

Abstract
Myeloperoxidase (MPO) is a hemoprotein, involved in the leukocyte mediated defense mechanism, and uses hydrogen peroxide (H2O2) and chloride (Cl-) to produce hypochlorous acid. In human saliva and hypochloremic alkalosis syndrome occurring in breast fed infants, the MPO-H2O2 system functions in lower Cl- concentration (10-70 mM) compared to plasma levels (100 mM) as part of the antibacterial defense system. The impact of low Cl- concentration and exposure to high peroxynitrite (ONOO-) synthesized from cigarette smoke or oxidative stress on MPO function is still unexplored. Rapid mixing of ONOO- and MPO caused immediate formation of a transient intermediate MPO Compound II which then decayed to MPO-Fe (III). Double mixing of MPO with ONOO- followed by H2O2 caused immediate formation of Compound II followed by MPO heme depletion, a process that occured independent of ONOO- concentration. Peroxynitrite/H2O2-mediated MPO heme depletion was confirmed by HPLC analysis and in-gel heme staining showing 60-70% less heme content compared to the control. A non-reducing denaturing SDS PAGE showed no fragmentation or degradation of protein. Myeloperoxidase heme loss was completely prevented by pre-incubation of MPO with saturated amounts of Cl-. Chloride binding to the active site of MPO constrains ONOO- binding by filling the space directly above the heme moiety or by causing a protein conformational change that constricts the distal heme pocket, thus preventing ONOO- from binding to MPO heme iron. Peroxynitrite interaction with MPO may serve as a novel mechanism for modulating MPO catalytic activity, influencing the regulation of local inflammatory and infectious events in vivo.
Keywords: Hydrogen peroxidase, hypohalous acid, inflammation, mammalian peroxidase, smoking
Introduction
Chloride (Cl-) is a major intracellular anion of living organisms due to its role in regulating electrogenic cation transport across intracellular and plasma membranes, membrane potential, cell volume, and intracellular pH [1,2]. The intracellular concentration of Cl- ranges from 1 to 60 mM while the extracellular concentration of Cl- is 100 mM (± 10 mM) [3-5]. Low serum Cl- levels have been observed in diseases associated with the loss of gastric fluids (vomiting and gastric suction), renal insufficiency, use of diuretics, chronic respiratory acidosis, and adrenocortical disorders [6,7]. The lowest level of Cl- (30–70 mM) is reported in hypochloremic alkalosis syndrome occurring in breast fed infants [6]. Defect of transmembrane Cl- channel is also associated with chronic inflammatory diseases such as cystic fibrosis [8]. Human saliva has low Cl- concentration (10-50 mM) and myeloperoxidase-hydrogen peroxide (MPO-H2O2) as parts of the oral antibacterial defense system [9,10]. The impact of low Cl- concentration and exposure to high peroynitrite (ONOO-) (from cigarette smoke or oxidative stress) on myeloperoxidase (MPO) function is still unexplored.
Myeloperoxidase catalyzes the formation of potent oxidants implicated in the pathogenesis of various diseases including atherosclerosis, asthma, arthritis, and cancer [11-13]. Myeloperoxidase uses H2O2 to oxidize Cl- to a potent oxidant, hypochlorous acid (HOCl). The enzyme first interacts with H2O2 to form a ferryl porphyrin Π cation radical [Fe(IV)=O•+Π], Compound I, which is capable of oxidizing Cl- through a 2e- transition to HOCl [12,14,15]. Hypochlorous acid is the active component of bleach and has potent bactericidal and viricidal activities [16]. Alternatively, compound I can oxidize various organic and inorganic compounds by two consecutive 1e− transitions, yielding radical species and the peroxidase intermediates compound II [Fe(IV) = O] and ground state, respectively [17-19].
Both MPO and ONOO- have emerged as potential participants in the promotion of reactive oxidants and diffusible radical species [20,21]. These oxidant species are capable of both initiating lipid peroxidation and promoting an array of post-translational modifications of target proteins, including halogenation, nitration, and oxidative cross-linking [20,21]. Peroxynitrite is a powerful oxidant that is generated through diffusion controlled reaction rates of nitric oxide (NO) with superoxide (O2•-) [21-23]. This reaction may be of particular importance wherever enhanced rates of NO and O2•- production occur. Peroxynitrite is a short-lived molecule and its concentrations are difficult to estimate in vivo because of its multiple pathways of production and reactions [22,23]. However, the rate of ONOO- production in vivo in specific compartments has been estimated to be as high as 50–100 μM per min [23,24]. Myeloperoxidase and its reactive oxidants commonly participate in tissue injury in a large number of inflammatory conditions and are used as markers for atherosclerotic cardiovascular disease [11-13].
Several studies demonstrated the ability of ONOO- to bind to the heme moiety of several hemoproteins, such as globins [25], peroxidases [26], cytochromes P450 [27], nitric oxide synthase (NOS) [28], and cyclooxygenase-2 (COX-2) [29]. In each case, the catalytic site of these enzymes was shown to interact with OONO- accelerating its decomposition. Myeloperoxidase and inducible NOS (iNOS) are both co-localized and secreted from the primary granules of activated leukocytes [30]; hence, MPO typically performs its functions in environments where NO and ONOO- are formed. We hypothesize that MPO may function differently under these conditions. To test this hypothesis, we evaluated MPO function in the presence of ONOO- in a low Cl- concentration environment. Our current results suggest that ONOO- in combination with H2O2 inhibit MPO through a mechanism that involves heme depletion. This mechanism may contribute to vascular endothelial dysfunction that is induced by oxidative stress in various diseases.
Materials and Methods
Materials
ONOO- used was of the highest purity grades obtained from Sigma Chemical Co. (St. Louis, MO).
Enzyme purification
Myeloperoxidase was purified from detergent extracts of human leukocytes as described [31-34]. Trace levels of contaminating eosinophil peroxidase (EPO) were then removed by passage over a sulphopropyl Sephadex column [35]. Purity of isolated MPO was established by demonstrating a Reinheitzal (RZ) value of >0.85 (A430/A280); SDS PAGE analysis was achieved with Coomassie blue staining, and in-gel tetramethylbenzidine peroxidase staining to confirm absence of observable contaminating EPO activity [37]. Enzyme concentration was determined spectrophotometrically utilizing extinction coefficients of 89,000 M-1cm-1/heme of MPO [31-34].
H2O2 selective electrode measurements
H2O2 measurements were carried out using an H2O2-selective electrode (Apollo 4000 Free Radical Analyzer; World Precision Instruments, Sarasota, FL). Experiments were performed at 25 °C by immersing the electrode in 3 ml of 0.2 M sodium phosphate buffer, pH 7.0, under air. In addition to the high concentration of the phosphate buffer, the desired ONOO- solutions were prepared in 0.002 M sodium hydroxide (NaOH) solution before used to keep the pH of the reaction mixture unaltered after the addition of ONOO-. 20 μM H2O2 was added to a continuously stirred buffer solution containing 100 mM Cl- during which the rise and fall of H2O2 concentration was continuously monitored. Where indicated, an increasing concentration of ONOO- and 20 μl MPO (30 nM final) was simultaneously added to the reaction mixture. To determine the effect of ONOO- on H2O2 consumption by MPO, similar experiments were repeated by the spontaneous addition of increasing concentration of ONOO- and 20 μl of the enzyme solution (20 nM final) to a continuously stirred H2O2 solution in the absence of Cl-.
Absorbance measurements
Spectra were recorded with a Cary 100 Bio UV-visible spectrophotometer, at 25 °C, in phosphate buffer pH 7.0. Experiments were performed in a 1 ml cuvette containing MPO (0.7-1.0 μM) pre-incubated with increasing concentrations of ONOO- (0–100 μM, final), in the absence and presence of 100 mM Cl-. Concentrated volumes of H2O2 solution were added to the sample cuvette (20 μM final), and absorbance changes were recorded from 300 to 700 nm.
