

Abstract
Epithelial integrity is essential for homeostasis and poses a formidable barrier to pathogen entry. Major factors for viral entry into epithelial cells are the localization and abundance of the primary receptor. The coxsackievirus and adenovirus receptor (CAR) is a primary receptor for these two pathogenic groups of viruses. In polarized epithelia, a low-abundance, alternatively spliced eight-exon isoform of CAR, CAREx8, is localized apically where it can support viral infection from the air-exposed surface. Using biochemical, cell biology, genetic, and spectroscopic approaches, we show that the levels of apical CAREx8 are negatively regulated by the PDZ domain-containing protein MAGI-1 (membrane-associated guanylate kinase with inverted orientation protein-1) and that two MAGI-1 PDZ domains, PDZ1 and PDZ3, regulate CAREx8 levels in opposing ways. Similar to full-length MAGI-1, expression of the isolated PDZ3 domain significantly reduces cell surface CAREx8 abundance and adenovirus infection. In contrast, the PDZ1 domain is able to rescue CAREx8 and adenovirus infection from MAGI-1-mediated suppression. These data suggest a novel cell-based strategy to either suppress viral infection or augment adenovirus-based gene therapy.
INTRODUCTION
Understanding the development, maintenance, and composition of epithelial barriers is vital to human health and disease. Epithelial barriers segregate the microbe-infested external environment from the body's sterile internal environment. Mechanistic studies of pathogenic microbial penetration of epithelia provide insights into epithelial cell structure and regulation and may lead to novel therapeutic approaches. A major unanswered issue is how pathogenic viruses can initiate infection from the apical surface using receptors segregated on the basolateral membrane (12, 37, 39).
Two distinct groups of viruses, coxsackie B viruses and many adenoviruses (AdVs), interact with the coxsackievirus and adenovirus receptor (CAR) (1, 2, 34). CAR has several alternatively spliced transcripts, two of which encode transmembrane splice forms: one that terminates after the seventh exon (CAREx7) and one that splices from a cryptic splice donor site within the seventh exon and terminates after the eighth exon (CAREx8) (11, 32, 34). These two isoforms differ only in the last 26 (CAREx7) or 13 (CAREx8) amino acids (aa) of the cytoplasmic domain, suggesting distinct interactions and functional roles. In polarized epithelial cells, CAREx7 resides on the basolateral surface and is thus sequestered from potential viral interactions on the apical surface (38, 39). In contrast, although it is a less abundant isoform, CAREx8 can localize at the apical surface in primary airway epithelia where it can mediate adenovirus infection from the airway (11). The knowledge of the existence of an apical receptor results in a paradigm shift from the commonly held belief that there must be a transient or sustained break in the barrier for the virus to gain access to the basolateral receptor (3, 4, 36, 44). Furthermore, modulation of the expression levels and localization of CAREx8 presents an opportunity to change the susceptibility of an epithelium to these viruses.
The apical and basolateral surfaces of polarized epithelial cells are quite different in terms of lipid and protein composition due to intracellular sorting and trafficking (26, 30). Protein-sorting pathways have key molecules that recognize specific proteins, lipids, or posttranslationally modified sequences. One large class of protein-sorting molecules is the family of PSD95/Dlg/ZO-1 (PDZ)-domain-containing proteins (21, 35). The PDZ domain is a modular sequence, about 90 aa in length, which generally interacts with specific C-terminal motifs named PDZ-binding domains. Membrane-associated guanylate kinases (MAGUKs) contain one or several PDZ domains, along with other interacting domains, that allow diverse interactions and, often, multimerization (15). MAGI-1 (membrane-associated guanylate kinase with inverted orientation protein-1) is a member of the MAGUK family and has three alternatively spliced isoforms (MAGI-1a, -b and -c) (7, 24). MAGI-1 contains an inactive guanylate kinase domain, two WW domains, and up to six PDZ domains (PDZ0 to -5). MAGI-1 is present in the tight junctions of cultured epithelia. MAGI-1 binding partners include several important junction-associated proteins (9, 13, 17, 18, 41–43), ion channels (27, 31), the tumor suppressor PTEN (23), and viral oncogenes (16, 33), thus implicating it in epithelial junction composition and stability, epithelial-mesenchymal transition, and invasiveness.
Previous work has shown that MAGI-1 is able to suppress CAREx8 expression in nonpolarized cells (11). We hypothesized that MAGI-1 may also regulate the levels of CAREx8 in polarized epithelia and, thus, regulate apical adenoviral infection. Here, we show an inverse relationship between the levels of MAGI-1 and apical adenoviral infection of polarized epithelial cells. We also show that CAREx8 directly interacts with two MAGI-1 PDZ domains, each having distinct biologically relevant effects on adenovirus infection. While PDZ1 coexpression with MAGI-1 protects CAREx8 from MAGI-1-mediated loss and results in increased viral infection, interaction with PDZ3 sequesters CAREx8 within the cell and decreases viral infection. These results show promise for developing derivative molecules able to enhance or suppress the expression of CAREx8 in order to control the susceptibility of polarized epithelia to viral infection.
MATERIALS AND METHODS
Materials.
Anti-actin and RmcB (anti-CAR) were from Millipore (Bedford, MA), anti-FLAG was from Sigma-Aldrich (St. Louis, MO), mouse anti-green fluorescent protein (anti-GFP) was from Clontech (Mountainview, CA), anti-V5, anti-ZO-1, rabbit anti-GFP, and fluorescently labeled secondary antibodies (Abs) were from Invitrogen (Carlsbad, CA), anti-MAGI was from Novus Biologicals (Littleton, CO), and anti-myc was from Cell Signaling (Danvers, MA). Horseradish peroxidase-labeled secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). Rabbit anti-CAR-1605 and anti-CAREx8-5678 have previously been described (10, 11, 14, 28). Madin-Darby canine kidney (MDCK) epithelial and Chinese hamster ovary (CHO-K1) cells (ATCC, Rockville, MD) were cultured under standard conditions (minimum essential medium [MEM; Invitrogen] with 5% fetal bovine serum [FBS; PAA Laboratories, Dartmouth, MA] or Dulbecco's modified Eagle's medium [DMEM] with 10% FBS, 1% penicillin-streptomycin, 2 mM l-glutamine). Individual MAGI-1b PDZ domains (PDZ1, -2, -3, and -5) were cloned into pcDNA/V5/GW/D-TOPO vector (Invitrogen) for the in vitro pulldown assays. Plasmids for myc-tagged full-length MAGI-1c and all PDZ domains (PDZ0 to -5) were generously provided by Zhigang Xu (Shandong University, Shandong, China) and Stefan Heller (Stanford University School of Medicine, Stanford, CA) (42). For Fluorescence Resonance Energy Transfer (FRET) and binding assays, MAGI-1 PDZ1, PDZ2, and PDZ3 domains and the CAREx8 C terminus were cloned (In-Fusion; Clontech) into a pGEX-6p vector (GE Healthcare, Piscataway, NJ) modified to contain a His tag upstream of the glutathione transferase (GST) tag (pHH2). Xfect transfection reagent (Clontech) was used for plasmid transfection. Adenovirus vectors, AdV-CAREx7, AdV-CAREx8, AdV-GFP, and AdV–β-galactosidase (AdV-β-Gal) were from the University of Iowa Vector Core, Iowa City, IA.
Cell polarization, TER, and conductance.
For polarization studies, cells were seeded on 10-mm-diameter polyester 0.4-μm-pore-size Millicell semipermeable membranes (Millipore). Media on the apical surface of cells were removed every alternate day in order to establish and maintain an air-liquid interface. Transepithelial resistance (TER) was measured with a chopstick ohmmeter (World Precision Instruments, Sarasota, FL) every other day, as previously described (28), and reported as conductance (1/TER).
Immunocytochemistry.
