

Abstract
The Beijing family of Mycobacterium tuberculosis strains is part of lineage 2 (also known as the East Asian lineage). In clinical studies, we have observed that isolates from the sublineage RD207 of lineage 2 were more readily transmitted among humans. To investigate the basis for this difference, we tested representative strains with the characteristic Beijing spoligotype from four of the five sublineages of lineage 2 in the guinea pig model and subjected these strains to comparative whole-genome sequencing. The results of these studies showed that all of the clinical strains were capable of growing and causing lung pathology in guinea pigs after low-dose aerosol exposure. Differences between the abilities of the four sublineages to grow in the lungs of these animals were not overt, but members of RD207 were significantly more pathogenic, resulting in severe lung damage. The RD207 strains also induced much higher levels of markers associated with regulatory T cells and showed a significant loss of activated T cells in the lungs over the course of the infections. Whole-genome sequencing of the strains revealed mutations specific for RD207 which may explain this difference. Based on these data, we hypothesize that the sublineages of M. tuberculosis are associated with distinct pathological and clinical phenotypes and that these differences influence the transmissibility of particular M. tuberculosis strains in human populations.
INTRODUCTION
The emergence and spread of apparently new strains of Mycobacterium tuberculosis, including multiple and extensively drug-resistant (M/XDR) strains, have raised considerable concern (43). Of particular concern is the Beijing family of strains, a family that is thought to have enhanced pathogenicity, a predilection for drug resistance (15, 47), and a less effective response to M. bovis BCG-based vaccines (32, 35).
The Beijing family belongs to lineage 2 (also known as the East Asian lineage) of M. tuberculosis. This lineage is defined by the region of difference (RD) RD105, is monophyletic, and is divided into five sublineages based on their specific RDs: RD105, RD207, RD181, RD150, and RD142 (48). Strains from four of the five sublineages (RD207, RD181, RD150, and RD142) have the characteristic spoligotype that defines the Beijing family (21). In a recent population-based study in San Francisco, CA, we demonstrated that different sublineages of lineage 2 differed in the number of secondary cases of tuberculosis they caused (19). Based on this observation, we hypothesized that there are differences in the pathogenicities of these sublineages. To investigate this hypothesis, we used an animal model to examine the pathogenicities of four sublineages among lineage 2. We examined the capacity of a representative panel of clinical isolates to cause infection and grow in the guinea pig model (31) after low-dose aerosol exposure. The primary question posed was whether our model could demonstrate increased pathogenicity of RD207, the sublineage that was associated with more secondary cases than the other three sublineages in San Francisco. In addition, we performed analysis of whole-genome sequence data to explore the possible mechanisms that could explain the different epidemiological and pathological characteristics of the different sublineages. We also repeated the epidemiological analysis with a larger sample size to confirm the relationship between the sublineages and their ability to cause secondary cases in San Francisco.
MATERIALS AND METHODS
Study population.
We have been conducting a population-based study of the molecular epidemiology of tuberculosis in San Francisco since 1991 (9). For the current study, we used M. tuberculosis identified as belonging to lineage 2 isolated from incident cases of tuberculosis between January 1991 and December 2008. The protocols and procedures for the protection of human participants were approved by the University of California, San Francisco.
Genotyping.
For the molecular epidemiological assessment, we used insertion sequence (IS)6110 restriction fragment length polymorphism (RFLP) to determine the genotype of clinical isolates of M. tuberculosis. Strains with the same IS6110 genotype (identical numbers and molecular weights of the IS6110 bands) were defined as clustered (51). Patients within the cluster were considered to have an epidemiologic link and thus to be part of a chain of transmission. The initial case identified was considered to be the index case, and subsequent cases were considered secondary cases. Cases having isolates with no matching RFLPs (unique cases) were considered a result of reactivation of latent infection. The methods for determination of lineage and sublineage have been described previously (16, 49).
Animal model.
Female outbred Hartley guinea pigs (∼500 g in weight) were purchased from the Charles River Laboratories (North Wilmington, MA) and held under barrier conditions in a biosafety level III animal laboratory. The specific-pathogen-free nature of the guinea pig colonies was demonstrated by testing sentinel animals. All experimental protocols were approved by the Animal Care and Usage Committee of Colorado State University and comply with NIH guidelines.
Experimental infections.
Ten strains representing the four sublineages with the Beijing spoligotype were used in this study (Table 1). These consisted of strains 4233, 4588, and 4619 (RD142), 3376 and 3446 (RD150), 3393, 3507, and 4147 (RD181), and 4334 and 5097 (RD207).
Table 1.
Characteristics of M. tuberculosis strains included in the animal model
| Sublineage | Specimen | Site of disease | Initial chest X-ray | HIV status | IS6110 band no. | IS6110 RFLP pattern | Type of case | Cluster size |
|---|---|---|---|---|---|---|---|---|
| RD142 | 4233 | Pulmonary | Abnormal, no cavities | Unknown | 18 | Unique | Unique | |
| RD142 | 4588 | Pulmonary | Abnormal, no cavities | Unknown | 17 | Unique | Unique | |
| RD142 | 4619 | Lymphatic-cervical | Abnormal, no cavities | Unknown | 19 | Unique | Unique | |
| RD150 | 3376 | Pulmonary | Abnormal, no cavities | Unknown | 21 | 2090000 | Secondary case | 10 |
| RD150 | 3446 | Pulmonary | Cavities | Negative | 21 | 2010000 | Secondary case | 5 |
| RD181 | 3393 | Pulmonary | Abnormal, no cavities | Unknown | 21 | Unique | Unique | |
| RD181 | 3507 | Pulmonary | Cavities | Unknown | 21 | Unique | Unique | |
| RD181 | 4147 | Pulmonary | Cavities | Unknown | 19 | Unique | Unique | |
| RD207 | 4334 | Pulmonary | Abnormal, no cavities | Unknown | 8 | 4990000 | First case | 2 |
| RD207 | 5097 | Pulmonary | Abnormal, no cavities | Unknown | 9 | Unique | Unique |
All strains were grown in 7H9 broth containing 0.05% Tween 80. Thawed aliquots of frozen cultures were diluted in sterile water to the desired inoculum concentrations. A Madison chamber aerosol generation device was used to expose the animals to M. tuberculosis. This device was calibrated to deliver approximately 20 bacilli into the lungs. Lung bacterial counts on days 30 and 60 were determined by plating serial dilutions of tissue homogenates on nutrient 7H11 agar and counting CFU after 3 weeks of incubation at 37°C. The infection inoculum and day 1 lung bacterial counts were determined for all the bacterial strains tested by plating serial dilutions of inoculum or tissue homogenates on nutrient 7H11 agar and counting CFU after 3 weeks' incubation at 37°C. No significant differences in terms of the infection dose or the day 1 bacterial uptake values were seen among any of the strains tested.
Histological analysis.
Same-lung lobes from each guinea pig were fixed with 4% paraformaldehyde in phosphate-buffered saline. Paraffin-embedded sections from these tissues were stained using hematoxylin and eosin and examined microscopically. The concurrent progression of lung and lymph node lesions was evaluated using a histological grading system (37).
