasites are less likely to be recolonized by drug-sensitive parasites, reducing within-host competition.
Although a biological cost of resistance has been measured in in vitro experiments [52,53] and estimated from the field [11,12], reductions in the competitive ability of the parasite are not expected to be equal across different axes of competition. This has significant biological and epidemiological importance. On the basis of in vitro experiments in which CQ-resistant parasites had an estimated 25 per cent loss of fitness per generation [52], as well as murine models demonstrating slower growth of drug-resistant parasites in the mouse [54,55], it has been suggested that mutations conferring resistance decrease the reproductive efficiency of the parasite and slow growth [10]. The end result of the lower growth rate is assumed to be a decrease in the probability of a parasite being transmitted when a mosquito feeds on blood because of the lower relative numbers of gametocytes. A similar mechanism is also presumed to affect the duration of infection; however, evidence on the relative cost of resistance on clearance is lacking, particular in relation to transmissibility. We found that although the reductions in transmission probability were important, the ability to persist in an infection may be a more important measure of the resistant parasite's competitive ability, particularly in lower-transmission settings. These results can be partly explained by the vast time-scale differences between these processes. Because the time from infection of a susceptible mosquito to infection of a human is an order of magnitude faster than the duration of infection, the benefit of increasing the duration of infection is significantly greater than the benefit of increasing infectiousness within an infection. The result is that resistant parasites can invade even with significant fitness costs of transmission, but not when the fitness cost of clearance increases significantly.
Because the parasite has evolved an extremely long duration of infection to maximize transmission opportunities and can transmit efficiently at very low densities [56], it is not surprising that the effect of changes in the clearance rate is more pronounced on the viability of drug-resistant parasites than on reductions in transmission. However, the effect on attempts to control the spread of resistance is important. Recent evidence has shown the emergence of a delayed clearance phenotype to the drug artemisinin in western Cambodia [57], which has led to calls for implementing a resistance containment strategy. Our results suggest that interventions that can shorten the duration of infection for resistant parasites, such as mass drug administration or mass screening and treating, may be more beneficial than has been recognized by earlier models that ignored superinfection [58].
Within-host competition can also produce coexistence of the two different parasite phenotypes at the population level. Coexistence can occur when the relative advantage of the sensitive parasite, measured as transmission potential over recovery rate, is greater in mixed infections. However, the parameter range over which coexistence can occur is altered by the transmission rate. At low transmission rates, competition is lessened between different phenotypes as well as with the same phenotypes. Individuals with drug-resistant parasites can then be easily reinfected because they have only a few parasites. Thus, if drug-sensitive parasites have an advantage in mixed infections, the result for a fixed treatment rate will be a greater tendency to coexist. On the other hand, as the transmission rate increases, individuals who are treated with drugs are likely to harbour multiple drug-resistant parasite clones. In this case, the invasion capability of drug-sensitive parasites is reduced, making the range over which coexistence can occur smaller.
In terms of designing control strategies, the time until resistance reaches a critical level, which is critically influenced by the transmission and treatment rates, may be a more important measure than the final equilibrium level of resistance. Currently, there is no standardized methodology for assessing the frequency of resistance, though it is generally measured as the proportion of a type specimen isolated from a clinical infection that did not adequately respond to treatment [46]. However, screening surveys of the human population using in vitro tests to determine resistance of a type specimen has also been suggested as a means to ascertain the frequency of resistance in the population [59]. These sampling mechanisms are not identical when individuals have complex infections, and it is important to understand how these sampling differences may end up measuring radically different quantities. In high-transmission areas, when the frequency of resistance is low, complex infections will primarily consist of drug-sensitive parasites. Because sampling is imperfect, the probability of false negatives (i.e. at least one resistant parasite is present but was not detected) is high [45], which suggests that random screening surveys as a measure of resistance in higher-transmission areas may result in biased results. This is probably also true when withdrawing a drug. For instance, even though the detection of CQ-resistant mutations has dropped to undetectable levels in Malawi [11,60], the complexity of malaria infections suggests that drug-resistant parasites may still be present at low frequencies and that the reintroduction of CQ may be followed by a rapid resurgence in resistant infections.