Cells seeded on Millicell membranes or collagen-coated glass slides were subjected to immunocytochemistry as previously described (11, 28). Briefly, cells were fixed with methanol containing 1% paraformaldehyde, blocked with 2% bovine serum albumin (BSA) in SuperBlock blocking buffer (Pierce, Rockford, IL), and incubated with primary Abs, followed by Alexa Fluor-labeled secondary Abs (Invitrogen). Millicell membranes were mounted onto glass slides using Vectashield mounting media with DAPI (4′,6′diamidino-2-phenylindole; Vector Laboratories, Burlingame, CA). The CAREx8 C-terminal peptide consisted of the unique terminal 13 aa. Staining was evaluated using a laser-scanning confocal microscope (Olympus FV1000) with a 60× oil immersion lens; images are shown as either single x-y or single x-z optical sections.
AdV-β-Gal infection and β-galactosidase assay.
CHO-K1 and MDCK cells were infected with AdV-β-Gal at a multiplicity of infection of 100 for 1 h at 37°C. Virus inoculums were removed and cells washed 2 times and incubated for 24 h prior to lysis. The protein concentration was determined by a Bio-Rad protein assay (Hercules, CA), and β-galactosidase expression was determined with a Galacto-Light Plus system (Applied Biosystems, Carlsbad, CA) as previously described (11).
IP and WB.
Cells were lysed, immunoprecipitated (IP), and blotted as previously described (11, 28). Briefly, ice-chilled cells were washed and lysed in buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, protease inhibitors [leupeptin and aprotinin at 20 μg/ml, pepstatin at 10 μg/ml, and phenylmethylsulfonyl fluoride at 17.4 μg/ml]). Cells were scraped, sonicated, and centrifuged. The protein concentration was estimated by a Bio-Rad protein assay. For IP, primary Ab was added to equal amounts of protein for 2 h up to overnight at 4°C. Proteins were isolated with protein G Sepharose (GE Healthcare) and eluted with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer, followed by SDS-PAGE and Western blotting (WB). Protein bands were detected with ECL reagents (Pierce, Rockford, IL) and imaged on a Fuji LAS 4000 imaging system and/or developed in an X-ray medical-film processor (Konica SRX 101).
Cell surface biotinylation.
Polarized cells were subjected to cell surface biotinylation as previously described (10). Briefly, the apical surface of cells was incubated with Sulfo-NHS-SS-biotin (Thermo Fisher Scientific, Waltham, MA) at 1 mg/ml, free Sulfo-NHS-SS-biotin was quenched with 100 mM glycine, and cells were lysed as described above. Equal amounts of protein were incubated with NeutrAvidin beads (Pierce) to isolate Sulfo-NHS-SS-biotin-labeled proteins prior to SDS-PAGE and WB.
MAGI-1 siRNA.
Two different Stealth small interfering RNAs (siRNAs) (Invitrogen; A [5′-AAAUCUUGCUCCUGUGUUGCUUGGG] and B [5′-UAGAGGUCCAUGUUAUACUCUCGGC]) or a control siRNA (C [5′-AUUUGAAGCCACGUCCGCUGGGCUG]) were transfected into MDCK cells with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. Briefly, a siRNA duplex was transfected into MDCK cells at the time of seeding. Cells were seeded to achieve 100% confluence 48 h after plating.
RNA isolation and real-time RT-PCR.
Total RNA was isolated with TRIzol (Invitrogen) according to the manufacturer's protocol. Reverse transcription (RT) was performed using 1 μg of RNA and a Quanta First Strand kit (Quanta BioSciences, Gaithersburg, MD) prior to quantitative PCR according to the manufacturer's protocol. Quantitative PCR was performed using SYBR green with low ROX (Quanta) in Stratagene's real-time PCR system (Agilent Technologies, Santa Clara, CA) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the internal standard. The primers used were as follows: for MAGI-1F, 5′-GCTGAACAAGGACCTGCGACATTT; for MAGI-1R, 5′-AGTTATAGTCCACGCCAGGCACTT. The relative levels of expression of target genes were quantified by comparative threshold cycle (CT) analysis using Mx4000p software v5 for data analysis.
Yeast two-hybrid assay.
Yeast two-hybrid (Y2H) screening was performed using a Matchmaker kit (Clontech) according to the manufacturer's protocols. Briefly, the cytoplasmic C terminus of CAREx8 was subcloned into the pGBKT7 bait vector. The known prey MAGI-1 PDZ domains (PDZ0 to -5) were individually cloned into the PGADT7 prey vector, pretransformed into yeast Y187 cells, and screened by separate matings with Y2H yeast cells carrying each bait plasmid and plating onto high-stringency selective synthetic dropout (SD)-/-Leu/-Trp media. Colonies that grew in 3 to 6 days were picked and rescreened on SD-Ade/-His/-Leu/-Trp plates containing X-α-Gal (5-bromo-4-chloro-3-indolyl-β-α-d-galactopyranoside).
In vitro pulldown assay.
A TNT T7 quick-coupled transcription/translation system (Promega Corporation, Madison, WI) was used to synthesize l-[35S]methionine (PerkinElmer, Waltham, MA)-labeled MAGI-1 PDZ domains according to the manufacturer's protocol. COS-7 cells were transfected with the FLAG-tagged CAREx8 plasmid and subjected to IP. In vitro-translated MAGI-1 PDZ domain proteins were mixed with CAR-IP lysates at 4°C for 2 h, followed by washing, centrifugation, and elution with 2× denaturing buffer. Labeled proteins were separated by SDS-PAGE and either visualized by autoradiography of dried gels or transferred to polyvinylidene difluoride (PVDF) for autoradiography followed by WB.
MAGI-1 PDZ domains and CAREx8 C-terminal protein purification.
pHH2-PDZ and -CAREx8 plasmids transformed into Rosetta (BL21) Escherichia coli cells (EMD Chemicals, Gibbstown, NJ) were grown in LB cultures to an optical density at 600 nm (OD600) of 1.5 to 1.9, induced with 0.1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG), and incubated for an additional 5 h at 37°C. Cells were harvested by centrifugation, and pellets were resuspended in 2× L&C buffer (40 mM Tris [pH 8.0], 0.6 mM NaCl, 20% glycerol)–2× protease inhibitor cocktail (Sigma-Aldrich)–2 mM EDTA–2 mM dithiothreitol (DTT). The resuspended cells were sonicated on ice for 6 min with 30-s pulses and centrifuged at 9,500 rpm at 4°C for 20 min. Filtered supernatant (Whatman filters; GE Healthcare) (1.2-μm pore size) was pumped through a Fractogel GST-bind cartridge (EMD Millipore, Billerica, MA) according to the manufacturer's protocol. The column was washed with 1× L&C buffer, followed by ATP wash buffer (2× L&C buffer, 10 mM ATP, 50 mM MgCl2) and then 1× L&C buffer containing 1 mM DTT and 1 mM EDTA. GST-tagged protein was eluted with reduced glutathione after washing. To obtain protein without any tag, PreScission protease mix (1 ml of L&C buffer containing DTT and EDTA and 50 to 80 μg of protease) was circulated through the column for 5 h. L&C buffer containing DTT and EDTA was pumped through the column to collect fractions containing tag-free proteins, which were quantified using the Bio-Rad protein assay, and the quality was verified using SDS-PAGE.
Protein labeling.
Proteins were dialyzed with 10 mM HEPES (pH 8.0), 0.1 mM EDTA, 0.4 mM DTT, 400 mM KCl, and 5% glycerol at 4°C for a minimum of 16 h using a Slide-a-lyzer dialysis cassette (Thermo Scientific). Purified MAGI-1 PDZ domain proteins without the GST tag were labeled with a FluoroLink Ab Cy3-labeling kit (GE Healthcare, Piscataway, NJ), while purified CAREx8 C-terminal protein was labeled with a FluoroLink Ab Cy5-labeling kit according to the manufacturer's recommendations. Absorbance was used to determine protein concentrations and dye-to-protein ratios. The labeling ratios were approximately one dye per peptide.
FRET.