Organ digestion.
To prepare single-cell suspensions, the same lungs, lymph nodes, and spleens were perfused with 20 ml of a solution containing phosphate-buffered saline (PBS) and heparin (50 U/ml; Sigma-Aldrich, St. Louis, MO) through the pulmonary artery. The caudal lobe was aseptically removed from the pulmonary cavity, placed in medium, and dissected. The dissected lung tissue was incubated with complete Dulbecco's modified Eagle medium (cDMEM) containing collagenase XI (0.7 mg/ml; Sigma-Aldrich) and type IV bovine pancreatic DNase (30 μg/ml; Sigma-Aldrich) for 30 min at 37°C. The digested lungs were further disrupted by gently pushing the tissue twice through a cell strainer (BD Biosciences, Lincoln Park, NJ). Red blood cells were lysed with ACK buffer, washed, and resuspended in cDMEM. Total cell numbers were determined by flow cytometry using BD Liquid Counting Beads, as described by the manufacturer (BD PharMingen, San Jose, CA).
Flow cytometric analysis of cell surface markers.
Single-cell suspensions from each individual guinea pig were first incubated as previously described (30, 31) with antibodies to CD4, CD8, pan T cell, CD45, MIL4, B cell, macrophage, and class II at 4°C for 30 min in the dark after washing the cells with PBS containing 0.1% sodium azide (Sigma-Aldrich). The anti-guinea pig macrophage MR-1 antibody is an intracytoplasmic antigen, and therefore cell membranes were permeabilized using Leucoperm (Serotec Inc., Raleigh, NC) according to the manufacturer's instructions prior to intracellular staining. Data acquisition and analysis were done using a FACSCalibur flow cytometer (BD Biosciences, Mountain View, CA) and the CellQuest software program (BD Biosciences, San Jose, CA). Compensation of the spectral overlap for each fluorochrome was done using CD4, MIL4, or CD3 antigen from cells gated in the forward-scattered light (FSClow) versus side-scattered light (SSClow), FSCmid/high versus SSCmid/high, or SSClow versus MIL4pos/neg, SSChigh versus MIL4neg, and SSChigh versus the MIL4pos region, respectively. Analyses were performed with an acquisition of at least 100,000 total events.
RT-PCR analysis.
Expression of mRNA encoding the cytokines gamma interferon (IFN-γ), interleukin 12p40 (IL-12p40), tumor necrosis factor alpha (TNF-α), transforming growth factor β (TGF-β), IL-17, and the regulatory T cell-associated intracellular marker Foxp3 was quantified using real-time reverse transcription-PCRs (RT-PCRs). The same lobe from each guinea pig (n = 5) lung was added to 1 ml of TRIzol RNA reagent (Invitrogen), homogenized, and frozen immediately, and total RNA was extracted according to the manufacturer's protocol. RNA samples from each group and each time point were reverse transcribed using the reverse transcriptase enzyme (Moloney murine leukemia virus [MMLV] reverse transcriptase; Invitrogen). Four-microliter samples of cDNA were then amplified using the iQ SYBR green supermix (Bio-Rad), following the manufacturer's protocol, on the iQ5 iCycler amplification detection system (Bio-Rad). A negative control using ultrapure Molecular Biology water as the template and a nontemplate control (NTC) were run to confirm that the signals were derived from RNA and not due to contaminating genomic DNA. In order to ensure that only the correct gene was amplified and not primer-dimer or nonspecific secondary products, a melting curve analysis was performed for each run. Fold induction of mRNA was determined by analyzing cycle threshold (CT) values normalized for hypoxanthine phosphoribosyltransferase (HPRT) (CT) expression. The primer sequences for guinea pig IFN-γ, IL-12p40, TNF-α, TGF-β1 and 18S were previously published (3, 26). The primer sequences for guinea pig Foxp3 were determined with assistance from Anand Damodaran (Genotypic Technology, Bangalore, India): forward, 5′ AGAAAGCACCCTTTCAAGCA 3′; reverse, 5′ GAGGAAGTCCTCTGGCTCCT 3′; and forward, 5′ TTCTTCCAAACACAGGATCAGC 3′; reverse, 5′ TCATTTCCGATAGGGCTTGG 3′. Primer sequences used for IL-17 were as follows: forward, 5′ CTCTGCAGGACCATCTC 3′; reverse, 5′ TTACTCGGGCTGTGTCAATG 3′; and forward, 5′ AGTCGTGTGTGATGGGAGTG 3′; reverse, 5′ TCAAGTTCCTGCTGCTGTTG 3′.
Whole-genome sequencing.
Illumina technology was used to sequence the whole genome of the 10 M. tuberculosis strains. Briefly, DNA was fragmented, end repaired, A′ tagged, ligated to adaptors, size selected, and enriched with 18 PCR cycles. Between 305 and 542 Mb of paired-end 51 cycle sequence data were generated on each library.
Mapping and SNP calling.
The software program BWA was used to map the reads from the 10 strains against the M. tuberculosis complex reference genome, which is a reconstructed ancestor that is H37Rv-like in its structure, but H37Rv alleles were replaced by those present in the inferred common ancestor of all M. tuberculosis complex lineages (12). BWA outputs were analyzed using the SAM tools software program (23, 24). We applied heuristic filters to remove problematic positions and a threshold of the probability of difference from the reference base. We set 200 as the maximum read depth to call a single-nucleotide polymorphism (SNP), and Phred-scaled probability was set as 20. SNP lists for individual strains were combined in a single nonredundant data set, and the corresponding base call was recovered for each strain. We excluded SNPs in PE/PPE genes, genes described as integrase, transposase, or phage, and SNPs for which at least one strain showed an ambiguous SNP call. The software program MEGA4 (46) was used to reconstruct a neighbor-joining phylogeny, using the number of differences for the 10 lineage 2 strains sequenced for this study, 23 sequences from different M. tuberculosis complex lineages previously published (11), and the sequence of an M. tuberculosis strain from the RD105 sublineage from our collection of strains. SNPs were mapped to the tree using the software program Mesquite (25), and specific sublineage SNPs were identified.
Prediction of functional effects of nsSNPs.
The SIFT (sorting intolerant from tolerant) algorithm (28) was used to predict the mutations most likely to affect protein function. SIFT searches for homologs of the gene of interest in other bacteria and does the following: (i) scores the conservation of the positions where mutations are found, and (ii) weights this score by the nature of the amino acid change. These measures were then used as a proxy for the impact of a specific mutation on protein function. Mutated positions with normalized probabilities less than 0.05 were predicted to have an impact on the protein, and those greater than or equal to 0.05 were predicted to have no impact (28). We used the nonredundant protein sequence database downloaded from NCBI on 6 June 2012. This database combines entries from GenPept, Swissprot, PIR, PDF, PDB, and NCBI RefSeq. Only nonsynonymous SNPs (nsSNPs) for monophyletic groups (observed in all its descendants) were included in the analysis.