Rapid kinetic measurements
The kinetic measurements of MPO Compound I and/or Compound II formation and decay in the absence and presence of different ONOO- and/or Cl- concentrations were performed using a diode array dual syringe stopped-flow instrument obtained from Hi-Tech, Ltd. (Model SF-61), at 10 °C. Diode array sequential mixing is a variation of stopped-flow where two reactants are mixed in a pre-mixer and then flow into an aging loop in the flow line to permit a delay to allow the reaction completion. After a specified period of time, that mixture is forced to mix with a third reactant and the reaction studied is followed by gathering the change in absorbance at single or multiple wavelengths as a function of time. The stopped-flow instrument was designed to collect multiple numbers of complete spectra (200-800 nm) at specific time ranges. The detector was automatically calibrated relative to a holmium oxide filter, as it has spectral peaks at 360.8, 418.5, 446.0, 453.4, 460.4, 536.4, and 637.5 nm, which were used by the software to correctly align pixel positions with wavelength. Experiments were initially performed under conditions identical to those recently reported for MPO and other related hemoproteins to facilitate comparison [31-34]. Double mixing stopped-flow experiments, involve pre-incubating solutions of 4 μM MPO with increasing Cl- concentrations following rapid mixing of equal volumes of an H2O2-containing buffer solution (80 μM H2O2) and/or different ONOO- concentrations (7-200 μM). After the reaction was completed, ONOO--treated and non-treated samples were collected to determine MPO heme content using the HPLC analysis. Reactions were monitored at both 432 and 450 nm. The time course of the absorbance change was fit to a single-exponential, (Y = -Ae-kt + C), or a double-exponential (Y = Ae-k1t + Be-k2t + C) function as indicated. Signal-to-noise ratios for all kinetic analyses were improved by averaging at least six to eight individual traces.
HPLC analysis and in-gel staining of heme continent in MPO–ONOO--treated and non-treated samples
10 μL of the ONOO--treated samples collected from the double mixing stopped-flow studies and identical concentration of untreated MPO were individually injected onto a Alltech, 5μm particle size, 4.6 × 150 mm reverse-phase octadecylsilica (C18) HPLC column. The column was eluted at a flow rate of 1.0 mL/min with linear gradients of buffers A and B (buffer A = 0.1% trifluoroacetic acid (TFA) in H2O; buffer B = 100% acetonitrite (CH3CN) containing 0.1% v/v TFA). The solvent gradient was 0% B at t = 0-5 min, then increasing from 40% to 60% B at t = 10-20 min. This composition was maintained until t = 25 min, before being reduced to the initial 0% B composition [36]. Under these conditions, the hemoprotein was completely resolved and eluted at 15 min, and was detected using a Shimadzu UV spectrophotometer set at 400 nm. Each sample was analyzed in duplicate or triplicate.
The ONOO-/H2O2-treated samples and the control were then individually concentrated using Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-50 membrane (from Millipore) with 30 kD cutoff by centrifuging at 5000 rpm for 30 min. After concentrating, protein content was quantified by Bradford assay using BSA as a standard. Twenty μg of MPO was incubated with Laemmli buffer [37] containing 63 mM Tris-HCl (pH 6.8), 2% (w/v) SDS, 10% (w/v) glycerol, and 0.0025% (w/v) bromophenol blue. 2-Mercaptoethanol/DTT, which interfered with the heme-staining method described below, was omitted from the Laemmli buffer. A non-reducing denaturing gel electrophoresis was performed for 4 h at a constant voltage of 60V on discontinuous 15% SDS-gels. Gels and buffers, which had been prepared according to Laemmli [37] were equilibrated at 4 °C prior to electrophoresis, and electrophoresis was performed at the same temperature. The gels were then either stained for protein with Coomassie Blue or for heme with O-dianisidine/H2O2 following a slight modification of a published method [38]. Gels were washed for 10 min in methanol/sodium acetate (0.25 M, pH 5.0) = 3:7 (v/v) and, subsequently, incubated in the dark for 20 min in a freshly prepared solution, containing 7 parts of 0.25 M sodium acetate, pH 5.0, and 3 parts of 12 mM O-dianisidine in methanol. Gels were developed by adding H2O2 to a final concentration of 1.25 M, (bands developed immediately) and washed for 30 min in H2O/methanol/acetic acid = 8:1:1 (v/v/v). The gels were scanned. Relative amounts of protein or heme were estimated by densitometric analysis of the scanned image using Image J software from NIH.
Solution preparation
Peroxynitrite solutions were prepared by diluting needed volume in 0.002 M NaOH solution. The concentration of the working solutions was determined spectrophotometrically by measuring abosorbance at 302 nm (extinction coefficient of 1,705 M-1cm-1). During preparation, all the solutions were kept on ice to minimize any decomposition. No changes in the pH of the reaction mixture were detected after mixing the basic ONOO- solution with the enzyme solutions.
Results
Peroxynitrite is an effective inhibitor of MPO
The ability of ONOO- to reduce the rate of H2O2 consumption by MPO in the absence and presence of plasma levels of Cl- suggests that ONOO- may serve as an effective inhibitor of MPO. To test our hypothesis, we sought to examine whether the rate of H2O2 consumption was reduced during MPO steady-state catalysis using an H2O2-selective electrode. Following addition of H2O2 solution (10 μl, 20 μM final) to the continuously stirred buffer solution containing plasma levels of Cl- (100 mM) (3 ml final), the H2O2 signal rose rapidly, achieved a maximum after ∼30 s, and fell gradually as H2O2 was depleted by autoreduction (2H2O2 → 2H2O +O2) (Fig. 1A). Addition of nanomolar levels of MPO to the reaction mixture caused a rapid decay in the level of free H2O2 (Fig. 1A), indicating that H2O2 is catalytically consumed by the peroxidase. Addition of ONOO- to the H2O2 solution in the absence of MPO had no significant effect on the rate of H2O2 decay. Simultaneous addition of increasing concentrations of ONOO- (0, 3, 6, 9, 12, 20 μM) and a fixed amount of MPO (20 nM) to the stirred H2O2 (20 μM) solution demonstrated a significant deceleration in H2O2 consumption (Fig. 1A). Thus, ONOO- down-regulates the rate of H2O2 consumption under conditions likely to be physiological in the phagolysosome or at sites of inflammation.
Fig. 1.
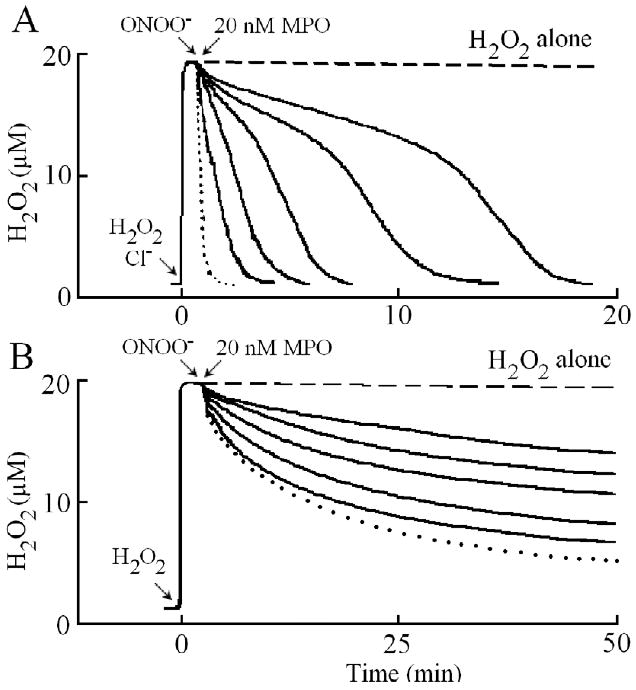
Peroxynitrite inhibits H2O2 consumption by MPO in the presence (Panel A) and absence (Panel B) of plasma levels of Cl-. Panel A, a typical recording by an H2O2-selective electrode demonstrating the autoreduction of H2O2 (20 μM) following its addition to a stirred 0.2 M phosphate buffer (pH 7.0) supplemented with 100 mM Cl-, at 25 °C (dashed line). Addition of 20 μl (30 μM in 3ml solution, final) of MPO solution to the H2O2/Cl- stirred buffer causes the rapid removal of H2O2 from the solution (dotted line). Simultaneous addition of fixed amounts of MPO (20 nM) and increasing concentrations of ONOO- (0, 3, 6, 9, 12, 20 μM, solid lines from left to right, respectively) caused slower H2O2 consumption. Panel B, a typical recording by an H2O2 -selective electrode demonstrating the auto-reduction of H2O2 (20 μM) following its addition to a stirred phosphate buffer (dashed line). Addition of MPO (300 nM) to a buffered solution supplemented with 20 μM H2O2 showed a rapid consumption of H2O2 followed by a slower decay (dotted line). Simultaneous addition of fixed amounts of MPO (300 nM) and increasing concentrations of ONOO- (3, 6, 9, 12, 20 μM, solid line from bottom to top, respectively) to H2O2 solution caused slower H2O2 consumption. The experiment shown is representative of three.