Fluorescence Resonance Energy Transfer (FRET) from each Cy3-labeled MAGI-1 PDZ domain to each Cy5-labeled CAR isoform was performed as described previously (19, 20) with a PC1 photon-counting spectrofluorometer (ISS Inc., Champaign, IL). Briefly, emission spectra (560 to 700 nm) were obtained from 30 nM donor (Cy3-labeled PDZ proteins)–phosphate-buffered saline (PBS) at 25°C upon excitation at 550 nm with increasing concentrations of acceptor (Cy5-labeled CAREx8 C-terminal protein). The spectra were corrected for background (buffer only and acceptor only), and the maximal intensities were measured using Vinci 1.5 software (ISS Inc., Champaign, IL). The protein binding affinity and the energy transfer efficiency were calculated using the amount of sensitized acceptor fluorescence, and the intermolecular distance was calculated according to the Förster equation as described previously (19, 20).
Direct ligand binding assay.
Binding assays were performed using fluorescently labeled peptide and a nonlabeled peptide. Increasing concentrations of purified unlabeled peptide were added to 30 nM Cy-labeled protein–2 ml of phosphate-buffered saline (pH 7.4). Fluorescence emission spectra (for Cy3, 560 to 650 nm; for Cy5, 660 to 700 nm) were obtained at 25°C with a PC1 photon-counting spectrofluorometer (ISS Inc., Champaign, IL) upon excitation of either Cy3-labeled MAGI-1 PDZ protein at 550 nm or Cy5-labeled CAR at 650 nm and corrected for background (buffer only) and maximal intensities measured. The dissociation constant and the number of binding sites were obtained by calculation of a double-reciprocal plot of the fluorescence intensity of the labeled protein. The saturation curves were fitted to a Hill plot to confirm the number of binding sites using SigmaPlot software (20).
Statistical analysis.
All experiments were performed independently at least three times. Data were expressed as either the results of a representative experiment or the means ± standard errors of the means of the results of at least three separate experiments. Significant differences were analyzed using Student's t test and two-tailed distribution. Results were considered to be statistically significant when P ≤ 0.05.
RESULTS
Localization of CAREx8 is distinct from that of CAREx7 and MAGI-1 in polarized epithelial cells.
Previous investigations demonstrated that endogenous CAREx7 and CAREx8 have distinct localizations in polarized primary human airway epithelia (11). Moreover, CAREx7 and CAREx8 have distinct interactions with MAGI-1 in nonpolarized cells (COS-7 and CHO), such that CAREx7 recruits MAGI-1 to cell-cell junctions whereas MAGI-1 is able to suppress the cellular levels of CAREx8 (11, 13). Thus, we asked whether MAGI-1 was a major regulator of CAREx8 in polarized epithelia. Since primary airway epithelia are resistant to genetic modification and currently available antibodies to human MAGI-1 are unsuitable for WB, MDCK cells were chosen for this study as model epithelial cells. MDCK cells polarize rapidly, express detectable endogenous MAGI-1, express CAREx7 and CAREx8 isoforms at a ratio similar to that seen with primary airway epithelia (12:1; data not shown), and are amenable to genetic manipulation.
Considering the distinctly different localization of each CAR isoform in polarized primary human airway cells, we first asked whether endogenous CAR and MAGI-1 colocalized in polarized MDCK cells (Fig. 1A to C). MDCK cells were seeded onto semipermeable membranes for 4 days and then fixed and stained with Abs for MAGI-1 (green) and total CAR (red, Fig. 1A) or, specifically, CAREx8 (red, Fig. 1B). Abs against both MAGI-1 and total CAR showed overlapping labeling at cell boundaries (Fig. 1A, merge image; see yellow), consistent with residence in the junctional adhesion complex. In contrast, immunocytochemistry for endogenous CAREx8 protein was low and localization rarely overlapped with that of MAGI-1 (Fig. 1B). To confirm the specificity of the CAREx8 immunocytochemistry, CAREx8-specific Ab binding was competed with purified CAREx8 C-terminal peptide and revealed low to no background staining (Fig. 1C). No reduction in junctional CAR staining was observed when Ab for total CAR was incubated with CAREx8 C-terminal peptide (Fig. 1D). Consistent with primary human airway epithelia, the majority of CAR within the polarized epithelium was CAREx7.
Fig 1.

Localization of CAREx8 is distinct from CAREx7 and MAGI-1 localization in polarized epithelial cells, and CAREx8 expression increases apical adenovirus infection. (A) Immunocytochemistry for endogenous MAGI-1 (green) and total CAR (red, Ab 1605p) in polarized MDCK cells. Overlapping staining was present at cell junctions (yellow, MERGE). (B) Endogenous CAREx8 expression (red, Ab 5678p) is low in MDCK cells and does not overlap MAGI-1 (green). (C and D) Peptide competition for anti-CAREx8 binding (red, Ab 5678p) demonstrates specificity of immunocytochemistry for CAREx8 (C) but not total CAR (D) (red, Ab 1605p). (E) Immunocytochemistry for exogenous expression of GFP (green), CAREx8 (red), or CAREx7 (red) in polarized MDCK cells. Nuclei are stained with DAPI (blue). (F) Time course of transepithelial conductance in MDCK cells expressing exogenous GFP, CAREx8, or CAREx7 (representative). (G) Apical AdV-β-Gal transduction of 4-day-old polarized MDCK cells expressing exogenous GFP, CAREx8, or CAREx7 (data represent averages of the results of 3 separate experiments; n = 4 to 6 replicates per experiment). Representative single optical x-y or x-z plane confocal images are shown (60× oil immersion); dashed lines in panel E represent Millicell membrane. Bars = 20 μm. *, P < 0.05.
To further verify the localization and activity of each CAR isoform, MDCK cells were transduced with adenovirus encoding CAREx7, CAREx8, or GFP and investigated for immunocytochemistry, transepithelial electrical conductance, and apical adenovirus infection. Consistent with immunocytochemistry in similarly transduced primary airways (11), GFP and exogenous CAREx8 were diffuse throughout the cytoplasm whereas exogenous CAREx7 localized primarily at the basolateral junctions (Fig. 1E). The transepithelial conductance of the epithelium was measured over several days in order to monitor cell polarization (Fig. 1F). There was no significant difference in conductance between cells expressing CAREx7, CAREx8, or GFP, indicating that these cells were polarizing at similar rates. To investigate whether exogenous expression of CAREx7, CAREx8, or GFP would alter the susceptibility of polarized MDCK cells to adenovirus infection, adenovirus carrying the gene for β-galactosidase (AdV-β-Gal) was added to the apical surface of polarized MDCK cells for 1 h and analyzed for β-galactosidase activity 24 h later. Significantly more viral transduction was found in cells overexpressing CAREx8 than under any other conditions (Fig. 1G). This indicates that overexpression of CAREx8, in the absence of altered conductance, augments apical adenoviral infection in polarized MDCK cells.
MAGI-1 regulates CAREx8 expression in polarized epithelial cells.
We previously demonstrated that CAREx8 protein expression was significantly reduced upon cotransfection with MAGI-1 in nonpolarized cells (11). To investigate whether this also occurs in polarized cells, MDCK cells were cotransfected with MAGI-1-GFP and each of CAREx7, CAREx8, or empty pcDNA3.1 plasmid and polarized for 4 days, followed by immunocytochemistry or adenovirus infection. Cells transfected with MAGI-1 alone (green, Fig. 2A) were costained with Ab for the tight junction protein ZO-1 (red, Fig. 2A). MAGI-1 staining appeared at the basolateral junctions and diffusely within the cell. Patterns of MAGI-1 staining were similar in cells cotransfected with MAGI-1 and CAREx7 (Fig. 2B). Staining for CAREx7 was largely at the tight and basolateral junctions, where it overlapped with MAGI-1 staining (Fig. 2B, MERGE panel; yellow). CAREx8 and MAGI-1 were rarely expressed in same cells when cotransfected (Fig. 2C; the arrow identifies a cell expressing CAREx8 juxtaposed to a cell expressing MAGI-1 [arrowhead]). This was in contrast to CAREx7 coexpressed with MAGI-1, where abundant colocalization was observed at the basolateral surface in the majority of the cells (Fig. 2B). These data are consistent with the MAGI-1-mediated reduction of CAREx8 observed in nonpolarized cells (11).
Fig 2.