Statistical analysis.
To determine the association between the sublineages and secondary cases, univariate analyses were performed using the χ2 test of proportions and the 2-tailed Fisher exact test. Univariate and multivariate odds ratios (ORs) were calculated using logistic regression with the secondary case status as the dependent variable. The independent variable of primary interest was the specific lineage 2 sublineage, and the analysis was controlled for place of birth (United States born versus foreign born), smear positivity, and cavitary disease. Statistical analyses were performed using the software program SAS version 9.2 (SAS Institute Inc., Cary, NC). The guinea pig data are representative of one experiment. Each experiment consisted of 5 guinea pigs infected with one of the M. tuberculosis strains for each of the time points 0, 30, and 60 days (total of 15 guinea pigs per strain). Mean values were calculated from results for individual guinea pigs within each group (n = 5) ± standard errors of the means (SEM). Student's t test was used to compare the statistical differences in numbers of bacilli, flow cytometric data, and RT-PCR values between different groups.
RESULTS
Relationship between sublineages and ability to cause secondary cases.
From January 1991 to December 2008, 3,847 cases of tuberculosis were reported in San Francisco, of which 3,311 (86%) had a positive culture for M. tuberculosis. RFLPs and lineage data were available for 2,361 (71%) of all culture-positive cases. There were 648 (27%) patients with M. tuberculosis from lineage 2, and 593 (92%) had sublineage data. We excluded 7 index cases with solely extrapulmonary disease, since their likelihood of transmitting M. tuberculosis is low, and assigned index case status to the next pulmonary case in sequence. The clinical characteristics were similar among patients with and those without sublineage data. Based on RFLP genotyping, there were 114 secondary cases associated with 65 index cases and 407 cases with unique isolates. Univariate analysis demonstrated that patients born in the United States were more likely to be secondary cases (OR, 6.61; 95% confidence interval [CI], 3.88 to 11.2; P < 0.001), as were patients with isolates from sublineage RD207 compared with the other sublineages (OR, 2.04; 95% CI, 1.02 to 4.08; P = 0.04) (Table 2). The multivariate analysis was based on 92 clustered cases (of 114) in 481 observations (sputum smear status was not available in several cases). It showed that the only independently significant risk factor for being a secondary case was being born in the United States (OR, 5.22; 95% CI, 2.89 to 9.42; P < 0.001). The adjusted odds of sublineage RD207 being a secondary case were 1.98 (95% CI, 0.91 to 4.29; P = 0.08) (Table 2), which is higher than we previously published (19) and supports the previous observation that in San Francisco, sublineage RD207 may be more likely than other sublineages to be transmitted.
Table 2.
Univariate and multivariate odds of being a secondary case
| Risk factor | Category | Univariate |
Multivariate |
||
|---|---|---|---|---|---|
| No. (%) of secondary cases | Odds ratioa (95% CI), P value | No. (%) of secondary cases | Odds ratiod (95% CI), P value | ||
| Sublineage | RD207 | 13 (32) | 2.04 (1.02–4.08), 0.043 | 13 (35) | 1.98 (0.91–4.29), 0.083 |
| Other | 101 (19) | 79 (18) | |||
| Birthplace | USA | 37 (54) | 6.61 (3.88–11.2), <0.001 | 29 (50) | 5.22 (2.89–9.42), <0.001 |
| Foreign | 77 (15) | 63 (15) | |||
| Cavitiesb | Yes | 10 (17) | 0.83 (0.41–1.69), 0.609 | 9 (16) | 0.72 (0.32–1.60), 0.416 |
| No | 103 (20) | 83 (20) | |||
| Sputum smear statusc | Positive | 40 (22) | 1.29 (0.81–2.04), 0.278 | 40 (22) | 1.28 (0.77–2.11), 0.335 |
| Negative | 53 (18) | 52 (17) | |||
Excluding 7 extrapulmonary index cases: 114 secondary cases in 586 observations.
Excluding 5 missing data on cavitary status: 113 secondary cases in 581 observations.
Excluding 103 missing data on smear status: 93 secondary cases in 483 observations.
Excluding 7 extrapulmonary index cases and 105 with missing data: multivariate model composed of 92 secondary cases in 481 observations.
Capacity of clinical isolates to grow after low-dose aerosol exposure.
The course of infection in the lungs of guinea pigs harboring each isolate is shown in Fig. 1. All 10 strains grew progressively for the first 30 days, with the members of the RD142, RD150, and RD207 sublineages growing well and with more modest growth seen for the three RD181 isolates.
Fig 1.

Course of infection following exposure of guinea pigs to approximately 20 viable M. tuberculosis bacilli representing four sublineages of lineage 2. The panel shows the bacterial growth in the lungs from guinea pigs receiving a low-dose aerosol of M. tuberculosis laboratory strains assayed on days 30 and 60 after infection. Results are expressed logarithmically as the mean log10 bacillus CFU (n = 5); SEM did not exceed 0.35.
All 10 infections caused moderate to severe pathology in the lungs over the course of the infection (Fig. 2 and 3). In all cases the infections induced extensive mixed inflammation and necrosis, but this was particularly pronounced in the case of infections with the two RD207 sublineage strains 4334 and 5097 (Fig. 3G to J), which established multiple large highly necrotic lesions in the lungs by day 30, resulting in marked consolidation by day 60. Milder degrees of pathology at day 30 were seen in the other groups, particularly RD150 and RD181. By day 60, lesions in all the animals infected with the clinical isolates showed severe increases in secondary lesion progression, characterized by multiple foci of extensive inflammation coalescing within the pulmonary parenchyma (Fig. 2 and 3), whereas lung involvement in the cases of the two RD207 sublineage strains was especially severe (Fig. 3G to J). These progressive changes in tissue lesions were further reflected by lesion score analysis, revealing that the RD207 sublineage strains showed statistically significantly more extensive damage in the lungs than all the other sublineages over the course of the infections (Fig. 4A and B).
Fig 2.

Granulomatous responses from guinea pigs infected with sublineage RD142 and RD150 strains of M. tuberculosis. The panel shows representative photomicrographs from sections of paraformaldehyde-fixed and paraffin-embedded guinea pig tissues from the same lungs which were collected on days 30 and 60 after infection with RD142 (A to F) and RD150 (G to J) strains. In all animals, there were coalescing foci of inflammation that tracked along airways. Inflammatory lesions had central regions of necrosis surrounded by histiocytic cells and peripheral lymphocytes. By day 60, there was often mineral present within the lesions. The severity of inflammation and pulmonary consolidation was greater for RD142 than for RD150. Hematoxylin-and-eosin staining; total magnification (A to J), ×10.
Fig 3.

Granulomatous responses from guinea pigs infected with sublineage RD181 and RD207 strains of M. tuberculosis. The panel shows representative photomicrographs from sections of paraformaldehyde-fixed and paraffin-embedded guinea pig tissues from the same lungs, which were collected on days 30 and 60 after infection with RD181 (A to F) and RD207 (G to J) strains. Pulmonary pathology was similar to that seen with the RD142 and RD150 strains (Fig. 2). The extent of lung involvement was notably greater with the RD207 strains than with the RD181 strains and was very severe by day 60 (G to J). Hematoxylin-and-eosin staining; total magnification (A to J), ×10.