One important methodological consideration was to ensure that the extent of nitrite (NO2-)/nitrate (NO3-), the ONOO- decay products, was minimal so as not to contribute to H2O2 consumption by MPO. At the levels of ONOO- that produce dramatic effects on H2O2 consumption by the enzyme, the levels of NO2-/NO3- present are nominal. At the levels of NO2-/NO3- contaminating these preparations (0.03-3.0 μM), no significant effect are observed on rates of MPO compound II formation or decay (data not shown). Thus, MPO inhibition observed in Fig. 1A is due to the ONOO- interaction with MPO.
In the absence of Cl-, addition of MPO to the reaction mixture caused an immediate rapid decay in the level of free H2O2, followed by a slower decay indicating that H2O2 was consumed as a substrate by MPO during catalysis (Fig. 1B), as previously reported [31]. The first step occurred immediately after the enzyme addition and was attributed to the formation of MPO Compound I. Formation of Compound I requires multiple cycles of H2O2 consumption before it converts to Compound II, the inactive form of the enzyme. The second step was much slower and was limited by the conversion of MPO compound II to MPO-Fe(III). This interpretation is based on the previously reported spectral changes which showed that, addition of H2O2 to MPO-Fe(III) in the absence of co-substrates leads to the accumulation of compound II via rapid initial formation of compound I, and subsequent spontaneous one e- heme reduction [19]. Compound II, possesses a characteristic Soret absorbance peak at 455 nm that is easily distinguished from the Soret absorbance peaks of MPO-Fe(III) and the MPO Compound I. MPO compound II is unstable and converted gradually to the ground state, MPO-Fe(III), within minutes of initiating the reaction. Simultaneous addition of increasing concentrations of ONOO- (0, 3, 6, 9, 12, 20 μM) and fixed amount of MPO (12 μl, 20 nM final) to continuously stirred H2O2 (20 μM) solution decreased the rate of H2O2 consumption (Fig. 1B), indicating that ONOO- significantly prevented MPO catalysis. This indicates that ONOO- in the absence of Cl- may have served as an inhibitor of MPO under these experimental conditions.
Peroxynitrite modulates the catalytic activity of MPO
Peroxynitrite might theoretically serve as a ligand for MPO-Fe(III) and as a substrate for MPO Compounds I and/or II. In order to assess the physiological relevance of ONOO- binding to MPO, we determined the rates of ONOO- binding to the MPO-Fe(III). We first utilized diode array double mixing stopped-flow techniques to evaluate ONOO- interactions with MPO-Fe(III). The experiments were carried out at four different conditions: mixing ONOO- with MPO in the absence and presence of plasma levels of Cl-, and mixing MPO first with ONOO- in the absence and presence of Cl- and the mixtures then received equal concentration of H2O2. Under these conditions, the MPO-Fe(III) to Compound II transition can be followed as a shift in MPO Soret absorbance from 432 nm to 452 nm. MPO heme depletion can be followed by the loss of extinction at 452 nm. Due to the instability of ONOO-, we first mixed the basic ONOO- in an aging loop in the flow line to permit a delay to allow ONOO- solution to be neutralized, and the reaction mixture was rapidly mixed with MPO solution. MPO rapidly bound to ONOO-, and was directly converted to MPO Compound II without any sign of Compound I accumulation (Fig. 2A). Compound II was unstable and converted gradually to MPO-Fe(III), within minutes of initiating the reaction (Fig. 2B). Peroxynitrite-dependent formation of Compound II and complex decay reflect the use of ONOO- as a 1e- substrate for MPO Compounds I and II. The decay of Compound II was relatively fast and best fit to a single exponential process, generating a rate constant of 0.027 s-1.
Fig. 2.
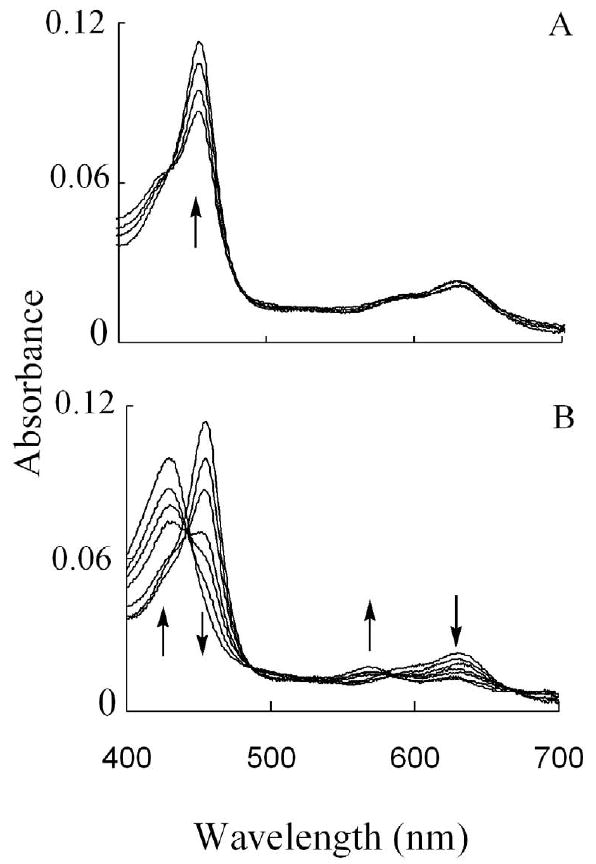
The interaction between MPO and ONOO- causes the transient formation of MPO Compound II which then decays to MPO-Fe(III). (Panel A) Absorbance spectra of MPO Compound II recorded by diode array double mixing stopped-flow at 10 °C, after mixing ONOO- solution (24 μM) with the phosphate buffer solution (0.1 mM, pH 7) in the aging loop, and the mixture was allowed to react with a buffer solution supplemented with 2 μM MPO. Spectra were collected at 0.002, 0.004, 0.067, and 0.149 s after initiating the reaction. (Panel B) Shows spectra traces for the same reaction collected at 14.2, 89, 116, 127, 134, 149, and 449 s after initiating the reaction. Arrows indicate the direction of spectral change over time. The experiment shown is representative of three.
To investigate the mechanism by which ONOO- and Cl- exert their effects, we utilized diode array stopped-flow spectroscopy to examine the buildup and decomposition of MPO Compound II. Investigations were carried out by rapidly mixing a ONOO- solution with MPO-Fe(III) incubated with increasing concentration of Cl-, after 1 s delay time for the reaction completion, the reaction mixtures received equal volume of H2O2. In the absence of Cl-, we observed a rapid accumulation of Compound II followed by heme depletion as judged by the significant loss in absorbance at 452 nm (Fig. 3A). Fig. 3A shows spectra collected at 5, 95, 295, 445, and 575 s of initiating the reaction indicating that MPO heme depletion was a slow process. A further slight absorbance increase was observed after 30 min at 432 nm, indicating that MPO heme depletion is a permanent process with slight recovery (Fig. 3A inset). Similar kinetics were observed in the MPO sample that was first equilibrated with 25 mM Cl-, except that the percentage of MPO-Fe(III) recovery monitored at 432 nm was slightly higher over the same time period (Fig 3B). Fig. 3B shows spectra collected at 35, 65, 105, 145, and 215 s of initiating the reaction. The inset of this panel shows recovery of MPO-Fe(III) over the next 30 min of initiating the reaction. Incubation of MPO with 50 mM Cl- was associated with partial conversion to Compound II with 75% recovery of MPO-Fe(III) (Fig. 3C). Fig. 3C shows spectra collected at 5, 15, 55, 85, 120 and 220 s of initiating the reaction. Pre-incubation of MPO with 100 mM Cl-, caused a slight rapid decrease in absorbance at 432 nm followed by immediate recovery similar to that previously observed in the absence of ONOO- where the enzyme generates HOCl. These results may suggest that, under these experimental conditions, the formation of MPO Compound I is slower than its decay, therefore, MPO compound I cannot be accumulated. These results may also indicate that plasma levels of Cl- prevent MPO heme depletion.