Increased MAGI-1 expression suppresses apical CAREx8 and adenovirus infection in polarized epithelia. (A to C) Immunofluorescence staining of exogenously expressed MAGI-1 (green) and (A) the tight junction protein ZO-1 (red; endogenous), (B) CAREx7 (red; exogenous), or (C) CAREx8 (red; exogenous, white arrow). The arrowhead indicates a MAGI-1-expressing cell. Nuclei were stained with DAPI (blue). (D) Time course of conductance in MDCK cells expressing exogenous GFP and MAGI-1, CAREx7 and MAGI-1, or CAREx8 and MAGI-1 (representative). (E) Apical AdV-β-Gal transduction of 4-day-old polarized MDCK cells cotransfected with GFP, MAGI-1, CAREx7, and CAREx8. (F) Apical biotinylation of cotransfected polarized MDCK cells blotted for total CAR (1605p). A representative Western blot and quantification of three separate experiments are shown. All transfections were balanced to contain equal amounts of plasmid with empty parental plasmid pcDNA3.1. Representative single optical x-z plane confocal images are shown (60× oil immersion). Bars = 10 μm. *, P < 0.05.
We next asked whether CAREx7 or CAREx8 coexpression with MAGI-1 would alter apical adenovirus infection. Transepithelial conductance was investigated first, and no significant difference was found between cells cotransfected with MAGI-1 and GFP, CAREx8, or CAREx7 (Fig. 2D). To test the susceptibility of cotransfected cells to adenovirus infection, AdV-β-Gal was added to the apical surface of cotransfected 4-day-old cultures (Fig. 2E). No difference in transduction was observed in cells transfected with GFP and MAGI-1. Similar to the single-transfection conditions (Fig. 1G), CAREx8 cotransfection with GFP resulted in approximately 8.2 and 2.4 times more transduction than cotransfection with GFP/MAGI-1 and CAREx7, respectively (Fig. 2E). However, there was a significant reduction in apical adenovirus infection in cells cotransfected with MAGI-1 and CAREx8, and it was reduced to a level similar to that seen with CAREx7 with or without MAGI-1.
To test whether transduction reflected a change in the levels of apical CAR, polarized cultures were subjected to apical biotinylation. Labeled apical proteins were isolated with Neutravidin beads. Western blots (WB) were probed with the Ab for total CAR (Fig. 2F), which detects both endogenous CAR (dog; upper band) and exogenous CAR (human; lower band). Total apical CAR (both bands) was quantified from three separate experiments and is shown relative to epithelia expressing CAREx8 cotransfected with empty pcDNA plasmid. In agreement with the apical transduction data, significantly more apical CAR was detected under the CAREx8 conditions relative to CAREx8 cotransfected with MAGI-1, which reduced the apical surface expression of CAR to that observed with CAREx7. Taken together, these data strongly suggest that MAGI-1 is a major regulator of CAREx8, and thus of adenovirus infection, in polarized MDCK cells.
MAGI-1 knockdown increases apical adenovirus infection in polarized epithelia.
We next asked whether a reduction of endogenous MAGI-1 expression would lead to increased adenovirus infection. MDCK cells were transfected with either of two different MAGI-1 siRNAs (A or B) and compared to MDCK cells transfected with a control siRNA (C) or nontransfected siRNA (N) 48 h posttransfection. MAGI-1 RNA levels were significantly reduced by siRNA duplexes A and B with respect to control cells (Fig. 3A). Although siRNA C appeared to increase MAGI-1 mRNA levels, it did not alter MAGI-1 protein levels in comparison to nontransfected controls (Fig. 3B and C). WB and immunocytochemistry (Fig. 3D and E; data for siRNA B shown) showed that MAGI-1 protein expression was reduced by siRNAs A and B. Analysis of conductance indicated that the junctional barriers were similarly intact (data not shown). MAGI-1 knockdown with siRNA A or B resulted in a significant increase in apical AdV-β-Gal infection, in comparison to siRNA C or nontransfected cell results (Fig. 3F). Although the sensitivity of our CAREx8-specific Ab was insufficient to give a quantitative increase of endogenous CAREx8 as observed by WB or immunocytochemistry, these data indicate that endogenous MAGI-1 plays a role in suppressing apical adenovirus infection.
Fig 3.
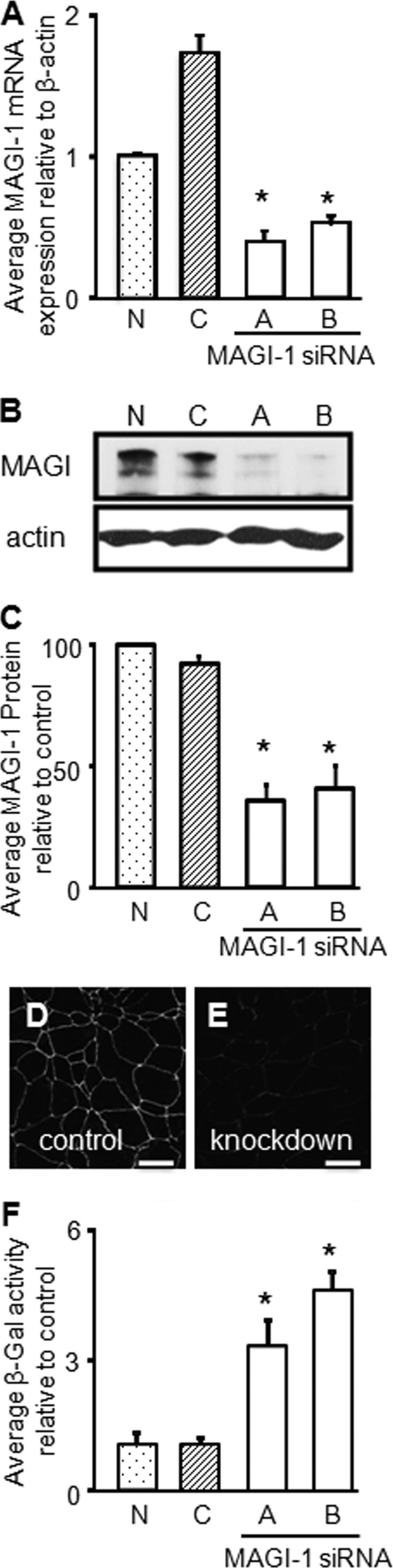
siRNA-mediated knockdown of MAGI-1 increases apical adenovirus infection in polarized MDCK cells. (A) Quantitative RT-PCR for MAGI-1 mRNA expression after MAGI-1 siRNA A or B knockdown, relative to control siRNA (C) or nontransfected cells (N). (B) WB analysis of MAGI-1 protein expression after MAGI-1 siRNA knockdown. (C) Quantification of MAGI-1 protein levels, relative to actin, with respect to nontransfected cells (100%) (n = 3). (D and E) Immunocytochemistry for endogenous MAGI-1 levels in (D) control siRNA- and (E) MAGI-1 siRNA B-treated cells. (F) Apical AdV-β-Gal transduction of polarized MDCK cells transfected with MAGI-1 siRNA A or B, control siRNA (C), or nontransfected siRNA (N). Representative confocal microscopy images are shown (60× oil immersion). Bars = 20 μm. Graphs represent averages of the results of 3 separate experiments. *, P < 0.05.
CAREx8 interacts with MAGI-1 PDZ1 and PDZ3 domains.
MAGI-1 appears to play a critical role in the abundance of CAREx8 and apical viral infection in polarized cells. Previous work indicated that the interaction requires a PDZ-based interaction (11, 13). We therefore dissected the CAR-MAGI-1 interaction using isolated individual MAGI-1 PDZ domains to determine which domain(s) specifically interacts directly with CAREx8.
Each individual MAGI-1 PDZ domain was fused with the GAL4 activation (prey) domain, and the whole intracellular C terminus of CAREx8 was fused with the GAL4 DNA-binding domain (bait) for yeast-2-hybrid (Y2H) analysis. Interaction between these domains was investigated by Y2H. An interaction (blue color) occurred between the CAREx8 C terminus and both PDZ1 and PDZ3 (Fig. 4A).
Fig 4.

