

Abstract
Background. Evidence from genotype-phenotype studies suggests that genetic diversity in pathogens have clinically relevant manifestations that can impact outcome of infection and epidemiologic success. We studied 5 closely related Mycobacterium tuberculosis strains that collectively caused extensive disease (n = 862), particularly among US-born tuberculosis patients.
Methods. Representative isolates were selected using population-based genotyping data from New York City and New Jersey. Growth and cytokine/chemokine response were measured in infected human monocytes. Survival was determined in aerosol-infected guinea pigs.
Results. Multiple genotyping methods and phylogenetically informative synonymous single nucleotide polymorphisms showed that all strains were related by descent. In axenic culture, all strains grew similarly. However, infection of monocytes revealed 2 growth phenotypes, slower (doubling ∼55 hours) and faster (∼25 hours). The faster growing strains elicited more tumor necrosis factor α and interleukin 1β than the slower growing strains, even after heat killing, and caused accelerated death of infected guinea pigs (∼9 weeks vs 24 weeks) associated with increased lung inflammation/pathology. Epidemiologically, the faster growing strains were associated with human immunodeficiency virus and more limited in spread, possibly related to their inherent ability to induce a strong protective innate immune response in immune competent hosts.
Conclusions. Natural variation, with detectable phenotypic changes, among closely related clinical isolates of M. tuberculosis may alter epidemiologic patterns in human populations.
Mycobacterium tuberculosis, the etiologic agent of tuberculosis, is a highly successful human pathogen. Initial genomic studies of M. tuberculosis revealed high species-level conservation, with little evidence for horizontal gene transfer [1–4]. More recently, a number of single nucleotide polymorphism (SNP)–based studies have delineated up to 9 discrete lineages [1, 5, 6]. Molecular epidemiologic studies, using biomarkers such as IS6110-based restriction fragment length polymorphisms (RFLPs) and spoligotyping, have revealed further diversity, facilitating a range of investigations including transmission studies [7]. Furthermore, using comparative sequence analysis of 89 genes in 108 strains, Hershberg et al [8] predicted that an average pair of M. tuberculosis complex strains would have roughly 300 functional differences due to SNPs alone. Thus, there is mounting evidence of greater genetic diversity than previously thought.
Accumulating evidence from genotype-phenotype studies suggests that genetic diversity among M. tuberculosis isolates may have clinically relevant manifestations that could impact on outcome of infection [9–12]. Strain-dependent variations in replication rates, immunogenicity, pathogenesis, survival, and transmission potential have been described elsewhere [9, 13]. Recent reports have associated strain-specific microbial factors that alter the host immune response with enhanced virulence [14, 15]. For example, W-Beijing strain HN878 induces a low proinflammatory response and is hypervirulent in animals [10, 13]. This phenotype is associated with production of an immune suppressive phenolic glycolipid [16]. In another report, hypoimmunogenic strain CH, responsible for an outbreak in Leicester, was linked to a specific deletion [14]. An in vitro study in human macrophages, comparing phylogenetically defined “ancient” and “modern” lineages, noted reduced cytokine responses in the latter group [17]. In a study by Homolka et al [18], genetic diversity appeared to have functional consequences during intracellular infection of bone marrow–derived macrophages, where transcriptomic profiles were lineage specific. Thus, it appears that genetically diverse M. tuberculosis clinical isolates can differ in their phenotypic characteristics.
Studies have identified predominant or emerging M. tuberculosis clones in human populations [7]. However, it remains unclear whether there is a microbial basis to explain why some variants cause widespread disease and other closely related strains remain limited in spread. Previously, we described M. tuberculosis strains BE, H6, and C28, all descendants of a single clone [19]. We identified 2 additional closely related strains. Collectively, these 5 strains were predominant among the US-born tuberculosis patients in our study population. Here, we used these 5 genetically related M. tuberculosis variants as a model to understand the evolution of M. tuberculosis and its spread in populations. First, we carried out a population-based analysis of M. tuberculosis isolates from New York City (NYC) and the state of New Jersey, allowing us to define the prevalence of clones or variants in our population and to characterize the molecular events that demonstrate relatedness and distinguish our model strains. Next, we examined axenic and intracellular growth and cytokine-inducing capacity of each variant in naive human monocytes. Finally, survival of aerosol-infected guinea pigs was evaluated. Our results provide evidence of phenotypic diversity associated with microevolutionary events and suggest an underlying biologic basis for the differences in epidemiologic success of the 5 groups of variants.
METHODS
Study Population
Isolates from culture-positive tuberculosis patients counted by the NYC Department of Health and Mental Hygiene, Bureau of Tuberculosis Control since 1995 were included. The collection is based on convenient sampling until 2000, when universal genotyping was introduced [20]. From 2001 to 2008, basic demographic data were available from the tuberculosis registry. Additionally, patient isolates collected as part of a population-based study in New Jersey (1996–2000) and convenient samples from 2001 to 2009 were included.
Molecular Profiling
All study isolates were subjected to IS6110-based RFLP, spoligotyping, and 12-loci MIRU-VNTR [21–23]. Representative M. tuberculosis strains included H, C, BE, H6, and C28. In all experiments, the closely related strain CDC1551 was used as a control.
Principal Genetic Group and Synonymous SNP Analysis
To establish relatedness and broader phylogenetic relationships we performed principal genetic group (PGG) analysis [4]. For further resolution, we examined phylogenetically informative synonymous SNPs (sSNPs) [6].
IS6110 Flanking Regions or Insertion-Site Mapping
The conventional inverse polymerase chain reaction (PCR) method was modified to identify the location of IS6110 insertion(s) in the chromosome [19, 24–26].
Direct Repeat Fine Mapping
PCR was performed using BioRad MyCycler (BioRad), Choice Blue Master mix (Denville), primers (designed based on spacer sequences from van Embden et al [27]) and amplification program (94°C for 4 minutes, 35 cycles of 94°C for 30 seconds, 62°C for 30 seconds, 72°C for 50 seconds, and 72°C for 2 minutes).
