

Abstract
The lack of a suitable in vitro hepatitis B virus (HBV) infectivity model has limited examination of the early stages of the virus-cell interaction. In this study, we used an immortalized cell line derived from human primary hepatocytes, HuS-E/2, to study the mechanism of HBV infection. HBV infection efficiency was markedly increased after dimethyl sulfoxide (DMSO)-induced differentiation of the cells. Transmission electron microscopy demonstrated the presence of intact HBV particles in DMSO-treated HBV-infected HuS-E/2 cells, which could be infected with HBV for up to at least 50 passages. The pre-S1 domain of the large HBsAg (LHBsAg) protein specifically interacted with clathrin heavy chain (CHC) and clathrin adaptor protein AP-2. Short hairpin RNA knockdown of CHC or AP-2 in HuS-E/2 cells significantly reduced their susceptibility to HBV, indicating that both are necessary for HBV infection. Furthermore, HBV entry was inhibited by chlorpromazine, an inhibitor of clathrin-mediated endocytosis. LHBsAg also interfered with the clathrin-mediated endocytosis of transferrin by human hepatocytes. This infection system using an immortalized human primary hepatocyte cell line will facilitate investigations into HBV entry and in devising therapeutic strategies for manipulating HBV-associated liver disorders.
INTRODUCTION
In humans, hepatitis B virus (HBV) causes acute and chronic infections, which are often associated with severe liver diseases, including cirrhosis and hepatocellular carcinoma (HCC) (12). It is estimated that approximately 350 million individuals worldwide suffer from chronic HBV infection, despite the availability of an effective vaccine for more than 25 years. Epidemiological studies have demonstrated an approximately 100-fold increase in the relative risk of HCC among HBV carriers compared to noncarriers (2). Although HBV infection is the main cause of liver disorders in many regions of the world, investigations on HBV morphogenesis have been impeded by the lack of a suitable in vitro HBV infectivity model. Consequently, knowledge of the molecular events involved in HBV infection of hepatocytes is limited.
HBV is a small DNA virus consisting of a nucleocapsid, which protects the 3.2-kb viral genome, surrounded by an envelope (43). The large, middle, and small HBV envelope proteins bear different isoforms of the HBV surface antigen (HBsAg), referred to, respectively, as LHBsAg, MHBsAg, and SHBsAg, which are encoded in a single open reading frame with three in-phase start codons. SHBsAg contains the S domain, and MHBsAg has a 55-amino-acid extension of the S domain, known as the pre-S2 domain, while LHBsAg contains a further 108-amino-acid region that extends from the pre-S2 domain, making up the pre-S1 domain. LHBsAg plays pivotal roles in the HBV infection process, during which the externally exposed highly conserved pre-S1 sequence of LHBsAg mediates the binding of virion to a putative cellular receptor (7, 24, 29, 38, 46). However, the detailed mechanism of HBV entry into host cells remains unclear.
The attachment of HBV to hepatocytes during infection has long been proposed to be a potential target for antiviral intervention. Although a few host proteins have been demonstrated to interact with HBV particles or viral surface antigens, the exact identity of the cell surface HBV receptor remains elusive (8, 16, 45, 48). Insights into the early infection events of human HBV are limited because of the lack of a cell culture system supporting the full replication cycle. To date, two cell types have been shown to be susceptible to HBV infection. One is the human hepatoma cell line HepaRG, which becomes infectible after dimethyl sulfoxide (DMSO)-induced differentiation (18), while the other cell type, the normal human primary hepatocyte, is readily infected by HBV (4, 19), but the limited lifetime of the cells in vitro and the lack of a consistent source severely restrict their further application.
In this study, we utilized an immortalized human primary hepatocyte cell line, HuS-E/2, transduced with human telomerase reverse transcriptase (hTERT) and human papillomavirus E6E7 (HPV/E6E7), which is phenotypically similar to primary hepatocytes (1). Our data show that, after treatment with DMSO, HuS-E/2 cells can be infected by HBV and that the HBV genome is replicated in these cells. Important steps in HBV morphogenesis are being investigated, particularly the interaction between host proteins and HBsAg.
MATERIALS AND METHODS
Plasmids. (i) p1.3HBcl.
p1.3HBcl, which contains a 1.3-fold HBV genome of the ayw subtype (11) on a modified pUC13 vector backbone, in which the transcription of pregenomic RNA is controlled by the virus's own core promoter and enhancer I and II regulatory elements, has been described previously (6).
(ii) pcDNA3.0-HA-LHBsAg, pcDNA3.0-HA-MHBsAg, and pcDNA3.0-HA-SHBsAg.
For the construction of plasmids pcDNA3.0-HA-LHBsAg, pcDNA3.0-HA-MHBsAg, and pcDNA3.0-HA-SHBsAg, cDNA fragments encoding large, middle, and small HBsAg (amino acid residues 1 to 389, 108 to 389, and 163 to 389, respectively) were generated by PCR from p1.3HBcl using the respective forward primer LHBsAg(1)F (5′-CCAAGCTTAGC2850ATGGGGCAGAATCTTTCC2867-3′), MHBsAg(1)F (5′-CCAAGCTTAGC3174ATGCAGTGGAAT3-3′), or SHBsAg(1)F (5′-CCAAGCTTAGC157ATGGAGAACATC168-3′) and the reverse primer HBsAg(389)R (5′-837TAGCGGCCGCTTAAATGTAT828-3′). The PCR products were cloned into plasmid pCRII-TOPO (Invitrogen) to generate pCRII-LHBsAg, pCRII-MHBsAg, and pCRII-SHBsAg, and the HindIII and NotI fragments from these plasmids were then subcloned into the BamHI and XbaI sites of plasmid pcDNA3.0-HA (Invitrogen) following a blunt-end reaction.
(iii) pGEX-6p-1-LHBsAg(1-111), pGEX-6p-1-LHBsAg(111-274), and pGEX-6p-1-LHBsAg(274-389).
To generate plasmids pGEX-6p-1-LHBsAg(1-111), pGEX-6p-1-LHBsAg(111-274), and pGEX-6p-1-LHBsAg(274-389), coding for glutathione S-transferase (GST)–LHBsAg1-111, GST–LHBsAg111-274, and GST–LHBsAg274-389, respectively, a KpnI/EcoRI fragment (394 bp), EcoRI/BamHI fragment (489 bp), or BamHI/ApaI fragment (323 bp) was obtained from pcDNA3.0-HA-LHBsAg and subcloned, respectively, into the SmaI, SalI, or BamHI/SmaI site of plasmid pGEX-6P-1 (GE Healthcare Biosciences) following a blunt-end reaction.
(iv) pLKO.1-shCHC, pLKO.1-shAP1, pLKO.1-shAP2, and pLKO.1-shLuc.