Fig 4.

The sublineage RD207 shows increased lesion scores in the lungs. Lung lesion scores for the guinea pigs on day 30 (A) or day 60 (B) after infection with the various sublineage strains. The histopathology was characterized using a lesion scoring system that showed the significant extent of lung disease compared to that of the other organs during chronic infection (n = 5; *, Student's t test, P < 0.05). Please note that the calcification/necrosis ratio was zero for all strains in panel A.
Immune responses to clinical isolates.
We tracked the expression of the TH1 cytokines IFN-γ, IL-12p40, and TNF-α (Fig. 5) and compared this information based on the closer phylogenetic relationship between the respective sublineages to levels of proinflammatory IL-17 and Foxp3 and TGF-β (Fig. 6), markers associated with downregulation of immunity. All 10 strains generated appreciable levels of the cytokines IFN-γ, IL-12p40, and TNF-α, indicating generation of a TH1 response (Fig. 5A to C). Whereas strong signals were observed for IFN-γ (Fig. 5A), these waned significantly by day 60 in animals infected with the RD150 and RD207 strains.
Fig 5.
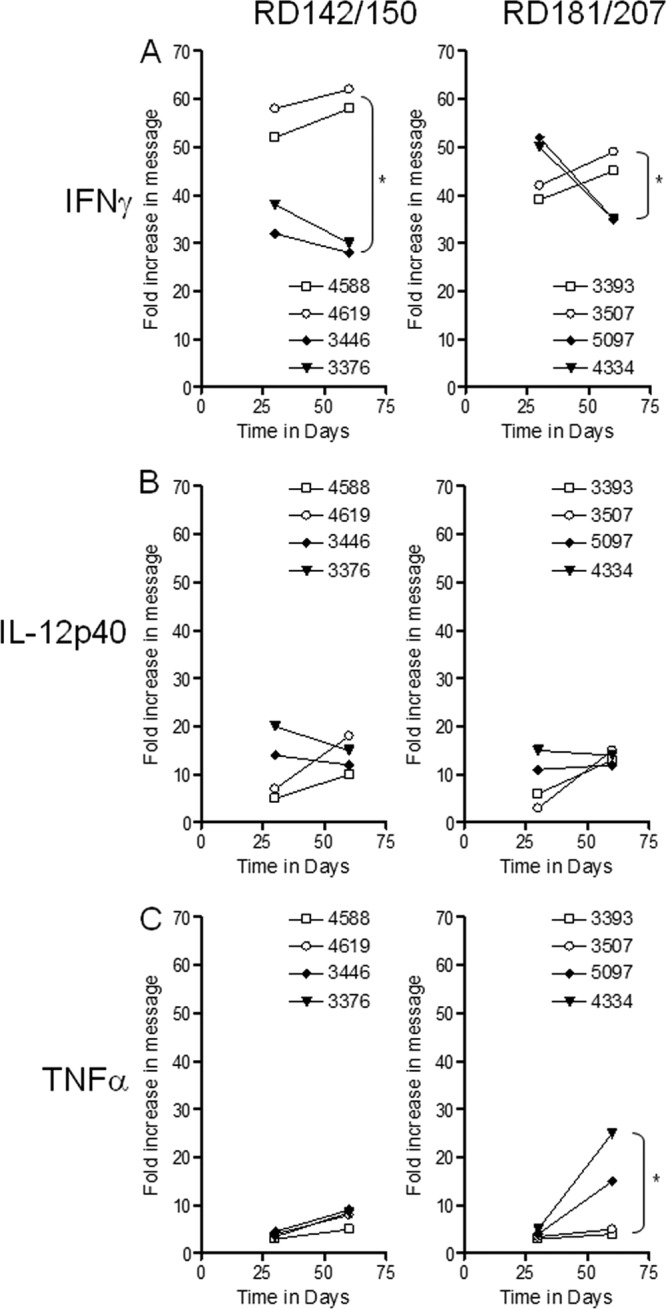
RT-PCR analysis of the fold increase in expression of cytokines associated with the TH1 response in the lungs of guinea pigs following exposure to representative strains of the four sublineages of lineage 2. Panel A shows IFN-γ, panel B shows IL-12p40, and panel C shows TNF-α expression in the lungs on days 30 and 60 from guinea pigs exposed to a low dose of RD142 (4588 and 4619), RD150 (3446 and 3376), RD181 (3507 and 3393), and RD207 (5097 and 4334) sublineage strains. Cytokine mRNA expression was quantified using real-time RT-PCR. Fold induction of mRNA was calculated from the threshold cycle (CT) normalized to HPRT CT values using the values of the uninfected guinea pig lung cells. Results are expressed as the average (n = 4) of the fold induction in each group (SEM did not exceed 0.30). *, Student's t test: for IFN-γ, RD142/RD150, P < 0.01, and RD181/RD207, P < 0.04; for TNF-α for RD181/RD207, P < 0.03.
Fig 6.
