

Abstract
Infections with cardiotrophic viruses and immune-mediated responses against the heart have been suggested to play a dominant role in the pathogenesis of idiopathic dilated cardiomyopathy (DCM). Furthermore, immune-mediated inflammatory diseases (IMIDs) may result in DCM. It has not previously been assessed whether DCM patients with and without an IMID have different prevalences and quantities of cardiotrophic viruses in the heart. Therefore, we compared the profiles of cardiotrophic viruses in heart tissue of DCM patients with and without an IMID. Serum and myocardial tissue samples were obtained from 159 consecutive patients with DCM and 20 controls. Patients were subdivided into three groups, the first two based on the presence (n = 34) or absence (n = 125) of an IMID and the third being a control group. The parvovirus B19 virus genome was detected in equal quantities in the non-IMID DCM patients (100/125) and the control group (15/20) but in lower quantities in the IMID patients (21/34, P = 0.02). Both the non-IMID and IMID DCM patients demonstrated increased myocardial inflammation compared to controls: 12.5 ± 1.8 and 14.0 ± 3.2 CD45-positive inflammatory cells, respectively, versus 5.1 ± 0.7 for the controls (P < 0.05 for both). Importantly, significantly higher parvovirus B19 copy numbers could be amplified in non-IMID than in IMID patients (561 ± 97 versus 191 ± 92 copies/μg DNA, P < 0.001) and control subjects (103 ± 47 copies/μg DNA, P < 0.001). The present study shows decreased parvovirus B19 prevalence and copy numbers in hearts of DCM patients with an IMID compared to those without an IMID. These findings may suggest that DCM patients with an IMID have a different pathophysiologic mechanism from that which is present in the virus-induced form of DCM.
INTRODUCTION
Idiopathic dilated cardiomyopathy (DCM) is a myocardial disease characterized by dilatation and impaired contraction of the myocardium, not caused by ischemia or abnormal loading conditions (30). DCM can manifest at any age but is most common between 30 and 50 years, causing considerable morbidity and mortality. Currently, DCM is the leading cause of cardiac transplantation in young adults (7). DCM is a disease that may result from a variety of underlying pathologies. Recent reports revealed both inflammatory processes related to infections with cardiotrophic viruses and autoimmune mechanisms as major predisposing factors in the pathogenesis of this disease (5, 11, 22, 24, 31–33, 44). It would seem important to distinguish an immune-mediated inflammatory disorder from infectious cardiac disease, since there is growing evidence that for selected patients, specific treatment strategies may be effective only if based on their immunological and virological characterization (12, 16, 21).
To date, there have been only limited data available on the differences of cardiotrophic viruses present in the hearts of patients with DCM with and without the presence of an immune-mediated inflammatory disease (IMID). Therefore, we determined the prevalence and quantity of cardiotrophic viruses using quantitative PCR in a well-defined cohort of patients with DCM in both the presence and absence of an IMID.
MATERIALS AND METHODS
Study population. (i) DCM subjects.
Between April 2005 and October 2010, consecutive patients with clinically diagnosed DCM were enrolled in the University Hospital of Maastricht. To elucidate a possible myocardial origin of their disease, these patients underwent endomyocardial biopsy (EMB) after angiographic and echocardiographic exclusion of significant coronary artery disease (>50% stenosis) and other possible causes of cardiac dysfunction (familial cardiomyopathy, valvular disease, hypertension, or ethyl abuses). The clinical diagnosis of DCM was suspected in all patients who presented with unexplained left ventricular (LV) dysfunction and/or dilated LV in association with longer existing symptoms of heart failure (New York Heart Association [NYHA] functional classes II to IV) in spite of conventional supportive heart failure medication. For each patient, echocardiographic, LV, end-diastolic diameter (EDD), and end-systolic diameter (ESD) were obtained in the standard parasternal, apical, and subxiphoid views (Sonos 5500; Philips Medical Systems, Best, Netherlands) according to the recommendations of the American Society of Echocardiography (17). The left ventricle ejection fraction (LVEF) was calculated in a standard manner and was used to assess LV systolic function (17).
(ii) IMID subjects.
IMIDs involve an immune response that is inappropriate or excessive and is caused or accompanied by a dysregulation of the normal cytokine milieu. They also cause acute and chronic inflammatory injury in any organ system leading to end organ damage (9, 10, 14, 34, 43). Patients were seen via a multidisciplinary approach, consisting of consultations with both a cardiologist (R.D. and S.H.) and a clinical immunologist (P.V.P. and J.W.C.T.). Some IMIDs lack established clinical criteria, while others overlap significantly in symptom manifestations. To address this issue, IMIDs were classified into subgroups that had disparate organ system involvement, were physiologically distinct, and presented with contrasting symptoms (37). Patients were classified as IMID subjects in the presence of a previously diagnosed IMID or newly discovered systemic disorder comprising the following: vasculitides, connective tissue diseases, rheumatologic diseases, and others (Table 1).