M. tuberculosis Growth In Vitro
Mycobacteria were grown from stocks on 7H10 plates (with OADC), resuspended in 7H9 broth (to optical density [OD] 540 nm 0.05 absorbance units) and rotated in glass tubes at 37°C for 2 weeks. OD 540 nm was measured and serial dilutions plated on 7H10 every 2–3 days; colony-forming units (CFUs) were enumerated.
Stimulation of Human Monocytes and Cytokine Detection
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Paque separation [28] from blood from anonymous healthy donors (New Jersey Blood Center), plated at 5 × 106 per well in 24-well plates (Corning), and allowed to adhere for 2 hours [29]. Adherent monocytes were infected with live M. tuberculosis strains at multiplicity of infection (MOI) of 1:5 (bacilli:monocytes). Intracellular growth was determined by disrupting monocytes by probe sonication and plating serial dilutions on 7H10 [29]. Doubling times were calculated from day 0 to day 3 postinfection. In some experiments, M. tuberculosis strains were heat-killed at 80°C for 10 minutes before monocyte stimulation. Supernatants from stimulated or unstimulated monocytes collected at 24–48 hours postinfection were stored at −80°C and batch analyzed using a multiplex human cytokine Luminex panel for interleukin 1β (IL-1β), interleukin 1 receptor antagonist (IL-Ra), interleukin 6 (IL-6), interleukin 10 (IL-10), granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon-inducible protein 10 (IP-10), macrophage inflammatory protein 1α (MIP-1α), macrophage inflammatory protein 1β (MIP-1β), RANTES (regulated on activation, normal T cell expressed and secreted), tumor necrosis factor α (TNF-α), vascular endothelial growth factor (VEGF), and interleukin 12p70 (IL-12p70) (Bio-Rad). Results were read using a Bio-Plex 200 system; data and statistical analyses were carried out using GraphPad Prism, version 4 (GraphPad).
Low-Dose Aerosol Infection of Guinea Pigs
Out-bred female Hartley guinea pigs weighing 450–500 g (Charles River Laboratories) were maintained in an ABSL-3 in isolator cages (Thoren). Bacterial suspensions (1 × 106 CFU in the nebulizer) were used to infect 14 guinea pigs per strain (BE, C28, H6, C, or H) (experiment 1) or 18 animals/strain (CDC1551, BE, H6, or C28; 1 × 107 CFU) (experiment 2) by low aerosol method with a Madison chamber [30]. The Madison chamber has been calibrated so that when 1 × 106 CFU are placed into the nebulizer chamber it will deposit approximately 10–20 CFU into the lungs. In both experiments, 5 guinea pigs per group were killed at day 30 postinfection to enumerate CFUs in lungs and spleen; the remaining animals were followed for survival. Differences in survival curves were based on log rank using a Holm-Sidak method for multiple comparisons. A standardized histological scoring system was used to evaluate pathology (Supplementary Methods). All experimental procedures were approved by the Colorado State University Institutional Animal Care and Use Committee.
RESULTS
Genetic Relatedness of M. tuberculosis Clinical Isolates
We previously described strains BE, H6, and C28, members of PGG2 that share an identical spoligotype and have 1–3 copies of IS6110, acquired sequentially (Figures 1A and 2A) [19]. Analysis using 36-informative sSNPs that group M. tuberculosis into 9 lineages classified the 3 strains into group IV, within the Euro-American lineage [5, 6]. To identify precursor and descendant strains, we analyzed sSNP group IV isolates in our population-based collections by IS6110 RFLP and spoligotyping. CDC1551, a member of PGG2 and sSNP group V, was used as a reference. We identified 2 groups of strains H and C in PGG2 and sSNP group IV that are closely related to BE, H6, and C28 (Figure 1). Insertion-site mapping (ISM) identified a common IS6110 insertion in the E region (Rv0403c, mmpS1) of BE, H6, C28, H, C, and CDC1551. H, C, and CDC1551 share an additional IS6110 insertion in the DR region that is found in most M. tuberculosis strains but absent in BE, H6, and C28. The C strain has a third insertion in Rv0797, a hot-spot region for insertion-site elements, known as the insertion preferential locus (ipl) [26]. ISM data suggested that IS6110 elements were gained and lost in strain H to beget C and BE, respectively.
Figure 1.

IS6110-based restriction fragment length polymorphism and insertion-site mapping. A, Southern blot hybridization of Mycobacterium tuberculosis strains showing PvuII-restricted M. tuberculosis chromosomal DNA blot hybridized with BamHI–SalI fragment of IS6110. B, IS6110 insertion site mapping. Arrows indicate the hybridization bands corresponding to the IS6110 insertions in 5 M. tuberculosis chromosomal regions denoted by Rv numbers. Triangles schematically demonstrate the position and orientation of IS6110 [25]. Abbreviation: STD, molecular weight standards.
Figure 2.

Spacer oligonucleotide typing (spoligotyping) and the organization of the direct repeat (DR) locus (Rv2813–Rv2816c). A, Binary depiction of spoligotyping of Mycobacterium tuberculosis strains. Positive hybridization (probe specific to 43 standard spacers) is denoted by black squares. Row 1, H37Rv, spoligotype S00001; row 2, CDC1551, S00089; row 3, strain C, S00030; row 4, strain H, S00009; row 5, S75 group, S00075. B, The organization of the DR locus. C, Primer design for DR locus analysis (based on sequences in [27]). Boxes and numbers represent the presence and position of each discrete direct variant repeat (DVR). Boxes outlined in bold represent DVR duplications. Dashed lines indicate the loss of DVRs. Triangles depict the location of IS6110 insertions.
