

Abstract
Foot-and-mouth disease is a highly contagious viral illness of wild and domestic cloven-hoofed animals. The causative agent, foot-and-mouth disease virus (FMDV), replicates rapidly, efficiently disseminating within the infected host and being passed on to susceptible animals via direct contact or the aerosol route. To survive in the host, FMDV has evolved to block the host interferon (IFN) response. Previously, we and others demonstrated that the leader proteinase (Lpro) of FMDV is an IFN antagonist. Here, we report that another FMDV-encoded proteinase, 3Cpro, also inhibits IFN-α/β response and the expression of IFN-stimulated genes. Acting in a proteasome- and caspase-independent manner, the 3Cpro of FMDV proteolytically cleaved nuclear transcription factor kappa B (NF-κB) essential modulator (NEMO), a bridging adaptor protein essential for activating both NF-κB and interferon-regulatory factor signaling pathways. 3Cpro specifically targeted NEMO at the Gln 383 residue, cleaving off the C-terminal zinc finger domain from the protein. This cleavage impaired the ability of NEMO to activate downstream IFN production and to act as a signaling adaptor of the RIG-I/MDA5 pathway. Mutations specifically disrupting the cysteine protease activity of 3Cpro abrogated NEMO cleavage and the inhibition of IFN induction. Collectively, our data identify NEMO as a substrate for FMDV 3Cpro and reveal a novel mechanism evolved by a picornavirus to counteract innate immune signaling.
INTRODUCTION
Foot-and-mouth disease (FMD) severely compromises livestock production, resulting in high economic losses and international restrictions on the export of animals and animal products (22). The etiological agent, FMD virus (FMDV) (genus Aphthovirus, family Picornaviridae), possesses a positive-polarity, single-stranded RNA genome of 8.5 kb that encodes a single polyprotein. Upon cleavage by three virus-encoded proteinases, leader (Lpro), 2A, and 3Cpro (35), the polyprotein is processed into intermediate precursors and fully cleaved individual structural and nonstructural proteins that execute distinct functions in the viral life cycle. To propagate rapidly and efficiently at the initial site of infection, FMDV has developed the ability to counteract the host innate immune response, which is the first line of defense combating viral invasion or replication before more specific protection by the adaptive immune system is established (21, 23, 46).
Innate immune responses are activated through host pattern recognition receptors (PRRs), which recognize molecular structures called pathogen-associated molecular patterns (PAMPs) that are structurally conserved within large groups of pathogens. Upon engagement of PAMPs, PRRs initiate signaling pathways, ultimately triggering the production of type I interferons (IFNs) and inflammatory cytokines and the expression of IFN-stimulated genes (ISGs). These effector proteins collectively suppress the replication of invading pathogens and facilitate the development of adaptive immune responses (28). Sensing of picornavirus is mediated primarily by melanoma differentiation-associated gene 5 (MDA5) (20, 25, 27), but recent evidence has suggested a role for retinoic acid-inducible gene I (RIG-I), as well (39). After sensing cytoplasmic viral RNAs, RIG-I and/or MDA5 interacts with IFN-beta promoter stimulator 1 (IPS-1, also known as MAVS/VISA/Cardif) via the caspase activation and recruitment domain (CARD)-like domains. Subsequently, through a process facilitated by an essential adaptor, nuclear transcription factor kappa B (NF-κB) essential modulator (NEMO) and inhibitors of κB kinase (IKK)-related kinases, such as IKK-α, -β, and -ε, as well as TANK-binding kinase 1 (TBK1), are activated, leading to the activation of various latent transcription factors, including interferon regulatory factor 3 (IRF-3) and NF-κB, through phosphorylation-dependent mechanisms. These transcription factors migrate into the nucleus and directly activate promoters of type I IFNs and inflammatory cytokines/chemokines (26, 58). During coevolution with their hosts, many viruses have acquired mechanisms to circumvent these host innate immune responses. Of note, a number of cysteine proteases encoded by different RNA viruses whose primary functions are to process viral polyprotein have been found to antagonize the induction of type I IFNs (8, 10, 14, 30, 32, 33, 38, 40, 44, 45, 47, 56, 61). Prime examples are the coronaviral papain-like proteases (PLpro), such as severe acute respiratory syndrome coronavirus (SARS-CoV) PLpro (14), human coronavirus NL63 (HCoV-NL63) PLP2 (10), and mouse hepatitis virus A59 (MHV-A59) PLP2 (61); the arteriviral cysteine proteases, nonstructural protein 1 (nsp1) and nonstructural protein 2 (nps2) of the porcine reproductive and respiratory syndrome virus (PRRSV) (30, 44, 45, 47); and the 3Cpro cysteine proteases of several picornaviruses, such as coxsackievirus B3 (CVB3) (38), enterovirus 71 (EV71) (32, 33), and hepatitis A virus (HAV) (40, 56). It remains unclear whether the 3Cpro of FMDV antagonizes the innate immune signaling pathways.
Here, we provide evidence that FMDV 3Cpro impairs RIG-I/MDA5 signaling by cleaving a downstream essential adaptor protein, NEMO. We found that infection of cells with FMDV led to 3Cpro-mediated proteolysis of NEMO. We identified the 3Cpro cleavage site within NEMO and revealed that the cleavage products were severely impaired for signaling to either IRF-3 or NF-κB. These data uncover a novel mechanism evolved by FMDV to disarm host innate immune responses.
MATERIALS AND METHODS
Cells, virus, and chemicals.
Porcine kidney (PK-15) cells were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum (FCS), 100 U/ml penicillin, and 10 μg/ml streptomycin sulfate at 37°C with 5% CO2 in a humidified incubator. Porcine kidney (IBRS-2) cells were cultured in Eagle's minimal essential medium (MEM) supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin, and 10 μg/ml streptomycin sulfates. FMDV (strain O/ES/2001) was propagated in IBRS-2 cells using standard virology techniques, and the supernatants of infected cells were clarified and stored at −80°C. Poly(I·C) (Sigma-Aldrich), a surrogate for viral double-stranded RNA (dsRNA), was used to stimulate the cells. The broad caspase inhibitor z-VAD-FMK and the proteasome inhibitor MG132 were obtained from Beyotime (China).
Plasmids.
The luciferase reporter plasmids IFN-α1-Luc, IFN-β-Luc, 4× PRDIII/I-Luc, and 4× PRDII-Luc have been described previously (50, 52). The luciferase reporter plasmids 4× PRDIII/I-Luc (referred to as IRF-3-Luc) and 4× PRDII-Luc (referred to as NF-κB-Luc) contained four copies of the IRF- or NF-κB-binding motif of the porcine IFN-β promoter, respectively, upstream of the firefly luciferase reporter gene. cDNA expression constructs encoding porcine IPS-1, MDA5, RIG-I, and a constitutively activated form of RIG-I (RIG-I-N) have been described previously (50–52). cDNA encoding porcine NEMO was amplified by standard reverse transcription (RT)-PCR from total RNA extracted from porcine peripheral blood mononuclear cells (PBMCs) and cloned into pCMV-Tag 2B (Stratagene). NEMO-K277A, a constitutively activated porcine NEMO mutant, was generated in a manner analogous to that described for mouse NEMO (7) and cloned into pCMV-Tag 2B. The cDNA expression constructs encoding various FMDV proteins and intermediate precursors used in this study have been described previously (51). Mutagenesis of NEMO (K277A, Q348R, E349R, E354R, E362R, Q365R, Q383R, E390R, E391R, and Q401R) and FMDV 3Cpro (H46Y, C163G, and H181Y) constructs was carried out by overlap extension PCR using specific mutagenic primers (available upon request). All constructs were validated by DNA sequencing.