Plasmids pLKO.1-shCHC, pLKO.1-shAP1, pLKO.1-shAP2, and pLKO.1-shLuc were obtained from the National RNAi Core Facility (Academia Sinica, Taiwan). pLKO.1-shCHC, pLKO.1-shAP1, and pLKO.1-shAP2 transcribe the small-hairpin RNAs (shRNAs) 5′-GCCAAUGUGAUCUGGAACUUA-3′, 5′-GCAGGAAGUUAUGUUCGUGAU-3′, and 5′-CCUCAUCAACAACGCCAUCAA-3′, which were used for CHC, AP-1, and AP-2 RNA interference, respectively, whereas pLKO.1-shLuc transcribes shRNA 5′-CAAAUCACAGAAUCGUCGUAU-3′ for luciferase RNA interference.
Cell lines and DNA transfection.
HuS-E/2 cells at passage 20, kindly provided by Kunitada Shimotohno (Kyoto University, Japan), were grown as described previously (1) and passaged every 3 days using a 1:4 dilution. For transient DNA transfection, cells were cultured and seeded at 20% confluence 18 h prior to transfection. DNA transfection was performed with Lipofectin (Invitrogen) according to the manufacturer's procedure. For DMSO treatment, HuS-E/2 cells were seeded at 60% confluence for 18 h and DMSO was added to a final concentration of 2% (vol/vol), and then the DMSO-containing culture medium was changed every 3 days for 10 to 12 days, when the differentiated HuS-E/2 cells were used for HBV infection experiments in the continued presence of 2% DMSO. Cells of the cell lines Huh7 (a human hepatoma cell line), HepG2 (a human hepatocellular carcinoma cell line), and 293 (a human embryonic kidney cell line) were maintained as described previously (54).
Preparation of HBV particles and HBV infection of cell cultures.
Continuous HBV proliferation can be achieved in HepG2.2.15 cells stably transfected with the HBV genome of the adw2 subtype (47). HepG2.2.15 cells were used because of the unlimited supply and constant quality and were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented with 5% heat-inactivated fetal bovine serum (FBS; Thermo) plus 100 units of penicillin and 100 μg of streptomycin per ml (both from Invitrogen), and the culture supernatant was collected every 4 days and concentrated 100-fold as described previously (51), with modifications. In brief, the culture medium was clarified by centrifugation at 1,000 × g at 4°C for 10 min, and then the supernatant was layered on top of a 20% sucrose cushion (20% sucrose, 20 mM HEPES, pH 7.4, 0.1% bovine serum albumin [BSA]) and centrifuged at 197,000 × g for 3 h at 4°C to pellet the HBV particles, which were resuspended in DMEM containing 10% FBS and stored at −80°C. The HBV titer was normally about 1 × 108 HBV genome equivalents per milliliter, as determined by real-time PCR using serial dilutions of a known amount of plasmid p1.3HBcl to generate a standard curve.
HBV infectious particles were also obtained from HBV-transgenic mice (line Tg[HBV1.3]24-3), which contain a 1.3-times-overlong HBV genome of the ayw subtype on the Institute for Cancer Research (ICR) mouse background (5). The HBV-transgenic mouse produces high levels of HBV replicative DNA, viral mRNAs, and proteins in the liver, a situation which mimics that in patients with progressive chronic HBV infection. To obtain a high concentration of HBV, serum was collected from 6- to 8-week-old male HBV-transgenic mice with HBV titers of >5 × 107 genome copies per ml.
In HBV infection experiments, 106 untreated or DMSO-treated HuS-E/2 cells were incubated for 20 h with 100 μl of HBV (108 particles/ml) in 1 ml of culture medium supplemented with 4% polyethylene glycol 8000 (Sigma), and then the cells were washed three times with phosphate-buffered saline (PBS) and incubated in fresh medium containing 2% DMSO for the indicated time. To test the effects of inhibitors on HBV endocytosis, cells were incubated for 1 h prior to and during HBV infection (20 h) with 1 μM brefeldin A (BFA; Sigma), 10 mM methyl-β-cyclodextrin (MCD; Sigma), or 10 μg/ml of chlorpromazine (CPZ; Sigma), and then the medium was replaced with medium containing 2% DMSO and the cultures were incubated for 12 days.
Antibodies and fluorescent reagents.
The antibodies used were mouse monoclonal anti-clathrin heavy chain (anti-CHC; Transduction Laboratories) or anti-AP-1 or anti-AP-2 (BD Biosciences), goat polyclonal (Western blot assays; Dako) or mouse monoclonal (immunofluorescence studies, Chemicon) anti-HBsAg, goat polyclonal anti-GST (Sigma), mouse monoclonal anti-HBV core antigen (Abcam) or anti-hemagglutinin (anti-HA; Roche), rabbit polyclonal anti-Ki-67 (Thermo Scientific), and mouse monoclonal anti-actin (Millipore) antibodies. Alexa 594-conjugated transferrin was from Molecular Probes. Gold-conjugated (electron microscopy [EM] studies), horseradish peroxidase-conjugated (Western blots), and Alexa 488- and Cy3-conjugated (immunofluorescence studies) secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc.
TEM.
For transmission electron microscopy (TEM), DMSO-treated HuS-E/2 cells were seeded on TMX coverslips (Nunc) in a 12-well plate, infected with HBV, and incubated for 10 days. The cells were then fixed for 10 min in PBS containing 4% paraformaldehyde, permeabilized by incubation for 15 min with 0.5% Triton X-100, and incubated for 1 h at room temperature with PBS containing 1% BSA. They were then incubated overnight at 4°C with mouse monoclonal antibody against HBsAg in PBS containing 1% BSA, washed three times in PBS, and incubated for 1 h at room temperature with 18-nm colloidal gold-conjugated goat anti-mouse antibody in PBS. The samples were then washed, postfixed with 1% osmium tetroxide, dehydrated in a graded series of ethanol, and embedded in Epon resin (Sigma). Ultrathin sections (70 nm) were collected on copper grids and stained with 2% uranyl acetate before examination on an Hitachi H-7100 transmission electron microscope at an accelerating voltage of 100 kV.
DNA and RNA isolation, reverse transcription, and real-time PCR.
Total DNA was extracted using a genomic DNA isolation kit (Nexttec Biotechnologie, Germany). Total RNA was isolated from cultured cells using TRIzol reagent (Invitrogen). Reverse transcription was performed with the RNA templates, avian myeloblastosis virus reverse transcriptase (Roche), and oligo(dT) primer. The products were subjected to real-time PCR using primer sets for specific genes and SYBR green PCR master mix (Bio-Rad). The primer sets used were QHBVsF (5′-CTCCACCAATCGCCAGTC-3′) and QHBVsR (5′-ATCCTCGAGAAGATTGACGATAAG-3′) for HBV HBsAg (5), QHBVcF (5′-CGTTTTTGCCTTCTGACTTCTTTC-3′) and QHBVcR (5′-ATAGGATAGGGGCATTTGGTGGTC-3′) for HBV core (5), cccHBVF (5′-CCTCTGCCGATCCATACTGCGGAAC-3′) and cccHBVR (5′-GCTTGCCTGAGTGCAGTATGG-3′) for HBV covalently closed circular DNA (cccDNA) (22), albuminF (5′-AGTTTGCAGAAGTTTCCAAGTTAGTG-3′) and albuminR (5′-AGGTCCGCCCTGTCATCAG-3′) for albumin (1), and AFPF (5′-TGCAGCCAAAGTGAAGAGGGAAGA-3′) and AFPR (5′-ATAGCGAGCAGCCCAAAGAAGAA-3′) for alpha-fetoprotein (AFP) (52). The primer set used in parallel as the internal control was 5′-TGTGTCCGTCGTGGATCTGAC-3′ and 5′-GATGCCTGCTTCACCACCTTC-3′ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Results were analyzed using an iCycler iQ real-time PCR detection system (Bio-Rad). A series of 10-fold dilutions (2 × 104 to 2 × 109 copies/ml) of plasmid p1.3HBcl was prepared to generate a standard curve in parallel PCRs.