Spoligotyping indicated that BE (S00075; ST38) arose from H (S00009; ST137), following deletion of direct variable repeats (DVR) 20–25, including the loss of the IS6110 element in DR (Figure 2). Although the spoligotype of C (S00030; ST197) indicated relatedness to H [31], this pattern likely arose as a result of multiple DVR deletion events (Figure 2). Sequencing of the entire DR region indicated that DVR17 is duplicated in tandem among BE, H6, C28, C, H, and CDC1551, unlike other unrelated M. tuberculosis strains. Similarly, DVR25 is duplicated and translocated between DVRs 31 and 32 in C, H, and CDC1551. Although BE, H6, and C28 have DVRs 20–25 that were deleted, the translocated copy of DVR25 was found between DVRs 31 and 32 (Figure 2B), which suggests that the deletion involving IS6110 occurred after the DVR25 duplication. These DVR duplications are likely evolutionary scars that occurred prior to divergence of sSNP groups IV and V. Interestingly, this DVR25 duplication is shared among Mycobacterium africanum (GM041182; www.tbdb.org), Mycobacterium bovis (AF2122/97), and selected M. tuberculosis strains from sSNP groups II.A, IV, and V. The MIRU-VNTR profile of BE, H6, and C28 indicates homogeneity (224326153324) and close relatedness to H (224325153323). MIRU-VNTR pattern for C (222325143223) is closer to H compared with the other strains, supporting interstrain relatedness. The 5 representative M. tuberculosis clinical isolates characterized in these genetic studies and CDC1551 were used for phenotypic studies.
Growth of Strains in Broth and Human Monocytes
We compared the growth properties of the study strains to CDC1551 in axenic culture and in human monocytes. All strains grew similarly in broth culture over 2 weeks (Figure 3A). Human monocytes were infected at a 1:5 MOI, and intracellular growth was observed for 72 hours. H (doubling time: 52.8 ± 7.4) and C (57.2 ± 11.5) grew similarly and were considerably slower than BE (30.7 ± 6), H6 (25.5 ± 5.4), and C28 (24.8 ± 5) (Figure 3B). In another experiment, BE grew similarly to CDC1551 (Figure 3B, inset).
Figure 3.
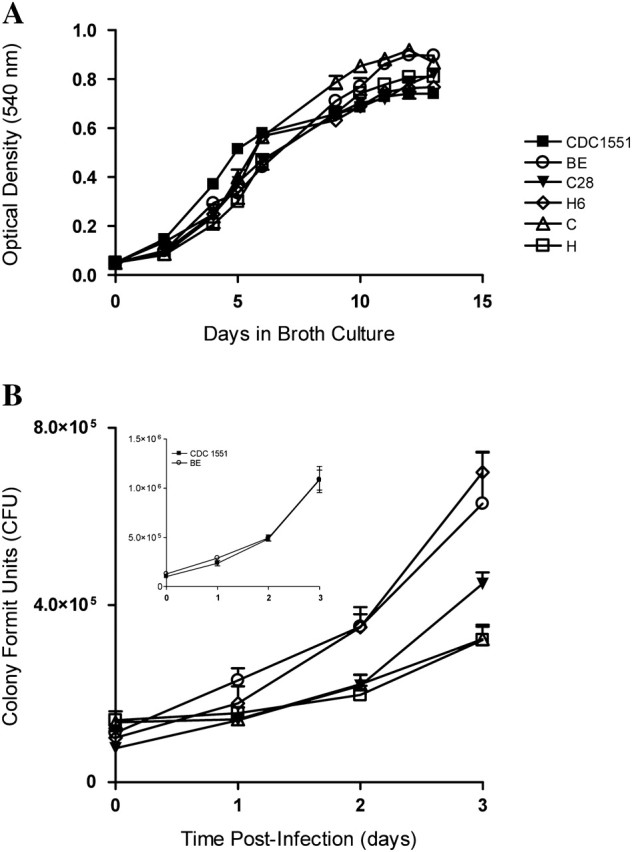
In vitro growth. A, Growth in 7H9 broth. B, Intracellular growth in human monocytes. Monocytes were infected with Mycobacterium tuberculosis clinical isolates at multiplicity of infection (MOI) 1:5 (bacilli:monocyte), for each strain, colony-forming units (CFUs) were enumerated at 24, 48, and 72 h postinfection. Results are the mean ± standard error of the mean (SEM) of 4 independent experiments (independent donors) done in duplicate. Significant difference was detected between BE, H6, and C28 compared with H (P < .001).
Effect of 5 Strains on Cytokine Production in Human Monocytes
To examine whether our slower growing (H and C) and faster growing (BE, H6, and C28) M. tuberculosis strains have a differential capacity to induce cytokine/chemokines, we infected adherent human monocytes and collected supernatants at 24 and 48 hours postinfection. BE, H6, and C28 induced significantly higher levels of IL-1β and TNF-α than did C and H (Figure 4A). Similar TNF-α levels were induced at 24 hours postinfection by BE and CDC1551 (Figure 4A, insert). To determine whether the differences in growth rates, and consequently the intracellular bacillary load, were responsible for the differential proinflammatory cytokine response, we stimulated monocytes with similar numbers of heat-killed H, C, BE, H6, or C28 (Figure 4B). Although the overall levels of induced cytokines were lower, the cytokine profile was similar to that induced by live organisms, indicating that the growth rates did not fully account for the cytokine differences. All viable and heat-killed strains induced similar amounts of the other cytokines/chemokines measured (not shown).
Figure 4.

Effect of live or heat-killed Mycobacterium tuberculosis clinical isolates on cytokine induction by human monocytes. Adherent monocytes were exposed to live (A) or heat-killed (B) M. tuberculosis at a multiplicity of infection (MOI) of 1:5 (bacilli:monocyte). Results are mean ± SD of 7 (A) or 4 (B) independent experiments (independent donors) done in duplicate. A 2-tailed paired t test was used for statistical analysis (*P < .05 and **P < .001 [in comparison to H]). Abbreviations: IL-1β, interleukin 1β; TNF-α, tumor necrosis factor α.