Table 1.
Baseline characteristics of study groupsa
| Characteristic | Control (n = 20) | Non-IMID (n = 125) | IMID (n = 34) |
|---|---|---|---|
| Age (yr) | 65 ± 3 | 53 ± 2 | 54 ± 3 |
| Sex (no. of males: no. of females) | 19: 1 | 80: 45 | 16: 14 |
| No. of yrs since heart failure diagnosis | NA | 3 ± 1 | 4 ± 1 |
| Medications at enrollment, no. (%) | |||
| Maintenance corticosteroid therapy | 0 (0) | 0 (0) | 22 (65)b |
| Beta blocker | 18 (90) | 98 (78) | 25 (74) |
| Angiotensin receptor blocker or angiotensin converting enzyme inhibitor | 9 (45) | 104 (83) | 24 (71) |
| Aldosterone antagonist | 0 (0) | 74 (59) | 19 (56) |
| Systemic autoimmune disorder, no. (%) | |||
| Vasculitides | |||
| Churg-Strauss syndrome | 9 (26) | ||
| Wegener's granulomatosis | 2 (6) | ||
| Connective tissue diseases | |||
| Dermatomyositis/polymyositis | 2 (6) | ||
| Lupus erythematosus | 1 (3) | ||
| Sjögren's syndrome | 1 (3) | ||
| Other rheumatologic diseases | |||
| Rheumatoid arthritis | 5 (15) | ||
| Ankylosing spondylitis | 1 (3) | ||
| Other diseases | |||
| Sarcoidosis | 4 (12) | ||
| Graves' disease | 3 (9) | ||
| Psoriasis | 3 (9) | ||
| Crohn's disease | 2 (6) | ||
| Autoimmune hypothyroidism | 1 (3) |
Values are means ± SEM. Non-IMID, DCM patients without an immune-mediated inflammatory disease; IMID, DCM patients with an immune-mediated inflammatory disease; NA, not applicable.
P < 0.001 compared to controls and non-IMID.
(iii) Control subjects.
The control group consisted of subjects undergoing elective coronary artery bypass surgery (CABG) for coronary artery disease. Patients were included as controls in the case of a normal systolic and diastolic function preoperatively assessed by a complete cardiac analysis and the absence of an IMID. All patients gave written, informed consent before blood and cardiac tissue samples were obtained. The Human Research Committee of the University Hospital Maastricht approved the protocol.
Endomyocardial biopsy.
In the DCM population, diagnostic right ventricular endomyocardial biopsy specimens from the right interventricular septum were obtained using a transcatheter bioptome (Cordis). A total of 6 biopsy specimens were taken per patient; two for (immuno)histochemical analysis and four for the detection of virus genomes. For patients undergoing CABG, transmural biopsy specimens were taken from the left ventricular anterior wall. A total of 3 samples were acquired following the application of the cross-clamp using a Tru-Cut biopsy needle (Cardinal Health). The subendocardial part was used for further virus detection and (immuno)histology.
Immunohistological analysis.
For (immuno)histochemical analysis of myocardial inflammation, formalin-fixed, paraffin-embedded EMBs were analyzed according to the Dallas classification (2) and infiltrating inflammatory cells were specifically quantified as defined by the task force of the World Heart Federation's Council on Cardiomyopathies (25). Tissue sections of 4-μm thickness were subjected to (immuno)histochemical stainings. Inflammatory cells were stained using CD45 and CD68 antibodies (Dako, Glostrup, Denmark). The total tissue area of the myocardial biopsy specimens on the histological slide was determined by morphometrical analysis (using morphometrical software Leica Qwin, version 3, Cambridge, United Kingdom). The number of immunohistochemically positive cells was determined using a light microscope and expressed per square millimeters as described before (19). The amount of collagen was quantified as the percentage of Sirius red-stained area per total myocardial tissue area, excluding perivascular and endocardial fibrosis. The analysis was performed by one experienced investigator (R.J.V.S.) blinded to patient details.
Preparation and detection of virus genomes.
DNAs or RNAs of parvovirus B19, human herpesvirus 6, adenovirus, Epstein-Barr virus, and enterovirus were identified and quantified with quantitative PCR as described previously (12). DNA was extracted from endomyocardial biopsy specimens using a QIAamp DNA blood minikit (Qiagen, Venlo, Netherlands). RNA was isolated using automated MagnaPure total nucleic acid extraction (Roche Diagnostics GmbH, Mannheim, Germany). Extractions were performed according to the manufacturer's instructions. DNA concentrations were determined using a nanodrop instrument (Thermo Fisher Scientific, Wilmington, DE). Real-time PCR was performed with primers and a TaqMan probe as described by Watzinger et al. (42a). The PCR mix consisted of 20 μl isolated DNA or DNA, final concentrations of 600 nM each primer and 200 nM probe and 1× Absolute QPCR mix (Abgene, Epsom, United Kingdom). All real-time PCRs were performed using an ABI prism 7000 (Applied Biosystems, Foster City, CA) and quantified using a standard curve. The PCR assay used has a linear quantitative range from 10E7 copies to 1 × 10E2 with a detection probability above 95%. Below this range, semiquantitative detection is performed by extrapolation of the standard curve. We evaluated the variation for each DNA concentration in the standard curve over a period of 3 months. Our results showed that for all concentrations higher than 500 copies/ml the standard deviation was 0.15 log or less. Only for the two lowest concentrations (500 and 200 copies/ml) were standard deviations measured to be up to a maximum of 0.38 log. The quality of the assays was ensured by positive and negative controls as well as by a test on amplification inhibition in each sample by an external amplification control. For quantification of viral loads, standard curves were included in each run.