In this model of malaria superinfection, which was extended to incorporate competition between drug-sensitive and drug-resistant parasites, our results are limited to examining only the axis of competition between drug-resistant and drug-sensitive phenotypes. The evolution of antimalarial drug resistance is obviously a complicated process involving superinfection and many interacting processes, including immunity [25], the patterns of drug use and heterogeneous biting [49]. The results of this simple model clearly demonstrate that within-host competition is a significant component in the emergence and spread of drug resistance even at lower transmission rates. Models that examine the best ways to control or contain the emergence of resistance must take account of superinfection and a variable degree of within-host competition across the spectrum of transmission. In particular, the assumption of a blanket fitness cost of resistance is less reasonable when the duration of infection dominates the probability that resistance will spread. Future studies in this area will include how heterogeneous biting by the mosquito vector changes the dynamics of competition, particularly in low-transmission areas, and how host immunity interacts with virulence to change the dynamics of the emergence of drug resistance.
Acknowledgements
This work was funded by Princeton University's Health Grand Challenges Program. This publication was also made possible by grant no. 1R01GM100471-01 from the National Institute of General Medical Sciences (NIGMS) at the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS. E.Y.K. was supported by Princeton University (Harold W. Dodds Fellowship). D.L.S. is funded by the RAPIDD program of the Science and Technology Directorate, Department of Homeland Security, and the Fogarty International Center, National Institutes of Health.
References
- 1.Ehrlich P. 1913. Address in pathology, ON CHEMIOTHERAPY. Delivered before the Seventeenth International Congress of Medicine. Br. Med. J. 2, 353–359 10.1136/bmj.2.2746.353 (doi:10.1136/bmj.2.2746.353) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levin S., Pimentel D. 1981. Selection of intermediate rates of increase in parasite–host systems. Am. Nat. 117, 308–315 10.1086/283708 (doi:10.1086/283708) [DOI] [Google Scholar]
- 3.Nowak M. A., May R. M. 1994. Superinfection and the evolution of parasite virulence. Proc. R. Soc. Lond. B 255, 81–89 10.1098/rspb.1994.0012 (doi:10.1098/rspb.1994.0012) [DOI] [PubMed] [Google Scholar]
- 4.van Baalen M., Sabelis M. W. 1995. The dynamics of multiple infection and the evolution of virulence. Am. Nat. 146, 881. 10.1086/285830 (doi:10.1086/285830) [DOI] [Google Scholar]
- 5.Macdonald G. 1950. The analysis of infection rates in diseases in which superinfection occurs. Trop. Dis. Bull. 47, 907–915 [PubMed] [Google Scholar]
- 6.McKenzie F. E., Smith D. L., O'Meara W. P., Riley E. M., Rollinson D., Hay S. I. 2008. Strain theory of malaria: the first 50 years. Adv. Parasitol. 66, 1–46 10.1016/S0065-308X(08)00201-7 (doi:10.1016/S0065-308X(08)00201-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paul R., Hackford I., Brockman A., Muller-Graf C., Price R., Luxemburger C., White N., Nosten F., Day K. 1998. Transmission intensity and Plasmodium falciparum diversity on the northwestern border of Thailand. Am. J. Trop. Med. Hyg. 58, 195–203 [DOI] [PubMed] [Google Scholar]
- 8.White N. J. 2004. Antimalarial drug resistance. J. Clin. Invest. 113, 1084–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felger I., Beck H.-P. 2008. Fitness costs of resistance to antimalarial drugs. Trends Parasitol. 24, 331–333 10.1016/j.pt.2008.05.004 (doi:10.1016/j.pt.2008.05.004) [DOI] [PubMed] [Google Scholar]
- 10.Babiker H. A., Hastings I. M., Swedberg G. 2009. Impaired fitness of drug-resistant malaria parasites: evidence and implication on drug-deployment policies. Expert Rev. Anti-Infect. Ther. 7, 581–593 10.1586/eri.09.29 (doi:10.1586/eri.09.29) [DOI] [PubMed] [Google Scholar]