CAREx8 interacts with MAGI-1 PDZ1 and PDZ3. (A) Yeast Y187 cells were transformed with pairs of pGBKT7-derived bait vector (CAREx8 C terminus) and pGADT7-derived prey vectors (PDZ domains) and plated on selective media containing X-α-Gal. Blue colonies indicate interaction. (B) In vitro pulldown assay. Individual [35S]methionine-labeled MAGI-1 PDZ domains were added to immunoprecipitated FLAG-tagged CAREx8, followed by WB for CAR and autoradiography for PDZ domain binding. In vitro translation for all PDZ domains was confirmed by autoradiography (total IVT) and CAREx8 expression in lysates by WB. (C) Co-IP of myc-tagged MAGI-1 PDZ domains with FLAG-tagged CAREx8 by mouse α-CAR RmcB from cotransfected lysates. Blots were probed with rabbit α-FLAG (CAREx8; ∼46 kDa) and rabbit α-myc (MAGI-1 PDZ domains; 20 to 28 kDa). WB of total lysates confirmed expression of all PDZ domains, CAR, and equal loading (actin). N, no PDZ domain.
To confirm the Y2H results, an in vitro pulldown assay was performed. Individual V5-tagged MAGI-1 PDZ domains were generated in vitro in the presence of [35S]methionine. FLAG-CAREx8 from lysates of transfected COS-7 cells was immunoprecipitated (IP) with anti-FLAG Ab and mixed with aliquots of synthesized 35S-labeled MAGI-1 PDZ domains. PDZ domains specifically binding to CAREx8 were analyzed by autoradiography or WB. Although CAREx8 was detected under all conditions, CAREx8 pulled down only 35S-labeled MAGI-1 PDZ1 and PDZ3 (Fig. 4B). Expression of all PDZ domains and CAR C termini was verified by autoradiography and anti-1605 CAR Ab analysis, respectively (Fig. 4B, lower and side panels).
Coimmunoprecipitation (Co-IP) further confirmed the CAREx8-MAGI-1 PDZ interactions. FLAG-tagged CAREx8 plasmid was cotransfected with each individual myc-tagged MAGI-1 PDZ domain plasmid in COS-7 cells and incubated for 48 h. CAR was immunoprecipitated with monoclonal anti-CAR-RmcB Ab, which recognizes the extracellular domain of CAR, followed by WB analysis with Abs that recognize CAR (FLAG or 1605p) and PDZ domains (myc). Similar to the results described above, CAREx8 pulled down PDZ1 and PDZ3 (Fig. 4C). Transfection of CAR and PDZ plasmids was confirmed by blotting total lysate (Fig. 4C, lower panels).
CAREx8 has stronger affinity for MAGI-1 PDZ3 than PDZ1.
The results described above show qualitative interactions between MAGI-1 PDZ1 and PDZ3 domains and CAREx8. We next asked whether there was a difference in affinity between PDZ1 or PDZ3 and CAREx8. Cy3/Cy5 spectroscopy fluorescence resonance energy transfer (FRET) was used to establish direct binding and to calculate the intermolecular distance between purified Cy3-labeled MAGI-1 PDZ1 or PDZ3 domains and the Cy5-labeled CAREx8 C terminus (Fig. 5). The Cy3-labeled MAGI-1 PDZ2 domain was used as a control for these experiments. Cy3/Cy5 labels form a common donor/acceptor pair for FRET, and FRET occurs only if the labeled proteins are in very close proximity (less than 100 Å apart) (19). The occurrence of FRET was observed as a decrease in Cy3 fluorescence intensity (seen as a decrease at 570 nm) and a subsequent increase in Cy5 fluorescence intensity (seen as an increase at approximately 670 nm) (Fig. 5; emission spectra for CAREx8 with PDZ1 [Fig. 5A] and PDZ3 [B] but not with PDZ2 [C]). The maximum peak values were recorded at 570 nm for Cy3 and 670 nm for Cy5 (data not shown) for each titration and used to create binding curves (Fig. 5). The change in fluorescence intensity as a function of PDZ concentration yielded saturable hyperbolic ligand binding curves, indicating high-affinity binding for PDZ1-CAREx8 and PDZ3-CAREx8 with binding affinities (Kd values) of 7.0 ± 0.7 nM for PDZ1-CAREx8 and 0.4 ± 0.2 nM for PDZ3-CAREx8 (Fig. 5A and B). FRET for each reacting combination yielded intermolecular distances of 54.2 and 56.6 Å, respectively, further indicating direct interactions. Linear plots of data demonstrate a single binding site (shown as a single straight line) for CAREx8 with both PDZ1 and PDZ3. In contrast, no interaction was observed between CAREx8 and PDZ2 (Fig. 5C; Kd > 500 nM), as expected.
Fig 5.

Fluorescence resonance energy transfer (FRET) from donor Cy3-labeled MAGI-1 PDZ domains to acceptor Cy5-labeled CAREx8 C terminus. Emission spectra, binding curves (average change in maximal fluorescence intensity at 570 nm [F0-F] of Cy3-PDZ domains and Cy5 CAREx8 C terminus upon excitation of Cy3 at 550 nm with increasing concentrations of Cy5 CAREx8 C terminus [0 to 500 nM]), and double-reciprocal linear plot of binding curves are shown. (A) Cy3-PDZ1 and Cy5 CAREx8. (B) Cy3-PDZ3 and Cy5-CAREx8. (C) Cy3-PDZ2 and Cy5-CAREx8 (no interaction). Data represent averages of the results of 3 separate experiments.
Cy3-labeled PDZ domains were titrated with nonlabeled CAR (Fig. 6), while Cy5-labeled CAR was titrated with nonlabeled PDZ (data not shown) in direct binding assays designed to account for conformational changes that could occur during an interaction and alter FRET-derived binding affinities. Cy3 fluorescence on the PDZ domains was quenched with increasing concentrations of the nonlabeled CAREx8 C terminus and yielded saturable binding curves. High-affinity binding was evident, with PDZ3-CAREx8 > PDZ1-CAREx8 (Kd = 2.6 to 42 nM; Fig. 6A and B). Although the affinities detected by the direct binding assay were slightly weaker than those determined by FRET, potentially due to the greater sensitivity of FRET (19), the trends were consistent with high-affinity binding, as well as with CAREx8 having higher affinity for PDZ3 than PDZ1. Consistent with the FRET studies, no detectable binding was found between PDZ2 and the CAREx8 C terminus (Fig. 6C). These data confirm that the C terminus of CAR is sufficient to interact directly with the PDZ1 and PDZ3 domains of MAGI-1, and vice versa (data not shown), with high affinity.
Fig 6.

Fluorescent ligand binding assays of MAGI-1 PDZ domains and CAREx8 C terminus. Changes in fluorescence intensity of the Cy3-PDZ domain at 570 nm upon excitation at 550 nm and titration with CAR are shown. (A) Cy3-PDZ1 by CAREx8 C terminus. (B) Cy3-PDZ3 by CAREx8 C terminus. (C) Cy3-PDZ2 by CAREx8 C terminus (no interaction). Binding curve values represent the means ± standard errors of the means (SE); n = 5.
MAGI-1 PDZ3 decreases viral infection, while PDZ1 inhibits MAGI-1-mediated CAREx8 suppression to allow adenovirus infection.
Since CAREx8 interacted with PDZ3 of MAGI-1 with a higher affinity than with PDZ1, we hypothesized that PDZ3 would be the domain of MAGI-1 that is responsible for the suppression of CAREx8. The effect of coexpressing CAREx8 with full-length MAGI-1, PDZ1, or PDZ3 domains on adenovirus infection was investigated in CHO-K1 cells that do not express endogenous CAR. CHO-K1 cells transfected with equal amounts of CAREx8, MAGI-1, PDZ1, PDZ3, or empty pcDNA3.1 plasmid were infected with AdV-β-Gal 48 h posttransfection. As expected, expression of CAREx8 significantly increased adenovirus transduction whereas expression of MAGI-1, PDZ1, or PDZ3 did not cause any significant change in adenovirus infection compared to pcDNA3.1 plasmid-transfected control cells (Fig. 7A).
Fig 7.