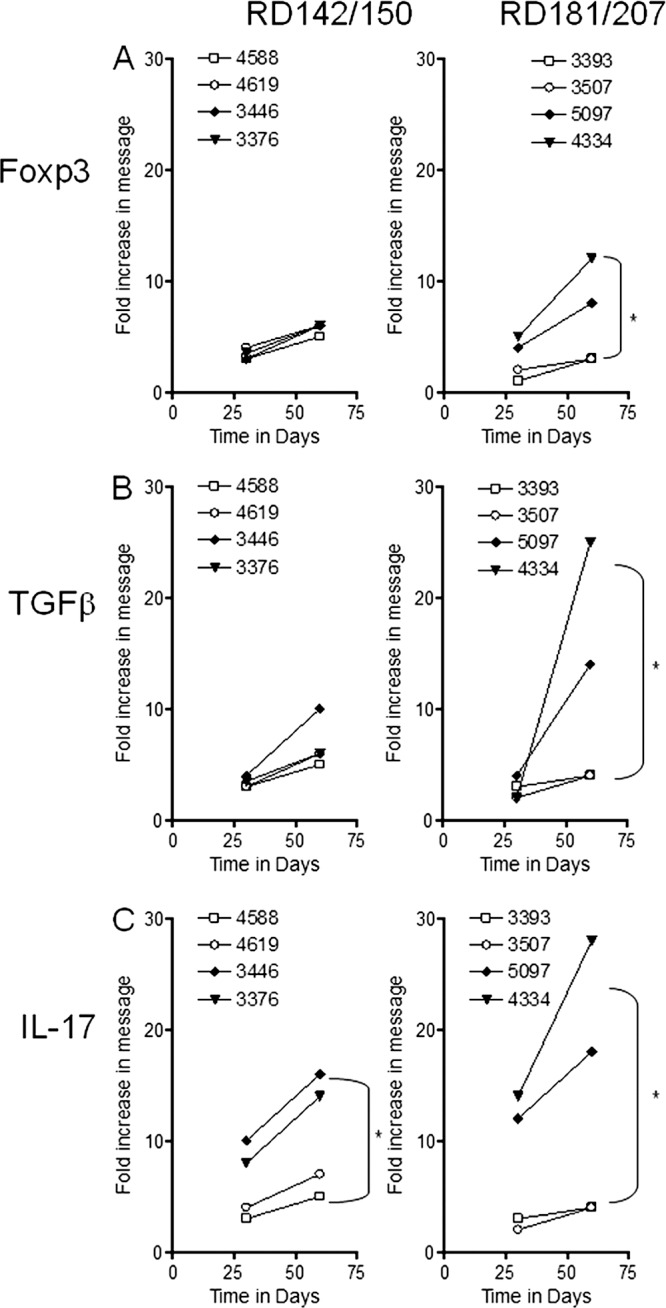
RT-PCR analysis of the fold increase in expression of proteins associated with regulatory T cell and TH17 T cell responses in the lungs of guinea pigs following exposure to representative strains of the four sublineages of lineage 2. Panel A shows Foxp3, panel B shows TGF-β, and panel C shows IL-17 expression in the lungs on days 30 and 60 from guinea pigs exposed to a low dose of RD142 (4588 and 4619), RD150 (3446 and 3376), RD181 (3507 and 3393), or RD207 (5097 and 4334) sublineage strains. Cytokine mRNA expression was quantified using real-time RT-PCR. Fold induction of mRNA was calculated from the threshold cycle (CT) normalized to HPRT CT values and then to values for uninfected guinea pig lung cells. Results are expressed as the average (n = 4) of the fold induction in each group (SEM did not exceed 0.30). *, Student's t test: for Foxp3, RD181/RD207, P < 0.04; for TGF-β, RD181/RD207, P < 0.02; for IL-17, RD142/RD150, P < 0.04; for RD181/RD207, P < 0.01.
We also examined the induction of regulatory molecules, given our earlier observations (32, 44) that this seems to be a common property of many of the Beijing strains. As the infections progressed, increases in message for Foxp3 were seen in all strains (Fig. 6A) and most significantly for the two RD207 strains, suggesting the arrival in the lungs of regulatory T cells. In addition, a very large increase in TGF-β expression (Fig. 6B) in animals infected with the two RD207 strains was observed during this time. Finally, increased expression of IL-17 message was observed (Fig. 6C), again very prominently in response to the two RD207 and RD150 strains. This observation is consistent with the high levels of lung consolidation seen, presumably driven by IL-17-mediated local chemokine release.
We used flow cytometric protocols (31) to further define the cellular response in the guinea pig lungs. As shown in Fig. 7A, we observed substantially increased numbers of activated CD4+ CD45hi T cells in the lungs of guinea pigs infected with the RD207 and RD142 sublineage strains on day 30 after infection. However, as the infection progressed, the numbers of these cells dropped precipitously by day 60 (Fig. 7B).
Fig 7.

Flow cytometric analysis of CD4+ and CD45hi T cell subsets accumulating in the lungs over the course of the infection with representative strains of the four sublineages of lineage 2. Panel A shows representative flow cytometric analysis after 30 days of infection of the percentages of CD4+ and CD45hi T cell cells in RD142, RD150, RD181, and RD207 sublineages. Panel B shows the numbers of cells in the lungs on days 30 and 60 for RD142 strains 4588 and 4619, RD150 strains 3446 and 3376, RD181 strains 3507 and 3393, and RD207 strains 5097 and 4334. Cell numbers are expressed as total cells (×107) expressing each phenotype per 1.0 g of each tissue (n = 4) (SEM did not exceed 0.35). *, Student's t test for RD150 for day 30 to day 60, P < 0.03, and RD207 for day 30 to day 60, P < 0.01.
Whole-genome sequence analysis.
Analysis of the 10 whole-genome sequences identified a total of 1,534 SNPs specific for lineage 2. We inferred 51 nsSNPs specific for the RD207 sublineage, 24 for the RD142 sublineage, and 28 for the RD150 sublineage. There were no mutations exclusive for the RD181 sublineage, since all the mutations observed in RD181 were also present in the RD150 and RD142 strains. The numbers of nsSNPs that were considered to have an impact on the gene function based on the SIFT analysis were 18 for RD207, 13 for RD142, and 16 for RD150. The list of the genes affected and their SIFT values are shown in Table 3.
Table 3.
Genes specific for each sublineage that contained an nsSNP that may have an impact on gene function based on SIFT analysis
| Category | Locus | Gene symbol | Description of gene producta | Amino acid change | SIFT value |
|---|---|---|---|---|---|
| Mutations in sublineage RD207 | Rv0327c | cyp135A1 | Cytochrome P450 135A1 | R220H | 0.05 |
| Rv0380c | RNA methyltransferase | R174P | 0.01 | ||
| Rv0411c | glnH | Glutamine-binding lipoprotein | M1I | 0 | |
| Rv0622 | Hypothetical protein | P219L | 0.01 | ||
| Rv0859 | fadA | Acetyl-CoA acetyltransferase | K17N | 0 | |
| Rv0892 | Monooxygenase | T177I | 0 | ||
| Rv0944 | Formamidopyrimidine-DNA glycosylase | Y50H | 0.01 | ||
| Rv0989c | grcC2 | Polyprenyl-diphosphate synthase | L257 M | 0 | |
| Rv1073 | Hypothetical protein | V113L | 0.02 | ||
| Rv1523 | Methyltransferase | V167A | 0 | ||
| Rv1557 | mmpL6 | Transmembrane transport | A158G | 0.01 | |
| Rv1894c | Hypothetical protein | V168 M | 0 | ||
| Rv1934c | fadE17 | Acyl-CoA dehydrogenase | E394K | 0.02 | |
| Rv2579 | dhaA | Haloalkane dehalogenase | T2K | 0.02 | |
| Rv2688c | Antibiotic ABC transporter ATP-binding protein | C213R | 0 | ||
| Rv2821c | Hypothetical protein | V207L | 0.02 | ||
| Rv2959c | Methyltransferase | I146 M | 0 | ||
| Rv3057c | Short-chain dehydrogenase | V93 M | 0.02 | ||
| Mutations in sublineage RD150 | Rv0426c | Hypothetical protein | P97L | 0.04 | |
| Rv0458 | Aldehyde dehydrogenase | V457A | 0 | ||
| Rv0528 | Transmembrane protein | D59G | 0 | ||
| Rv0577 | TB27.3 gene | Hypothetical protein | P74A | 0.01 | |
| Rv0610c | Hypothetical protein | E235A | 0 | ||
| Rv0634c | Glyoxalase II | Y7H | 0 | ||
| Rv1009 | rpfB | Resuscitation-promoting factor RpfB | V265 M | 0 | |
| Rv1140 | Hypothetical protein | G84D | 0.02 | ||
| Rv1207 | folP2 | Dihydropteroate synthase 2 FolP2 | R73G | 0.04 | |
| Rv1638 | uvrA | Excision nuclease ABC subunit A | D200G | 0.02 | |
| Rv1665 | pks11 | Chalcone synthase Pks11 | P55A | 0.01 | |
| Rv2416c | eis | Enhanced intracellular survival protein | V163I | 0 | |
| Rv2715 | Hydrolase | P263S | 0.01 | ||
| Rv3167c | TetR family transcriptional regulator | L162F | 0.01 | ||
| Rv3665c | dppB | Peptide ABC transporter | T194I | 0 | |
| Rv3886c | mycP2 | Hypothetical protein | P45T | 0 | |
| Mutations in sublineage RD142 | Rv0775 | Hypothetical protein | R86Q | 0.04 | |
| Rv0826 | Hypothetical protein | N79S | 0.02 | ||
| Rv0989c | grcC2 | Polyprenyl-diphosphate synthase | A182G | 0.04 | |
| Rv1152 | Regulatory protein GntR | G105A | 0.04 | ||
| Rv1295 | thrC | Threonine synthase | G237S | 0 | |
| Rv1317c | alkA | Ada regulatory protein | A11T | 0 | |
| Rv1502 | Hypothetical protein | R196C | 0.05 | ||
| Rv1811 | mgtC | Mg2+ transport ATPase C | A40 M | 0 | |
| Rv2124c | metH | Methionine synthase | Y1098D | 0 | |
| Rv2394 | ggtB | Gamma-glutamyltransferase | V545F | 0 | |
| Rv2510c | Hypothetical protein | Q351E | 0 | ||
| Rv3667 | acs | Acetyl-CoA synthetase | A44T | 0 | |
| Rv3774 | echA21 | Enoyl-CoA hydratase | G124D | 0 |
CoA, coenzyme A.