Fig. 3.
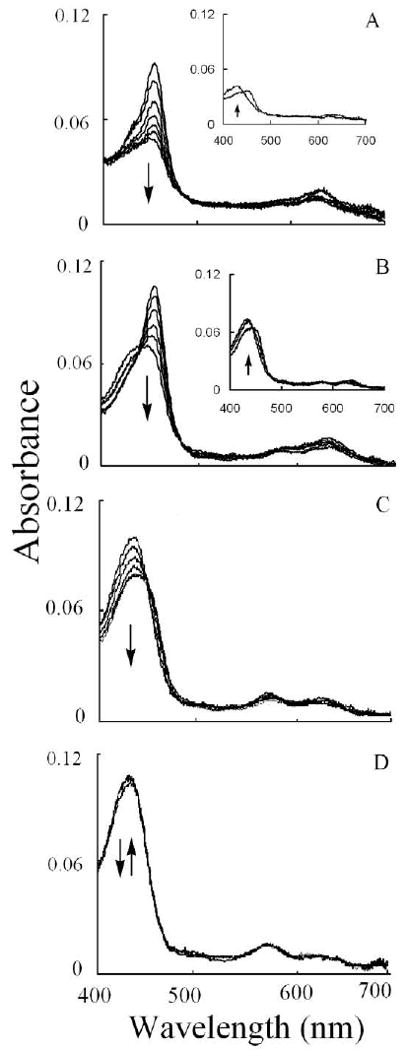
Chloride deficiency causes MPO heme depletion in the presence of ONOO-. Experiments were carried out by mixing a fixed amount of ONOO- solution with a phosphate buffer solution (0.2 M, pH 7) supplemented with a fixed amount of MPO and increasing Cl- concentrations in the aging loop, and the mixture was then allowed to react with a fixed amount of H2O2 solution using diode array double mixing stopped-flow at 10 °C, pH 7.0. The final MPO, ONOO-, and H2O2 concentrations used were 1, 6, and 20 μM, respectively, while the final Cl- concentrations used were 0 (Panel A), 25 (Panel B), 50 (Panel C), and 100 (Panel D) mM. Panel A shows spectra collected at 5, 95, 195, 295, 395, 445, and 575 s. The inset of this panel shows spectra collected at 575 and 800 s; Panel B shows spectra collected at 35, 65, 105, 145, 195, and 215 s. The inset of this panel shows spectra collect at 495, 995, and 1895 s; Panel C shows spectra collected at 5, 15, 55, 85, 120 and 220 s; while Panel D shows spectra collected at 0.01, 0.19, 0.69, 1.59, 3.39, and 5.59 s after initiating the reaction. Inserts of Panels A and B show the recovery of MPO-Fe (III). Arrows indicate the direction of spectral change over time. The experiment shown is representative of three.
As shown in Fig. 4 and summarized in the inset, the kinetics of the MPO heme depletion by ONOO- and H2O2 were slow with an observed rate constant of 0.002 s-1 and occur independent of ONOO- concentration.
Fig. 4.
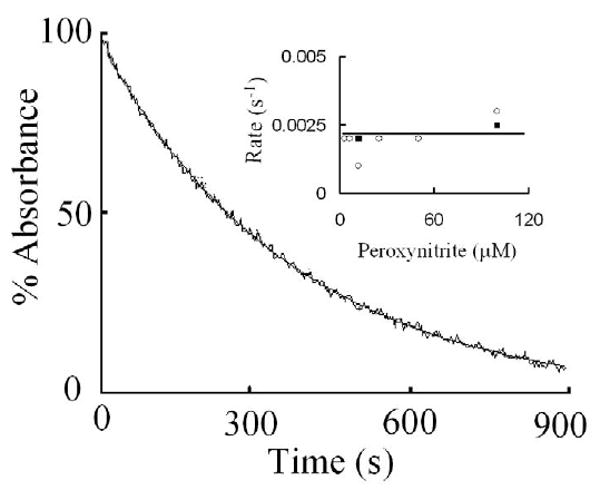
Myeloperoxidase heme depletion monitored at 452 nm is a relatively slow reaction. Experiments were carried out by mixing MPO solution with ONOO- in the absence of Cl-. The reaction mixture was then mixed using single wavelength double mixing stopped-flow methods. The final concentrations of MPO, ONOO-, and H2O2 were 4, 12, and 20 μM, respectively. The stopped-flow trace was best fitted to a single exponential function with a rate constant of 0.002 s-1. Insert shows rates of heme depletion as a function of ONOO- concentrations carried out in the presence of 25 (open circle) and 50 mM Cl- (closed circles). The data shown are representative of three similar experiments. The standard error for each individual rate constant has been estimated to be less than 8%.
We next investigated the ability of ONOO- to compete with Cl- and mediate MPO heme depletion. Fig. 5 compares the buildup of MPO Compound II over time after mixing MPO-Fe(III) pre-incubated with 25 mM Cl- with increasing ONOO- concentration and the reaction mixtures then received equal concentration of H2O2. There was a slight decrease in absorbance at 432 nm followed by instant recovery in the absence of ONOO- (Fig. 5A). A decrease in the Soret absorbance peak to a greater extent (25%) was observed in the presence of 3 μM ONOO-. In comparison, a direct conversion to Compound II to a maximum extent followed by heme depletion, as judged by the loss in the absorbance at 452 nm, was observed in the presence of 6 μM or higher ONOO-. In all cases, a 5-10% increase in absorbance was observed at 432 nm over a 90-min period, indicating that the MPO heme depletion is permanent, but with slight recovery.
Fig. 5.
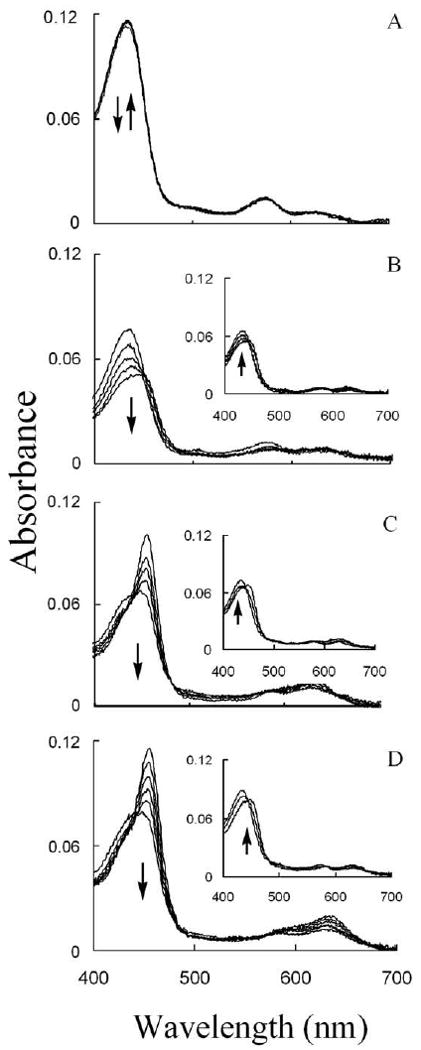
In the presence of sub-saturated amount of Cl-, MPO heme depletion depends on ONOO- bioavailability. Experiments were carried out by mixing ONOO- solution with a phosphate buffer solution (0.2 M, pH 7) supplemented with a fixed amount of MPO and fixed amount of Cl− in the aging loop, and the mixture was then allowed to react with fixed amount of H2O2 solution using diode array double-mixing stopped-flow methods at 10 °C. The final MPO, Cl-, and H2O2 concentrations used were 1 μM, 25 mM, and 20 μM, respectively. The final ONOO- concentration used were 0 (Panel A), 3 (Panel B), 6 (Panel C), and 12 (Panel D) μM. Panel A shows spectra collected at 1, 2, and 200 s; Panel B shows spectra collected at 0.05, 0.150, 0.55, 1.05, and 2.25 s; Panel C shows spectra collected at 0, 10, 15, 22, and 40 s; while Panel D shows spectra collected at 25, 55, 95, 145, 215, and 395 s after initiating the reactions. The insets show the recovery of MPO-Fe (III) over 90 min. Arrows indicate the direction of spectral change over time.