Luciferase reporter gene assay.
PK-15 cells grown in 24-well plates were cotransfected with 0.1 μg/well of a reporter plasmid, 0.05 μg/well of pRL-TK plasmid (Promega) (as an internal control for normalization of transfection efficiency), and the indicated expression plasmids or an empty control plasmid. Where indicated, cells were further transfected with poly(I·C) (1 μg/well) 24 h after the initial cotransfection. The cells were lysed 12 h later, and firefly luciferase and Renilla luciferase activities were determined using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer's protocol. The data represent relative firefly luciferase activity normalized to Renilla luciferase activity and are representative of three independently conducted experiments. Data are presented as means ± standard deviations (SD). Statistical P values of <0.05 were considered significant, and P values of <0.01 were considered highly significant.
RNA extraction and quantitative real-time RT-PCR.
To determine the effect of 3Cpro on the expression of 2′,5′-oligoadenylate synthetase (2′,5′-OAS); ISG54; IFN-inducible protein 10 (IP-10, also known as CXCL10); regulated upon activation, normal T-cell-expressed and -secreted (RANTES, also known as CCL5); tumor necrosis factor alpha (TNF-α); interleukin-6 (IL-6); and interleukin-1 beta (IL-1β), PK-15 cells in 24-well plates were transfected with 1 μg of empty vector or a plasmid encoding 3Cpro. Twenty-fours later, the cells were mock transfected or transfected with 1 μg of poly(I·C) for 12 h. Total RNA was extracted from the cells using TRIzol reagent (Invitrogen). One microgram of this total RNA was reverse transcribed to cDNA using avian myeloblastosis virus (AMV) reverse transcriptase (Toyobo, Japan), which (1 μl of 20 μl cDNA) was subsequently used in a SYBR green PCR assay (Applied Biosystems). The abundance of individual mRNA transcript in each sample was assayed three times and normalized to that of porcine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA (as an internal control). The primers were designed with Primer Express software v.3.0 (Applied Biosystems). The sequences of the primers are listed in Table S1 in the supplemental material.
Western blot analysis.
Briefly, PK-15 cells cultured in 60-mm dishes were transfected with the indicated plasmids. After 48 h, the cells were harvested by adding lysis buffer, and protein concentrations were measured in whole cellular extracts. Equal amounts of samples were then subjected to SDS-PAGE and analyzed for expression of RIG-I, MDA5, IPS-1, or NEMO protein by Western blotting using an anti-Flag antibody (Macgene, China). To confirm the expression levels of hemagglutinin (HA)-tagged wild-type (WT) and mutant 3Cpro, an anti-HA antibody (MBL, Japan) was used for immunoblotting. Expression of GAPDH was detected with an anti-GAPDH mouse monoclonal antibody (MAb) (Beyotime, China) to demonstrate equal protein sample loading.
Nucleotide sequence accession numbers.
GenBank accession numbers are as follows: FMDV strain O/ES/2001, AY686687; RIG-I, EU126659; MDA5, EU006039; IPS-1, EU082069; and NEMO, EU258760.
RESULTS
Identification of FMDV 3Cpro as a viral inhibitor of IFN-α/β induction.
To investigate how FMDV regulates type I IFN expression in porcine kidney cells, IBRS-2 cells were cotransfected with a luciferase reporter plasmid under the control of the porcine IFN-β promoter (IFN-β-Luc) and an internal control reporter plasmid (pRL-TK), followed by either mock infection or infection with FMDV at 0.1 PFU/cell. Transfection with poly(I·C), a widely used dsRNA surrogate, strongly activated the IFN-β promoter, indicating IBRS-2 cells harbor intact cytoplasmic pathways to respond to viral dsRNA. In contrast, IFN-β promoter-driven luciferase activity was barely detectable in FMDV-infected cells at any time points examined (Fig. 1A). This suggests that FMDV infection does not induce IFN-β expression and/or it actively inhibits IFN-β induction in IBRS-2 cells. This result was also consistent with those observed in other cell types (11). We and others have previously reported that Lpro (Lbpro or Labpro) of FMDV is an IFN antagonist (11–13, 49, 51, 52). To determine if other FMDV proteins or protein precursors have the ability to inhibit IFN-β expression, we constructed expression vectors encoding the 15 known FMDV proteins or processing intermediates and determined the impact of their ectopic expression on poly(I·C) activation of the IFN-β promoter in transiently transfected PK-15 cells (Fig. 1B). Overexpression of Lpro (Lbpro or Labpro) strongly suppressed poly(I·C)-stimulated IFN-β promoter activity compared to cells transfected with the empty vector (HA), as previously reported (52). Although their effects were less pronounced than that of Lpro, overexpression of 3Cpro or its intermediate precursors (3ABC, 3BC, and 3CD) substantially reduced IFN-β expression (Fig. 1B). In contrast, such an inhibitory effect was not observed in cells expressing 2AB, 2C, 3A, 3AB, 3B, or any of the 4 structural proteins (VP1 to -4). This implies that certain properties of 3Cpro may contribute to the ability of FMDV to inhibit induction of type I IFN transcription. Supporting this notion, we found that FMDV 3Cpro acted in a dose-dependent fashion to inhibit the activation of IFN-β (Fig. 1C) and IFN-α1 (Fig. 1D) promoters by poly(I·C). Furthermore, expression of FMDV 3Cpro also dose-dependently inhibited the upregulation of endogenous transcript for porcine IFN-β (Fig. 1E) and IFN-α1 (Fig. 1F) mRNAs. Collectively, these data demonstrate that FMDV 3Cpro is an antagonist of type I IFNs.
Fig 1.