Matrix metalloproteinase (MMP) activity assay by zymography.
The culture supernatant was mixed with 2× zymography sample buffer (0.125 M Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, and 0.005% bromophenol blue), incubated for 10 min at room temperature, and loaded onto SDS-polyacrylamide (10%) gels containing 0.1 mg/ml of gelatin. After electrophoresis, the gel was washed twice for 30 min each time with gentle agitation at room temperature in zymogram renaturing solution (2.5% Triton X-100 in water) to remove SDS and then incubated at 37°C for 18 h in reaction buffer (50 mM Tris-HCl, pH 7.9, 1 mM ZnCl2, and 5 mM CaCl2). After staining with Coomassie brilliant blue, gelatinase activity was identified as a clear zone on a blue background.
Immunofluorescence staining, coimmunoprecipitation, and Western blot analysis.
Immunofluorescence staining, coimmunoprecipitation, and Western blot analysis were performed as previously described (28). Immunoblots were developed with the ECL reagent (GE Healthcare Biosciences).
Expression and purification of recombinant fusion proteins and GST pulldown assay.
Expression and purification of recombinant fusion proteins and the GST pulldown assay were performed as described previously (20). Briefly, expression of GST fusion proteins in Escherichia coli BL21(DE3) was induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside, and then the bacterial cells were lysed by sonication at 4°C in PBS containing 1% Triton X-100 (PBST) and separated into soluble and insoluble fractions by centrifugation at 13,800 × g for 10 min at 4°C. To perform GST pulldown assays, the soluble fraction of bacterial lysates containing GST-fusion proteins was incubated for 3 h at 4°C with glutathione-Sepharose 4B beads (GE Healthcare Biosciences), and then the beads were washed 3 times with PBST and incubated at 4°C overnight with HuS-E/2 cell lysate prepared by lysis in PBST containing a protease inhibitor cocktail (1 mM phenylmethylsulfonyl fluoride, 10 μg/ml of aprotinin, 1 μg/ml of pepstatin A, 1 μg/ml of leupeptin). The beads were then washed with PBST, resuspended in sample buffer (12.5 mM Tris-HCl, pH 6.8, 2% SDS, 20% glycerol, 0.25% bromphenol blue, 5% β-mercaptoethanol), and subjected to SDS-polyacrylamide gel electrophoresis and Western blot analysis.
Transferrin internalization assay.
At 48 h posttransfection, HuS-E/2 cells were starved in Opti-MEM I (Invitrogen) for 20 min at 37°C, and then Alexa 594-conjugated transferrin (50 μg/ml) was added to the cells, which were then incubated for 20 min at 37°C and examined on a fluorescence microscope. Quantitation of Alexa 594-conjugated transferrin images was performed on 10 transfected cells without changing the intensity setting for the laser and photomultiplier, and the mean intensity of the signal from each cell was quantified with the software provided with the ImageMaster TotalLab (version 1.00) analyzer (GE Healthcare Biosciences).
MTT assay.
DMSO-treated HuS-E/2 cells were seeded at 1 × 104 cells/well in a 12-well plate for 24 h and were then treated with 1 μM BFA, 10 mM MCD, or 10 μg/ml of CPZ for an additional 21 h, when the medium was changed to medium containing 2% DMSO and the cultures were incubated for 12 days. Cell viability was then examined using MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]. Briefly, MTT (Sigma Chemical Co.) was added to each well at a final concentration of 20 μM, the samples were incubated for 1 h at 37°C, and then the supernatant was aspirated, the MTT-formazan crystals formed by metabolically viable cells were dissolved in 500 μl of DMSO, and the absorbance at 550 nm was measured on a microplate reader.
Soft agar colony formation assay.
Equal volumes (0.75 ml) of a single-cell suspension of cells (1.3 × 105 cells/ml) and agarose dissolved in DMEM containing 10% FBS were mixed, and the mixture was plated on 6-cm culture dishes on top of a base layer of 0.7% agarose containing 10% fetal calf serum (FCS) and allowed to gel. After 14 days of incubation, colony formation was observed by light microscopy.
RESULTS
HBV infection of DMSO-treated HuS-E/2 immortalized primary human hepatocytes.
It has been reported that the use of DMSO in medium maintains the differentiation of normal hepatocytes in primary cultures (17, 26). In our study, DMSO treatment consisted of incubation of the HuS-E/2 cells with 2% DMSO for 10 to 12 days, followed by infection and culture for a further 10 to 12 days in the presence of 2% DMSO, with a change of medium every 3 days. We first assessed the HBV infectivity of untreated and DMSO-treated HuS-E/2 cells using serum from ayw-subtype HBV-transgenic mice (5) by analyzing the RNA isolated from the cells 12 days later for HBV gene replication by reverse transcription-PCR (RT-PCR). Core protein mRNA was detected in DMSO-treated HuS-E/2 cells but not in untreated cells (Fig. 1A), indicating increased cell susceptibility to HBV infection and replication. Next, HBV particles derived from HepG2.2.15 cells, which stably express the adw2-subtype HBV genome (47), were used to infect DMSO-treated HuS-E/2 cells, and the RNA isolated 12 days later was found to contain HBsAg RNA (Fig. 1B). RNA template with no reverse transcription was used as the negative control in the PCR, and plasmid p1.3HBcl, which contains a 1.3-fold HBV genome, was used as the positive control. Similar infection efficiency was achieved using HBV from HBV-transgenic mouse serum or from HepG2.2.15 cells, but HBV derived from HepG2.2.15 cells was used in subsequent experiments because of the stable and continuous production. To confirm HBV replication in virus-infected DMSO-treated HuS-E/2 cells, total DNA was extracted from the cells and analyzed for the presence of HBV replicative DNA intermediates. As shown in Fig. 1C, HBV core DNA was detected in the DMSO-treated cells but not the untreated cells. More importantly, cccDNA was also observed. An immunofluorescence study showed that HBV core protein was detected in both control and DMSO-treated HBV-infected HuS-E/2 cells, with the percentage of cells expressing core protein at day 12 after infection increasing from less than 1% for untreated cells to about 30% for DMSO-treated cells (Fig. 1D). Furthermore, to assess the levels of viral replication, the culture medium was collected after 4 days' growth of infected DMSO-treated HuS-E/2 cells, HepG2.2.15 cells (positive control), or HepG2 cells or noninfected DMSO-treated HuS-E/2 cells (negative controls), the HBV particles were concentrated 100-fold, and total DNA was extracted and analyzed for HBV genomic DNA. The HBV titer of the 100-fold-concentrated medium from HepG2.2.15 cells and infected DMSO-treated HuS-E/2 cells was about 1 × 108 and 5 × 106 HBV genome equivalents (GE) per milliliter, respectively, as determined by PCR using serial dilutions of a known amount of plasmid p1.3HBcl as the standard (Fig. 1E). The HBV titers in the nonconcentrated culture medium from HepG2.2.15 cells and infected DMSO-treated HuS-E/2 cells were therefore about 106 GE/ml and 5 × 104 GE/ml, respectively, with this 20-fold difference possibly resulting from the chromosomal integration of HBV DNA in the HepG2.2.15 cell line, which was selected for consistent expression of HBV (47). Taken together, these data show that the complete life cycle of HBV can be reproduced in DMSO-treated HuS-E/2 cells.