To investigate how the flattening in the Soret absorbance peak at 452 nm, in H2O2/ONOO- treated samples, is related to MPO heme depletion, we analyzed the heme content in both H2O2/ONOO- treated and non-treated samples by means of HPLC (Fig. 6) and in-gel heme staining (Fig. 7). HPLC analysis showed that there was approximately 60-70% less heme content in ONOO-/H2O2 treated samples compared to non-treated sample (Fig. 6). This was confirmed by heme staining of the gels by means of the O-dianisidine/H2O2 method (Fig. 7, right panel) which also showed 60-70% less heme content in the absence of Cl- compared to 5-10% less heme content in the presence of Cl- compared to control (non-treated MPO sample) (Fig. 7, left panel). In both cases, non-reducing denaturing SDS PAGE showed no fragmentation or degradation of protein (Fig. 7, right panel).
Fig. 6.
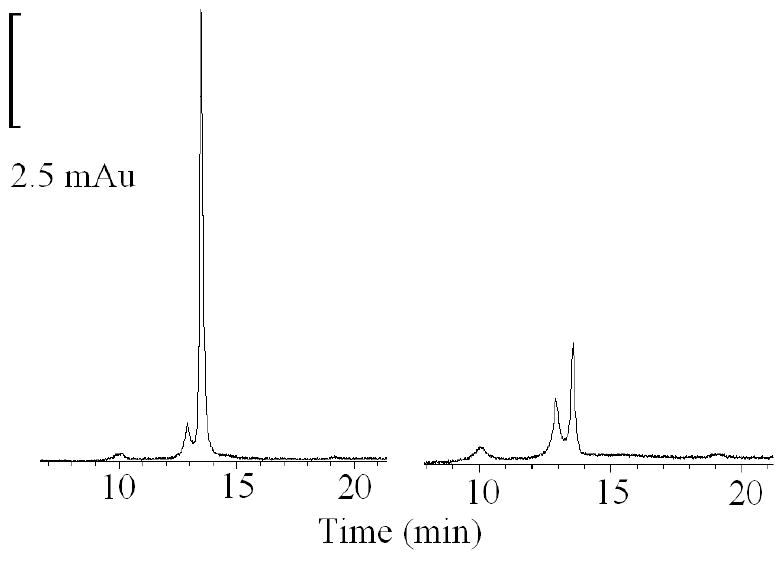
HPLC profiles of MPO and ONOO-treated MPO. Samples were taken from the reaction mixtures described in Fig. 4A and were analyzed by HPLC on a C18 reverse phase column (0.46 × 25 cm). Mobile phase was phosphate buffer with linear gradient from 0-60% of CH3CN with 0.1% TFA over 30 min at room temperature. Flow rate was 1 ml/min. The data shown are representative of three similar experiments.
Fig. 7.
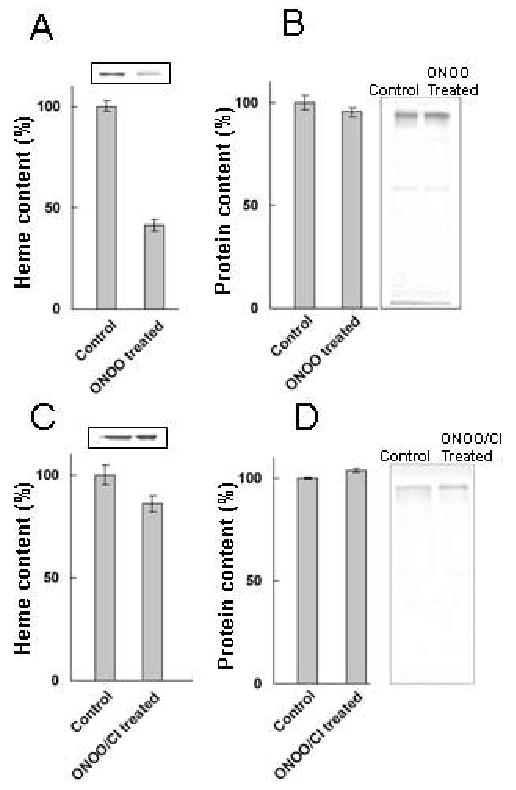
Effect of ONOO- and ONOO-/Cl- treatment on heme and protein content of MPO. Samples were taken from the reaction mixtures described in Fig. 4A and C and concentrated. Identical amounts of protein were run on a 15% non-reducing denaturing SDS PAGE and stained for heme (using O- dianisidine/H2O2) and for protein (using Coomassie Blue), as described in Materials and Methods. The bars represent ± SEM of heme and protein content respectively. Panel A and C are heme content in ONOO- treated MPO in the absence and presence of 100 mM Cl-, respectively. Panel B and D are Coomassie staining showing the protein content in the control and ONOO- treated MPO that carries out in the absence and presence of 100 mM Cl-, respectively. The data shown are representative of three similar experiments.
In addition, we have carried our experiments in the presence of bicarbonate and we did not notice any alterations in the kinetics of the reaction. CO2 presence has no influence on ONOO-/H2O2-mediated MPO heme depletion. Peroxynitrite reacts with CO2 to produce 1-carboxylato-2-nitrosodioxidane (ONOOCO2-). ONOOCO2- is extremely labile compound (half-life < 1μs), and rapidly decay to yield •NO2 and •CO3- [39]. No MPO modification has been observed in the range of ONOO- used. However, MPO protein nitration and protein fragmentation have been observed when higher ONOO- concentration was used (< 400 μ M) (data not shown).
Discussion
The results of the present study suggest that low Cl- concentrations (< 75 mM) may not be sufficient to protect ONOO-/H2O2 – mediated MPO heme depletion and iron release. Peroxidase-dependent bacterial killing requires active peroxidase and a physiological flux of the co-substrates H2O2 and Cl-. Thus, the impairment of MPO under increased ONOO- level and Cl--deficient environment may contribute to infection and inflammation. Consistent with this proposition, a number of human inflammatory diseases are characterized by low Cl- levels and free iron enhancement including cystic fibrosis, hypochloremic alkalosis, and gingivitis [6-10]. In addition, increased reactive oxygen species in oral fluid (where Cl- concentration is 50 mM) due to oxidative stress or smoking is a major cause of inflammation and microenvironmental tissue damage [40, 41]. Peroxynitrite interaction with MPO may serve as a novel mechanism for modulating MPO catalytic activity, influencing the regulation of local inflammatory and infectious events in vivo. In order to elucidate the negative impact of MPO and ONOO- interactions, the influence of ONOO- on MPO catalytic activity in low Cl- environment was investigated.
Our results elucidate the bi-directional relationship between the availability of ONOO-/Cl-/H2O2 and MPO catalytic activity and function. Using a combination of biochemical, physiological, and kinetic approaches, we showed that MPO may serve as a catalytic sink for ONOO-, limiting its bioavailability and function. ONOO- may modulate peroxidase catalytic activity by acting as both a ligand and a substrate. Fig. 8 shows a model that describes the interaction of MPO with Cl- and ONOO-. In this model, ONOO- may partition between two cycles during steady-state catalysis of MPO: cycle A that generates HOCl and cycle B that involves formation of a MPO-Fe(III)-ONOO complex. In the cycle A, the enzyme first interacts with H2O2 to form Compound I, which is capable of oxidizing Cl- through a 2e- transition to HOCl. The direct reaction between MPO Compound I and Cl- is extremely fast and occurs with second-order rate constants ranging from 2.2 × 106 to 4.7 × 106 M−1 s−1 [42, 43]. Because MPO Compound I formation rate is close in value to the 2e- oxidation of Cl-, Compound I accumulation cannot be detected during steady-state catalysis [31, 44, 45]. Alternatively, Compound I can oxidize ONOO- or its decomposed product, NO2-, by two consecutive 1e− transitions, yielding radical species and the peroxidase intermediates compound II and MPO-Fe(III), respectively. Utilizing double mixing stopped-flow methods, Furtmuller et al. have shown that ONOO- accelerates the formation of Compound II, when formed Compound I was rapidly mixed with ONOO-. A plot of ONOO- concentration versus rate of Compound I conversion to Compound II demonstrated linear kinetics and yielded a second order rate constant of 7.6 × 106 M-1s-1 [46]. Thus, the formation of the Fe(IV)=O•+Π complex prohibited ONOO- from binding to MPO heme iron, instead ONOO- served as a 1e- substrate for Compound I. In the same studies, the authors have shown that ONOO- stabilizes MPO Compound II but its decay product mediated the monophasic transitions of Compound II to MPO-Fe(III) with an observed second order rate constant of ∼ 440 M-1s-1 [46].