FMDV 3Cpro independently inhibits type I IFN promoter activation. (A) IBRS-2 cells were cotransfected with IFN-β-Luc and pRL-TK, followed by FMDV infection at 0.1 PFU/cell. As positive controls, 24 h later, the cells were transfected with poly(I·C). Cells were harvested 12 h later and subjected to a dual-luciferase assay. (B) PK-15 cells were transfected with IFN-β-Luc, along with pRL-TK plasmid and the indicated vectors expressing the FMDV proteins (0.8 μg), using Lipofectamine 2000. Twenty-four hours after the initial transfection, the cells were transfected with poly(I·C). Luciferase assays were performed 12 h after the second transfection. (C and D) PK-15 cells were transfected with IFN-β-Luc or IFN-α1-Luc, along with pRL-TK plasmid and increasing quantities (0, 0.125, 0.25, or 0.5 μg) of plasmid encoding 3Cpro, using Lipofectamine 2000. Twenty-four hours after the initial transfection, the cells were transfected with poly(I·C). Luciferase assays and real-time RT-PCR were performed 12 h after the second transfection. The results represent the means and standard deviations of three independent experiments. The firefly luciferase activity was normalized to the Renilla reniformis luciferase, and the untreated empty-vector control value was set to 1. (E and F) RNAs were extracted to measure porcine IFN-β (E) and IFN-α1 (F) mRNA by real-time RT-PCR.
FMDV 3Cpro substantially attenuates the expression of IFN-stimulated genes and inflammatory cytokines/chemokines.
Since FMDV 3Cpro inhibited poly(I·C)-induced IFN-α1/β transcription, we asked whether 3Cpro also attenuates expression of dsRNA-induced ISGs and cytokines/chemokines. To this end, PK-15 cells were transfected with a plasmid encoding FMDV 3Cpro (HA-3Cpro) or the empty control vector. Twenty-four hours posttransfection, the cells were further transfected with poly(I·C) or mock transfected. Total RNA was extracted from the cells and analyzed for the abundance of 2′,5′-OAS, ISG54, IP-10, RANTES, TNF-α, IL-6, and IL-1β mRNAs by SYBR green real-time RT-PCR. Poly(I·C) robustly induced the expression of 2′,5′-OAS (Fig. 2A), ISG54 (Fig. 2B), IP-10 (Fig. 2C), RANTES(Fig. 2D), TNF-α (Fig. 2E), IL-6 (Fig. 2F), and IL-1β (Fig. 2G) in cells transfected with control vector but was substantially less effective in cells expressing 3Cpro. These results show that 3Cpro efficiently impairs intracellular dsRNA-induced IFN-stimulated genes and inflammatory cytokines/chemokines.
Fig 2.

3Cpro significantly reduced the transcription of ISGs and inflammatory cytokines/chemokines. PK-15 cells were transfected with 1 μg of plasmid encoding 3Cpro (HA-3Cpro) or an empty vector (pCMV-HA), and 24 h later, the cells were transfected with 1 μg of poly(I·C). Twelve hours after the second transfection, total RNA was extracted and the expression of 2′,5′-OAS (A), ISG54 (B), IP-10 (C), RANTES (D), TNF-α (E), IL-6 (F), and IL-1β (G) and GAPDH genes was evaluated by quantitative real-time RT-PCR. The results are expressed as increases in mRNA levels relative to those in cells transfected in the absence of poly(I·C) and were normalized by using GAPDH housekeeping gene expression. The results are representative of those from three independent experiments. The error bars indicate standard deviations.
Protease activity governs the ability of FMDV 3Cpro to suppress dsRNA-induced IFN response.
As with the 3Cpro of other picornaviruses, FMDV 3Cpro is a cysteine proteinase responsible for most cleavages within the viral polyprotein (31, 48). For FMDV 3Cpro, residues His-46 and Cys-163 are part of the catalytic triad and His-181 is part of the binding pocket (6, 24). Mutations at any of these residues (Tyr for His-46 [H46Y], Gly for Cys-163 [C163G], or Tyr for His-181 [H181Y]) eliminated the capacity of FMDV 3Cpro to catalytically cleave P1, P2, or P3 substrates in trans (24). We tested the abilities of these 3Cpro mutants to inhibit IFN induction in transiently transfected PK-15 cells. As shown in Fig. 3, compared with control vector-expressing cells, WT 3Cpro strongly suppressed poly(I·C)-induced activation of the IFN-β promoter. In contrast, all three 3Cpro mutants (H46Y, C163G, and H181) were without effect. Thus, 3Cpro depends on its protease activity to disrupt the IFN antiviral response.
Fig 3.
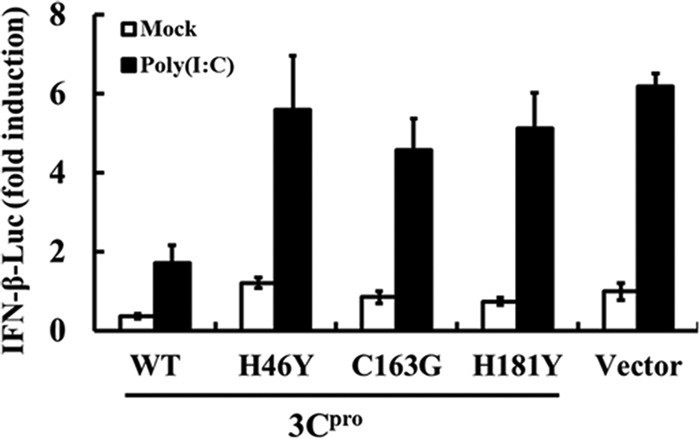
The protease activity of 3Cpro is required to suppress dsRNA-mediated IFN-β induction. PK-15 cells were cotransfected with IFN-β-Luc, along with pRL-TK plasmid, and the designated 3Cpro expression plasmids (1 μg). An empty vector (pCMV-HA) was used as a control. Twenty-four hours after the initial transfection, the cells were transfected with poly(I·C). Luciferase assays were performed 12 h after the second transfection. The error bars indicate standard deviations.
FMDV 3Cpro disrupts RIG-I/MDA5 signaling for activation of both IRFs and NF-κB.
Picornaviruses are sensed primarily by MDA5, but RIG-I may also be involved under some circumstances (20, 25, 27, 39). Given the pivotal role of RIG-I/MDA5 in mediating viral induction of IFN-α/β (26, 58), we further investigated whether overexpression of 3Cpro inhibits RIG-I/MDA5-mediated signaling. Overexpression of a constitutively activated RIG-I mutant (RIG-I-N) or MDA5 significantly activated the IFN-β and IFN-α1 promoters compared with that of the empty-plasmid control (Fig. 4A and B). However, activation of both promoters by RIG-I-N or MDA5 was significantly inhibited in the presence of 3Cpro. Similar results were obtained when these promoters were activated by overexpression of IPS-1, the downstream adaptor for RIG-I/MDA5 (29, 36, 43, 54). Transcription of type I IFNs is dependent on coordinated activation of the latent transcription factors IRF-3 and NF-κB (28). We found that activation of the IRF-3-dependent 4× PRDIII/I promoter and the NF-κB-dependent 4× PRDII promoters by overexpression of RIG-I-N, MDA5, or IPS-1 was significantly reduced by 3Cpro (Fig. 4C and D), suggesting that 3Cpro likely targets a step in the RIG-I/MDA5 pathway prior to the signaling bifurcation into IRFs and NF-κB. Alternatively, 3Cpro may act on two or more signaling proteins in the IRF and NF-κB branches, respectively.
Fig 4.