MAGI-1 PDZ3 decreases viral infection, while PDZ1 inhibits MAGI-1-mediated CAREx8 suppression to allow adenovirus infection. (A to C) CAR-deficient CHO-K1 cells were (A) single, (B) double, or (C) triple transfected with CAREx8, MAGI-1 (black bars), PDZ1 (dotted bars), or PDZ3 (white bars) and balanced with empty pcDNA3.1 plasmid, followed by AdV-β-Gal (multiplicity of infection [MOI], 100) transduction.(D and E) CHO-K1 cells were double transfected with CAREx8 and pcDNA3.1, MAGI-1, PDZ3, or PDZ1 and analyzed for (D) total CAREx8 or (E) cell-surface biotinylated CAREx8. Quantification of at least three individual experiments is shown in all bar graphs. *, P < 0.05.
In order to investigate the effect of the interacting PDZ domains on adenovirus infection, equal amounts of full-length MAGI-1 or each of the interacting MAGI-1 domain constructs, PDZ1 or PDZ3, were cotransfected with CAREx8, followed by adenovirus infection and a β-galactosidase assay. All transfections were balanced with empty pcDNA3.1 plasmid. As previously shown, coexpression of CAREx8 and MAGI-1 reduced adenovirus infection in comparison to the results seen with cells transfected with CAREx8 alone (Fig. 7B) (11). PDZ1 cotransfected with CAREx8 did not significantly alter adenovirus infection (dotted bar) in comparison to CAREx8 alone. However, coexpression of PDZ3 with CAREx8 reduced adenovirus infection to a degree similar to that seen with coexpression of MAGI-1 with CAREx8, suggesting that the PDZ3 domain is the domain contributing to CAREx8 suppression (white bar, Fig. 7B).
We then asked whether there would be any competition between PDZ1 or PDZ3 with MAGI-1 to increase or prevent the suppression of CAREx8 and thereby alter adenovirus infection. CHO-K1 cells were triple transfected with CAREx8, MAGI-1, and each PDZ domain. As expected, CAREx8 alone increased adenovirus infection and cotransfection of CAREx8 with MAGI-1 reduced adenovirus infection (Fig. 7C). Surprisingly, PDZ1 rescued adenovirus infection when triple transfected with CAREx8 and MAGI-1, suggesting that PDZ1 can inhibit or prevent MAGI-1-mediated CAREx8 suppression. Coexpression of PDZ3 with CAREx8 at the same time as full-length MAGI-1 significantly reduced infection; however, it was not to the same degree as was seen with MAGI-1 alone or PDZ3 alone, suggesting that these proteins are not additive and additional mechanisms may be at work. An intermediate reduction of adenovirus infection was obtained when CAREx8 was transfected with both PDZ1 and PDZ3, suggesting that there may be a competition between these domains (Fig. 7C).
Given these surprising results, and the fact that the amount of CAREx8 and, more specifically, CAREx8 at the cell surface is the major factor in adenovirus infection, total cellular CAREx8 and cell surface expression of CAREx8 were investigated. CHO-K1 cells were transfected with CAREx8 along with pcDNA plasmid (control), MAGI-1, isolated PDZ1, or PDZ3. Two days later, total cell lysates were subjected to WB to detect MAGI-1 (GFP Ab), CAREx8 (anti-FLAG Ab), or the PDZ domains (anti-myc Ab) (Fig. 7D). Blots were also probed for β-actin expression to control for protein loading. There was a significant reduction in total CAREx8 after cotransfection with MAGI-1. In contrast, there was no difference in the levels of total CAREx8 cotransfected with PDZ1 or PDZ3, indicating that PDZ3 does not decrease the cellular level of CAREx8. The cell surface levels of CAREx8 in CHO cells cotransfected with CAREx8 and MAGI-1 or isolated PDZ1 or PDZ3 were then investigated by cell surface biotinylation. Cotransfection of CAREx8 with PDZ1 did not reduce cell surface CAREx8 relative to CAREx8 alone (Fig. 7E). However, a significant and similar decrease in surface levels of CAREx8 in the presence of full-length MAGI-1 or PDZ3 was observed, suggesting that, in contrast to the loss of CAREx8 when coexpressed with MAGI-1, PDZ3 prevents cell surface localization of CAR. Taken together, our findings indicate that CAREx8 interacts with MAGI-1 PDZ1 and PDZ3 domains. Whereas PDZ3 holds CAREx8 within the cell, PDZ1 can inhibit MAGI-1-mediated CAREx8 loss, thereby allowing adenovirus infection.
DISCUSSION
This report describes the molecular interaction between the PDZ domain containing protein MAGI-1 and a specific isoform of CAR that comprises a novel mechanism able to regulate adenoviral infection from the apical surface of polarized epithelia (Fig. 8). We have previously shown that MAGI-1 can negatively regulate the protein expression of CAREx8 in nonpolarized cells (11). In accordance with the hypothesis that MAGI-1 regulates the levels of CAREx8 and hence adenovirus infection, we now show that knockdown of MAGI-1 increases apical adenovirus infection in polarized cells and that increasing the expression of MAGI-1 decreases apical infection. On a molecular level, CAREx8 binds to the MAGI-1 PDZ3 domain with high affinity and also binds to the PDZ1 domain with slightly lower affinity. PDZ3 selectively causes a reduction in cell surface CAREx8 and hence decreases adenovirus infection. However, in contrast to full-length MAGI-1, the reduction occurs without causing a decrease in total cellular CAREx8. Finally, we have also demonstrated that PDZ1 can protect CAREx8 from suppression in the presence of full-length MAGI-1.
Fig 8.

Schematic representation of MAGI-1 and isolated MAGI-1 PDZ domain interactions with CAREx8 and the effect on adenovirus infection. MAGI-1 suppresses apical CAREx8 protein expression and adenovirus infection. The PDZ3 domain of MAGI-1 also suppresses cell surface expression of CAREx8 and adenovirus infection. In contrast, expression of the MAGI-1 PDZ1 domain can rescue CAREx8 from MAGI-1-mediated suppression and allow adenovirus infection.
The current model for initiation of viral infection requires a breach in the tight junction barrier, allowing the virus access to the basolateral receptor (3, 4, 36, 38, 44). There was no significant difference in transepithelial conductance in polarized cultures overexpressing CAREx8, with or without MAGI-1, suggesting that the tight junction barrier was intact (Fig. 1E and 2D). It is known that the requirement for junctional penetration may be overcome by apical CAR expression, as experimentally shown by substituting the transmembrane and C terminus of CAR with a glycophosphatidylinositol (GPI) tail (40). Interestingly, although the GPI-linked CAR is almost exclusively present on the apical surface of airway, adenovirus infection is increased only approximately 3-to-4-fold. A similar magnitude was observed in the experimental manipulations of CAREx8 levels previously (11) and here (Fig. 1G and 2E) and is consistent with a direct pathway for apical adenovirus binding and entry into polarized airway epithelial cells. Interestingly, adenovirus binding and entry through apically localized CAR (albeit of undetermined isoform identity) appear to occur upon stimulation with macrophage-secreted cytokines, particularly interleukin-8 (IL-8), and they occur in differentiated epithelial cells (22, 25).
Similar to observations in primary human airway epithelia (11), endogenous CAREx7 localizes to the basolateral surface of polarized MDCK cells and primarily remains basolateral upon increased (exogenous) expression. Adenovirus infection in CAREx7-transduced cells was higher than in control cells, which reflects a portion being mislocalized, as shown by apical surface biotinylation (Fig. 2F), and may have been due to saturation of the apical 1b clathrin adapter complex (6). Although the normal biological function is unclear, a low level of endogenous CAREx8 is expressed within polarized MDCK cells. Expression of exogenous CAREx8 reveals strong localization within the apical and subapical regions, resulting in significantly increased adenovirus-mediated gene transfer. The difference in localization between CAREx8 and CAREx7 clearly demonstrates a difference in trafficking between the two isoforms and is under investigation. Exogenous coexpression of MAGI-1 and CAREx7 did not alter apical adenovirus infection. In contrast, coexpression of MAGI-1 with CAREx8 significantly suppressed cell surface CAREx8 and adenovirus infection, confirming that MAGI-1 plays a role in modulating the levels of adenovirus infection and can reverse the effect of CAREx8 overexpression (Fig. 2E and F).