Fig 1.

HBV infection of DMSO-treated HuS-E/2 immortalized primary human hepatocytes. (A) HuS-E/2 cells were cultured for 12 days with 2% DMSO or were left untreated and were then incubated for 20 h with sera from HBV-transgenic mice (Tgm HBV infection); controls were DMSO-untreated cells not incubated with HBV (not treated [NT]). Infection was performed at a multiplicity of infection of about 5 HBV genome equivalents per cell. After removal of nonbound HBV, the cells were incubated for a further 12 days in the presence of 2% DMSO, and then RNA was isolated and amplified to detect the presence of HBV core protein mRNA. Control RT-PCRs were performed for endogenous GAPDH. The DNA markers are shown as molecular masses in 100-bp increments on the left. (B) DMSO-differentiated HuS-E/2 cells were incubated for 20 h with HBV concentrated from the culture medium of HepG2.2.15 cells grown for 12 days. Infection was performed at a multiplicity of infection of 10. RNA was then isolated and subjected to reverse transcription to generate cDNA, and PCR was performed to detect the presence of HBV HBsAg mRNA. Lane 1, PCR results for the RNA sample; lane 2, PCR results for the cDNA sample; lane 3, DNA markers; lane 4, positive control of plasmid p1.3HBcl (+). (C) Total DNA was isolated from HBV-infected DMSO-treated cells (lanes 1 and 4) or DMSO-untreated cells (lanes 2 and 5) and subjected to PCR to detect the presence of HBV cccDNA (lanes 1 and 2) or core protein DNA (lanes 4 and 5). Lanes 3 and 6, positive-control plasmid p1.3HBcl. (D) HuS-E/2 cells seeded on 18-mm coverslips were treated for 12 days with 2% DMSO or were left untreated and were then incubated with HBV for 20 h, washed, and incubated for an additional 12 days with or without 2% DMSO. Viral infection was then examined by indirect immunofluorescence staining using monoclonal anti-core protein antibody (left) and Hoechst 33258 (center), applied at the same time as the secondary antibody to label the nucleus. The stained cells were visualized by fluorescence microscopy. (E) Total DNA was isolated from the 100-fold-concentrated culture medium from HepG2.2.15 cells (lane 1), HepG2 cells (lane 2), HBV-infected DMSO-treated cells (lanes 3), or noninfected DMSO-treated cells (lane 4) and subjected to PCR to detect the HBV genomic DNA. Lanes 5 to 7, serial dilutions of a known amount of plasmid p1.3HBcl as standard.
Transmission electron microscopy of HBV-infected HuS-E/2 cells.
Next, we examined the distribution of HBsAg, the envelope protein of HBV, in infected DMSO-treated HuS-E/2 cells. After HBV infection, untreated and DMSO-treated cells were cultured for 10 days and were then subjected to immunogold staining for HBsAg and examined by TEM. In infected cells, HBsAg was localized in the endoplasmic reticulum (ER), near the perinuclear area (Fig. 2A), while noninfected cells gave no signal (Fig. 2C). HBV Dane particles, which were homogeneous in size (40 to 50 nm in diameter), were observed near the plasma membrane (Fig. 2B). In addition, in cells producing HBsAg protein, many intracellular vesicles were observed to be budding from the nuclear envelope or from the ER membrane (Fig. 2B). To collect the secreted viral particles, medium from infected DMSO-treated HuS-E/2 cells was collected every 3 days for 12 days, pooled, and centrifuged, and the presence of HBV particles containing large, middle, and small forms of HBsAg was confirmed by Western blot analysis (Fig. 2D). These results showed that intact HBV Dane particles can be produced and secreted into the medium by HBV-infected DMSO-treated HuS-E/2 cells.
Fig 2.

TEM analysis of HBV-infected DMSO-treated HuS-E/2 cells and collection of secreted HBV particles. (A to C) TEM images of DMSO-treated HuS-E/2 cells producing HBV particles. Twenty hours after infection with HBV, DMSO-treated HuS-E/2 cells were washed and then incubated for 10 days, when immunogold labeling of HBsAg was performed using polyclonal anti-HBsAg antibodies, followed by secondary antibody coupled to 18-nm-diameter gold particles, and images were obtained by TEM. Black arrows, HBsAg at the ER (A) and intact HBV particles at the plasma membrane (B); white arrows, ER region in noninfected cells (C). N, nucleus; ER, endoplasmic reticulum; Cy, cytoplasm; PM, plasma membrane. (D) Western blot analysis of HBV particles secreted from HBV-infected DMSO-treated HuS-E/2 cells. DMSO-treated HuS-E/2 cells were infected with HBV for 20 h, washed with PBS, and incubated for a further 12 days. The culture medium was changed and collected every 4 days, and the samples were pooled and centrifuged to concentrate the virus particles as described in Materials and Methods. Western blot analysis was performed using anti-HBsAg antibodies. Lane −, noninfected cells as a control.
Gradual loss of susceptibility to HBV infection in HuS-E/2 cells.