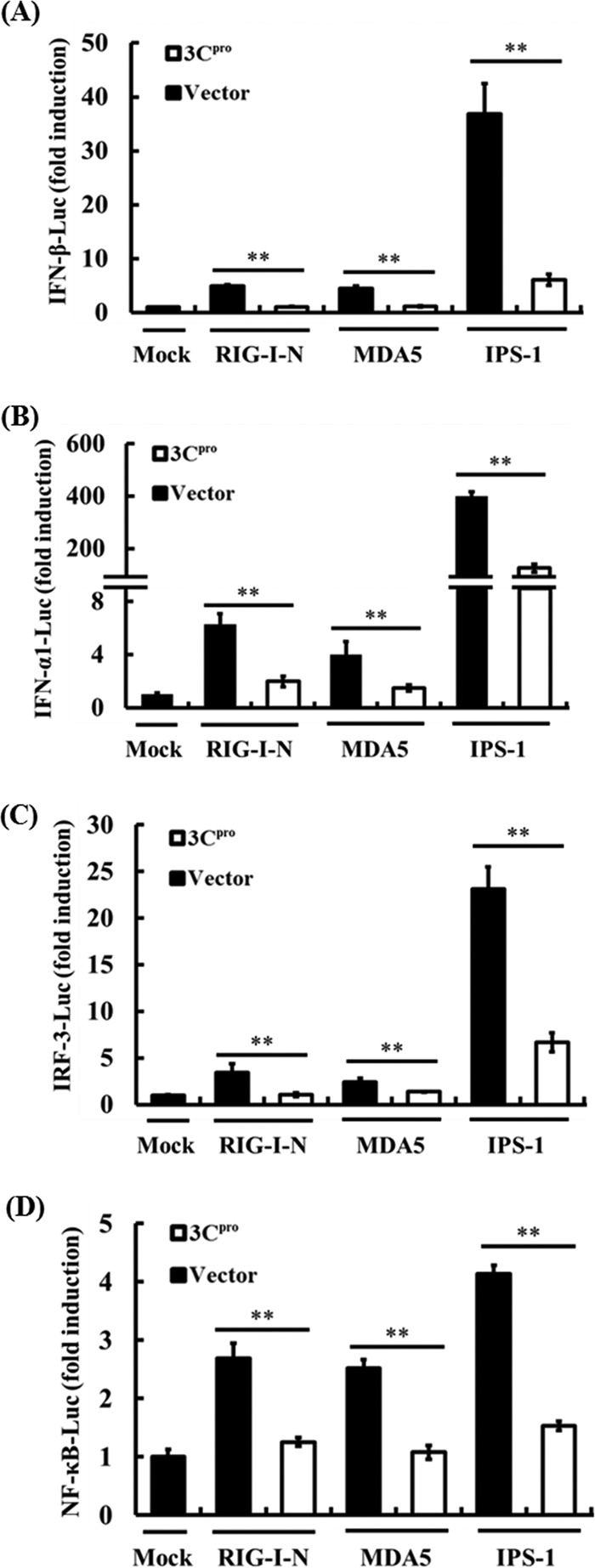
The FMDV 3Cpro protein disrupts RIG-I/MDA5 signaling. (A) PK-15 cells were cotransfected with IFN-β-Luc, pRL-TK plasmid, and 0.5 μg of plasmid encoding 3Cpro, together with the RIG-I-N, MDA5, or IPS-1 expression vector (0.5 μg). Twenty-four hours after the initial transfection, the cells were transfected with 1 μg of poly(I·C). Luciferase assays were performed 12 h after the second transfection. (B to D) The experiments were performed similarly to those in panel A, except that the IFN-α1-Luc (B), 4× PRDIII/I-Luc (C), or 4× PRDII-Luc (D) promoter reporter plasmid was used. **, P < 0.01. The error bars indicate standard deviations.
FMDV 3Cpro cleaves NEMO.
Our earlier finding that the protease activity was critical for FMDV 3Cpro-mediated inhibition of IFN response (Fig. 3) raised the possibility that 3Cpro may target a signaling component(s) of the IFN-inducing pathway(s) for cleavage. RIG-I/MDA5 signaling to IRFs and NF-κB diverges at NEMO in human and mouse cells (60). In agreement with this, we found that porcine NEMO is also required for NF-κB and IRF activation in PK-15 cells by small interfering RNA (siRNA) knockdown assays (see Table S2 and Fig. S1 in the supplemental material). Because 3Cpro disrupted activation of both IRFs and NF-κB via RIG-I/MDA5 (Fig. 4C and D), we investigated whether NEMO and its upstream signaling molecules, IPS-1, RIG-I, and MDA5, serve as substrates for 3Cpro. Since no commercial antibodies against porcine RIG-I, MDA5, IPS-1, and NEMO were available, we determined whether 3Cpro would cleave the ectopically expressed Flag–RIG-I, Flag-MDA5, Flag–IPS-1, or Flag-NEMO. The protein abundance of full-length Flag-NEMO was reduced in PK-15 cells coexpressing 3Cpro compared with those coexpressing the control vector (Fig. 5A, compare lanes 7 and 8, and B, compare lanes 2 and 3). This was accompanied by the appearance of a faster-migrating anti-Flag-reactive band of ∼48 kDa, presumably a NEMO cleavage product (Fig. 5A, lane 8, and B, lane 2). Importantly, the degree of NEMO cleavage was positively correlated with the expression level of 3Cpro (Fig. 5C). Furthermore, we found that 3Cpro formed a complex with NEMO in coimmunoprecipitation (co-IP) experiments (see Fig. S2 in the supplemental material), suggesting that FMDV 3Cpro associates with NEMO and induces NEMO cleavage. Interestingly, overexpression of 3Cpro also reduced the abundance of constitutively coexpressed Flag-RIG-I, Flag-MDA5, and Flag-IPS-1 (Fig. 5A, compare lanes 2, 4, and 6 to lanes 1, 3, and 5, respectively); however, no cleavage products were detected by an anti-Flag antibody. We also did not find evidence that 3Cpro interacted with RIG-I or MDA5 (see Fig. S2 in the supplemental material). The 3Cpro-mediated reduction in expression of Flag-RIG-I, Flag-MDA5, and Flag-IPS-1 was not investigated further in this study.
Fig 5.

FMDV 3Cpro cleaves NEMO. (A) PK-15 cells were transfected with Flag-tagged RIG-I, MDA5, IPS-1, or NEMO expression plasmid (2 μg), along with 1 μg of plasmid encoding 3Cpro, using Lipofectamine 2000. Cell lysates were prepared 48 h posttransfection and analyzed by Western blotting. (B) PK-15 cells were transfected with Flag-tagged NEMO expression plasmid (2 μg), along with 1 μg of plasmid encoding 3Cpro. Cell lysates were prepared 48 h posttransfection and analyzed by Western blotting. The lane with protein markers (Bio-Rad; catalog no. 161-0376) includes 250-, 150-, 100-, 75-, 50-, 37-, 25-, and 20-kDa molecular-mass bands. (C) PK-15 cells were transfected with Flag-tagged NEMO expression plasmid (2 μg), along with increasing quantities (0, 0.25, 0.5, 1, and 2 μg) of plasmid encoding 3Cpro. Cell lysates were prepared at 48 h posttransfection and analyzed by Western blotting. (D) IBRS-2 cells cultured in 60-mm dishes were transfected with Flag-tagged NEMO (Flag-NEMO) and infected with FMDV (1 PFU/cell) 24 h posttransfection. The cells were lysed 12 h postinfection and analyzed by Western blotting. Flag-NEMO-conjugated protein was verified with mouse anti-FLAG antibody. Rabbit anti-HA was used to confirm the expression of 3Cpro, and mouse anti-GAPDH antibody was used to detect GAPDH, which served as a protein-loading control.