Assessment of Virulence in the Guinea Pig Infection Model
Analysis of survival rates of infected guinea pigs revealed considerable variation. Guinea pigs infected with BE (median survival, 10.0 weeks postinfection), H6 (8.3 weeks), or C28 (9.1 weeks) had shorter survival rates (P < .01) than those infected with H (30.0 weeks); animals infected with BE (P < .05), H6, or C28 (P < .01) had shorter survival rates than those infected with C (18.4 weeks). H-infected animals had significantly longer (P < .001) survival rates, compared with those infected with C (Figure 5A). In another experiment, BE-infected animals survived longer than those infected with H6 (P < .01), C28 (P < .05), or CDC1551 (ns) (Figure 5B). In a separate experiment, the numbers of CFUs in the lungs and spleen were determined at day 30 postinfection for each strain. No statistically significant differences in the CFU of the different strains were observed in the lung and spleen (not shown).
Figure 5.
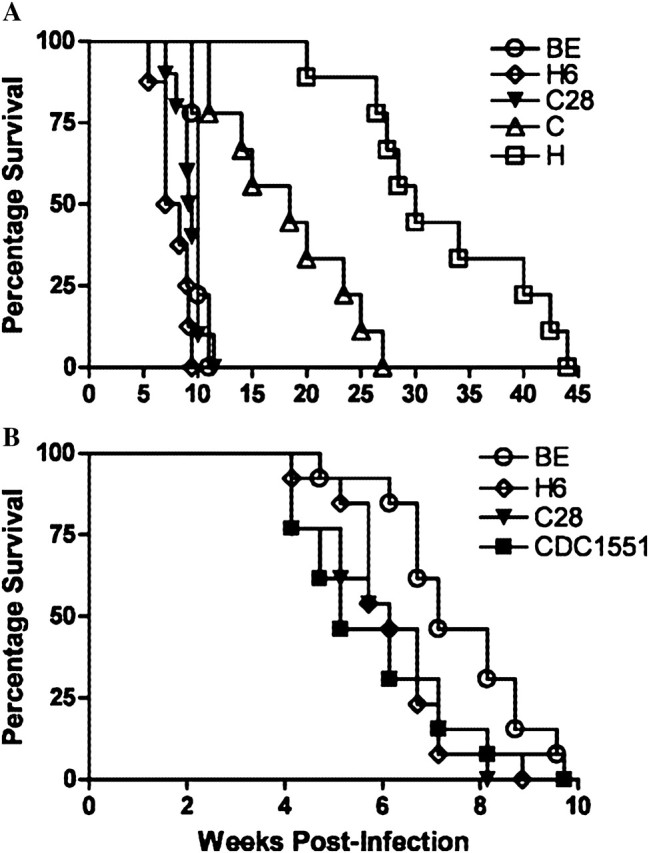
Survival of guinea pigs infected with Mycobacterium tuberculosis clinical isolates. Guinea pigs were infected by aerosol with average inoculum of 1 × 106 (range, 5.76–6.38 log) depositing 10–20 colony-forming units in the lungs. Guinea pig survival was plotted using the Kaplan-Meier method, and differences between curves were analyzed using the log-rank test. Results are from 2 representative experiments with 9 guinea pigs (A) and 13 guinea pigs (B) per group.
Using a standardized histologic score to evaluate total lung involvement and extent of necrosis in the granulomas, an increase (25%–35%) in pathology was noted in the lungs of guinea pigs infected with BE, H6, or C28 (not shown). Pairwise comparison of histologic scores within and between animals infected with faster or slower growing strains showed statistically significant differences. There were differences in fibrosis between guinea pigs infected with H vs BE (P = .005) or H6 (P = .006), in the extent of necrosis between animals infected with H vs H6 (P = .004) or C28 (P = .02) and the total lung score between animals infected with H vs BE (P = .009) or H6 (P = .03). Moreover, higher pathology scores in the spleen were noted between animals infected with H or C vs BE, H6, or C28 (P = .003–.037). Taken together, these results indicate that BE, H6, and C28 are more virulent in guinea pigs than are H and C.
Prevalence and Epidemiologic Characteristics
Collectively, these 5 strains were responsible for a significant proportion of tuberculosis cases in the NYC–New Jersey area (Figure 6). Among our NYC 2001–2008 population-based M. tuberculosis collection (N = 6260), the 5 strains accounted for an average 6.9% (SD ± 1.2) of all culture-positive cases, peaking at 9.5% in 2002. During this period, strains H, C, and BE caused a total of 420 cases, comprising 125, 207, and 88 cases, respectively, whereas only 1 H6 and no C28 cases were reported. Among US-born patients (n = 1787), the 5 strains accounted for an average 15.5% (SD ± 2.5) of tuberculosis cases, peaking at 21.2% in 2002. Additionally, population-based collection from 1996 to 2000 in New Jersey identified 133 cases (H, 42; C, 25; BE, 50; H6, 14; C28, 2), accounting for an average 7.1% (SD ± 2.4) of all tuberculosis cases, peaking at 10.5% in 1997 (Figure 6). Among US-born patients in New Jersey, these 5 strains accounted for an average 11.6% (SD ± 1.7) of tuberculosis cases, peaking at 12.5% in 1999. Finally, convenient collections from NYC and New Jersey identified an additional 221 cases and 88 cases, respectively (not shown).
Figure 6.
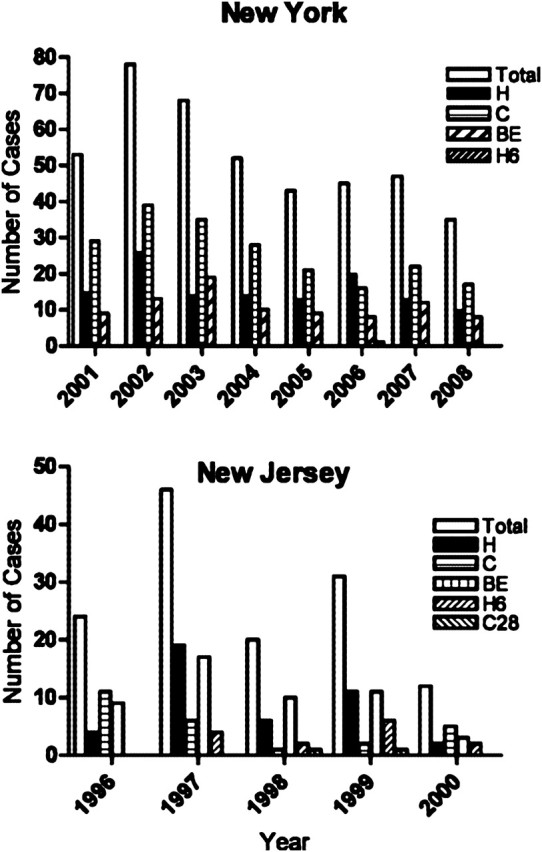
Number of culture-positive tuberculosis patients infected with strains H, C, BE, and H6 from population-based collection from New York City (NYC) between 2001 and 2008, and New Jersey between 1995 and 2000. Of note, no C28 cases were identified in NYC. Total bar refers to all H, C, BE, H6, and C28 cases.