Biochemical measurements. (i) sIL-2R.
The serum levels of soluble interleukin-2 receptor (sIL-2R) were analyzed with a commercially available Diaclone enzyme-linked immunosorbent assay (ELISA) kit (Sanquin, Amsterdam, Netherlands) (38). Serum samples were diluted 5-fold and analyzed in duplicate. Coefficient of variation (CV) values of duplicate analyses were <20%; otherwise, the samples were reanalyzed. Concentrations were calculated from a 6-point standard curve ranging from 69 to 2,200 pg/ml, taking into account the dilution factor. Concentrated samples (>2,200 pg/ml) were diluted further as required.
(ii) Neopterin.
Serum levels of neopterin were evaluated by the principle of a competitive ELISA, using a kit produced by IBL (DRG Instruments, Hamburg, Germany) (41, 42). Ten microliters of each sample was added to duplicate wells, coated with a goat anti-rabbit antibody of the microstrips together with the enzyme conjugate (1:201). CV values of duplicate analyses were <20%; otherwise, the samples were reanalyzed. Concentrations were calculated from a 6-point standard curve ranging from 0 to 28 ng/ml.
(iii) Anticardiac autoantibodies.
The presence of anticardiac autoantibodies was evaluated using monkey heart sections as the substrate (Scimedx corporation, Denville, NJ) by indirect immunofluorescence (6). Serum samples were diluted 1:20 in phosphate-buffered saline (PBS). Binding of autoantibodies was visualized with anti-human IgG conjugated to fluorescein isothiocyanate (FITC). The serum was considered positive when a striated, sarcolemmal-subsarcolemmal, intermyofibrillar, and/or intercalated disc pattern was detected.
(iv) Parvovirus B19-specific antibodies.
Parvovirus B19-specific immunoglobulin G (IgG) and IgM antibodies were detected in serum samples with a standardized ELISA (Biotrin International, Dublin, Ireland) according to the manufacturers' instructions.
Statistical analysis.
Data derived from the study subjects were entered into a database and analyzed using SPSS version 15.0 (SPSS Inc., Chicago, IL) statistical software. Group data are expressed as mean values ± standard errors of the means. Categorical variables were assessed using the chi-square test, and continuous variables were compared with nonparametric (un)paired Student's t test. Correlation calculations were performed using Pearson's correlation coefficient. The optimal cutoff point of sIL-2R and neopterin to predict IMID was calculated with receiving operating characteristics (ROC) analysis. The optimized point was obtained as the value that yielded the best sensitivity and specificity. All tests were 2-sided, and statistical significance was accepted at the 95% confidence interval (P < 0.05).
RESULTS
Population.
One hundred fifty-nine DCM patients and 20 controls (mean age, 53 ± 3 years) were enrolled in this study. Their main clinical characteristics at baseline are shown in Table 1. DCM patients had heart failure symptoms prior to enrollment for 3 ± 1 years and a reduced LVEF of 30% ± 1%. Thirty-four of the 159 (21%) DCM patients were classified as suffering from an IMID, categorized as vasculitides (33%), connective tissue diseases (12%), rheumatologic diseases (18%), and others (38%) (Table 1). The remaining 125 (79%) patients did not suffer from any immunologic disorder. Laboratory findings at baseline revealed elevated serum sIL-2R and neopterin levels in the IMID DCM patients (3,232 ± 168 pg/ml and 5.7 ± 2.0 ng/ml) compared to the non-IMID DCM patients (1,556 ± 77 pg/ml and 2.5 ± 0.5 ng/ml, P < 0.001 for both) and controls (1,431 ± 86 pg/ml and 1.5 ± 0.2 ng/ml, P < 0.05 for both), compatible with the fact that the IMID patients have more pronounced activation of their immune system. Circulating antiheart antibodies (AHA) were present in 5/34 (15%) IMID DCM patients, in 9/125 (7%, P = 0.31) non-IMID DCM patients, and in 1/20 (5%, P = 0.40) controls.
All DCM patients received conventional supportive heart failure medication for at least 6 months. Twenty-two of 34 (65%) IMID patients were receiving maintenance corticosteroid therapy, with an average dose of <10 mg/day at the time of inclusion in the present study.