NEMO is cleaved in FMDV-infected cells.
To exclude the possibility that the observed 3Cpro-mediated NEMO cleavage was an artificial effect of plasmid overexpression in cell culture, we analyzed NEMO cleavage in the context of FMDV infection. To this end, we transfected IBRS-2 cells with the Flag-NEMO-encoding vector for 24 h and then infected them with FMDV or, as a control, UV light-inactivated FMDV (UV-FMDV). Compared with uninfected or UV-FMDV-infected cells, the expression level of full-length Flag-NEMO in FMDV-infected cells was significantly reduced, and concomitantly, a NEMO cleavage product appeared (Fig. 5D). These phenomena closely mirrored those observed in cells ectopically expressing 3Cpro (Fig. 5A, B, and C).
3Cpro inhibits NEMO-mediated type I IFN signaling by disrupting activation of IRFs and NF-κB.
To better understand the effect of 3Cpro-mediated NEMO cleavage in type I IFN signaling, we assessed whether expression of 3Cpro disrupts NEMO-dependent type I IFN signaling. However, expression of wild-type NEMO did not significantly activate IRFs and NF-κB to induce type I IFN (Fig. 6A). In previous studies, constitutively activated forms of components in the type I IFN signaling pathway, such as RIG-I (RIG-I-N) and IRF-3 [IRF-3(5D)], were extensively used to study the regulation of IFN signaling by viral protein (4, 42, 59). Thus, we transfected cells with a constitutively activated form of NEMO (NEMO-K277A) to assess whether 3Cpro disrupts NEMO-dependent type I IFN signaling. Compared with WT NEMO, NEMO-K277A was substantially more potent in activating the IFN-β promoter and synthetic IRF- and NF-κB-dependent promoters (Fig. 6A). The K277A substitution did not alter the susceptibility of NEMO to 3Cpro cleavage (Fig. 6B). When transfected into PK-15 cells, NEMO-K277A resulted in constitutive activation of the IFN-β promoter. However, this was inhibited by coexpression of 3Cpro in a dose-dependent manner (Fig. 6C). Similar results were obtained when the cells were transfected with 4× PRDII-Luc or 4× PRDIII/I-Luc in place of IFN-β-Luc (see Fig. S3 in the supplemental material). Taken together, these data show that 3Cpro-mediated cleavage of NEMO-K277A likely impairs the ability of the latter to induce type I IFN expression by disrupting activation of IRFs and NF-κB.
Fig 6.

FMDV 3Cpro disrupts NEMO-mediated type I IFN signaling. (A) PK-15 cells were cotransfected with the indicated promoter reporter plasmid, pRL-TK plasmid, and the wild-type NEMO, NEMO-K277A, or empty expression vector (1 μg). Luciferase assays were performed 36 h after the transfection. (B) PK-15 cells were transfected with Flag-tagged NEMO-K277A expression plasmid (2 μg), along with 1 μg of plasmid encoding 3Cpro. Cell lysates were prepared 48 h posttransfection and analyzed for Flag-NEMO-K277A- and HA-3Cpro-conjugated proteins by Western blotting. Anti-GAPDH antibody was used to detect GAPDH, which served as a protein-loading control. (C) PK-15 cells were transfected with IFN-β-Luc, pRL-TK plasmid, and Flag-tagged NEMO-K277A expression plasmid (0.5 μg), along with increasing quantities (0, 0.02, 0.1, and 0.5 μg) of plasmid encoding 3Cpro. Luciferase assays were performed at 36 h after the transfection. (D) PK-15 cells cultured in 60-mm dishes were transfected with Flag-tagged NEMO expression plasmid (2 μg), along with the indicated 3Cpro expression plasmids (1 μg). Cell lysates were prepared 48 h posttransfection and analyzed for Flag-NEMO- and HA-3Cpro-conjugated proteins by Western blotting. Anti-GAPDH antibody was used to detect beta actin, which served as a protein-loading control. (E) PK-15 cells were cotransfected with Flag-tagged NEMO expression plasmid (2 μg) and 1 μg of plasmids encoding 3Cpro or empty vector. Forty hours after transfection, MG132 or zVAD-FMK was added to a final concentration of 20 nM. Cell lysates were prepared 8 h after treatment and analyzed by Western blotting. (F) PK-15 cells were cotransfected with IFN-β-Luc, pRL-TK plasmid, and Flag-tagged NEMO-K277A expression plasmid (0.5 μg), along with the indicated 3Cpro expression plasmids (0.5 μg). An empty vector (pCMV-HA) was used as a control. Cell extracts were collected 36 h after transfection and analyzed for firefly and Renilla luciferase expression. **, P < 0.01 compared with empty vector plus NEMO-K277A. The error bars indicate standard deviations.
The protease activity of 3Cpro is responsible for NEMO cleavage and the subversion of NEMO signaling.
We next determined whether 3Cpro cleaves NEMO by means of its protease activity. To this end, the abilities of the WT and various catalytically inactive mutants (H46Y, C163G, and H181Y) of 3Cpro to cleave ectopically coexpressed Flag-NEMO-K277A were compared (Fig. 6D). While WT 3Cpro induced NEMO cleavage, none of the 3Cpro mutants was able to do so. Thus, NEMO is a proteolytic substrate for 3Cpro.
A previous study showed that MDA5 is cleaved in poliovirus-infected cells in a way that is dependent on caspases, as well as functional proteasome (2). We considered such a possibility for FMDV 3Cpro-induced cleavage of NEMO. However, neither the broad caspase inhibitor z-VAD-FMK nor the proteasome inhibitor MG132 inhibited 3Cpro-mediated NEMO cleavage (Fig. 6E), indicating that the cleavage of NEMO observed in 3Cpro-expressing cells was independent of the apoptosis and proteasome pathways.
Importantly, all three 3Cpro mutants (H46Y, C163G, and H181Y) no longer inhibited signaling for activation of the IFN-β promoter by ectopically expressed NEMO-K277A (Fig. 6F), nor were they capable of suppressing activation of IRF- and NF-κB-dependent promoters (see Fig. S4 in the supplemental material). This result was consistent with the inability of these 3Cpro mutants to cleave NEMO (Fig. 6D), which may explain our earlier observation that the catalytically inactive 3Cpro mutants lost their capacity to inhibit dsRNA activation of IFN-β transcription (Fig. 3).