Differences in the epidemiologic/demographic characteristics of patients infected with BE, H6, or C28 strains vs those with C or H infections in NYC during 2001–2008 highlighted further differences between these strain groups (Table 1). BE, H6, and C28-infected patients were younger and more likely to have well-established risk factors for tuberculosis, including human immunodeficiency virus (HIV) positivity, history of homelessness, and substance and alcohol abuse. Moreover, BE-, H6-, and C28-infected patients were more likely to have acid-fast bacilli (AFB) smear-positive sputum, compared with those infected with C or H. Drug resistance was more frequent among C or H strains, compared with the other 3 strains.
Table 1.
Demographic, Epidemiologic, and Clinical Characteristics of the Study Population From New York City, 2001–2008
| Characteristics | C and H, No. (%) (n = 284) | BE and H6,a No. (%) (n = 75) | P Valueb |
| Average age, years (IQRc) | 47.1 (16.5) | 43.7 (14) | .038d |
| Sex | |||
| Male | 200 (70.4) | 24 (68.0) | .684 |
| Female | 84 (29.6) | 51 (32.0) | |
| Racee | |||
| Asian | 15 (5.3) | 2 (2.7) | .542 |
| Hispanic | 176 (62.0) | 47 (62.7) | .912 |
| Non-Hispanic black | 64 (22.5) | 22 (29.3) | .220 |
| Non-Hispanic white | 29 (10.2) | 4 (5.3) | .262 |
| Country of origin | |||
| US-born | 217 (76.4) | 61 (81.3) | .364 |
| Non-US-born | 67 (23.6) | 14 (18.7) | |
| Years in United States | .153f | ||
| <1 | 4 (4.8) | 2 (9.1) | |
| 1–5 | 8 (9.7) | 5 (22.7) | |
| >5–10 | 5 (6.0) | 2 (9.1) | |
| >10 | 66 (79.5) | 13 (59.1) | |
| Homeless | |||
| Yes | 91 (32.0) | 39 (52.0) | .001 |
| No/unknown | 193 (68.0) | 36 (48.0) | |
| Substance use | |||
| Yes | 84 (30.2) | 37 (50.0) | .002 |
| No/unknown | 194 (69.8) | 37 (50.0) | |
| Alcohol abuse | |||
| Yes | 96 (34.5) | 36 (48.7) | .026 |
| No/unknown | 182 (65.5) | 38 (51.3) | |
| Site of disease | .388 | ||
| Pulmonary only | 198 (69.7) | 57 (76.0) | |
| Extrapulmonary only | 39 (13.7) | 6 (8.0) | |
| Both sites | 47 (16.6) | 12 (16.0) | |
| Cavitary lesions, among patients with pulmonary disease | |||
| Yes | 141 (75.0) | 44 (81.5) | .323 |
| No | 47 (25.0) | 10 (18.5) | |
| HIV serostatusg | |||
| Positive | 92 (39.2) | 51 (68.0) | <.001 |
| Negative | 143 (60.8) | 23 (30.7) | |
| Unknown | 49 (17.2) | 1 (1.3) | |
| Prior tuberculosis disease | |||
| Yes | 12 (4.2) | 5 (6.7) | .376 |
| No | 272 (95.8) | 70 (93.3) | |
| Respiratory AFB smear-positive | |||
| Yes | 156 (54.9) | 56 (74.7) | .007 |
| No | 111 (39.1) | 18 (24.0) | |
| Unknown | 17 (6.0) | 1 (1.3) | |
| Highest smear grade in first 30 d, among respiratory AFB smear-positiveh | .083 | ||
| 1 | 22 (7.8) | 15 (20.0) | |
| 2 | 18 (6.3) | 8 (10.7) | |
| 3 | 43 (15.1) | 14 (18.7) | |
| 4 | 48 (16.9) | 10 (13.3) | |
| Unknown | 153 (53.9) | 28 (37.3) | |
| Drug resistance | |||
| Drug-susceptible | 231 (81.6) | 73 (97.3) | <.001 |
| Any resistance | 38 (13.4) | 2 (2.7) | .008 |
| Mulitdrug resistance | 14 (4.9) | 0 (0.0) | .05 |
Abbreviations: AFB, acid-fast bacilli; HIV, human immunodeficiency virus; IQR, interquartile range.
No cases of disease with strain C28 were reported from New York City.
χ2 or 2-tailed Fisher exact test as appropriate.
Range between 25th and 75th percentiles.
P value for comparison of each race group with all other race groups.
Wilcoxon rank-sum test.
Freeman-Halton extension of the Fisher exact test.
Comparison with known HIV status (positive and negative).
Comparison with known smear grade (ie, 1, 2, 3, and 4).
DISCUSSION
In this study, we provide evidence that closely related M. tuberculosis strains can diversify sufficiently to lead to recognizably altered phenotypes, which appear to be associated with epidemiologic characteristics. The process of evolution/adaptation of M. tuberculosis in heterogeneous host populations is complicated by the pathogen’s obligate intracellular lifestyle, requiring coexistence with the host to survive. Thus, strain-specific changes in the bacterial population potentially involve a combination of pathogen characteristics, properties of the host, and environmental factors. Here, we show that subtle genetic changes in strain H gave rise to BE, a strain with altered phenotypic properties in both tissue culture and guinea pig infection models. Interestingly, the phenotypes appear to correlate with our empirical epidemiologic observation(s).