- 11.Laufer M. K., Thesing P. C., Eddington N. D., Masonga R., Dzinjalamala F. K., Takala S. L., Taylor T. E., Plowe C. V. 2006. Return of chloroquine antimalarial efficacy in Malawi. N Engl. J. Med. 355, 1959–1966 10.1056/NEJMoa062032 (doi:10.1056/NEJMoa062032) [DOI] [PubMed] [Google Scholar]
- 12.De-quan L., et al. 1995. Changes in the resistance of Plasmodium falciparum to chloroquine in Hainan, China. Bull. World Health Organ. 73, 483–486 [PMC free article] [PubMed] [Google Scholar]
- 13.Levin S. A. 1983. Some approaches to the modelling of coevolutionary interactions. In Coevolution (ed. Ntecki M.), pp. 21–65 Chicago, IL: University of Chicago Press [Google Scholar]
- 14.Levin S. A. 1983. Coevolution. Lect. Notes Biomath. 52, 328–334 10.1007/978-3-642-87893-0_41 (doi:10.1007/978-3-642-87893-0_41) [DOI] [Google Scholar]
- 15.Daubersies P., Sallenave-Sales S., Magne S., Trape J. F., Contamin H., Fandeur T., Rogier C., Mercereau-Puijalon O., Druilhe P. 1996. Rapid turnover of Plasmodium falciparum populations in asymptomatic individuals living in a high transmission area. Am. J. Trop. Med. Hyg. 54, 18–26 [DOI] [PubMed] [Google Scholar]
- 16.Arnot D. 1998. Clone multiplicity of Plasmodium falciparum infections in individuals exposed to variable levels of disease transmission. Trans. R. Soc. Trop. Med. Hyg. 92, 580–585 10.1016/S0035-9203(98)90773-8 (doi:10.1016/S0035-9203(98)90773-8) [DOI] [PubMed] [Google Scholar]
- 17.Taylor L. H., Walliker D., Read A. F. 1997. Mixed-genotype infections of malaria parasites: within-host dynamics and transmission success of competing clones. Proc. R. Soc. Lond. B 264, 927–935 10.1098/rspb.1997.0128 (doi:10.1098/rspb.1997.0128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snounou G., Jarra W., Viriyakosol S., Wood J. C., Brown K. N. 1989. Use of a DNA probe to analyse the dynamics of infection with rodent malaria parasites confirms that parasite clearance during crisis is predominantly strain- and species-specific. Mol. Biochem. Parasitol. 37, 37–46 10.1016/0166-6851(89)90100-x (doi:10.1016/0166-6851(89)90100-x) [DOI] [PubMed] [Google Scholar]
- 19.De Roode J., Read A., Chan B., Mackinnon M. 2003. Rodent malaria parasites suffer from the presence of conspecific clones in three-clone Plasmodium chabaudi infections. Parasitology 127, 411–418 10.1017/S0031182003004001 (doi:10.1017/S0031182003004001) [DOI] [PubMed] [Google Scholar]
- 20.Dietz K. 1988. Mathematical models for transmission and control of malaria. In Malaria: principles and practice of malariology (eds Wernsdorfer W. H., McGregor I. A.), pp. 1091–1134 Edinburgh, UK: Churchill Livingstone [Google Scholar]
- 21.Fine P. 1975. Superinfection: a problem in formulating a problem. Bur. Hyg. Trop. Dis. 72, 475–488 [Google Scholar]
- 22.Bailey N. T. J. 1982. The biomathematics of malaria. London, UK: C. Griffin & Co [Google Scholar]
- 23.Dietz K., Molineaux L., Thomas A. 1974. A malaria model tested in the African savannah. Bull. World Health Organ. 50, 347–357 [PMC free article] [PubMed] [Google Scholar]
- 24.Walton G. 1947. On the control of malaria in Freetown, Sierra Leone: 1. Plasmodium falciparum and Anopheles gambiae in relation to malaria occurring in infants. Ann. Trop. Med. Parasitol. 41, 380–407 [DOI] [PubMed] [Google Scholar]
- 25.Klein E. Y., Smith D. L., Boni M. F., Laxminarayan R. 2008. Clinically immune hosts as a refuge for drug-sensitive malaria parasites. Malar. J. 7, 67. 10.1186/1475-2875-7-67 (doi:10.1186/1475-2875-7-67) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aneke S. J. 2002. Mathematical modelling of drug resistant malaria parasites and vector populations. Math. Methods Appl. Sci. 25, 335–346 10.1002/mma.291 (doi:10.1002/mma.291) [DOI] [Google Scholar]
- 27.Esteva L., Gumel A. B., de León C. V. 2009. Qualitative study of transmission dynamics of drug-resistant malaria. Math. Comput. Model 50, 611–630 10.1016/j.mcm.2009.02.012 (doi:10.1016/j.mcm.2009.02.012) [DOI] [Google Scholar]
- 28.Koella J., Antia R. 2003. Epidemiological models for the spread of anti-malarial resistance. Malar. J. 2, 3. 10.1186/1475-2875-2-3 (doi:10.1186/1475-2875-2-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slatkin M. 1974. Competition and regional coexistence. Ecology 55, 128–134 10.2307/1934625 (doi:10.2307/1934625) [DOI] [Google Scholar]