3Cpro cleaves NEMO at glutamine 383.
We next examined the sequence of porcine NEMO for potential 3Cpro cleavage sites. Previous studies on 3Cpro substrate specificity documented a preference for glutamine (Gln, Q) or glutamic acid (Glu, E) at the P1 position (6). As the cleavage of N-terminally Flag-tagged NEMO by 3Cpro yielded a slightly shorter than full-length, ∼48-kDa product that could be detected by anti-Flag (Fig. 5B), we focused on a cluster of potential 3Cpro cleavage sites in the carboxyl-terminal region of NEMO (Fig. 7A). We constructed a series of NEMO mutants in which the invariant Gln or Glu at each potential P1 position was replaced with arginine (Arg, R) and examined their cleavage by 3Cpro. A specific mutation, Q383R, blocked the cleavage event, giving rise to only full-length NEMO protein in the presence of 3Cpro (Fig. 7B, lane 8). Escarmis et al. showed that a mutation in the FMDV capsid precursor may affect 3Cpro-mediated proteolytic cleavage at a distant site (17). Therefore, glutamine 383 may be the 3Cpro cleavage site per se, or mutation at that site may affect potential 3Cpro cleavage sites in NEMO. However, 3Cpro-mediated NEMO cleavage was not affected by the Q348R, E349R, E354R, E362R, Q365R, E390R, E391R, or Q401R mutation (Fig. 7B and C), supporting the notion that glutamine 383 is a unique cleavage site targeted by 3Cpro in NEMO. Further corroborating this notion, NEMO-K277A bearing the Q383R mutation also became resistant to 3Cpro cleavage (Fig. 7D).
Fig 7.

FMDV 3Cpro-mediated NEMO cleavage is involved in the inhibition of type I IFN induction. (A) Schematic representation of wild-type porcine NEMO and its derivatives. The domains of porcine NEMO were annotated analogously to those of human NEMO. (B to D) PK-15 cells were transfected with Flag-tagged wild-type NEMO or NEMO mutants as indicated, along with HA-3Cpro or empty vector. Cell lysates were prepared 48 h posttransfection and analyzed for NEMO by Western blotting with an anti-Flag antibody. Western blots with anti-HA antibody show expression of 3Cpro, and Western blots for GAPDH served as a protein-loading control. (E) Schematic representation of constitutively activated NEMO-K277A and its derivatives. (F) PK-15 cells were cotransfected with IFN-β-Luc, pRL-TK plasmid, and either 0.5 μg of plasmid encoding Flag-fused NEMO-K277A (Full), putative 3Cpro-induced cleavage fragments of NEMO-K277A, or empty vector. Cell extracts were collected at 36 h after transfection and analyzed for firefly and Renilla luciferase expression. *, P < 0.05; **, P < 0.01. The error bars indicate standard deviations.
3Cpro-induced NEMO cleavage fragments exhibit impaired type I IFN signaling.
Previous studies have indicated that the carboxyl-terminal zinc finger domain of NEMO is required for full NEMO activation (34, 55, 60). Because 3Cpro targeted NEMO at a specific residue (Q383) between the leucine zipper region and the zinc finger domain (Fig. 7E), we next determined whether either of the 3Cpro-generated cleavage fragments of NEMO remained active. PK-15 cells were transfected with IFN-β-Luc, together with DNA constructs encoding full-length NEMO-K277A or either of the two putative fragments that would result from 3Cpro cleavage (fragments encompassing residues 1 to 383 and 384 to 419, respectively). The N-terminal fragment (1 to 383, bearing the K277A substitution) induced IFN-β promoter activation at a significantly lower level than full-length NEMO-K277A, whereas the C-terminal fragment (384 to 419) was completely defective for such activity (Fig. 7F). Similar results were obtained when the cells were transfected with 4× PRDII-Luc or 4× PRDIII/I-Luc in lieu of IFN-β-Luc (see Fig. S5 in the supplemental material). These data demonstrate that 3Cpro-mediated proteolysis of NEMO results in disruption of innate immune signaling by reducing the availability of full-length NEMO and by generating fragments severely impaired in or deficient for signaling.
DISCUSSION
FMD is one of the most contagious diseases of cloven-hoofed animals. Like many other positive-strand RNA viruses, FMDV is very sensitive to the antiviral actions of IFNs (9, 15, 16, 37, 53, 57). To survive in the host, FMDV has evolved strategies to block IFN expression. Previous studies have revealed that FMDV Lpro, a papain-like proteinase (22), antagonizes the innate immune responses by inhibiting type I interferon production (11–13, 49, 52). Lpro performs this function by acting as a viral deubiquitinase (49), by degrading the NF-κB and IRF-3/7 proteins (12, 52), and by cleaving eIF-4G to shut off host protein synthesis (19). The Lpro-mediated immune evasion is thought to play an important role in FMDV pathogenesis and virulence. In this study, we present evidence that 3Cpro is another FMDV-encoded IFN antagonist. Furthermore, we demonstrate that 3Cpro adopts a mechanism distinct from that of Lpro, i.e., by proteolytically cleaving NEMO, an adaptor protein essential for viral activation of IRFs and NF-κB and, ultimately, IFN synthesis. Not only do our data highlight the multifaceted control of host innate immunity by FMDV, but they uncover a novel mechanism by which FMDV antagonizes innate immune signaling.
Host cells have developed highly specialized mechanisms to detect and combat invading viruses. Among these, the cytoplasmic RIG-I-like receptors (RLR) and membrane-associated Toll-like receptors (TLRs) represent two major classes of PRRs that sense virus infection and initiate signaling cascades culminating in the activation of IRFs and NF-κB and subsequent IFN antiviral response. Not surprisingly, many viruses have evolved countermeasures to disrupt signaling through these pathways, and picornaviruses are no exception. Among the known viral evasion strategies, cleavage of or interaction with receptors or adaptor molecules serves as a powerful means of inactivating antiviral signaling, which has the effect of suppressing common downstream targets of key innate immune signaling pathways. For example, poliovirus, rhinovirus type 1a, and encephalomyocarditis virus induce MDA5 degradation, which in the case of poliovirus is accomplished in a proteasome- and caspase-dependent manner (2). RIG-I is cleaved in cells infected with poliovirus, rhinoviruses, echovirus, and encephalomyocarditis virus (3, 39). Coxsackievirus B and HAV cleave the RIG-I/MDA5 adaptor protein IPS-1, as well as the Toll-like receptor 3 (TLR3) adaptor, Toll–IL-1 receptor domain-containing adaptor-inducing IFN-β (TRIF) (38, 40, 56). TRIF is also reported to be cleaved in EV71-infected cells (33). Most of these events are associated with the 3Cpro proteinase of picornaviruses (3, 33, 38, 40, 56). However, viral cleavage of a signaling adaptor proximal to the IKK complex has not been reported to date.