Prevalence and quantification of virus genome.
The parvovirus B19 virus genome could be amplified in the heart tissues of all groups, with a similar prevalence in the non-IMID DCM patients (100/125, 80%) and controls (15/20, 75%) but a significantly lower prevalence in the IMID DCM patients (21/34, 62%, P = 0.02) than in the non-IMID DCM patients and controls. Other viruses, such as human herpesvirus 6, adenovirus, Epstein-Barr virus, and enterovirus, revealed a similar distribution in all groups (Table 2). The presence of multiple cardiotrophic viruses was more frequently detected in the non-IMID DCM group than in the IMID DCM group and controls (33/125 [26%]) versus 2/20 [10%] and 2/34 [8%]; for both, P < 0.05). Importantly, significantly higher parvovirus B19 copy numbers could be amplified in non-IMID DCM patients than in IMID DCM patients (561 ± 97 versus 191 ± 92 copies/μg DNA, P < 0.001) and control subjects (103 ± 47 copies/μg DNA, P < 0.001), whereas other viruses did not reveal significant differences in virus copy numbers (Table 2).
Table 2.
Comparison of echocardiographic, virological, and (immuno)histological parameters in study groupsa
| Parameter | Control (n = 20) | Non-IMID (n = 125) | IMID (n = 34) |
|---|---|---|---|
| LVEF (%) | 57 ± 2 | 29 ± 1b | 31 ± 2b |
| LVESD (mm) | 51 ± 2 | 62 ± 1b | 61 ± 2c |
| LVEDD (mm) | 36 ± 2 | 53 ± 1b | 51 ± 2b |
| PVB19 | |||
| Prevalence, no. (%) | 15 (75) | 100 (80) | 21 (62)d |
| Quantification (copies/μg DNA) | 103 ± 47 | 561 ± 97e | 191 ± 92 |
| HHV-6 | |||
| Prevalence, no. (%) | 2 (10) | 22 (22) | 2 (7) |
| Quantification (copies/μg DNA) | 11 ± 10 | 23 ± 8 | 2 ± 2 |
| EV | |||
| Prevalence, no. (%) | 0 (0) | 2 (2) | 0 (0) |
| Quantification (copies/μg DNA) | NA | 1 ± 1 | NA |
| ADV | |||
| Prevalence, no. (%) | 0 (0) | 0 (0) | 0 (0) |
| Quantification (copies/μg DNA) | NA | NA | NA |
| EBV | |||
| Prevalence, no. (%) | 0 (0) | 4 (4) | 1 (3) |
| Quantification (copies/μg DNA) | 0 ± 0 | 1 ± 1 | 2 ± 1 |
Values are means ± SEM. Non-IMID, DCM patients without an immune-mediated inflammatory disease; IMID, DCM patients with an immune-mediated inflammatory disease; LVEF, left ventricular ejection fraction; LVESD, left ventricular end-systolic diameter; LVEDD, left ventricular end-diastolic diameter; PVB19, parvovirus B19; HHV-6, human herpesvirus 6; EV, enterovirus; ADV, adenovirus; EBV, Epstein-Barr virus.
P < 0.001 compared to controls.
P = 0.001 compared to controls.
P = 0.02 compared to non-IMID.
P < 0.001 compared to controls and IMID.
Using a cutoff value of 500 parvovirus B19 copies/μg DNA as a clinically relevant threshold (4), we found a 92% positive predictive value and 74% negative predictive value to differentiate IMID from non-IMID DCM patients.
Serum levels for parvovirus B19-specific antibodies were available from 99 patients (78 non-IMID and 21 IMID patients), revealing a seroprevalence in 62 (80%) of the non-IMID and in 16 (76%) of the IMID DCM patients. Among all parvovirus B19-seropositive patients, parvovirus B19 could be amplified in the heart in 60 (97%) of the non-IMID DCM patients but only in 11 (68%) of the IMID-DCM patients. IgM was not detected in any of the patients.
Endomyocardial inflammation and collagen content.
Conventional histology did not reveal any active or borderline myocarditis in any EMB according to the Dallas criteria (2). Both the IMID and non-IMID DCM patients revealed an increased number of CD45-positive lymphocytes (12.5 ± 1.8 and 14.0 ± 3.2 cells/mm2, respectively) and of CD68-positive macrophages (8.8 ± 3.1 and 10.2 ± 3.6 cells/mm2, respectively) and an increased amount of collagen content (7.3 ± 1.2 and 7.6 ± 0.6%, respectively) compared to the controls (5.1 ± 0.7 cells/mm2, 5.6 ± 0.7 cells/mm2, and 3.6 ± 0.3%, P < 0.05, P = 0.17, and P < 0.05, respectively) (Fig. 1).
Fig 1.