We show that the strain groups C and H are closely related to BE, H6, and C28 by descent and share key molecular signatures (Figures 1 and 2) [19]. Plasticity in the DR region, typically driven by homologous recombination between DRs, transposition of IS6110, and/or slippage during DNA replication, has been used to detail microevolutionary events [27, 32, 33]. Interestingly, some unrelated M. tuberculosis lineages and M. tuberculosis complex members, such as M. bovis [27], share the DVR25 duplication. It is possible, albeit unlikely, that the duplication events arose independently (convergence) or from random lineage-specific loss of the second copy of DVR25. Thus, the use of a combination of molecular markers with varying clock speeds can shed light on the order of events and group and distinguish closely related strains of M. tuberculosis.
Although the distribution of the 5 variants in our population may be anecdotal, it likely reflects the consequences of microevolutionary events modifying bacterial properties and/or host-pathogen interactions. Faster comparative M. tuberculosis growth, in vitro or in vivo, has been used as a proxy for virulence or bacterial fitness [34–36]. However, the correlation between growth rates and epidemiologic success is unclear [35–38]. One study examining growth and cytokine patterns among a large group of phylogenetically related strains did not find consistent profiles except within large clusters [39]. We found that strains that have caused extensive disease (C and H) grew significantly slower in human monocytes compared with other strains (Figure 3). Our intracellular studies suggest that the faster growing strains elicited significantly more TNF-α and IL-1β than these slower growing strains (Figure 4A). Previous studies have suggested that the cytokine-inducing capacity of M. tuberculosis is a property of bacillary load, which is determined by growth kinetics [35]. However, our results with heat-killed bacilli suggest that the differential cytokine profiles induced by these strains were independent of bacillary load. Previous studies have shown that early innate immune response is driven by intrinsic bacterial properties, independent of replication [15, 40]. In one report using human macrophage infections, overall levels of cytokine production appeared to track with phylogenetic lineages, although with some intralineage variation [17]. A recent study suggested that innate immune response and virulence, largely consistent within lineages, appears associated with distinct cell envelope lipid [41]. In contrast, a study by Wang et al [42] showed considerable variation among a diverse group of Beijing strains but did not find a distinct cytokine profile associated with successful sublineages.
Proteomic analysis of BE, H6, and C28 grown in broth cultures identified several differences in protein levels [43]. Strains H6 and C28 were found to produce higher levels of proteins involved in virulence, detoxification, metabolic pathways, and adaptation, and lower levels of proteins involved in cell wall and cell processes. Additionally, BfrB and Cfp29, B- and T-cell antigens, respectively [44, 45], were more abundant in H6 and C28 than in BE. The differential abundance of proteins may partly explain the subtle differentials in growth rates and cytokine induction in monocytes. These data suggest that, during the course of microevolution, M. tuberculosis can alter proteomic profiles, potentially affecting biomedically relevant properties.
Guinea pigs are relatively susceptible to low-dose aerosol infection and have been used extensively to examine virulence and pathogenesis of M. tuberculosis isolates [12]. Animals infected with strains BE, H6, or C28 had a significantly shorter median survival than those infected with C or H. In guinea pigs, survival is often proportional to levels of inflammation but not necessarily to bacillary burden and/or progressive pathology [46, 47]. In our study, the strains that induced higher levels of proinflammatory cytokines in vitro were associated with more inflammation in guinea pigs, as indicated by lung pathology, as well as more rapid mortality. In contrast, CDC1551 infection in mice has been associated with significantly longer survival than other M. tuberculosis strains, despite inducing high levels of proinflammatory cytokines [37].
The clinical strains in this report represent one of the largest groups of clustered strains circulating in the NYC region. In the early 1990s, the C strain was associated with an outbreak in a large homeless shelter in Manhattan and widespread dissemination in NYC, including 20% of all drug-susceptible tuberculosis cases reported in a large urban NYC hospital [48, 49]. The H strain and, to a lesser extent, BE have caused large clusters in the region for the last 15 years. Similarly, the most frequent drug-susceptible strain in San Francisco is closely related to H [33]. The high prevalence of our strains among US-born patients was largely consistent with previous reports [19, 49, 50].
Our study has demonstrated that natural variation, with detectable phenotypic changes, among clinical isolates may alter epidemiologic patterns in populations. Although our epidemiologic observation may be subject to confounding, it is possible that infection with M. tuberculosis strains that are good inducers of proinflammatory cytokines are controlled more efficiently in healthy individuals with an effective innate response. This may explain the limited number of secondary cases associated with the outbreak of immunogenic CDC1551 strain [36]. However, the spread of such strains may be unhindered in immunocompromised populations. Consistent with our findings, a separate study comparing tuberculosis cases infected with strains C and BE showed that BE cases were significantly associated with more respiratory AFB smear-positive status, HIV coinfection, and other traditional tuberculosis risk factors [50]. In contrast, M. tuberculosis strains that elicit a less protective innate immune response, such as the slower growing C and H strains, may more effectively cause active disease and promote spread in the general population. Thus, host factors can facilitate the selective spread of otherwise less epidemiologically fit organisms.
Numerous studies have highlighted large strain clusters and variants that have caused extensive disease. Reasons for spread have largely been attributed to individual- and ecological-level risk factors. Population-based studies are poised to identify variants and are capable of indicating M. tuberculosis microevolutionary processes within an epidemiologic context. Studies exacting the nature of other polymorphisms inherent in bacterial populations and their consequences by methods such as whole-genome sequencing are warranted. Our studies highlight the utility of examining strains that have, over the course of diversification, altered their epidemiology. Identifying such variants may be useful to study the natural evolution of virulence, pathogenesis, and bacterial fitness.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://www.oxfordjournals.org/our_journals/jid/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments.