Results from the current study demonstrate that the 3Cpro of FMDV inhibits innate antiviral signaling by proteolytically cleaving NEMO, an adaptor downstream of the RLR and TLR pathways bridging the activation of IRFs (by the IRF-activating kinases TBK1 and IKKε) and NF-κB (by the classical IKKs, IKKα and IKKβ) (60). We found NEMO cleavage was specifically attributable to the protease activity of 3Cpro and not a result of activation of cellular caspases or proteasome. Importantly, we demonstrated that porcine NEMO was cleaved by 3Cpro in FMDV-infected porcine (the natural host for FMDV) cells, confirming the biological relevance of this finding. Of note, evidence for NEMO as a prime target for microbial pathogens to disrupt host innate immunity is just emerging. The M45 protein of mouse cytomegalovirus interacts with NEMO and redirects it to autophagosomes for subsequent lysosomal degradation (18). The lpaH9.8 protein of Shigella bacteria acts as an E3 ubiquitin ligase to promote polyubiquitination and proteosomal degradation of NEMO (1). However, FMDV 3Cpro-mediated proteolysis of NEMO (revealed in the current study) represents the first example of a viral immune evasion mechanism that involves direct cleavage of NEMO by a viral protease.
The porcine NEMO protein exhibits a high degree (89%) of sequence conservation with its well-characterized human counterpart (41). The most prominent functional domains, including the two coiled-coil motifs (CC1 and CC2), a leucine zipper (LZ) motif, and a zinc finger (ZF) domain (41), are recognized in the porcine counterpart and arranged in the same order as in human NEMO (Fig. 7E). This evolutionary conservation is a strong indicator that human and porcine NEMO proteins likely employ common modes of type I IFN regulation. We found that FMDV 3Cpro specifically targeted the Gln 383 residue (Fig. 7B and D), which lies between the C-terminal LZ motif and ZF domain of NEMO. Thus, NEMO proteolysis by 3Cpro cleaves off the ZF domain, which plays a crucial role in full activation of NF-κB and IRFs that orchestrate immune and inflammatory responses (34, 55, 60). Indeed, we found the NEMO fragments resulting from 3Cpro cleavage were either severely impaired for or completely deficient in activation of NF-κB, IRF, and IFN induction (Fig. 7F; see Fig. S5 in the supplemental material).
Interestingly, we also observed that the abundances of ectopically expressed Flag–RIG-I, Flag-MDA5, and Flag–IPS-1 proteins were reduced in cells coexpressing 3Cpro (Fig. 5A). However, no cleavage products were observed, suggesting these proteins are unlikely to be proteolytic substrates for FMDV 3Cpro. While the nature of this observation remains to be studied, we speculate that it may result from inhibition of cellular translational machinery following 3Cpro-mediated cleavage of the translation initiation factor eIF-4A (5). Regardless, 3Cpro-mediated suppression of cellular protein synthesis, especially that of the antiviral proteins (RIG-I, MDA5, IPS-1, etc.), may contribute to the FMDV disruption of innate immunity. Unfortunately, it is impossible to dissect the activity of 3Cpro toward NEMO cleavage from that toward inhibiting host protein translation, as mutations disrupting the catalytic activity of 3Cpro abrogated not only NEMO cleavage, but also eIF-4A proteolysis (data not shown).
In summary, our data identify 3Cpro as a novel FMDV-encoded IFN antagonist and demonstrate that the 3Cpro-mediated proteolytic cleavage of NEMO is directly involved in the inhibition of type I IFN transcription by FMDV. This adds another layer of complexity to the immune evasion strategies evolved by this economically important viral pathogen. It should be noted, however, that apart from this novel viral countermeasure, 3Cpro may dampen host innate immunity by cleaving the translation initiation factor eIF-4A (5) and thereby shutting off host protein synthesis, resulting in reduced expression of IFN and antiviral ISG proteins. Furthermore, it will be interesting to investigate whether NEMO cleavage is a shared attribute among 3C proteinases of different picornaviruses, which may facilitate the survival of this family of viruses.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Natural Sciences Foundation of China (31121004) and the National Basic Research Program (973) of China (2005CB523200).
Footnotes
Published ahead of print 20 June 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Ashida H, et al. 2010. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat. Cell Biol. 12:66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barral PM, et al. 2007. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 81:3677–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barral PM, Sarkar D, Fisher PB, Racaniello VR. 2009. RIG-I is cleaved during picornavirus infection. Virology 391:171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Basler CF, et al. 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 77:7945–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belsham GJ, McInerney GM, Ross-Smith N. 2000. Foot-and-mouth disease virus 3C protease induces cleavage of translation initiation factors eIF4A and eIF4G within infected cells. J. Virol. 74:272–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Birtley JR, et al. 2005. Crystal structure of foot-and-mouth disease virus 3C protease. New insights into catalytic mechanism and cleavage specificity. J. Biol. Chem. 280:11520–11527 [DOI] [PubMed] [Google Scholar]
- 7. Bloor S, et al. 2008. Signal processing by its coil zipper domain activates IKK gamma. Proc. Natl. Acad. Sci. U. S. A. 105:1279–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Z, et al. 2007. Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366:277–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chinsangaram J, Piccone ME, Grubman MJ. 1999. Ability of foot-and-mouth disease virus to form plaques in cell culture is associated with suppression of alpha/beta interferon. J. Virol. 73:9891–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clementz MA, et al. 2010. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 84:4619–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Los Santos T, de Avila Botton S, Weiblen R, Grubman MJ. 2006. The leader proteinase of foot-and-mouth disease virus inhibits the induction of beta interferon mRNA and blocks the host innate immune response. J. Virol. 80:1906–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Los Santos T, Diaz-San Segundo F, Grubman MJ. 2007. Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J. Virol. 81:12803–12815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de los Santos T, Segundo FD, Zhu J, Koster M, Dias CC, Grubman MJ. 2009. A conserved domain in the leader proteinase of foot-and-mouth disease virus is required for proper subcellular localization and function. J. Virol. 83:1800–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Devaraj SG, et al. 2007. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 282:32208–32221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dias CC, Moraes MP, Segundo FD, de los Santos T, Grubman MJ. 2011. Porcine type I interferon rapidly protects swine against challenge with multiple serotypes of foot-and-mouth disease virus. J. Interferon Cytokine Res. 31:227–236 [DOI] [PubMed] [Google Scholar]
- 16. Diaz-San Segundo F, et al. 2011. Antiviral activity of bovine type III interferon against foot-and-mouth disease virus. Virology 413:283–292 [DOI] [PubMed] [Google Scholar]