Fig. 8.
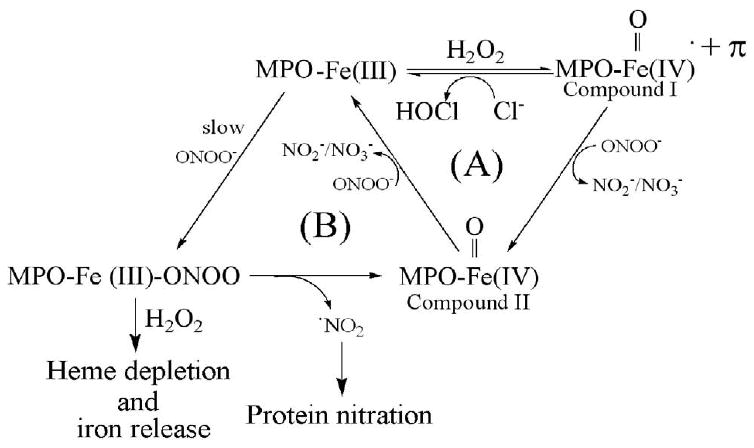
Proposed kinetic model showing the interaction of H2O2, Cl-, and ONOO- with MPO.
In a Cl- deficient atmosphere and in combination with H2O2 (cycle B), ONOO- may mediate MPO heme depletion. ONOO- can serve as a ligand for the enzyme and the presumed intermediate formed, MPO-Fe-ONOO, rendering it catalytically inactive. The ability of ONOO- to bind and generate a low-spin six-coordinate Fe-ONOO complex is consistent with the pocket being able to accommodate a large ligand like ONOO-. The intermediate formed is extremely labile and ONOO- undergoes a heme iron-catalyzed cleavage that leads to the accumulation of MPO Compound II and the release of a NO2 radical, a powerful nitrating specie. The subsequent stability of MPO Compound II depends on the experimental conditions. Particularly in the absence of H2O2 and through this sub-cycle, Compound II decays to MPO-Fe(III) with a relatively fast rate consistent with ONOO- or its decay product serving as a 1e- reductant for Compound II.
Myeloperoxidase heme destruction is relatively slow process and only achieves partial reactivation. Indeed, in the presence of H2O2 and ONOO-, only a small portion of the spectrum (5-10%) is converted to MPO-Fe(III) over a 1.5 h period. The MPO system described here may be the clearest example, to date, of MPO heme undergoing degradation. Thus, consumption of H2O2 and the formation of HOCl is not possible through this pathway. Significant conversion to MPO-Fe(III) is observed only under conditions where H2O2 is deficient. A similar result has been observed in the presence of sub-saturated amount of Cl-. Peroxynitrite is a relatively bulky molecule and its binding to the sixth coordinate position on the distal MPO heme group is limited by the close proximity of surrounding amino acids [47]. Previous studies by Bolscher and Wever have demonstrated that hydrogen bonding and co-substrate interaction play a contributory and even predominant role in ligand discrimination by MPO [48]. This MPO heme destruction is prevented to a large extent by prior Cl- binding to the enzyme, indicating an important role for Cl- in stabilizing the iron- nitrogen bond. Thus, Cl- binding to the active site of MPO may constrain ONOO- binding either by filling the space directly above the heme moiety or by causing a protein conformational change that constricts the distal heme pocket, thus preventing ONOO- from binding to the catalytic site of the enzyme which in this case is the heme moiety. This would explain the potential role of Cl- in preventing ONOO--mediated MPO heme depletion. The potential physiological relevance of this process was evaluated using rapid kinetic measurements and continuous monitoring of H2O2 levels with an H2O2-selective electrode. Thus, the order of addition (the enzyme and Cl- mixed first and ONOO- second, or the enzyme and ONOO- mixed first and Cl- second) may have a distinct effect on MPO catalytic activity and function.
The studies of the reaction of H2O2 with MPO-Fe(III)-ONOO complex suggest that the H2O2 plays a role in MPO heme depletion and perhaps in the iron release mechanism. Only the MPO-Fe(III)-ONOO Cl- deficient complex was degraded by H2O2. This state is spectrally distinguishable by the flattening in heme Soret region from 400 to 460 nm. Our stopped-flow diode array studies characterized the process. The flattening of the Soret peak of MPO can be explained by the oxidation of the heme prosthetic group that leads to heme degradation. The fact that this did not occur at all when the co-substrate, Cl-, was present indicates that this ion prevents the access of ONOO- to the MPO heme moiety. The ability of ONOO- to react with hemoproteins at nearly diffusion-controlled rates, promoting inhibition of many heme by interacting with their metal centers is well known [25-29]. Several studies have documented the ability of ONOO- to bind to the heme moiety of several hemoproteins, such as globins, peroxidases, cytochrome P450, NOS, and COX-2 [25-29]. In each case, the catalytic site of these enzymes was shown to interact with OONO- accelerating its decomposition. Since MPO heme depletion is mediated by a combination of both H2O2 and ONOO-, the MPO heme destruction mechanism that we are studying appears to be distinct from other hemoprotein model compounds.
Heme depletion was confirmed by HPLC and in-gel heme staining which clearly showed that there was 60-70% less heme content in peroxynitrite/H2O2-treated MPO compared to control. A mechanism that describes the involvement of both ONOO- and H2O2 in cytochrome P-450 heme degradation and could apply for MPO heme depletion was described by Schaefer et al. [49]. The free iron generated by MPO destruction may induce oxidative stress and make it highly toxic, as it can rapidly react with H2O2 and molecular oxygen to produce free radical [50]. Toxicity of free iron can damage blood vessels and produce vasodilation with increased vascular permeability, leading to hypotension and metabolic acidosis. Iron-related oxidative stress can promote lipid peroxidation, DNA strand breaks, and modification or degradation of biomolecules, eventually leading to cell death [51,52].
The bioavailability of Cl- and relative affinity of MPO-Fe(III) for either ONOO- or H2O2, and the relative concentrations of Cl- vs. ONOO-, are what govern the overall partitioning of ONOO- between the active (cycle A) and inactive cycles (cycle B) (Fig. 8). The ability of ONOO- to reduce the rate of H2O2 consumption by MPO in the presence of plasma levels of Cl- suggests that MPO displays higher affinity towards ONOO- vs Cl-. MPO reacts with ONOO- and is directly converted to MPO Compound II, an inactive form of the enzyme. This process limits MPO catalytic activity by the conversion of MPO Compound II to MPO-Fe(III). The transformation that is apparent from our H2O2-selective electrode measurements (Fig. 1A) may represent a switch in subcycles in which the first step in H2O2 consumption is limited by the conversion of MPO Compound II to MPO-Fe(III) or the recovery of MPO after heme depletion. In this case, the second step of H2O2 consumption is the peroxidation reaction after the enzyme's recovery following ONOO- treatment (Figs. 1 and 3). Under the later circumstances, the enzyme generates HOCl. Collectively, these studies suggest that ONOO- may function as a potent inhibitor of MPO at sites of inflammation, influencing its bioavailability and function. Since ONOO- competes directly with the co-substrate Cl- for the catalytic site of MPO, the ratio of the substrates to inhibitor is the critical factor determining the extent of the inhibition. When this ratio is high (see Figs. 2 and 3), the rate and the extent of this reaction approaches that of the uninhibited one. Thus, ONOO-/H2O2-mediated MPO heme destruction could occur at normal plasma levels but higher ONOO- concentration.