We would like to thank Dr Daniel Sinsimer, Dr. Liang Chen, and Blas Pleixoto for technical assistance and Dawn Verdugo for critical reading of the manuscript.
Financial support.
This work was supported by tuberculosis vaccine testing and research materials (HHSN266200091c) to Colorado State University and R01AI066046 (to G. K.).
Potential conflicts of interest.
All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Brosch R, Gordon SV, Marmiesse M, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A. 2002;99:3684–9. doi: 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [see comments] [published erratum appears in Nature 1998 Nov 12;396(6707):190] [DOI] [PubMed] [Google Scholar]
- 3.Fleischmann RD, Alland D, Eisen JA, et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184:5479–90. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sreevatsan S, Pan X, Stockbauer KE, et al. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci U S A. 1997;94:9869–74. doi: 10.1073/pnas.94.18.9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comas I, Homolka S, Niemann S, Gagneux S. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS One. 2009;4:e7815. doi: 10.1371/journal.pone.0007815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutacker MM, Mathema B, Soini H, et al. Single-nucleotide polymorphism-based population genetic analysis of Mycobacterium tuberculosis strains from 4 geographic sites. J Infect Dis. 2006;193:121–8. doi: 10.1086/498574. [DOI] [PubMed] [Google Scholar]
- 7.Mathema B, Kurepina NE, Bifani PJ, Kreiswirth BN. Molecular epidemiology of tuberculosis: current insights. Clin Microbiol Rev. 2006;19:658–85. doi: 10.1128/CMR.00061-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hershberg R, Lipatov M, Small PM, et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 2008;6:e311. doi: 10.1371/journal.pbio.0060311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dormans J, Burger M, Aguilar D, et al. Correlation of virulence, lung pathology, bacterial load and delayed type hypersensitivity responses after infection with different Mycobacterium tuberculosis genotypes in a BALB/c mouse model. Clin Exp Immunol. 2004;137:460–8. doi: 10.1111/j.1365-2249.2004.02551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manca C, Tsenova L, Bergtold A, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha /beta. Proc Natl Acad Sci U S A. 2001;98:5752–7. doi: 10.1073/pnas.091096998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caws M, Thwaites G, Dunstan S, et al. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog. 2008;4:e1000034. doi: 10.1371/journal.ppat.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palanisamy GS, DuTeau N, Eisenach KD, et al. Clinical strains of Mycobacterium tuberculosis display a wide range of virulence in guinea pigs. Tuberculosis (Edinb) 2009;89:203–9. doi: 10.1016/j.tube.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Tsenova L, Ellison E, Harbacheuski R, et al. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J Infect Dis. 2005;192:98–106. doi: 10.1086/430614. [DOI] [PubMed] [Google Scholar]
- 14.Newton SM, Smith RJ, Wilkinson KA, et al. A deletion defining a common Asian lineage of Mycobacterium tuberculosis associates with immune subversion. Proc Natl Acad Sci U S A. 2006;103:15594–8. doi: 10.1073/pnas.0604283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rengarajan J, Murphy E, Park A, et al. Mycobacterium tuberculosis Rv2224c modulates innate immune responses. Proc Natl Acad Sci U S A. 2008;105:264–9. doi: 10.1073/pnas.0710601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reed MB, Domenech P, Manca C, et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–7. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- 17.Portevin D, Gagneux S, Comas I, Young D. Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog. 2011;7:e1001307. doi: 10.1371/journal.ppat.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homolka S, Niemann S, Russell DG, Rohde KH. Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: delineation of conserved core and lineage-specific transcriptomes during intracellular survival. PLoS Pathog. 2010;6:e1000988. doi: 10.1371/journal.ppat.1000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathema B, Bifani PJ, Driscoll J, et al. Identification and evolution of an IS6110 low-copy-number Mycobacterium tuberculosis cluster. J Infect Dis. 2002;185:641–9. doi: 10.1086/339345. [DOI] [PubMed] [Google Scholar]
- 20.Clark CM, Driver CR, Munsiff SS, et al. Universal genotyping in tuberculosis control program, New York City, 2001–2003. Emerg Infect Dis. 2006;12:719–24. doi: 10.3201/eid1205.050446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamerbeek J, Schouls L, Kolk A, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14. doi: 10.1128/jcm.35.4.907-914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Embden JD, Cave MD, Crawford JT, et al. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J Clin Microbiol. 1993;31:406–9. doi: 10.1128/jcm.31.2.406-409.1993. [see comments] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol Microbiol. 2000;36:762–71. doi: 10.1046/j.1365-2958.2000.01905.x. [DOI] [PubMed] [Google Scholar]
- 24.Steinlein LM, Crawford JT. Reverse dot blot assay (insertion site typing) for precise detection of sites of IS6110 insertion in the Mycobacterium tuberculosis genome. J Clin Microbiol. 2001;39:871–8. doi: 10.1128/JCM.39.3.871-878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurepina NE, Sreevatsan S, Plikaytis BB, et al. Characterization of the phylogenetic distribution and chromosomal insertion sites of five IS6110 elements in Mycobacterium tuberculosis: non-random integration in the dnaA–dnaN region. Tuber Lung Dis. 1998;79:31–42. doi: 10.1054/tuld.1998.0003. [DOI] [PubMed] [Google Scholar]