- 30.Cohen J. E. 1970. A Markov contingency-table model for replicated Lotka–Volterra systems near equilibrium. Am. Nat. 104, 547–560 10.1086/282689 (doi:10.1086/282689) [DOI] [Google Scholar]
- 31.Wargo A. R., Huijben S., de Roode J. C., Shepherd J., Read A. F. 2007. Competitive release and facilitation of drug-resistant parasites after therapeutic chemotherapy in a rodent malaria model. Proc. Natl Acad. Sci. USA 104, 19 914–19 919 10.1073/pnas.0707766104 (doi:10.1073/pnas.0707766104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Meara W. P., Smith D. L., McKenzie F. E. 2006. Potential impact of intermittent preventive treatment (IPT) on spread of drug-resistant malaria. PLoS Med. 3, e141. 10.1371/journal.pmed.0030141 (doi:10.1371/journal.pmed.0030141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hastings I. M. 1997. A model for the origins and spread of drug-resistant malaria. Parasitology 115, 133–141 10.1017/S0031182097001261 (doi:10.1017/S0031182097001261) [DOI] [PubMed] [Google Scholar]
- 34.Hastings I. M. 2006. Complex dynamics and stability of resistance to antimalarial drugs. Parasitology 132, 615–624 10.1017/S0031182005009790 (doi:10.1017/S0031182005009790) [DOI] [PubMed] [Google Scholar]
- 35.Smith D. L., Battle K. E., Hay S. I., Barker C. M., Scott T. W., McKenzie F. E. 2012. Ross, Macdonald, and a theory for the dynamics and control of mosquito-transmitted pathogens. PLoS Pathog. 8, e1002588. 10.1371/journal.ppat.1002588 (doi:10.1371/journal.ppat.1002588) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macdonald G. 1957. The epidemiology and control of malaria. London, UK: Oxford University Press [Google Scholar]
- 37.Smith D. L., McKenzie F. E. 2004. Statics and dynamics of malaria infection in Anopheles mosquitoes. Malar. J. 3, 13. 10.1186/1475-2875-3-13 (doi:10.1186/1475-2875-3-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medica D. L., Sinnis P. 2005. Quantitative dynamics of Plasmodium yoelii sporozoite transmission by infected Anopheline mosquitoes. Infect. Immun. 73, 4363–4369 10.1128/IAI.73.7.4363-4369.2005 (doi:10.1128/IAI.73.7.4363-4369.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shute P. G. 1945. An investigation into the number of sporozoites found in the salivary glands of Anopheles mosquitoes. Trans. R. Soc. Trop. Med. Hyg. 38, 493–498 10.1016/0035-9203(45)90058-7 (doi:10.1016/0035-9203(45)90058-7) [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg R., Wirtz R. A., Schneider I., Burge R. 1990. An estimation of the number of malaria sporozoites ejected by a feeding mosquito. Trans. R. Soc. Trop. Med. Hyg. 84, 209–212 10.1016/0035-9203(90)90258-G (doi:10.1016/0035-9203(90)90258-G) [DOI] [PubMed] [Google Scholar]
- 41.Druilhe P., Daubersies P., Patarapotikul J., Gentil C., Chene L., Chongsuphajaisiddhi T., Mellouk S., Langsley G. 1998. A primary malarial infection is composed of a very wide range of genetically diverse but related parasites. J. Clin. Invest. 101, 2008–2016 10.1172/JCI119890 (doi:10.1172/JCI119890) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sinden R. E., Billingsley P. F. 2001. Plasmodium invasion of mosquito cells: hawk or dove? Trends Parasitol. 17, 209–211 10.1016/S1471-4922(01)01928-6 (doi:10.1016/S1471-4922(01)01928-6) [DOI] [PubMed] [Google Scholar]
- 43.Lipsitch M., Colijn C., Cohen T., Hanage W. P., Fraser C. 2009. No coexistence for free: neutral null models for multistrain pathogens. Epidemics 1, 2. 10.1016/j.epidem.2008.07.001 (doi:10.1016/j.epidem.2008.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garrett-Jones C. 1964. The human blood index of malaria vectors in relation to epidemiological assessment. Bull. World Health Organ. 30, 241–261 [PMC free article] [PubMed] [Google Scholar]
- 45.Hastings I. M., Nsanzabana C., Smith T. A. 2010. A comparison of methods to detect and quantify the markers of antimalarial drug resistance. Am. J. Trop. Med. Hyg. 83, 489–495 10.4269/ajtmh.2010.10-0072 (doi:10.4269/ajtmh.2010.10-0072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bloland P. B., Kazembe P. N., Oloo A. J., Himonga B., Barat L. M., Ruebush T. K. 1998. Chloroquine in Africa: critical assessment and recommendations for monitoring and evaluating chloroquine therapy efficacy in sub-Saharan Africa. Trop. Med. Int. Health 3, 543–552 10.1046/j.1365-3156.1998.00270.x (doi:10.1046/j.1365-3156.1998.00270.x) [DOI] [PubMed] [Google Scholar]