DISCUSSION
All the strains representing four sublineages of lineage 2 of M. tuberculosis with the Beijing spoligotype were capable of growing and causing lung pathology in guinea pigs exposed to low-dose aerosol infection. The ability of mycobacteria to grow in the lungs over time is the most conventional measure of strain virulence. While differences between the four sublineages were not overt, members of the RD207 sublineage, consisting of strains more likely to cause secondary clinical cases, caused more-severe pathology in these animals than members of the RD142 or RD181 sublineage, with the fourth sublineage, RD150, showing an intermediate pattern. All strains tested were capable of inducing TH1 immunity. In comparison with the RD207 sublineage, the RD142 strains showing the highest IFN-γ responses were associated with mild inflammation and reduced regulatory Foxp3+ expression. This finding suggests that the RD142 strains are more immunogenic, and it is consistent with the ability of the guinea pigs to control and contain these particular strains more quickly. In addition, all strains induced some degree of regulatory host molecules (Foxp3, TGF-β, and IL-17), which seems to be a general property of the Beijing strains analyzed in animal studies to date (32, 44). Members of the RD207 sublineage gave the highest signals.
Animals infected with the RD207 sublineage showed both a significant drop in activated CD4+ CD45hi T cells in the lungs as the infections progressed and a concomitant large rise in markers associated with regulatory T cell influx into the lungs. Together, these data indicate that different sublineages of lineage 2 do not behave in a comparable manner in the animal model but instead have some observable differences in their degree of immunopathology and capacity to generate protective and/or regulatory immunity. Hence, this supports the hypothesis, albeit cautiously made, that the RD207 sublineage of lineage 2 may be associated with distinct clinical and pathological properties and that these properties may influence the transmission capacity of isolates within patient communities.
To explore the possible mechanisms for differences observed across sublineages, we analyzed the whole-genome sequences of the strains included in this study. Each of the sublineages had specific mutations that, based on their SIFT values, suggested an impact on gene function. The strains from sublineage RD207 had a mutation in Rv0989c (grcC2), a diphosphate synthase required for cell wall biosynthesis (27). These strains also had a mutation in the gene Rv2959c, encoding a methyltransferase involved in the biosynthesis of phenolglycolipid, which is considered a virulence factor (39). It is tantalizing to speculate that a mutation in this gene may render the bacteria more virulent, as observed in the epidemiologic analysis (more secondary cases) and in the animal model (more necrosis and inflammation).
The strains from sublineage RD150 had mutations that may have an impact on the function of four interesting genes. Rv0577, a gene restricted to members of the M. tuberculosis complex (18), has been used for diagnostic purposes (45). The protein encoded by Rv0577 may regulate innate and adaptive immunity by interacting with Toll-like receptor 2 (8). Rv1009 (rpfB) is one of the most immunogenic resuscitation-promoting factors (40), and deletion of this gene has been associated with delayed reactivation from chronic tuberculosis in mouse (50). Rv1638 (uvrA) is part of the nucleotide excision repair system which counteracts the deleterious effects of DNA lesions (41) and is essential for Mycobacterium smegmatis to survive under conditions of hypoxia and low carbon source (13). Rv2416c (eis) encodes a secretory protein that enhances intracellular survival of M. tuberculosis in monocytes and contributes to its pathogenicity (53). A study demonstrated that Eis impaired the host defense against tuberculosis by disturbing the cross regulation of T cells, producing an imbalance between TH1 and TH2 responses, which could be a factor in the pathogenesis of tuberculosis (22).
The strains from sublineage RD142 have mutations in three interesting genes for which other polymorphisms have been described. Rv0989c (grcC2) had a different mutation in RD142 strains than in the RD207 strains discussed previously. Rv1811 (mgtC) encodes a virulence factor required to survive in macrophages and under conditions with low Mg2+ (6). Different mutations in this gene have been found in strains from the Euro-American lineage (spoligotype Haarlem) (2). Rv1317c (alkA) is part of the AdaA-AlkA adaptive response in M. tuberculosis, and multiple mutations have been found in strains from lineage 4 (Euro-American), lineage 2 (the same mutation has been found previously in the W-Beijing 210 strain, which belongs to the RD142 sublineage), and in Mycobacterium bovis (29). It has been suggested that a defective adaptive response by these genes will confer a selective advantage to M. tuberculosis (54).
The strains from RD181 are a paraphyletic group, defined as a group of organisms which includes the most recent common ancestor of all of its members but not all of the descendants of that most recent common ancestor. In this particular case, RD181 strains share an ancestor, but the group also includes RD150 and RD142 strains. This implies that SNPs shared by all RD181 strains are also present in RD150 and RD142 strains and that there were not common nsSNPs exclusive for all RD181 strains.