Although they are transduced with hTERT and HPV/E6E7, HuS-E/2 cells still maintain the characteristics of primary hepatocytes after 30 weeks in culture (1). We cultured untreated HuS-E/2 cells from passages 20 to 80 and examined their growth characteristics at passages 50 and 80. As shown in Fig. 3A, at passage 50, HuS-E/2 cells did not show any transformed-like characteristics (top left), whereas they became more three-dimensional and stretched out at passage 80 (bottom left). The proliferation-related Ki-67 antigen, which is associated with the clinical course of cancer (53), was expressed at a low level at passage 50 but at a much higher level at passage 80 (Fig. 3A, right). Also, a significant increase in MMP2 and MMP9 activity was observed on zymograms at passage 80 compared to passage 50 (Fig. 3B), indicating progression of tumorigenesis. To examine the hepatic characteristics of HuS-E/2 cells at different passages, levels of mRNAs coding for albumin, alpha-fetoprotein (AFP), and GAPDH in HuS-E/2 cells at passages 20, 50, and 80 were compared to those in 293 human embryonic kidney cells as a negative control (1) and HepG2 cells and Huh7 cells (human liver carcinoma cell lines; positive controls). Similar levels of mRNA coding for albumin, which is made specifically by the liver (25), were seen in all hepatic cell samples but not in the kidney cells (Fig. 3C). Expression of AFP, which correlates well with hepatocellular carcinoma growth and serves as a tumor marker (14), was seen in HepG2 cells, Huh7 cells, and HuS-E/2 cells at passages 50 and 80 but not passage 20 and was not seen in 293 cells. It has been reported that the ability of cells to form colonies in a semisolid medium is a marker of anchorage independence and is positively associated with metastatic potential (40). We therefore performed a soft agar colony formation assay to measure the colony formation ability of HuS-E/2 cells at different passages. After 2 weeks in culture, passage 80 HuS-E/2 cells showed a significant capacity for soft agar colony formation, similar to that of Huh7 cells, while passage 20 and 50 cells generated fewer colonies with a reduced colony size (Fig. 3D). In an HBV infection experiment, DMSO-treated HuS-E/2 cells could be infected by HBV at passage 50 but not at passage 80, as shown by the production of HBsAg mRNA (Fig. 3E). These data show that HuS-E/2 cells can be successfully infected by HBV before they turn cancerous.
Fig 3.
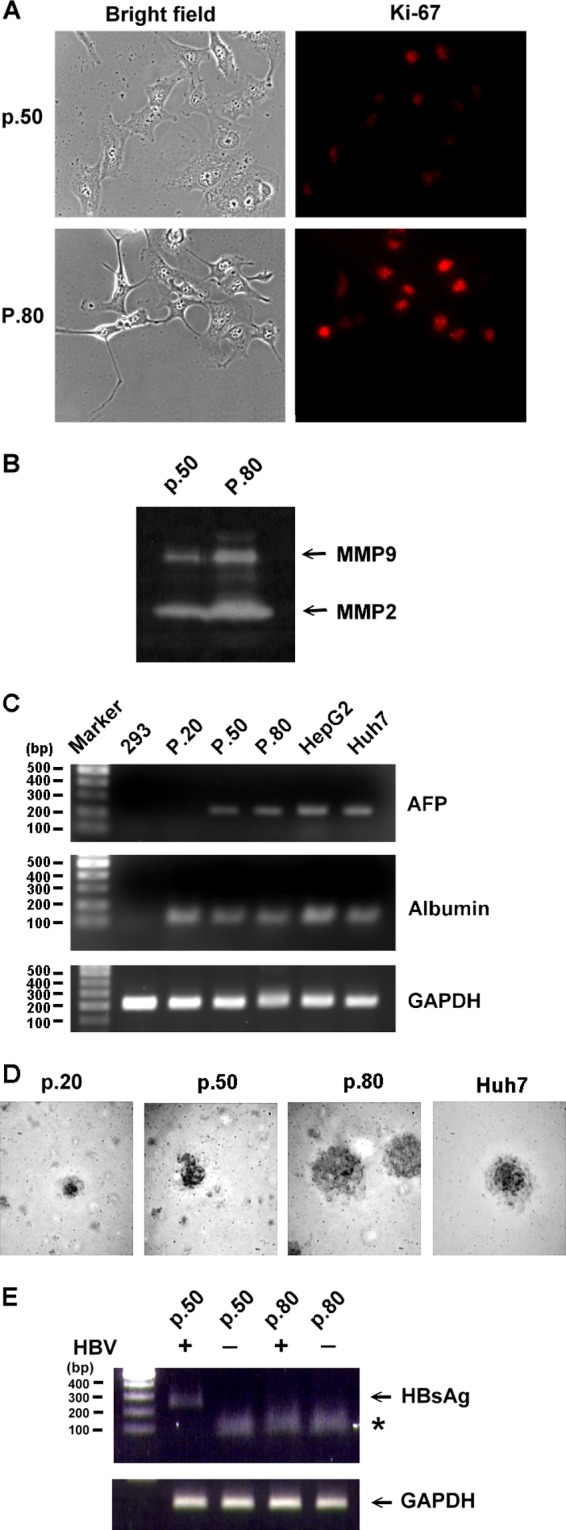
Characterization of HuS-E/2 cells at different passages and their susceptibility to HBV. (A) Immunofluorescence staining for Ki-67. HuS-E/2 cells at passage 50 (p.50) and passage 80 (P.80) were subjected to immunofluorescence staining with rabbit anti-Ki-67 antibodies and Cy3-conjugated goat anti-rabbit IgG antibodies (red) and visualized under a fluorescence microscope (right). Phase-contrast images are shown on the left. (B) Increase in MMP-2 and MMP-9 gelatinolytic activity in HuS-E/2 cells with increased numbers of passages. The zymographic assay was performed after the indicated number of passages. (C) Levels of mRNAs coding for AFP and albumin in 293 cells, HuS-E/2 cells at passages 20, 50, and 80, HepG2 cells, and Huh7 cells. RNA was isolated from cells and analyzed by RT-PCR. Control RT-PCRs were performed for endogenous GAPDH. DNA markers are shown as molecular masses in 100-bp increments. (D) Colony formation in soft agar of HuS-E/2 cells at passage 20, 50, or 80 and of Huh7 cells. Cells were incubated in 0.35% agarose containing 10% FCS on top of 0.7% agarose containing 10% FCS at 37°C for 14 days, and then colonies were photographed under a light microscope. (E) HuS-E/2 cells at passage 50 or 80 were cultured for 12 days with 2% DMSO and incubated with or without HBV for 20 h, and then nonbound HBV was removed and the cells were incubated for an additional 12 days, when RNA was isolated and subjected to reverse transcription and PCR analysis to detect the presence of HBV HBsAg mRNA. Control PCRs were performed for endogenous GAPDH. DNA markers are shown as molecular masses in 100-bp increments.
LHBsAg forms complexes with CHC and AP-2.
Clathrin-mediated endocytosis is a pathway commonly used for virus entry (31). Clathrin and its adaptor proteins (APs) constitute the major component of clathrin-coated vesicles. AP-1 is known to be the carrier for cargo proteins from the trans-Golgi network to endosomes, whereas AP-2 is important in endocytosis at the plasma membrane (27, 41). To determine whether clathrin, AP-1, and AP-2 were involved in HBV infection of human primary hepatocytes, a GST pulldown assay was performed on lysates of HuS-E/2 cells. cDNAs coding for amino acids 1 to 111, 111 to 274, and 274 to 389 of LHBsAg fused to GST were generated (Fig. 4A) and cloned into plasmid pGEX-6p-1, and their expression in E. coli was examined by Western blot analysis. As shown in Fig. 4A, GST–LHBsAg1-111 and GST–LHBsAg111-274 were successfully expressed. Some leaky expression of GST–LHBsAg1-111 and GST–LHBsAg111-274 was seen, possibly as a result of ER stress. However, GST–LHBsAg274-389 could not be expressed in E. coli. In the GST pulldown experiment, GST–LHBsAg1-111 pulled down both CHC and AP-2, but binding of AP-1 was very weak, whereas GST–LHBsAg111-274, like GST itself, did not interact with any of the three proteins (Fig. 4B). This shows that the region of LHBsAg consisting of amino acid residues 1 to 111, containing the pre-S1 domain, is necessary for interaction with CHC and AP-2. To examine whether LHBsAg formed complexes with CHC and AP-2, coimmunoprecipitation experiments using anti-HA or anti-CHC antibodies for immunoprecipitation were performed on HuS-E/2 cells transfected with plasmids encoding HA-tagged HBsAg. As shown in Fig. 4C, using anti-HA antibodies, endogenous AP-2 was coimmunoprecipitated with HA-LHBsAg but not with HA-MHBsAg or HA-SHBsAg. Little HA-SHBsAg was detected in cell lysates because of the high secretion efficiency. In addition, HA-LHBsAg but not HA-MHBsAg or HA-SHBsAg was coimmunoprecipitated with endogenous CHC using anti-CHC antibodies (Fig. 4D). These results demonstrate that the pre-S1 domain in LHBsAg specifically interacts with CHC and AP-2 in HuS-E/2 cells.