We next hypothesized that decreasing the endogenous levels of MAGI-1 would increase adenovirus infection. Knockdown of MAGI-1 was achieved with two MAGI-1-specific siRNAs and correlated with increased apical adenovirus transduction. Unfortunately, the endogenous level of CAREx8 in polarized MDCK cells is lower than the sensitivity of our Ab for WB analysis or for quantitative fluorescence analysis. Although we have not directly shown that knockdown of MAGI-1 increases apical CAREx8 in polarized cells, our data show that the expression level of MAGI-1 can alter the susceptibility of polarized epithelia to apical adenovirus transduction.
MAGI-1 contains six PDZ domains (PDZ0 to PDZ5), any of which may bind to the PDZ binding domain of CAREx8. The interactions between isolated MAGI-1 PDZ domains and CAREx8 were investigated in four different ways, including Y2H analysis, in vitro translation-interaction, coimmunoprecipitation from cells, and quantitative ligand binding and FRET assays. Our results show that CAREx8 interacts with both the PDZ1 and PDZ3 domains of MAGI-1. Binding assays and FRET experiments confirmed strong affinity and direct binding between both MAGI-1 PDZ1 and PDZ3 and the C terminus of CAREx8 and that the CAREx8-PDZ3 interaction shows approximately 20 times higher affinity than CAREx8-PDZ1. Moreover, analysis of FRET data confirms close (<60-Å) intermolecular proximity and suggests a single binding site. Taken together with investigations of canonical C-terminal PDZ binding domains (21, 35) and the fact that deletion of the last 4 amino acids of each CAR isoform ablates interactions (11, 13), these results show that the PDZ domain-CAREx8 binding domain interactions occur directly at the extreme end of the C terminus. The ability to bind two domains confirms the concept that PDZ domains may bind an array of consensus sequences (-X-S/T-X-Φ) or be very specific and that binding must be tested directly. Moreover, it confirms previous findings that the unique sequence upstream from the PDZ binding domain at the C terminus may affect binding and, in this case, appears to be responsible for the difference in affinity.
Triple-transfection experiments in CAR- and MAGI-1-deficient CHO cells provide significant insight into the mechanism behind the regulation of CAREx8 by MAGI-1 (Fig. 7). Coexpression of CAREx8 with PDZ3 reduced cell surface CAREx8 expression. Interestingly, the reduction in cell surface CAREx8 was not due to a decrease in total CAREx8 protein expression, as observed with full-length MAGI-1. This suggests that CAREx8 loss likely involves multiple MAGI-1 domains and potentially other proteins that interact with MAGI-1. Moreover, this suggests that inhibition by PDZ3 might be reversible if the interaction with PDZ3 could be controlled. Coexpression of PDZ1 with CAREx8 did not alter cell surface levels of CAREx8 or adenovirus infection. Surprisingly, however, coexpression of PDZ1 with full-length MAGI-1 and CAREx8 rescued CAREx8 levels and adenovirus infection. It is possible that the relatively small isolated PDZ1 domain may preferentially bind CAREx8 over full-length MAGI-1, thus preventing CAREx8 from interacting with the PDZ3 domain and subsequent loss. Alternatively, PDZ1 may prevent the recruitment of CAREx8, destabilizing proteins by blocking the binding of such proteins to full-length MAGI-1. Although competition between PDZ1 binding and PDZ3 binding likely occurs, future investigations will provide greater insight into the mechanism of regulation. Moreover, mimetics of PDZ1 or PDZ3 may be useful for manipulating the levels of CAREx8 at the apical surface and, hence, altering adenovirus infection.
MAGI-1 is an important cellular scaffolding protein that has been implicated in cell survival, apoptosis, polarity, and cancer. MAGI-1 interacts with an array of different proteins, and although the specific PDZ domain interaction has not been described for all of these interactions, several have been identified for PDZ1 and PDZ3. In particular, several other adhesion proteins interact with MAGI-1, including endothelial cell-specific adhesion molecule (ESAM; PDZ3) (41), junction adhesion molecule 4 (JAM4; PDZ1 and PDZ4) (18), nephrin (PDZ2 and PDZ3) (17), and Sidekick 1 and 2 (PDZ2 and PDZ3) (43). Interestingly, E4-ORF1 of adenovirus interacts with both MAGI-1 PDZ1 and -3 and causes the degradation of MAGI-1 (16). Although it is interesting to speculate that E4-ORF1 might alter CAREx8 protein levels during adenovirus replication, the benefit is unclear, considering that many viruses downregulate their receptor to prevent superinfection and aid in dispersion. Finally, brain-specific angiogenesis inhibitor-1 (BAI-1) interacts with PDZ3 (29) and Rho family nucleotide exchange factor mNet interacts with PDZ1 (8). Considering all of these interactions and the high likelihood that many additional interactions exist, future development of peptide and small-molecule inhibitors should proceed with a careful focus on specificity to disrupt only the MAGI-1-CAREx8 interaction and limit cellular toxicity.
In summary, MAGI-1 can regulate adenovirus infection of polarized epithelia from the apical surface. Our data suggest that this effect is through the regulation of CAREx8 protein levels and that the MAGI-1 activity can be both positive (PDZ1) and negative (PDZ3) (Fig. 8). The concept of targeting PDZ domain interactions as druggable targets has received significant attention recently (5) and can now be extended to altering the susceptibility of the epithelium to viral infection. Although we did not observe cellular toxicity in triple-transfected CHO-K1 cells (data not shown), it is possible that the interruption of other interactions, while targeting CAREx8-MAGI-1 interactions, may have deleterious effects on the cell and must be monitored closely. However, the development of small molecules that can transiently mimic or block CAREx8-MAGI-1 interactions may facilitate adenovirus-mediated gene transfer or, alternatively, reduce viral infection in susceptible populations or in the case of a lethal adenovirus outbreak and provide a significant advance in therapeutic options.
ACKNOWLEDGMENTS
We are grateful to Kathleen Frondorf, Sydney Wiltshire, and Raquel Mateus for technical help and to David Schaffer, Dawn Wooley, and Paula Bubulya for reading the manuscript.