- 17. Escarmis C, Perales C, Domingo E. 2009. Biological effect of Muller's Ratchet: distant capsid site can affect picornavirus protein processing. J. Virol. 83:6748–6756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fliss PM, et al. 2012. Viral mediated redirection of NEMO/IKKgamma to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 8:e1002517 doi:10.1371/journal.ppat.1002517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foeger N, Glaser W, Skern T. 2002. Recognition of eukaryotic initiation factor 4G isoforms by picornaviral proteinases. J. Biol. Chem. 277:44300–44309 [DOI] [PubMed] [Google Scholar]
- 20. Gitlin L, et al. 2006. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. U. S. A. 103:8459–8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Golde WT, Nfon CK, Toka FN. 2008. Immune evasion during foot-and-mouth disease virus infection of swine. Immunol. Rev. 225:85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grubman MJ, Baxt B. 2004. Foot-and-mouth disease. Clin. Microbiol. Rev. 17:465–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grubman MJ, Moraes MP, Diaz-San Segundo F, Pena L, de los Santos T. 2008. Evading the host immune response: how foot-and-mouth disease virus has become an effective pathogen. FEMS Immunol. Med. Microbiol. 53:8–17 [DOI] [PubMed] [Google Scholar]
- 24. Grubman MJ, Zellner M, Bablanian G, Mason PW, Piccone ME. 1995. Identification of the active-site residues of the 3C proteinase of foot-and-mouth disease virus. Virology 213:581–589 [DOI] [PubMed] [Google Scholar]
- 25. Husser L, Alves MP, Ruggli N, Summerfield A. 2011. Identification of the role of RIG-I, MDA-5 and TLR3 in sensing RNA viruses in porcine epithelial cells using lentivirus-driven RNA interference. Virus Res. 159:9–16 [DOI] [PubMed] [Google Scholar]
- 26. Kato H, Takahasi K, Fujita T. 2011. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol. Rev. 243:91–98 [DOI] [PubMed] [Google Scholar]
- 27. Kato H, et al. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105 [DOI] [PubMed] [Google Scholar]
- 28. Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137 [DOI] [PubMed] [Google Scholar]
- 29. Kawai T, et al. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988 [DOI] [PubMed] [Google Scholar]
- 30. Kim O, Sun Y, Lai FW, Song C, Yoo D. 2010. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells. Virology 402:315–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klump W, Marquardt O, Hofschneider PH. 1984. Biologically active protease of foot and mouth disease virus is expressed from cloned viral cDNA in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 81:3351–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lei X, et al. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 84:8051–8061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lei X, et al. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 85:8811–8818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Makris C, Roberts JL, Karin M. 2002. The carboxyl-terminal region of IkappaB kinase gamma (IKKgamma) is required for full IKK activation. Mol. Cell. Biol. 22:6573–6581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mason PW, Grubman MJ, Baxt B. 2003. Molecular basis of pathogenesis of FMDV. Virus Res. 91:9–32 [DOI] [PubMed] [Google Scholar]
- 36. Meylan E, et al. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172 [DOI] [PubMed] [Google Scholar]
- 37. Moraes MP, et al. 2007. Enhanced antiviral activity against foot-and-mouth disease virus by a combination of type I and II porcine interferons. J. Virol. 81:7124–7135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mukherjee A, et al. 2011. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 7:e1001311 doi:10.1371/journal.ppat.1001311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Papon L, et al. 2009. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 393:311–318 [DOI] [PubMed] [Google Scholar]
- 40. Qu L, et al. 2011. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 7:e1002169 doi:10.1371/journal.ppat.1002169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scheidereit C. 2006. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene 25:6685–6705 [DOI] [PubMed] [Google Scholar]
- 42. Sen A, Feng N, Ettayebi K, Hardy ME, Greenberg HB. 2009. IRF3 inhibition by rotavirus NSP1 is host cell and virus strain dependent but independent of NSP1 proteasomal degradation. J. Virol. 83:10322–10335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682 [DOI] [PubMed] [Google Scholar]
- 44. Shi X, et al. 2011. The nonstructural protein 1 papain-like cysteine protease was necessary for porcine reproductive and respiratory syndrome virus nonstructural protein 1 to inhibit interferon-beta induction. DNA Cell Biol. 30:355–362 [DOI] [PubMed] [Google Scholar]
- 45. Song C, Krell P, Yoo D. 2010. Nonstructural protein 1alpha subunit-based inhibition of NF-kappaB activation and suppression of interferon-beta production by porcine reproductive and respiratory syndrome virus. Virology 407:268–280 [DOI] [PubMed] [Google Scholar]
- 46. Summerfield A, Guzylack-Piriou L, Harwood L, McCullough KC. 2009. Innate immune responses against foot-and-mouth disease virus: current understanding and future directions. Vet. Immunol. Immunopathol. 128:205–210 [DOI] [PubMed] [Google Scholar]
- 47. Sun Z, Chen Z, Lawson SR, Fang Y. 2010. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J. Virol. 84:7832–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vakharia VN, Devaney MA, Moore DM, Dunn JJ, Grubman MJ. 1987. Proteolytic processing of foot-and-mouth disease virus polyproteins expressed in a cell-free system from clone-derived transcripts. J. Virol. 61:3199–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang D, et al. 2011. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 85:3758–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang D, et al. 2008. Molecular cloning and functional characterization of porcine IFN-beta promoter stimulator 1 (IPS-1). Vet. Immunol. Immunopathol. 125:344–353 [DOI] [PubMed] [Google Scholar]
- 51. Wang D, et al. 2011. Foot-and-mouth disease virus (FMDV) leader proteinase negatively regulates the porcine interferon-lambda1 pathway. Mol. Immunol. 49:407–412 [DOI] [PubMed] [Google Scholar]
- 52. Wang D, et al. 2010. Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem. Biophys. Res. Commun. 399:72–78 [DOI] [PubMed] [Google Scholar]
- 53. Xiong Y, Lin M, Yuan B, Yuan T, Zheng C. 2009. Expression of exogenous IFN-alpha by bypassing the translation block protects cells against FMDV infection. Antiviral Res. 84:60–66 [DOI] [PubMed] [Google Scholar]
- 54. Xu LG, et al. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727–740 [DOI] [PubMed] [Google Scholar]
- 55. Yang F, et al. 2004. The zinc finger mutation C417R of I-kappa B kinase gamma impairs lipopolysaccharide- and TNF-mediated NF-kappa B activation through inhibiting phosphorylation of the I-kappa B kinase beta activation loop. J. Immunol. 172:2446–2452 [DOI] [PubMed] [Google Scholar]
- 56. Yang Y, et al. 2007. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. U. S. A. 104:7253–7258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yao Q, Huang Q, Cao Y, Qian P, Chen H. 2008. Porcine interferon-gamma protects swine from foot-and-mouth disease virus (FMDV). Vet. Immunol. Immunopathol. 122:309–311 [DOI] [PubMed] [Google Scholar]
- 58. Yoneyama M, Fujita T. 2007. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 18:545–551 [DOI] [PubMed] [Google Scholar]
- 59. Yu S, et al. 2010. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J. Gen. Virol. 91:2080–2090 [DOI] [PubMed] [Google Scholar]
- 60. Zhao T, et al. 2007. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat. Immunol. 8:592–600 [DOI] [PubMed] [Google Scholar]
- 61. Zheng D, Chen G, Guo B, Cheng G, Tang H. 2008. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 18:1105–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.