Fig 4.

Interaction between LHBsAg and CHC or AP-2. (A) (Top) Schematic representation of the amino acid (a.a.) residues within the LHBsAg subdomains, designated pre-S1, pre-S2, and S; (bottom) Western blot results when LHBsAg cDNA fragments coding for amino acids 1 to 111, 111 to 274, or 274 to 389 were cloned into pGEX-6p-1 for expression in E. coli and purification as GST fusion proteins and antibodies against GST were used to detect the expression of the GST fusion proteins. Arrowheads, leaky expression of proteins. (B) GST pulldown assay. GST-LHBsAg fusion proteins or GST bound to glutathione-Sepharose 4B beads were incubated with lysates of HuS-E/2 cells, and then, after GST pulldown, Western blot analysis was performed using antibodies against CHC, AP-1, AP-2, or GST. The positions of molecular mass markers are shown on the left. (C and D) Coimmunoprecipitation and Western blot analysis. HuS-E/2 cells were transfected with plasmid pcDNA3.0-HA-LHBsAg, pcDNA3.0-HA-MHBsAg, or pcDNA3.0-HA-SHBsAg coding, respectively, for HA-tagged LHBsAg, MHBsAg, or SHBsAg, and then, at 2 days posttransfection, the cells were harvested and subjected to immunoprecipitation (IP) with antibodies specific for HA (C) or CHC (D), followed by Western blot analysis with antibodies against HA, AP-2, or CHC, as indicated. NT, nontransfected cells. The molecular mass markers are indicated on the left. Asterisks, proteins coimmunoprecipitated with CHC; arrowheads, nonspecific bands.
shRNA-mediated knockdown of CHC or AP-2 inhibits HBV infection.
To determine the roles of CHC, AP-1, and AP-2 in regulating infection by HBV, DMSO-treated HuS-E/2 cells were transfected with specific shRNAs directed against CHC, AP-1, or AP-2. The efficiency and specificity of the shRNAs in downregulating the expression of the corresponding endogenous proteins were demonstrated by Western blot analysis; as controls, transfection of cells with control shRNA against luciferase did not affect expression of CHC, AP-1, or AP-2 (Fig. 5A). These shRNA-knockdown HuS-E/2 cells were then infected with HBV, and the effect of shRNA treatment on HBV core protein mRNA levels was examined. As shown in Fig. 5B, using shRNA specific for CHC or AP-2, HBV core protein mRNA levels dropped to 4% or 19%, respectively, of control levels, while AP-1 shRNA and control luciferase shRNA had little effect. Cells with no shRNA treatment and no HBV infection were used as negative controls, and p1.3HBcl was used as the positive control. These data show that CHC and AP-2 play critical roles in HBV morphogenesis.
Fig 5.
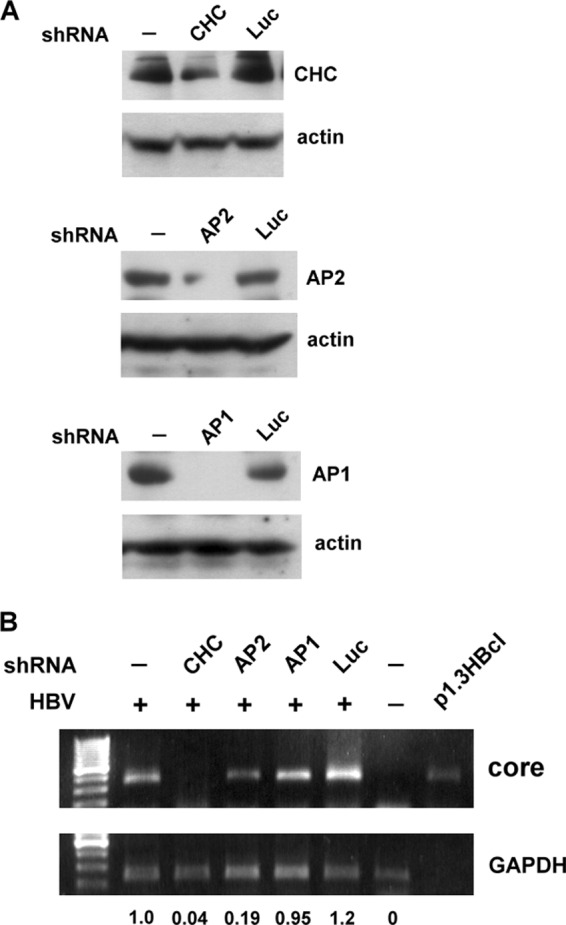
shRNA-mediated knockdown of CHC or AP-2 inhibits HBV infection. (A) shRNA knockdown of CHC, AP-2, or AP-1. DMSO-treated HuS-E/2 cells were transfected with plasmids expressing shRNAs specific for CHC (top), AP-2 (center), or AP-1 (bottom); cells transfected with plasmid containing shRNA against luciferase (Luc) served as controls. At 2 days posttransfection, Western blot analysis was performed using antibodies against CHC, AP-2, or AP-1, as indicated, with actin as the internal control. (B) Effect of CHC, AP-2, or AP-1 knockdown on HBV infection. DMSO-treated HuS-E/2 cells were transfected with shRNAs against CHC, AP-2, or AP-1 as described for panel A, and then at 2 days posttransfection were subjected to HBV infection or left untreated. At 12 days postinfection, RNA was isolated from the infected cells and subjected to reverse transcription and PCR analysis to detect HBV core protein mRNA. Plasmid p1.3HBcl served as the positive control. Relative expression levels of core mRNA are shown.
Clathrin-dependent endocytosis is critical for HBV infection.