Although some of these mutations may explain the pathological and immunological differences observed, there are a multitude of factors that can influence the transmission and pathogenic capabilities of a given isolate, such as host and environmental factors (5, 20, 27, 38), HIV coinfection (42), and the concentration of organisms in environmental air (17). Until recently, the only bacterial factor considered was the presence of drug resistance; some studies have suggested that M. tuberculosis resistant to isoniazid is less transmissible (52) and less pathogenic than fully susceptible organisms (7, 11), although an earlier study in our laboratory (33) investigating the virulence of multidrug-resistant isolates did not show much evidence of loss of virulence of these strains. More recent studies suggest that different groups of isolates of M. tuberculosis may contribute to different clinical outcomes (14). As noted recently (52), the application of new molecular typing techniques has increased both our knowledge of bacterial factors and the identification of separate lineages of isolates. It is overly optimistic to expect that the myriad of factors can be modeled in animals such as the guinea pig used here, but such models can provide clues. Like humans, the guinea pig undergoes a process of granulomatous inflammation and necrosis when infected with M. tuberculosis, and the differing degrees to which this occurs may be an indicator of the virulence of the infecting isolate (36, 37). Moreover, by applying new flow cytometric techniques (30, 31) and RT-PCR methods, one can detect differences in the expression of protective immunity (RD142 strains clearly generated the strongest response, suggesting that they are of increased immunogenicity), as well as the induction of signals consistent with the generation of regulatory T cells (which we found here to be highest in animals infected with the RD207 strains).
Most studies to date of the pathogenicity of strains with the guinea pig model have tended to focus on the Beijing strains, and far less is known about other lineages or families of strains. There is a growing concern that the newly emerging isolates of M. tuberculosis in general are more pathogenic, and this may have a serious impact on vaccine effectiveness. Not only is there a suspicion that BCG may actually have selected for the more virulent strains (1), but recent data (32) show that BCG is only transiently protective against Beijing strains and cannot overcome the induction of regulatory T cells. Since some new-generation vaccines are also based on BCG (4), this raises the real possibility that such vaccines will not work (32, 34, 35). While as yet unproven, the induction of regulatory T cells by these pathogenic strains, coupled with dampening or loss of protective immunity but continuance of TH17 responses (as seen here), may drive the degree of severity of lung pathology, which in turn will enable bacilli to escape the lungs and then potentially be transmitted.
One of the primary public health strategies to control tuberculosis is the evaluation of persons in close contact with an infectious tuberculosis patient (contact investigation) to identify secondary cases of active tuberculosis and latent tuberculosis infection. The bacterial factors governing transmissibility and pathogenicity of M. tuberculosis are poorly understood. Therefore, additional clinical and animal studies such as ours may serve to identify factors (like the features of the exposure or the immune status of the exposed person) that suggest a situation in which there is a greater risk of developing active tuberculosis. In these cases, the evaluation of contacts should be undertaken with great urgency. Also, we have discovered mutations likely to be functional in genes that are currently being used for diagnostic purposes (Rv0577 and Rv1009) (10, 45) or as candidates for subunit vaccines (Rv1009). These polymorphisms may limit their efficacy as diagnostic or vaccine targets.
In conclusion, the current molecular method-based sublineage classification appears to be associated biologically with clinical and pathological consequences, and differences between sublineages, particularly in the context of loss of protective immunity and increased lung damage, may favor or influence the capacity of these isolates to be transmitted within communities.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (grant numbers AI083856, AI081959, AI070456, AI092002, AI034238, AI090928, and HHSN266200700022C; National Institutes of Health Innovation Award [grant number 1DP2OD006450]), American Recovery and Reinvestment Act funds, and the Swiss National Science Foundation (PP00A-119205). Further support was provided by the College of Veterinary Medicine and Biomedical Sciences, Colorado State University.
We thank M. Shin and J. Nguyen for library preparation and sequence data generation.
Footnotes
Published ahead of print 20 June 2012
REFERENCES
- 1. Abebe F, Bjune G. 2006. The emergence of Beijing family genotypes of Mycobacterium tuberculosis and low-level protection by bacille Calmette-Guerin (BCG) vaccines: is there a link? Clin. Exp. Immunol. 145:389–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alix E, Godreuil S, Blanc-Potard AB. 2006. Identification of a Haarlem genotype-specific single nucleotide polymorphism in the mgtC virulence gene of Mycobacterium tuberculosis. J. Clin. Microbiol. 44:2093–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allen SS, et al. 2008. Altered inflammatory responses following transforming growth factor-beta neutralization in experimental guinea pig tuberculous pleurisy. Tuberculosis (Edinb.) 88:430–436 [DOI] [PubMed] [Google Scholar]
- 4. Beresford B, Sadoff JC. 2010. Update on research and development pipeline: tuberculosis vaccines. Clin. Infect. Dis. 50(Suppl 3):S178–S183 [DOI] [PubMed] [Google Scholar]
- 5. Borgdorff MW, et al. 2010. Progress towards tuberculosis elimination: secular trend, immigration and transmission. Eur. Respir. J. 36:339–347 [DOI] [PubMed] [Google Scholar]
- 6. Buchmeier N, et al. 2000. A parallel intraphagosomal survival strategy shared by Mycobacterium tuberculosis and Salmonella enterica. Mol. Microbiol. 35:1375–1382 [DOI] [PubMed] [Google Scholar]
- 7. Burgos M, DeRiemer K, Small PM, Hopewell PC, Daley CL. 2003. Effect of drug resistance on the generation of secondary cases of tuberculosis. J. Infect. Dis. 188:1878–1884 [DOI] [PubMed] [Google Scholar]
- 8. Byun EH, et al. 2012. Mycobacterium tuberculosis Rv0577, a novel TLR2 agonist, induces maturation of dendritic cells and drives Th1 immune response. FASEB J. 26:2695–2711 [DOI] [PubMed] [Google Scholar]
- 9. Cattamanchi A, et al. 2006. A 13-year molecular epidemiological analysis of tuberculosis in San Francisco. Int. J. Tuberc. Lung Dis. 10:297–304 [PubMed] [Google Scholar]
- 10. Chegou NN, et al. 2012. Potential of novel Mycobacterium tuberculosis infection phase-dependent antigens in the diagnosis of TB disease in a high burden setting. BMC Infect. Dis. 12:10 doi:10.1186/1471-2334-12-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cohen T, Sommers B, Murray M. 2003. The effect of drug resistance on the fitness of Mycobacterium tuberculosis. Lancet Infect. Dis. 3:13–21 [DOI] [PubMed] [Google Scholar]