Differences in histological staining of collagen content and cardiac inflammation between subgroups, revealing increased collagen content and inflammatory cells in both the IMID and non-IMID groups of DCM patients. Non-IMID, DCM patients without an immune-mediated inflammatory disease; IMID, DCM patients with an immune-mediated inflammatory disease; HE, hematoxylin and eosin stain; SR, Sirius red stain; CD45, CD45-staining lymphocytes.
Increased inflammation (>14 positive lymphocytes/mm2) was detected in 23 (18%) of the non-IMID DCM patients and 10 (34%, P = 0.16) of the IMID DCM patients, whereas none of the controls demonstrated increased inflammation (P < 0.05 compared to both IMID and non-IMID DCM patients).
Collagen content significantly correlated with number of CD45-positive lymphocytes (R = 0.35, P < 0.001) but not with CD68 macrophages (R = 0.16, P = 0.46).
A significant correlation between the number of CD45-positive lymphocytes and parvovirus B19 copy numbers was observed in the non-IMID group (R = 0.27, P = 0.008) but not in the IMID group (R = 0.14, P = 0.52). In the overall population, increased numbers of CD45-positive inflammatory cells showed a trend toward a lower LVEF (R = 0.28, P = 0.07).
The IMID patients showed no significant correlation between sIL-2R or neopterin levels and the number of CD45-positive lymphocytes or CD68-positive macrophages, parvovirus B19 copy numbers, or cardiac function.
ROC analysis showed an area under the curve of 0.814 for sIL-2R to predict IMID DCM (95% confidence interval [CI], 0.71 to 0.92) (Fig. 2). An optimized cutoff point of 1,942 pg/ml showed 81% sensitivity, 73% specificity, and 94% negative predictive value. Neopterin was less sensitive and specific, as can be seen on the ROC curves, with an area under the curve of 0.624 (Fig. 2).
Fig 2.
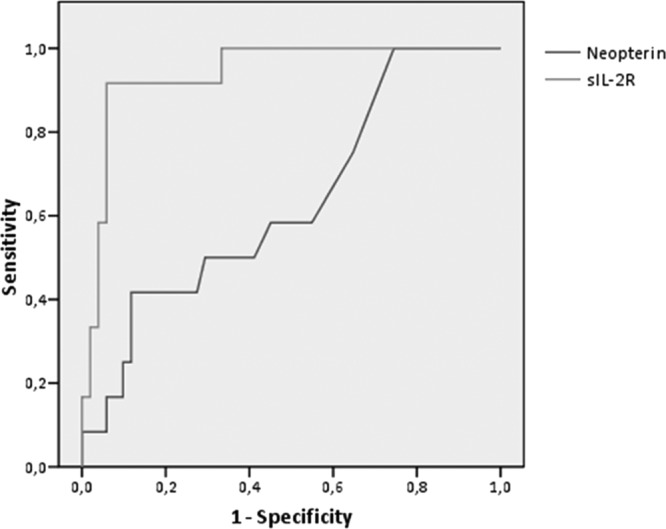
ROC analysis shows the power of neopterin and sIL-2R levels in differentiating IMID from non-IMID DCM patients. sIL-2R, soluble interleukin-2 receptor. The area under the ROC curve for sIL-2R is 0.814 (95% CI, 0.71 to 0.92) and for neopterin is 0.624 (95% CI, 0.64 to 0.79).
DISCUSSION
The present study evaluated the prevalence and quantity of cardiotrophic viruses in heart tissue of DCM patients with and without an IMID in comparison to control hearts without DCM. We observed significantly higher parvovirus B19 prevalence in non-IMID DCM patients compared to IMID DCM patients. In addition, parvovirus B19 copy numbers were higher in non-IMID than in IMID DCM patients. These findings suggest differences in pathophysiologic mechanisms between DCM in IMID patients and virus-induced forms of DCM. This may be important, since treatment strategies for the two forms of DCM may differ (12, 16, 21).
Our current understanding of inflammation-mediated pathogenesis in heart disease is largely derived from a variety of animal models, each focusing on different aspects of inflammatory heart disease. The pathogenesis has been described as a “triphasic process” with distinct mechanisms and clinical manifestation in each phase (3). The first phase is dominated by myocardial infection leading to direct destruction of the cardiomyocytes by direct virus-mediated lysis linked to the innate immune response. The second phase is set in motion by the subsequent immunologic response as virus peptide fragments are processed in the host cell and presented to the CD8+ lymphocytes (36). These subsequently primed T cells are capable of detecting the virus antigen and destroy the infected cardiac cells through Fas/Fas ligand, tumor necrosis factor (TNF)-related apoptosis-inducing ligand, cytokine, or perforin pathways. In addition, host myocardial cellular antigens can share epitopic similarities (molecular mimicry) with virus antigens and can therefore induce an autoimmune trait that can sustain the inflammatory response even after virus elimination and hence the chronic inflammatory phase (15, 39). Finally, in the third phase, the pathological signs of myocardial inflammation generally disappear and biventricular dilatation with cardiac failure can be observed, which has been linked to either chronic immune-mediated inflammation, virus persistence, or latent endomyocardial replication. These findings indicate that inflammatory cardiomyopathy is a unique disease for the progression of which both the primary antigen and subsequent autoimmune disease may be responsible.