- 26.Fang Z, Kenna DT, Doig C, et al. Molecular evidence for independent occurrence of IS6110 insertions at the same sites of the genome of Mycobacterium tuberculosis in different clinical isolates. J Bacteriol. 2001;183:5279–84. doi: 10.1128/JB.183.18.5279-5284.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Embden JD, van Gorkom T, Kremer K, Jansen R, van Der Zeijst BA, Schouls LM. Genetic variation and evolutionary origin of the direct repeat locus of Mycobacterium tuberculosis complex bacteria. J Bacteriol. 2000;182:2393–401. doi: 10.1128/jb.182.9.2393-2401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molloy A, Meyn PA, Smith KD, Kaplan G. Recognition and destruction of bacillus Calmette-Guerin-infected human monocytes. J Exp Med. 1993;177:1691–8. doi: 10.1084/jem.177.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manca C, Paul S, Barry CE, 3rd, Freedman VH, Kaplan G. Mycobacterium tuberculosis catalase and peroxidase activities and resistance to oxidative killing in human monocytes in vitro. Infect Immun. 1999;67:74–9. doi: 10.1128/iai.67.1.74-79.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izzo A, Brandt L, Lasco T, Kipnis AP, Orme I. NIH pre-clinical screening program: overview and current status. Tuberculosis (Edinb) 2005;85:25–8. doi: 10.1016/j.tube.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 31.Brudey K, Driscoll JR, Rigouts L, et al. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23. doi: 10.1186/1471-2180-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warren RM, Streicher EM, Sampson SL, et al. Microevolution of the direct repeat region of Mycobacterium tuberculosis: implications for interpretation of spoligotyping data. J Clin Microbiol. 2002;40:4457–65. doi: 10.1128/JCM.40.12.4457-4465.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aga RS, Fair E, Abernethy NF, et al. Microevolution of the direct repeat locus of Mycobacterium tuberculosis in a strain prevalent in San Francisco. J Clin Microbiol. 2006;44:1558–60. doi: 10.1128/JCM.44.4.1558-1560.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312:1944–6. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 35.Theus SA, Cave MD, Eisenach KD. Intracellular macrophage growth rates and cytokine profiles of Mycobacterium tuberculosis strains with different transmission dynamics. J Infect Dis. 2005;191:453–60. doi: 10.1086/425936. [DOI] [PubMed] [Google Scholar]
- 36.Valway SE, Sanchez MP, Shinnick TF, et al. An outbreak involving extensive transmission of a virulent strain of Mycobacterium tuberculosis. N Engl J Med. 1998;338:633–9. doi: 10.1056/NEJM199803053381001. [DOI] [PubMed] [Google Scholar]
- 37.Manca C, Tsenova L, Barry CE, 3rd, et al. Mycobacterium tuberculosis CDC1551 induces a more vigorous host response in vivo and in vitro, but is not more virulent than other clinical isolates. J Immunol. 1999;162:6740–6. [PubMed] [Google Scholar]
- 38.North RJ, Izzo AA. Mycobacterial virulence: virulent strains of Mycobacteria tuberculosis have faster in vivo doubling times and are better equipped to resist growth-inhibiting functions of macrophages in the presence and absence of specific immunity. J Exp Med. 1993;177:1723–33. doi: 10.1084/jem.177.6.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Theus S, Eisenach K, Fomukong N, Silver RF, Cave MD. Beijing family Mycobacterium tuberculosis strains differ in their intracellular growth in THP-1 macrophages. Int J Tuberc Lung Dis. 2007;11:1087–93. [PubMed] [Google Scholar]
- 40.Murray RA, Mansoor N, Harbacheuski R, et al. Bacillus Calmette Guerin vaccination of human newborns induces a specific, functional CD8+ T cell response. J Immunol. 2006;177:5647–51. doi: 10.4049/jimmunol.177.8.5647. [DOI] [PubMed] [Google Scholar]
- 41.Krishnan N, Malaga W, Constant P, et al. Mycobacterium tuberculosis lineage influences innate immune response and virulence and is associated with distinct cell envelope lipid profiles. PLoS One. 2011;6:e23870. doi: 10.1371/journal.pone.0023870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C, Peyron P, Mestre O, et al. Innate immune response to Mycobacterium tuberculosis Beijing and other genotypes. PLoS One. 2010;5:e13594. doi: 10.1371/journal.pone.0013594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehaffy C, Hess A, Prenni JE, Mathema B, Kreiswirth B, Dobos KM. Descriptive proteomic analysis shows protein variability between closely related clinical isolates of Mycobacterium tuberculosis. Proteomics. 2010;10:1966–84. doi: 10.1002/pmic.200900836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenkrands I, Rasmussen PB, Carnio M, Jacobsen S, Theisen M, Andersen P. Identification and characterization of a 29-kilodalton protein from Mycobacterium tuberculosis culture filtrate recognized by mouse memory effector cells. Infect Immun. 1998;66:2728–35. doi: 10.1128/iai.66.6.2728-2735.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sartain MJ, Slayden RA, Singh KK, Laal S, Belisle JT. Disease state differentiation and identification of tuberculosis biomarkers via native antigen array profiling. Mol Cell Proteomics. 2006;5:2102–13. doi: 10.1074/mcp.M600089-MCP200. [DOI] [PubMed] [Google Scholar]
- 46.Lasco TM, Cassone L, Kamohara H, Yoshimura T, McMurray DN. Evaluating the role of tumor necrosis factor-alpha in experimental pulmonary tuberculosis in the guinea pig. Tuberculosis (Edinb) 2005;85:245–58. doi: 10.1016/j.tube.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 47.Palanisamy GS, Smith EE, Shanley CA, Ordway DJ, Orme IM, Basaraba RJ. Disseminated disease severity as a measure of virulence of Mycobacterium tuberculosis in the guinea pig model. Tuberculosis (Edinb) 2008;88:295–306. doi: 10.1016/j.tube.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Driver CR, Kreiswirth B, Macaraig M, et al. Molecular epidemiology of tuberculosis after declining incidence, New York City, 2001–2003. Epidemiol Infect. 2007;135:634–43. doi: 10.1017/S0950268806007278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Friedman CR, Quinn GC, Kreiswirth BN, et al. Widespread dissemination of a drug-susceptible strain of Mycobacterium tuberculosis. J Infect Dis. 1997;176:478–84. doi: 10.1086/514067. [see comments] [DOI] [PubMed] [Google Scholar]
- 50.Macaraig M, Agerton T, Driver CR, et al. Strain-specific differences in two large Mycobacterium tuberculosis genotype clusters in isolates collected from homeless patients in New York City from 2001 to 2004. J Clin Microbiol. 2006;44:2890–6. doi: 10.1128/JCM.00160-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.