- 12. Comas I, et al. 2010. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 42:498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cordone A, Audrain B, Calabrese I, Euphrasie D, Reyrat JM. 2011. Characterization of a Mycobacterium smegmatis uvrA mutant impaired in dormancy induced by hypoxia and low carbon concentration. BMC Microbiol. 11:231 doi:10.1186/1471-2180-11-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Jong BC, et al. 2008. Progression to active tuberculosis, but not transmission, varies by Mycobacterium tuberculosis lineage in The Gambia. J. Infect. Dis. 198:1037–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ebrahimi-Rad M, et al. 2003. Mutations in putative mutator genes of Mycobacterium tuberculosis strains of the W-Beijing family. Emerg. Infect. Dis. 9:838–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gagneux S, et al. 2006. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 103:2869–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Houk VN, Baker JH, Sorensen K, Kent DC. 1968. The epidemiology of tuberculosis infection in a closed environment. Arch. Environ. Health 16:26–35 [DOI] [PubMed] [Google Scholar]
- 18. Huard RC, et al. 2003. The Mycobacterium tuberculosis complex-restricted gene cfp32 encodes an expressed protein that is detectable in tuberculosis patients and is positively correlated with pulmonary interleukin-10. Infect. Immun. 71:6871–6883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kato-Maeda M, et al. 2010. Differences among sublineages of the East-Asian lineage of Mycobacterium tuberculosis in genotypic clustering. Int. J. Tuberc. Lung Dis. 14:538–544 [PMC free article] [PubMed] [Google Scholar]
- 20. Kik SV, et al. 2008. Tuberculosis outbreaks predicted by characteristics of first patients in a DNA fingerprint cluster. Am. J. Respir. Crit. Care Med. 178:96–104 [DOI] [PubMed] [Google Scholar]
- 21. Kremer K, et al. 2004. Definition of the Beijing/W lineage of Mycobacterium tuberculosis on the basis of genetic markers. J. Clin. Microbiol. 42:4040–4049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lella RK, Sharma C. 2007. Eis (enhanced intracellular survival) protein of Mycobacterium tuberculosis disturbs the cross regulation of T-cells. J. Biol. Chem. 282:18671–18675 [DOI] [PubMed] [Google Scholar]
- 23. Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maddison WP, Maddison DR. 2010. Mesquite: a modular system for evolutionary analysis. Version 2.73. http://mesquiteproject.org
- 26. McMurray DN, et al. 2005. Vaccine-induced cytokine responses in a guinea pig model of pulmonary tuberculosis. Tuberculosis (Edinb.) 85:295–301 [DOI] [PubMed] [Google Scholar]
- 27. Mitruka K, Oeltmann JE, Ijaz K, Haddad MB. 2011. Tuberculosis outbreak investigations in the United States, 2002–2008. Emerg. Infect. Dis. 17:425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ng PC, Henikoff S. 2001. Predicting deleterious amino acid substitutions. Genome Res. 11:863–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nouvel LX, Dos Vultos T, Kassa-Kelembho E, Rauzier J, Gicquel B. 2007. A non-sense mutation in the putative anti-mutator gene ada/alkA of Mycobacterium tuberculosis and M. bovis isolates suggests convergent evolution. BMC Microbiol. 7:39 doi:10.1186/1471-2180-7-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ordway D, et al. 2008. Influence of Mycobacterium bovis BCG vaccination on cellular immune response of guinea pigs challenged with Mycobacterium tuberculosis. Clin. Vaccine Immunol. 15:1248–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ordway D, et al. 2007. The cellular immune response to Mycobacterium tuberculosis infection in the guinea pig. J. Immunol. 179:2532–2541 [DOI] [PubMed] [Google Scholar]
- 32. Ordway DJ, et al. 2011. Mycobacterium bovis BCG-mediated protection against W-Beijing strains of Mycobacterium tuberculosis is diminished concomitant with the emergence of regulatory T cells. Clin. Vaccine Immunol. 18:1527–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ordway DJ, Sonnenberg MG, Donahue SA, Belisle JT, Orme IM. 1995. Drug-resistant strains of Mycobacterium tuberculosis exhibit a range of virulence for mice. Infect. Immun. 63:741–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Orme IM. 2010. The Achilles heel of BCG. Tuberculosis (Edinb.) 90:329–332 [DOI] [PubMed] [Google Scholar]
- 35. Orme IM. 2011. Development of new vaccines and drugs for TB: limitations and potential strategic errors. Future Microbiol. 6:161–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palanisamy GS, et al. 2009. Clinical strains of Mycobacterium tuberculosis display a wide range of virulence in guinea pigs. Tuberculosis (Edinb.) 89:203–209 [DOI] [PubMed] [Google Scholar]
- 37. Palanisamy GS, et al. 2008. Disseminated disease severity as a measure of virulence of Mycobacterium tuberculosis in the guinea pig model. Tuberculosis (Edinb.) 88:295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pena MJ, et al. 2003. Epidemiology of tuberculosis on Gran Canaria: a 4 year population study using traditional and molecular approaches. Thorax 58:618–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perez E, et al. 2004. Molecular dissection of the role of two methyltransferases in the biosynthesis of phenolglycolipids and phthiocerol dimycoserosate in the Mycobacterium tuberculosis complex. J. Biol. Chem. 279:42584–42592 [DOI] [PubMed] [Google Scholar]
- 40. Romano M, et al. 2012. Potential of Mycobacterium tuberculosis resuscitation-promoting factors as antigens in novel tuberculosis sub-unit vaccines. Microbes Infect. 14:86–95 [DOI] [PubMed] [Google Scholar]
- 41. Rossi F, et al. 2011. The biological and structural characterization of Mycobacterium tuberculosis uvrA provides novel insights into its mechanism of action. Nucleic Acids Res. 39:7316–7328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Selwyn PA, et al. 1989. A prospective study of the risk of tuberculosis among intravenous drug users with human immunodeficiency virus infection. N. Engl. J. Med. 320:545–550 [DOI] [PubMed] [Google Scholar]
- 43. Shah NS, et al. 2007. Worldwide emergence of extensively drug-resistant tuberculosis. Emerg. Infect. Dis. 13:380–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shang S, et al. 2011. Increased Foxp3 expression in guinea pigs infected with W-Beijing strains of M. tuberculosis. Tuberculosis (Edinb.) 91:378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sobral LF, et al. 2011. Identification of Mycobacterium bovis among mycobacterial isolates from human clinical specimens at a university hospital in Rio de Janeiro, Brazil. J. Bras. Pneumol. 37:664–668 [DOI] [PubMed] [Google Scholar]
- 46. Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1605 [DOI] [PubMed] [Google Scholar]
- 47. Tsenova L, et al. 2005. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J. Infect. Dis. 192:98–106 [DOI] [PubMed] [Google Scholar]
- 48. Tsolaki AG, et al. 2005. Genomic deletions classify the Beijing/W strains as a distinct genetic lineage of Mycobacterium tuberculosis. J. Clin. Microbiol. 43:3185–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tsolaki AG, et al. 2004. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc. Natl. Acad. Sci. U. S. A. 101:4865–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tufariello JM, et al. 2006. Deletion of the Mycobacterium tuberculosis resuscitation-promoting factor Rv1009 gene results in delayed reactivation from chronic tuberculosis. Infect. Immun. 74:2985–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Embden JD, et al. 1993. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J. Clin. Microbiol. 31:406–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Verhagen LM, et al. 2011. Mycobacterial factors relevant for transmission of tuberculosis. J. Infect. Dis. 203:1249–1255 [DOI] [PubMed] [Google Scholar]
- 53. Wu S, et al. 2009. Activation of the eis gene in a W-Beijing strain of Mycobacterium tuberculosis correlates with increased SigA levels and enhanced intracellular growth. Microbiology 155:1272–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang M, et al. 2011. The ada operon of Mycobacterium tuberculosis encodes two DNA methyltransferases for inducible repair of DNA alkylation damage. DNA Repair (Amst.) 10:595–602 [DOI] [PubMed] [Google Scholar]