In this respect, MPO may serve as a source of free iron under oxidative stress when both NO and O2•- are elevated. Recent studies have shown that catalytically active MPO and its oxidative species are present in human atherosclerotic lesions [13]. This implicates the enzyme involvement in low-density lipoprotein (LDL) oxidation in vivo [13]. It has also been shown that iron accumulates in atherosclerotic lesions in a catalytically active form. The source of this iron is still unclear, but it is thought to be hemoglobin released from damaged red cells at sites of vascular turbulence or in hemorrhagic atheromatous plaques [51,52]. Additionally, we suspect that exposure to high ONOO- from cigarette smoke can result in significant reduction of oral peroxidase levels via MPO heme depletion and interruption of the antimicrobial protective mechanisms of the respiratory and digestive systems. A defective oral peroxidase system can affect an integral part of the immunoprotective mechanisms of the respiratory and alimentary tracts and predispose them to chronic inflammation and infections [9,53]. This may explain the development of gingivitis and chronic periodental disease in smokers since smoking is the most common cause of these disorders in adults in the United States. A synergistic, deleterious interaction between cigarette or tobacco smoking and saliva has been reported [9,53]. This relationship may lead to rapid destruction of biological macromolecules such as enzymes and proteins as part of the pathogenesis and development of oral squamous cell carcinoma [9]. This lethal synergistic interaction of cigarette smoke and saliva is probably based on the reaction between redox-active metals in saliva and low reactive free radicals in cigarette smoke.
In related studies, we have shown that mammalian peroxidases may serve as a pathway for catalytic removal of NO at sites of inflammation by their ability to utilize NO as a 1e- substrate for both Compounds I and II [17,18,54]. Consistent with such an effect, MPO impairs NO-dependent vasodilation in isolated arterial and tracheal rings, and decreases NO bioavailability in cultured cells [54]. Myeloperoxidase-H2O2 system up-regulates the catalytic activity of iNOS by scavenging NO, and preventing feedback inhibition attributed to the formation of Fe-NO complex [18]. We have provided kinetic evidence for the existence of two distinct binding sites for halides which have a distinct effect on MPO heme iron microenvironment [55]. More recently, we have shown that melatonin and tryptophan are potent inhibitors of MPO [32,45,56].
Understanding the properties of MPO reaction in Cl- deficiency and under oxidative stress environment is important because major parts of MPO activity and its heme contents are reduced. Since this depletion is permanent in the presence of low Cl- and/or higher ONOO- concentration, cells that maintain sufficient concentrations of Cl- would be expected to avoid MPO heme depletion and subsequent enzyme inactivation as described in this report.
Acknowledgments
This work was supported by the National Institutes of Health grant RO1 HL066367 and a grant from the Children's Hospital of Michigan (to H. M. A.-S.); and by an award from the American Heart Association (S. G.).
Abbreviations
- Cl-
chloride
- MPO
myeloperoxidase
- H2O2
hydrogen peroxidase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Estévez R, Jentsch TJ. CLC chloride channels: correlating structure with function. Curr Opin Struct Biol. 2002;12:531–539. doi: 10.1016/s0959-440x(02)00358-5. [DOI] [PubMed] [Google Scholar]
- 2.Foskett K. ClC and CFTR chloride channel gating. Annu Rev Physiol. 1998;60:689–717. doi: 10.1146/annurev.physiol.60.1.689. [DOI] [PubMed] [Google Scholar]
- 3.McCormick DA. Membrane properties and neurotransmitter actions. In: Shepherd GM, editor. The synaptic organization of brain. New York: Oxford UP; 1990. pp. 32–66. [Google Scholar]
- 4.Wills MR. Value of plasma chloride concentration and acid-base status in the differential diagnosis of hyperparathyroidism from other causes of hypercalcaemia. J Clin Path. 1971;24:219–227. doi: 10.1136/jcp.24.3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia L, Rigoulet M, Georgescauld D, Dufy B, Sartor P. Regulation of intracellular chloride concentration in rat lactotrophs: possible role of mitochondria. FEBS Lett. 1997;400:113–118. doi: 10.1016/s0014-5793(96)01365-8. [DOI] [PubMed] [Google Scholar]
- 6.Harrington JT. Metabolic alkalosis. Kidney Int. 1984;26:88–97. doi: 10.1038/ki.1984.139. [DOI] [PubMed] [Google Scholar]
- 7.Wack RP, Roscelli JD. Chloride deficiency syndrome in older exclusively breast-fed infants. Arch Pediatr Adolesc Med. 1994;148:438–441. doi: 10.1001/archpedi.1994.02170040104023. [DOI] [PubMed] [Google Scholar]
- 8.Fischer H, Schwarzer C, Illek B. Vitamin C controls the cystic fibrosis transmembrane conductance regulator chloride channel. Proc Natl Acad Sci U S A. 2004;101:3691–3696. doi: 10.1073/pnas.0308393100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashby MT. Inorganic chemistry of defensive peroxidases in the human oral cavity. J Dent Res. 2008;87:900–914. doi: 10.1177/154405910808701003. [DOI] [PubMed] [Google Scholar]
- 10.Vilja P, Lumikari M, Tenovuo J, Sievers G, Tuohimaa P. Sensitive immunometric assays for secretory peroxidase and myeloperoxidase in human saliva. J Immunol Methods. 1991;141:277–284. doi: 10.1016/0022-1759(91)90154-8. [DOI] [PubMed] [Google Scholar]
- 11.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 12.Davies MJ, Hawkins CL, Pattison DI, Rees MD. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10:1199–234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 13.Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 14.Hurst JK. Myeloperoxidase-active site structure and catalytic mechanisms. In: Everse J, Everse K, Grisham MB, editors. Peroxidases in chemistry and biology. 1st. Boca Raton: CRC Press; 1991. pp. 37–62. [Google Scholar]
- 15.Harrison JE, Schultz J. Studies on the chlorinating activity of myeloperoxidase. J Biol Chem. 1976;251:1371–1374. [PubMed] [Google Scholar]
- 16.Albrich JM, McCarthy CA, Hurst JK. Biological reactivity of hypochlorous acid: implications for microbicidal mechanisms of leukocyte myeloperoxidase. Proc Natl Acad Sci U S A. 1981;78:210–214. doi: 10.1073/pnas.78.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abu-Soud HM, Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. J Biol Chem. 2000;275:37524–37532. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 18.Galijasevic S, Saed GM, Diamond MP, Abu-Soud HM. Myeloperoxidase up-regulates the catalytic activity of inducible nitric oxide synthase by preventing nitric oxide feedback inhibition. Proc Natl Acad Sci U S A. 2003;100:14766–14771. doi: 10.1073/pnas.2435008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kettle AJ, Winterbourn CC. Myeloperoxidase: a key regulator of neutrophil oxidant production. Redox Report. 1997;3:3–15. doi: 10.1080/13510002.1997.11747085. [DOI] [PubMed] [Google Scholar]
- 20.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 21.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271(5 Pt 1):C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 22.Kelm M, Dahmann R, Wink D, Feelisch M. The nitric oxide/superoxide assay. Insights into the biological chemistry of the NO/O2•- interaction. J Biol Chem. 1997;272:9922–9932. doi: 10.1074/jbc.272.15.9922. [DOI] [PubMed] [Google Scholar]