Research was supported by grants from the National Institute of Allergy and Infectious Diseases (R15AI090625) and Wright State University Women in Science Giving Circle to K.J.D.A.E. and the Wright State University BARE Award to A.O.K. This work was supported in part by the USPHS National Institutes of Health grants DK77573 (H.A.H.). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 20 June 2012
REFERENCES
- 1. Bergelson JM, et al. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323 doi:10.1126/science.275.5304.1320 [DOI] [PubMed] [Google Scholar]
- 2. Carson SD, Chapman NN, Tracy SM. 1997. Purification of the putative coxsackievirus B receptor from HeLa cells. Biochem. Biophys. Res. Commun. 233:325–328 [DOI] [PubMed] [Google Scholar]
- 3. Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119–131 doi:10.1016/j.cell.2005.10.035 [DOI] [PubMed] [Google Scholar]
- 4. Coyne CB, Shen L, Turner JR, Bergelson JM. 2007. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2:181–192 doi:10.1016/j.chom.2007.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dev KK. 2004. Making protein interactions druggable: targeting PDZ domains. Nat. Rev. Drug Discov. 3:1047–1056 doi:10.1038/nrd1578 [DOI] [PubMed] [Google Scholar]
- 6. Diaz F, et al. 2009. Clathrin adaptor AP1B controls adenovirus infectivity of epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 106:11143–11148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dobrosotskaya I, Guy RK, James GL. 1997. MAGI-1, a membrane-associated guanylate kinase with a unique arrangement of protein-protein interaction domains. J. Biol. Chem. 272:31589–31597 [DOI] [PubMed] [Google Scholar]
- 8. Dobrosotskaya IY. 2001. Identification of mNET1 as a candidate ligand for the first PDZ domain of MAGI-1. Biochem. Biophys. Res. Commun. 283:969–975 [DOI] [PubMed] [Google Scholar]
- 9. Dobrosotskaya IY, James GL. 2000. MAGI-1 interacts with beta-catenin and is associated with cell-cell adhesion structures. Biochem. Biophys. Res. Commun. 270:903–909 [DOI] [PubMed] [Google Scholar]
- 10. Excoffon KJ, Gansemer N, Traver G, Zabner J. 2007. Functional effects of coxsackievirus and adenovirus receptor glycosylation on homophilic adhesion and adenoviral infection. J. Virol. 81:5573–5578 doi:10.1128/JVI.02562-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Excoffon KJ, et al. 2010. Isoform-specific regulation and localization of the coxsackie and adenovirus receptor in human airway epithelia. PLoS One 5:e9909 doi:10.1371/journal.pone.0009909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Excoffon KJ, et al. 2008. Reovirus preferentially infects the basolateral surface and is released from the apical surface of polarized human respiratory epithelial cells. J. Infect. Dis. 197:1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Excoffon KJ, Hruska-Hageman A, Klotz M, Traver GL, Zabner J. 2004. A role for the PDZ-binding domain of the coxsackie B virus and adenovirus receptor (CAR) in cell adhesion and growth. J. Cell Sci. 117:4401–4409 doi:10.1242/jcs.01300 [DOI] [PubMed] [Google Scholar]
- 14. Excoffon KJ, Traver GL, Zabner J. 2005. The role of the extracellular domain in the biology of the coxsackievirus and adenovirus receptor. Am. J. Respir. Cell Mol. Biol. 32:498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Funke L, Dakoji S, Bredt DS. 2005. Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annu. Rev. Biochem. 74:219–245 doi:10.1146/annurev.biochem.74.082803.133339 [DOI] [PubMed] [Google Scholar]
- 16. Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R. 2000. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 19:5270–5280 doi:10.1038/sj.onc.1203906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hirabayashi S, et al. 2005. MAGI-1 is a component of the glomerular slit diaphragm that is tightly associated with nephrin. Lab. Invest. 85:1528–1543 [DOI] [PubMed] [Google Scholar]
- 18. Hirabayashi S, et al. 2003. JAM4, a junctional cell adhesion molecule interacting with a tight junction protein, MAGI-1. Mol. Cell. Biol. 23:4267–4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hostetler HA, et al. 2011. Acyl-CoA binding proteins interact with the acyl-CoA binding domain of mitochondrial carnitine palmitoyl transferase I. Mol. Cell. Biochem. 355:135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hostetler HA, Petrescu AD, Kier AB, Schroeder F. 2005. Peroxisome proliferator-activated receptor alpha interacts with high affinity and is conformationally responsive to endogenous ligands. J. Biol. Chem. 280:18667–18682 [DOI] [PubMed] [Google Scholar]
- 21. Kennedy MB. 1995. Origin of PDZ (DHR, GLGF) domains. Trends Biochem. Sci. 20:350. [DOI] [PubMed] [Google Scholar]
- 22. Kesisoglou F, Schmiedlin-Ren P, Fleisher D, Zimmermann EM. 2010. Adenoviral transduction of enterocytes and M-cells using in vitro models based on Caco-2 cells: the coxsackievirus and adenovirus receptor (CAR) mediates both apical and basolateral transduction. Mol. Pharm. 7:619–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kotelevets L, et al. 2005. Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J. 19:115–117 [DOI] [PubMed] [Google Scholar]
- 24. Laura RP, Ross S, Koeppen H, Lasky LA. 2002. MAGI-1: a widely expressed, alternatively spliced tight junction protein. Exp. Cell Res. 275:155–170 doi:10.1006/excr.2002.5475 [DOI] [PubMed] [Google Scholar]
- 25. Lütschg V, Boucke K, Hemmi S, Greber UF. 2011. Chemotactic antiviral cytokines promote infectious apical entry of human adenovirus into polarized epithelial cells. Nat. Commun. 2:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mellman I, Nelson WJ. 2008. Coordinated protein sorting, targeting and distribution in polarized cells. Nat. Rev. Mol. Cell Biol. 9:833–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ridgway LD, Kim EY, Dryer SE. 2009. MAGI-1 interacts with Slo1 channel proteins and suppresses Slo1 expression on the cell surface. Am. J. Physiol. Cell Physiol. 297:C55–C65 doi:10.1152/ajpcell.00073.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sharma P, Kolawole AO, Wiltshire SM, Frondorf K, Excoffon KJ. 2012. Accessibility of the coxsackievirus and adenovirus receptor and its importance in adenovirus gene transduction efficiency. J. Gen. Virol. 93:155–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shiratsuchi T, et al. 1998. Cloning and characterization of BAI-associated protein 1: a PDZ domain-containing protein that interacts with BAI1. Biochem. Biophys. Res. Commun. 247:597–604 [DOI] [PubMed] [Google Scholar]
- 30. Shivas JM, Morrison HA, Bilder D, Skop AR. 2010. Polarity and endocytosis: reciprocal regulation. Trends Cell Biol. 20:445–452 doi:10.1016/j.tcb.2010.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tanemoto M, Toyohara T, Abe T, Ito S. 2008. MAGI-1a functions as a scaffolding protein for the distal renal tubular basolateral K+ channels. J. Biol. Chem. 283:12241–12247 [DOI] [PubMed] [Google Scholar]
- 32. Thoelen I, et al. 2001. Identification of alternative splice products encoded by the human coxsackie-adenovirus receptor gene. Biochem. Biophys. Res. Commun. 287:216–222 [DOI] [PubMed] [Google Scholar]
- 33. Thomas M, Glaunsinger B, Pim D, Javier R, Banks L. 2001. HPV E6 and MAGUK protein interactions: determination of the molecular basis for specific protein recognition and degradation. Oncogene 20:5431–5439 doi:10.1038/sj.onc.1204719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tomko RP, Xu R, Philipson L. 1997. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. U. S. A. 94:3352–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Ham M, Hendriks W. 2003. PDZ domains—glue and guide. Mol. Biol. Rep. 30:69–82 [DOI] [PubMed] [Google Scholar]
- 36. Vermeer PD, et al. 2009. MMP9 modulates tight junction integrity and cell viability in human airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 296:L751–L762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vogelmann R, Amieva MR, Falkow S, Nelson WJ. 2004. Breaking into the epithelial apical-junctional complex—news from pathogen hackers. Curr. Opin. Cell Biol. 16:86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walters RW, et al. 2002. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 110:789–799 doi:10.1016/S0092-8674(02)00912-1 [DOI] [PubMed] [Google Scholar]
- 39. Walters RW, et al. 1999. Basolateral localization of fiber receptors limits adenovirus infection from the apical surface of airway epithelia. J. Biol. Chem. 274:10219–10226 [DOI] [PubMed] [Google Scholar]
- 40. Walters RW, et al. 2001. Apical localization of the coxsackie-adenovirus receptor by glycosyl-phosphatidylinositol modification is sufficient for adenovirus-mediated gene transfer through the apical surface of human airway epithelia. J. Virol. 75:7703–7711 doi:10.1128/JVI.75.16.7703-7711.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wegmann F, Ebnet K, Du Pasquier L, Vestweber D, Butz S. 2004. Endothelial adhesion molecule ESAM binds directly to the multidomain adaptor MAGI-1 and recruits it to cell contacts. Exp. Cell Res. 300:121–133 doi:10.1016/j.yexcr.2004.07.010 [DOI] [PubMed] [Google Scholar]
- 42. Xu Z, Peng AW, Oshima K, Heller S. 2008. MAGI-1, a candidate stereociliary scaffolding protein, associates with the tip-link component cadherin 23. J. Neurosci. 28:11269–11276 doi:10.1523/JNEUROSCI.3833-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamagata M, Sanes JR. 2010. Synaptic localization and function of Sidekick recognition molecules require MAGI scaffolding proteins. J. Neurosci. 30:3579–3588 doi:10.1523/JNEUROSCI.6319-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zabner J, et al. 2003. Histamine alters E-cadherin cell adhesion to increase human airway epithelial permeability. J. Appl. Physiol. 95:394–401 [DOI] [PubMed] [Google Scholar]