- 47.White N. J. 1998. Preventing antimalarial drug resistance through combinations. Drug Resist. Updates 1, 3–9 10.1016/S1368-7646(98)80208-2 (doi:10.1016/S1368-7646(98)80208-2) [DOI] [PubMed] [Google Scholar]
- 48.White N. J., Pongtavornpinyo W. 2003. The de novo selection of drug-resistant malaria parasites. Proc. R. Soc. Lond. B 270, 545–554 10.1098/rspb.2002.2241 (doi:10.1098/rspb.2002.2241) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith D. L., Dushoff J., Snow R. W., Hay S. I. 2005. The entomological inoculation rate and Plasmodium falciparum infection in African children. Nature 438, 492–495 10.1038/nature04024 (doi:10.1038/nature04024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith D., Hay S. 2009. Endemicity response timelines for Plasmodium falciparum elimination. Malar J 8, 87. 10.1186/1475-2875-8-87 (doi:10.1186/1475-2875-8-87) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sama W., Owusu-Agyei S., Felger I., Dietz K., Smith T. 2006. Age and seasonal variation in the transition rates and detectability of Plasmodium falciparum malaria. Parasitology 132, 13–21 10.1017/S0031182005008607 (doi:10.1017/S0031182005008607) [DOI] [PubMed] [Google Scholar]
- 52.Hayward R., Saliba K. J., Kirk K. 2005. pfmdr1 mutations associated with chloroquine resistance incur a fitness cost in Plasmodium falciparum. Mol. Microbiol. 55, 1285–1295 10.1111/j.1365-2958.2004.04470.x (doi:10.1111/j.1365-2958.2004.04470.x) [DOI] [PubMed] [Google Scholar]
- 53.Peters J. M., Chen N., Gatton M., Korsinczky M., Fowler E. V., Manzetti S., Saul A., Cheng Q. 2002. Mutations in cytochrome b resulting in atovaquone resistance are associated with loss of fitness in Plasmodium falciparum. Antimicrob. Agents Chemother. 46, 2435–2441 10.1128/aac.46.8.2435-2441.2002 (doi:10.1128/aac.46.8.2435-2441.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chawira A. N., Warhurst D. C., Peters W. 1986. Qinghaosu resistance in rodent malaria. Trans. R. Soc. Trop. Med. Hyg. 80, 477–480 10.1016/0035-9203(86)90351-2 (doi:10.1016/0035-9203(86)90351-2) [DOI] [PubMed] [Google Scholar]
- 55.Walliker D., Hunt P., Babiker H. 2005. Fitness of drug-resistant malaria parasites. Acta Trop. 94, 251–259 10.1016/j.actatropica.2005.04.005 (doi:10.1016/j.actatropica.2005.04.005) [DOI] [PubMed] [Google Scholar]
- 56.Barnes K. I., White N. J. 2005. Population biology and antimalarial resistance: the transmission of antimalarial drug resistance in Plasmodium falciparum. Acta Trop. 94, 230–240 10.1016/j.actatropica.2005.04.014 (doi:10.1016/j.actatropica.2005.04.014) [DOI] [PubMed] [Google Scholar]
- 57.Dondorp A. M., Yeung S., White L., Nguon C., Day N. P. J., Socheat D., von Seidlein L. 2010. Artemisinin resistance: current status and scenarios for containment. Nat. Rev. Microbiol. 8, 272–280 10.1038/nrmicro2331 (doi:10.1038/nrmicro2331) [DOI] [PubMed] [Google Scholar]
- 58.Pongtavornpinyo W., Yeung S., Hastings I., Dondorp A., Day N., White N. 2008. Spread of anti-malarial drug resistance: mathematical model with implications for ACT drug policies. Malar. J. 7, 229. 10.1186/1475-2875-7-229 (doi:10.1186/1475-2875-7-229) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Picot S., Olliaro P., de Monbrison F., Bienvenu A. L., Price R. N., Ringwald P. 2009. A systematic review and meta-analysis of evidence for correlation between molecular markers of parasite resistance and treatment outcome in falciparum malaria. Malar. J. 8, 89. 10.1186/1475-2875-8-89 (doi:10.1186/1475-2875-8-89) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laufer M. K., Takala-Harrison S., Dzinjalamala F. K., Stine O. C., Taylor T. E., Plowe C. V. 2010. Return of chloroquine-susceptible falciparum malaria in Malawi was a reexpansion of diverse susceptible parasites. J. Infect. Dis. 202, 801–808 10.1086/655659 (doi:10.1086/655659) [DOI] [PMC free article] [PubMed] [Google Scholar]