- 23.Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 24.Alvarez MN, Piacenza L, Irigoín F, Peluffo G, Radi R. Macrophage-derived peroxynitrite diffusion and toxicity to Trypanosoma cruzi. Arch Biochem Biophys. 2004;432:222–232. doi: 10.1016/j.abb.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 25.Herold S, Fago A. Reactions of peroxynitrite with globin proteins and their possible physiological role. Comp Biochem Physiol Mol Integr Physiol. 2005;142:124–129. doi: 10.1016/j.cbpb.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Floris R, Piersma SR, Yang G, Jones P, Wever R. Interaction of myeloperoxidase with peroxynitrite. A comparison with lactoperoxidase, horseradish peroxidase and catalase. Eur J Biochem. 1993;215:767–775. doi: 10.1111/j.1432-1033.1993.tb18091.x. Erratum in: Eur. J. Biochem 216:881. [DOI] [PubMed] [Google Scholar]
- 27.Mehl M, Daiber A, Herold S, Shoun H, Ullrich V. Peroxynitrite reaction with heme proteins. Nitric Oxide. 1999;3:142–152. doi: 10.1006/niox.1999.0217. [DOI] [PubMed] [Google Scholar]
- 28.Maréchal A, Mattioli TA, Stuehr DJ, Santolini J. Activation of peroxynitrite by inducible nitric-oxide synthase: a direct source of nitrative stress. J Biol Chem. 2007;282:14101–14112. doi: 10.1074/jbc.M609237200. [DOI] [PubMed] [Google Scholar]
- 29.Deeb RS, Hao G, Gross SS, Lainé M, Qiu JH, Resnick B, Barbar EJ, Hajjar DP, Upmacis RK. Heme catalyzes tyrosine 385 nitration and inactivation of prostaglandin H2 synthase-1 by peroxynitrite. J Lipid Res. 2006;47:898–911. doi: 10.1194/jlr.M500384-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Evans TJ, Buttery LD, Carpenter A, Springall DR, Polak JM, Cohen J. Cytokine-treated human neutrophils contain inducible nitric oxide synthase that produces nitration of ingested bacteria. Proc Natl Acad Sci U S A. 1996;93:9553–9558. doi: 10.1073/pnas.93.18.9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tahboub YR, Galijasevic S, Diamond MP, Abu-Soud HM. Thiocyanate modulates the catalytic activity of mammalian peroxidases. J Biol Chem. 2005;280:26129–26136. doi: 10.1074/jbc.M503027200. [DOI] [PubMed] [Google Scholar]
- 32.Galijasevic S, Abdulhamid I, Abu-Soud HM. Melatonin is a potent inhibitor for myeloperoxidase. Biochemistry. 2008;47:2668–2677. doi: 10.1021/bi702016q. [DOI] [PubMed] [Google Scholar]
- 33.Galijasevic S, Proteasa G, Abdulhamid I, Abu-Soud HM. The potential role of nitric oxide in substrate switching in eosinophil peroxidase. Biochemistry. 2007;46:406–415. doi: 10.1021/bi061177u. [DOI] [PubMed] [Google Scholar]
- 34.Abu-Soud HM, Raushel FM, Hazen SL. A novel multistep mechanism for oxygen binding to ferrous hemoproteins: rapid kinetic analysis of ferrous-dioxy myeloperoxidase (compound III) formation. Biochemistry. 2004;43:11589–11595. doi: 10.1021/bi049541h. [DOI] [PubMed] [Google Scholar]
- 35.Rakita RM, Michel BR, Rosen H. Differential inactivation of Escherichia coli membrane dehydrogenases by a myeloperoxidase-mediated antimicrobial system. Biochemistry. 1990;29:1075–1080. doi: 10.1021/bi00456a033. [DOI] [PubMed] [Google Scholar]
- 36.Aguilar MI. HPLC of peptides and proteins methods and protocols. In: Aguilar MI, editor. Methods in molecular biology. Vol. 251. Totowa, NJ: Humana Press Inc.; 2004. pp. 9–22. [Google Scholar]
- 37.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 38.Thomas PE, Ryan D, Levin W. Anal Biochem. 1976;75:168–76. doi: 10.1016/0003-2697(76)90067-1. [DOI] [PubMed] [Google Scholar]
- 39.Merényi G, Lind J. Thermodynamics of peroxynitrite and its CO2 adduct. Chem Res Toxicol. 1997;10:1216–20. doi: 10.1021/tx970101j. [DOI] [PubMed] [Google Scholar]
- 40.Chaushu G, Bercovici M, Dori S, Waller A, Taicher S, Kronenberg J, Talmi YP. Salivary flow and its relation with oral symptoms in terminally ill patients. Cancer. 2000;88:984–987. [PubMed] [Google Scholar]
- 41.Reznick AZ, Klein I, Eiserich JP, Cross CE, Nagler RM. Inhibition of oral peroxidase activity by cigarette smoke: in vivo and in vitro studies. Free Radic Biol Med. 2003;34:377–384. doi: 10.1016/s0891-5849(02)01297-2. [DOI] [PubMed] [Google Scholar]
- 42.Furtmüller PG, Obinger C, Hsuanyu Y, Dunford HB. Mechanism of reaction of myeloperoxidase with hydrogen peroxide and chloride ion. Eur J Biochem. 2000;267:5858–64. doi: 10.1046/j.1432-1327.2000.01491.x. [DOI] [PubMed] [Google Scholar]
- 43.Marquez LA, Dunford HB. Kinetics of oxidation of tyrosine and dityrosine by myeloperoxidase compounds I and II. Implications for lipoprotein peroxidation studies. J Biol Chem. 1995;270:30434–40. doi: 10.1074/jbc.270.51.30434. [DOI] [PubMed] [Google Scholar]
- 44.Galijasevic S, Saed GM, Hazen SL, Abu-Soud HM. Myeloperoxidase metabolizes thiocyanate in a reaction driven by nitric oxide. Biochemistry. 2006;45:1255–1262. doi: 10.1021/bi051438k. [DOI] [PubMed] [Google Scholar]
- 45.Galijasevic S, Abdulhamid I, Abu-Soud HM. Potential role of tryptophan and chloride in the inhibition of human myeloperoxidase. Free Radic Biol Med. 2008;44:1570–1577. doi: 10.1016/j.freeradbiomed.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Furtmüller PG, Jantschko W, Zederbauer M, Schwanninger M, Jakopitsch C, Herold S, Koppenol WH, Obinger C. Peroxynitrite efficiently mediates the interconversion of redox intermediates of myeloperoxidase. Biochem Biophys Res Commun. 2005;337:944–954. doi: 10.1016/j.bbrc.2005.09.138. [DOI] [PubMed] [Google Scholar]
- 47.Fiedler TJ, Davey CA, Fenna RE. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 A resolution. J Biol Chem. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964. [DOI] [PubMed] [Google Scholar]
- 48.Bolscher BG, Wever R. A kinetic study of the reaction between human myeloperoxidase, hydroperoxides and cyanide. Inhibition by chloride and thiocyanate. Biochim Biophys Acta. 1984;788:1–10. doi: 10.1016/0167-4838(84)90290-5. [DOI] [PubMed] [Google Scholar]
- 49.Schaefer WH, Harris TM, Guengerich FP. Characterization of the enzymatic and nonenzymatic peroxidative degradation of iron porphyrins and cytochrome P-450 heme. Biochemistry. 1985;24:3254–3263. doi: 10.1021/bi00334a027. [DOI] [PubMed] [Google Scholar]
- 50.Wiseman JS, Nichols JS, Kolpak MX. Mechanism of inhibition of horseradish peroxidase by cyclopropanone hydrate. J Biol Chem. 1982;257:6328–6332. [PubMed] [Google Scholar]
- 51.Ong WY, Halliwell B. Iron, atherosclerosis, and neurodegeneration: a key role for cholesterol in promoting iron-dependent oxidative damage. Ann N Y Acad Sci. 2004;1012:51–64. doi: 10.1196/annals.1306.005. [DOI] [PubMed] [Google Scholar]
- 52.Trinder D, Fox C, Vautier G, Olynyk JK. Molecular pathogenesis of iron overload. Gut. 2002;51:290–295. doi: 10.1136/gut.51.2.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ihalin R, Loimaranta V, Tenovuo J. Origin, structure, and biological activities of peroxidases in human saliva. Arch Biochem Biophys. 2006;445:261–268. doi: 10.1016/j.abb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 54.Abu-Soud HM, Khassawneh MY, Sohn JT, Murray P, Haxhiu MA, Hazen SL. Peroxidases inhibit nitric oxide (NO) dependent bronchodilation: development of a model describing NO-peroxidase interactions. Biochemistry. 2001;40:11866–11875. doi: 10.1021/bi011206v. [DOI] [PubMed] [Google Scholar]
- 55.Proteasa G, Tahboub YR, Galijasevic S, Raushel FM, Abu-Soud HM. Kinetic evidence supports the existence of two halide binding sites that have a distinct impact on the heme iron microenvironment in myeloperoxidase. Biochemistry. 2007;46:398–405. doi: 10.1021/bi0609725. [DOI] [PubMed] [Google Scholar]
- 56.Lu T, Galijasevic S, Abdulhamid I, Abu-Soud HM. Analysis of the mechanism by which melatonin inhibits human eosinophil peroxidase. Br J Pharmacol. 2008;154:1308–1317. doi: 10.1038/bjp.2008.173. [DOI] [PMC free article] [PubMed] [Google Scholar]