Several autoimmune diseases have been related to heart failure (13, 28, 40). Furthermore, increased immune markers such as sIL-2R and neopterin as well as the presence of specific cardiac autoantibodies have been identified in patients with DCM (1, 6, 8, 23, 29). Increased levels of sIL-2R and neopterin have been shown to be more prevalent in patients with DCM than in those with ischemic heart failure or controls and have also been linked to disease severity (1, 23).
Our approach differed from previous studies by careful differentiation of DCM patients with and without an IMID. In addition, we investigated both immune markers and myocardial inflammation. We found increased levels of sIL-2R and neopterin within the IMID DCM subgroup only, compatible with the fact that these patients had more pronounced activation of their immune system. However, this increased systemic immune activation did not relate to the degree of myocardial inflammation, fibrosis, or cardiac function. The use of immunosuppressive therapy in the majority of IMID patients may have influenced both systemic and myocardial inflammation, possibly affecting these results.
In the present study, we found equal myocardial virus persistence, especially for parvovirus B19, in up to 80% of the non-IMID DCM patients and controls, comparable with results from previous reports (20, 35). Interestingly, IMID DCM patients showed a lower virus persistence and significantly lower virus copy numbers. While all DCM patients are equally infected with parvovirus B19 once in their lives, it may be suggested that IMID DCM patients with an increased T-cell activation, as reflected by increased serum sIL-2R levels, might have a better parvovirus B19 clearance. Additionally, the non-IMID subgroup demonstrated increased parvovirus B19 copy numbers in the heart, which correlated with an increased degree of myocardial inflammation, suggesting that parvovirus B19 may play an essential role in the pathogenesis of heart disease in patients without an IMID, while for IMID DCM patients the systemic inflammation might play the central role in the eventual development of DCM (Fig. 3).
Fig 3.
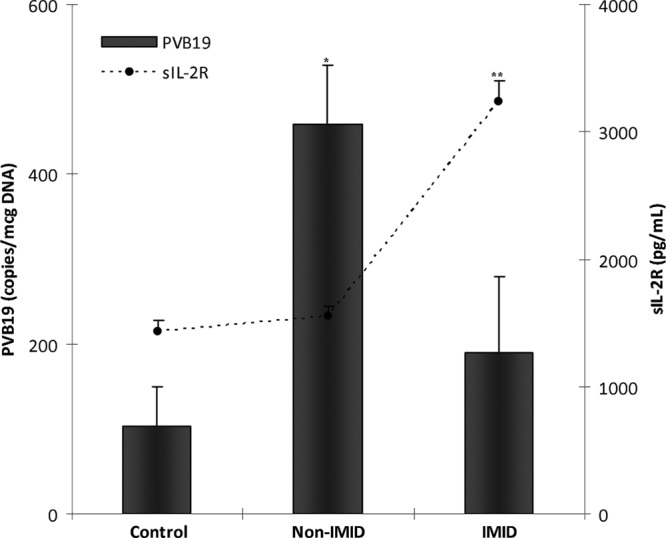
Differences in parvovirus B19 copy numbers and serum sIL-2R levels between subgroups. Values are means ± standard errors of the means (SEM). PVB19, parvovirus B19; sIL-2R, soluble interleukin-2 receptor; non-IMID, DCM patients without an immune-mediated inflammatory disease; IMID, DCM patients with an immune-mediated inflammatory disease; *, P < 0.001 compared to controls and IMID patients; **, P < 0.001 compared to controls and non-IMID patients.
Classification of DCM allows for specific therapeutic approaches targeting pathogenic pathways of the disease, since the same method of treatment will not be equally beneficial for both forms of inflammatory heart disease (18, 32). Several immunomodulatory treatment strategies aiming at either attenuation of chronic inflammation in DCM patients without virus persistence or virus elimination by antiviral approaches have demonstrated beneficial effects in selected DCM patients based on endomyocardial biopsy findings (12, 21, 26, 27). Determination of sIL-2R in addition to virus quantification in endomyocardial biopsy specimens may further help in the etiopathogenic differentiation of the disease.
In conclusion, decreased parvovirus B19 prevalence and copy numbers in the hearts of DCM patients with an underlying IMID compared to those without an autoimmune disease and controls suggest different pathophysiologic mechanisms in virus- and autoimmune-induced forms of DCM.
ACKNOWLEDGMENTS
We gratefully acknowledge Jos Austen, Jan Damoiseaux, and Steven Arends (Department of Internal Medicine, Division of Clinical and Experimental Immunology, Maastricht University Medical Centre, The Netherlands) for their expert assistance.