To examine whether HBV infection of HuS-E/2 cells was dependent on the clathrin-mediated endocytosis pathway, inhibitors that block the pathway at different steps were used and their effect on HBV morphogenesis was investigated. The inhibitors used were (i) BFA, which induces changes in Golgi structure and inhibits recruitment of cytosolic clathrin adaptors onto Golgi membranes and clathrin-mediated exocytosis (50); (ii) CPZ, which affects the assembly of clathrin-coated pits at the plasma membrane and is an inhibitor of clathrin-mediated endocytosis (21); and (iii) MCD, which depletes membrane cholesterol and is an inhibitor of caveola-mediated endocytosis (42). DMSO-treated HuS-E/2 cells were incubated with or without the inhibitor for 1 h before and during the 20 h of HBV infection and were then incubated in fresh medium containing 2% DMSO for 12 days, when real-time PCR was used to detect HBsAg mRNA as an index of efficiency of HBV infection. As shown in Fig. 6A, HBsAg mRNA was almost eliminated when cells were treated with CPZ, BFA had no effect, and MCD treatment resulted in decreased levels of HBsAg mRNA, implying a minor role of caveolae in HBV infection of primary human hepatocytes. The real-time PCR data showed that HBsAg mRNA levels in CPZ-treated cells were about 7% of those in control cells, while those in MCD-treated cells were about 62% of control levels (Fig. 6B). To examine the effect of BFA, CPZ, and MCD on cell viability, DMSO-treated HuS-E/2 cells were treated with the inhibitor for 21 h and were then cultured in the absence of the inhibitor for 12 days, when the MTT assay was performed to examine cell viability. As shown in Fig. 6C, none of the three inhibitors had any significant effect at the concentrations used, showing that the inhibition of HBV infection was not due to toxicity of the inhibitors. These data suggest that clathrin-mediated endocytosis facilitates hepatitis B virus entry.
Fig 6.
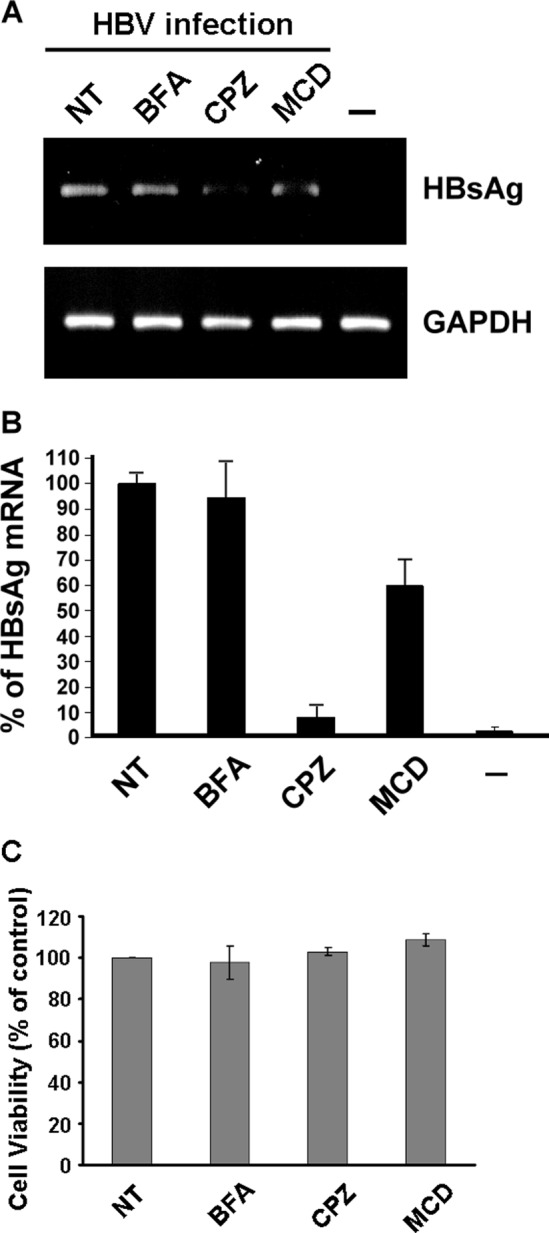
Effect of BFA, CPZ, or MCD on HBV infection. (A) DMSO-treated HuS-E/2 cells were treated with 1 μM BFA, 10 μg/ml of CPZ, or 10 mM MCD for 1 h at 37°C prior to and during HBV infection for 20 h, and then the cells were washed and incubated for 12 days, when RNA was isolated and subjected to reverse transcription and PCR to detect the presence of HBV HBsAg mRNA. Control PCRs were performed for endogenous GAPDH. NT, cells without drug treatment; −, noninfected control. (B) Real-time PCR analysis of HBV gene replication in DMSO-treated HuS-E/2 hepatocytes after inhibitor treatment. The data shown are the means and standard deviations for three independent experiments. (C) Lack of effect of BFA, CPZ, or MCD on the viability of DMSO-treated HuS-E/2 cells. Cells were seeded for 24 h and then treated with 1 μM BFA, 10 μg/ml of CPZ, or 10 mM MCD for 21 h at 37°C. The cells were then washed and incubated for an additional 12 days, when cell viability was measured by the MTT assay. The number of viable cells after treatment is expressed as a percentage of that in the nontreated control (NT). The data are the mean ± standard deviation for three independent experiments.
LHBsAg interferes with clathrin-mediated endocytosis of transferrin.
Since transferrin is a well-studied ligand that is endocytosed through clathrin-coated pits (34), a transferrin uptake assay was performed to directly assess whether LHBsAg interfered with clathrin-mediated endocytosis. HuS-E/2 cells were transfected with LHBsAg- or MHBsAg-expressing plasmids and then incubated with Alexa 594-conjugated transferrin and subjected to immunofluorescence staining with antibodies against HBsAg. Figure 7A shows transferrin uptake in the transfected cells, indicated by the arrows. Transferrin uptake was as efficient as that in nontransfected cells in cells expressing MHBsAg but was decreased to about 15% of control levels in cells expressing LHBsAg (Fig. 7B). These results demonstrate that the interaction between LHBsAg and the clathrin/AP-2 complex interferes with the clathrin-mediated endocytosis of transferrin by human hepatocytes.
Fig 7.
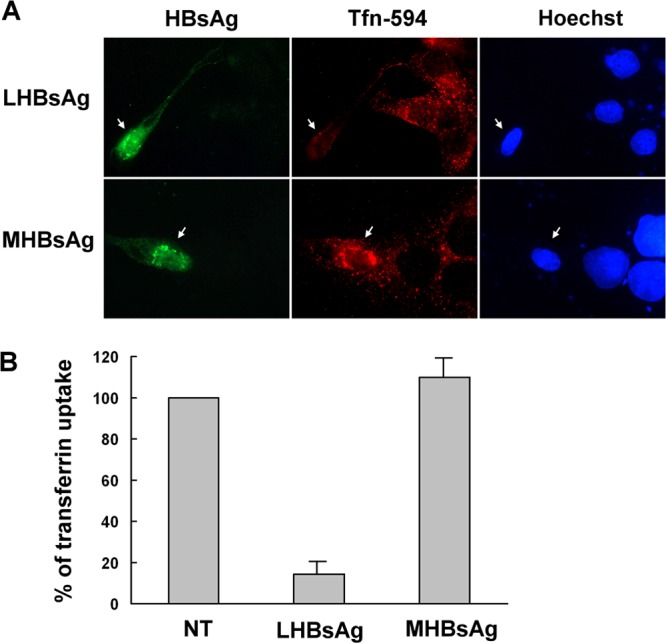
Effect of LHBsAg on internalization of transferrin. (A) Uptake of Alexa 594-conjugated transferrin in the presence of HBsAgs. Two days after transfection with plasmids encoding LHBsAg and MHBsAg, untreated HuS-E/2 cells were incubated with Alexa 594-conjugated transferrin (red) for 20 min and were then fixed, immunostained with mouse monoclonal anti-HBsAg antibodies and Alexa 488-conjugated goat anti-mouse IgG antibodies (green), and visualized by fluorescence microscopy. Hoechst 33258 (right) was applied at the same time as the secondary antibody to stain nuclei. Arrows, transfected cells. (B) Quantification of transferrin uptake. The intensity of the Alexa 594-conjugated transferrin signal was quantified for 10 transfected cells in each set, and the mean intensity was calculated and normalized against that of the nontransfected cells (NT). The graph shows the means and standard deviations for three independent experiments.