This study was supported by research grants from the Netherlands Heart Foundation (NHS, 2005B082 and 2007B036) and a VIDI grant from the Netherlands Organization for Scientific Research (NWO) to S.H.
Footnotes
Published ahead of print 13 June 2012
REFERENCES
- 1. Abbate A, et al. 2003. Plasma concentrations of interleukin-2 soluble receptor in mild ischaemic left ventricular dysfunction. Eur. J. Heart Fail. 5:23–25 [DOI] [PubMed] [Google Scholar]
- 2. Aretz HT. 1987. Myocarditis: the Dallas criteria. Hum. Pathol. 18:619–624 [DOI] [PubMed] [Google Scholar]
- 3. Ayach B, Fuse K, Martino T, Liu P. 2003. Dissecting mechanisms of innate and acquired immunity in myocarditis. Curr. Opin. Cardiol. 18:175–181 [DOI] [PubMed] [Google Scholar]
- 4. Bock CT, Klingel K, Kandolf R. 2010. Human parvovirus B19-associated myocarditis. N. Engl. J. Med. 362:1248–1249 [DOI] [PubMed] [Google Scholar]
- 5. Caforio AL, et al. 2001. Elevated serum levels of soluble interleukin-2 receptor, neopterin and beta-2-microglobulin in idiopathic dilated cardiomyopathy: relation to disease severity and autoimmune pathogenesis. Eur. J. Heart Fail. 3:155–163 [DOI] [PubMed] [Google Scholar]
- 6. Caforio AL, et al. 1997. Circulating cardiac-specific autoantibodies as markers of autoimmunity in clinical and biopsy-proven myocarditis. The Myocarditis Treatment Trial Investigators. Eur. Heart J. 18:270–275 [DOI] [PubMed] [Google Scholar]
- 7. Caforio AL, Iliceto S. 2008. Genetically determined myocarditis: clinical presentation and immunological characteristics. Curr. Opin. Cardiol. 23:219–226 [DOI] [PubMed] [Google Scholar]
- 8. Caforio AL, Mahon NJ, Tona F, McKenna WJ. 2002. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur. J. Heart Fail. 4:411–417 [DOI] [PubMed] [Google Scholar]
- 9. Cohen Tervaert J, et al. 2006. Principles and methods for assessing autoimmunity associated with exposure to chemicals. Environmental health criteria, vol 236 World Health Organization Press, Geneva, Switzerland [Google Scholar]
- 10. Davidson A, Diamond B. 2001. Autoimmune diseases. N. Engl. J. Med. 345:340–350 [DOI] [PubMed] [Google Scholar]
- 11. Dennert R, Crijns HJ, Heymans S. 2008. Acute viral myocarditis. Eur. Heart J. 29:2073–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dennert R, et al. 2010. Intravenous immunoglobulin therapy for patients with idiopathic cardiomyopathy and endomyocardial biopsy-proven high PVB19 viral load. Antivir. Ther. 15:193–201 [DOI] [PubMed] [Google Scholar]
- 13. Dennert RM, et al. 2010. Cardiac involvement in Churg-Strauss syndrome. Arthritis Rheum. 62:627–634 [DOI] [PubMed] [Google Scholar]
- 14. Eaton WW, Rose NR, Kalaydjian A, Pedersen MG, Mortensen PB. 2007. Epidemiology of autoimmune diseases in Denmark. J. Autoimmun. 29:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feldman AM, McNamara D. 2000. Myocarditis. N. Engl. J. Med. 343:1388–1398 [DOI] [PubMed] [Google Scholar]
- 16. Frustaci A, Russo MA, Chimenti C. 2009. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur. Heart J. 30:1995–2002 [DOI] [PubMed] [Google Scholar]
- 17. Gottdiener JS, et al. 2004. American Society of Echocardiography recommendations for use of echocardiography in clinical trials. J. Am. Soc. Echocardiogr. 17:1086–1119 [DOI] [PubMed] [Google Scholar]
- 18. Heymans S. 2007. Myocarditis and heart failure: need for better diagnostic, predictive, and therapeutic tools. Eur. Heart J. 28:1279–1280 [DOI] [PubMed] [Google Scholar]
- 19. Heymans S, et al. 2005. Increased cardiac expression of tissue inhibitor of metalloproteinase-1 and tissue inhibitor of metalloproteinase-2 is related to cardiac fibrosis and dysfunction in the chronic pressure-overloaded human heart. Circulation 112:1136–1144 [DOI] [PubMed] [Google Scholar]
- 20. Kuethe F, et al. 2009. Prevalence of parvovirus B19 and human bocavirus DNA in the heart of patients with no evidence of dilated cardiomyopathy or myocarditis. Clin. Infect. Dis. 49:1660–1668 [DOI] [PubMed] [Google Scholar]
- 21. Kuhl U, et al. 2003. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 107:2793–2798 [DOI] [PubMed] [Google Scholar]
- 22. Kuhl U, et al. 2005. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 112:1965–1970 [DOI] [PubMed] [Google Scholar]
- 23. Limas CJ, Goldenberg IF, Limas C. 1995. Soluble interleukin-2 receptor levels in patients with dilated cardiomyopathy. Correlation with disease severity and cardiac autoantibodies. Circulation 91:631–634 [DOI] [PubMed] [Google Scholar]
- 24. Limas CJ, et al. 2003. Prognostic significance of soluble interleukin-2 receptor levels in patients with dilated cardiomyopathy. Eur. J. Clin. Invest. 33:443–448 [DOI] [PubMed] [Google Scholar]
- 25. Maisch B, Portig I, Ristic A, Hufnagel G, Pankuweit S. 2000. Definition of inflammatory cardiomyopathy (myocarditis): on the way to consensus. A status report. Herz 25:200–209 [DOI] [PubMed] [Google Scholar]
- 26. Mason JW, et al. 1995. A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N. Engl. J. Med. 333:269–275 [DOI] [PubMed] [Google Scholar]
- 27. McNamara DM, et al. 2001. Controlled trial of intravenous immune globulin in recent-onset dilated cardiomyopathy. Circulation 103:2254–2259 [DOI] [PubMed] [Google Scholar]
- 28. Nussinovitch U, Shoenfeld Y. 2009. Autoimmunity and heart diseases: pathogenesis and diagnostic criteria. Arch. Immunol. Ther. Exp. (Warsz.) 57:95–104 [DOI] [PubMed] [Google Scholar]
- 29. Okazaki T, et al. 2003. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 9:1477–1483 [DOI] [PubMed] [Google Scholar]
- 30. Richardson P, et al. 1996. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 93:841–842 [DOI] [PubMed] [Google Scholar]
- 31. Rose NR. 2002. Mechanisms of autoimmunity. Semin. Liver Dis. 22:387–394 [DOI] [PubMed] [Google Scholar]
- 32. Rose NR. 2009. Myocarditis: infection versus autoimmunity. J. Clin. Immunol. 29:730–737 [DOI] [PubMed] [Google Scholar]
- 33. Rose NR. 2000. Viral damage or ‘molecular mimicry’—placing the blame in myocarditis. Nat. Med. 6:631–632 [DOI] [PubMed] [Google Scholar]
- 34. Rose NR, Bona C. 1993. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol. Today 14:426–430 [DOI] [PubMed] [Google Scholar]
- 35. Schenk T, Enders M, Pollak S, Hahn R, Huzly D. 2009. High prevalence of human parvovirus B19 DNA in myocardial autopsy samples from subjects without myocarditis or dilative cardiomyopathy. J. Clin. Microbiol. 47:106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seko Y, et al. 2002. Role of Fas/FasL pathway in the activation of infiltrating cells in murine acute myocarditis caused by Coxsackievirus B3. J. Am. Coll. Cardiol. 39:1399–1403 [DOI] [PubMed] [Google Scholar]
- 37. Sloka S. 2002. Observations on recent studies showing increased co-occurrence of autoimmune diseases. J. Autoimmun. 18:251–257 [DOI] [PubMed] [Google Scholar]
- 38. Stegeman CA, Tervaert JW, Huitema MG, Kallenberg CG. 1993. Serum markers of T cell activation in relapses of Wegener's granulomatosis. Clin. Exp. Immunol. 91:415–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Streitz M, et al. 2008. NS1 specific CD8+ T-cells with effector function and TRBV11 dominance in a patient with parvovirus B19 associated inflammatory cardiomyopathy. PLoS One 3:e2361 doi:10.1371/journal.pone.0002361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tyndall AJ, et al. 2010. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis. 69:1809–1815 [DOI] [PubMed] [Google Scholar]
- 41. van Haelst PL, et al. 2003. Usefulness of elevated neopterin and C-reactive protein levels in predicting cardiovascular events in patients with non-Q-wave myocardial infarction. Am. J. Cardiol. 92:1201–1203 [DOI] [PubMed] [Google Scholar]
- 42. van Haelst PL, et al. 2004. Circulating monocytes in patients with acute coronary syndromes lack sufficient interleukin-10 production after lipopolysaccharide stimulation. Clin. Exp. Immunol. 138:364–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42a. Watzinger F, Ebner K, Lion T. 2006. Detection and monitoring of virus infections by real-time PCR. Mol. Aspects Med. 27:254–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Williams JP, Meyers JA. 2002. Immune-mediated inflammatory disorders (I.M.I.D.s): the economic and clinical costs. Am. J. Manag. Care 8:S664–S681; quiz S682–S685 [PubMed] [Google Scholar]
- 44. Yoshikawa T, Baba A, Nagatomo Y. 2009. Autoimmune mechanisms underlying dilated cardiomyopathy. Circ. J. 73:602–607 [DOI] [PubMed] [Google Scholar]