DISCUSSION
Although normal human hepatocytes are the ideal system in which to study HBV infectivity and the development of liver diseases, they are very hard to maintain and manipulate. In order to establish future cell-based therapies for liver diseases, the shortage of suitable cell sources must be resolved. To overcome this problem, we examined whether cells of HuS-E/2, an immortalized cell line derived from human primary hepatocytes, could be made susceptible to HBV infection and provided several lines of evidence that, when HuS-E/2 cells are induced to differentiate by DMSO, they become susceptible to HBV infection. Furthermore, intact HBV particles were produced in infected cells. As in HuS-E/2 cells (1), expression of hTERT and HPV/E6E protein has been shown to be useful for efficient immortalization of human primary hepatocytes, but these proteins might lead to tumor development. However, HuS-E/2 cells retain primary hepatocyte characteristics for at least 30 weeks in culture (1), and we found that they could be infected by HBV for up to at least 50 passages.
Pathogens often take advantage of host trafficking pathways to carry out their life cycle, including infection, assembly, and release. Despite a recent report on the role of chaperones (39), little is known about the network of host transport proteins needed for HBV morphogenesis. Several links between clathrin adaptor complexes and viral biogenesis, e.g., in influenza virus (44), reovirus (10), and vesicular stomatitis virus (36), have been clearly demonstrated. It has been demonstrated that AP-2, a major component of the clathrin-associated adaptor protein complex, is required for clathrin-mediated endocytosis of many cargo proteins (27, 41). AP-2 has also been shown to be involved in virus infections (37). Here, we demonstrated that both AP-2 and clathrin play roles in HBV infection. HBV entry into host cells is dependent on the pre-S1 domain of LHBsAg (15), and in the present study, this was shown to form complexes with CHC and AP-2 in HuS-E/2 cells. Moreover, knockdown of CHC or AP-2 or inhibition of clathrin-mediated endocytosis by CPZ inhibited HBV morphogenesis. We therefore hypothesize that AP-2 may help the clathrin-mediated endocytosis of HBV by interacting with LHBsAg. This information will aid in identifying further transport steps in hepatocytes that are involved in the life cycle of HBV.
Entry of viruses into host cells is a multistep process that involves a number of different cell factors. Although HBV entry relies on the pre-S1 domain of LHBsAg to promote attachment to a specific receptor at the surface of human hepatocytes (15), the nature of this receptor is uncertain. Many viruses use clathrin-mediated endocytosis for entry into cells, but in most cases, the attachment receptor is a different molecule (35). Thus, whether AP-2, which is located in the plasma membrane, could act as an HBV receptor or coreceptor deserves further study. HuS-E/2 cells should be a useful tool for determining the identity of the receptor by performing antibody-blocking experiments using receptor-specific antibodies. In addition, since AP-2 is required for the clathrin-mediated endocytosis of receptor-cargo complexes, it may mediate the early step of HBV infection via the HBV receptor, which might act as a cargo protein for clathrin-mediated endocytosis.
HBV is divided into four major serotypes (adr, adw, ayr, and ayw), based on the antigenic epitopes on the envelope protein, and into eight genotypes (A to H), based on variation of the nucleotide sequence in the genome. In our study, both adw2 subtype HBV (genotype A) from HBV-transgenic mouse serum and ayw subtype HBV (genotype D) from HepG2.2.15 cells were demonstrated to infect and replicate in DMSO-treated HuS-E/2 cells. The infectivity and replication of other HBV subtypes/genotypes could be examined using the HuS-E/2 cell system.
Although the properties of HuS-E/2 cells approximate those of human primary hepatocytes for up to 30 weeks (1), HuS-E/2 cells may undergo some genetic or metabolic changes after prolonged culture. We observed that HuS-E/2 cells showed transformed-like characters and overexpressed some tumor markers, such as Ki-67 and alpha-fetoprotein, at passage 80 and that cells at this stage could no longer be infected, even after DMSO induction. One possibility is that expression and presentation of the HBV receptor are lost when HuS-E/2 cells become tumorigenic. It has been reported that several HBV mutants show enhanced growth in fulminant hepatitis patients (23), and HBV isolates obtained from such patients could be tested for replication in HuS-E/2 cells.
Although cholesterol has been shown to be essential for infectivity of HBV particles (9), the requirement for cholesterol in the hepatocyte membrane for HBV infection remains controversial. Treatment with MCD, a chemical which depletes membrane cholesterol, or with filipin and genistein, inhibitors of the raft/caveola endocytosis pathway, does not affect the susceptibility of primary Tupaia hepatocytes to HBV (3). However, plasma membrane caveolae, which are enriched in cholesterol and sphingolipid, have been reported to play a role in HBV entry into HepaRG cells (30, 49). In our study, MCD treatment inhibited HBV infection to a moderate extent, so involvement of the clathrin-independent pathway in HBV entry cannot be excluded. Some viruses are known to use more than one pathway to penetrate into host cells (32). For example, a study tracking single particles of influenza virus in simian kidney epithelial cells showed that 60% of influenza virus entry depends on clathrin-coated pits, while 40% of the particles enter through a clathrin-independent pathway (44). Thus, depending on the virus and cell type, penetration reactions can involve many different mechanisms.
Several clinical studies have described abnormalities in serum iron biochemistry and liver iron deposits in HBV patients with chronic progressive liver disease. For example, high levels of serum ferritin and transferrin have been reported in these patients (13, 33), although the causes of the increased serum ferritin and transferrin saturation remain unclear. Our observation that LHBsAg inhibited transferrin uptake by human hepatocytes provides a clue to the cause of the disordered metabolism of iron ions. Microarray and proteomic analysis of HBV-infected and noninfected HuS-E/2 human primary hepatocytes could be performed to analyze differentially expressed genes and proteins responsible for abnormalities of iron metabolism.
HBV is a major etiological factor in the development of hepatocellular carcinoma. Our infection system using HuS-E/2 immortalized human primary hepatocytes will be useful in testing the infectivity of HBV strains and in screening anti-HBV infection agents. Our data provide several lines of evidence that HBV LHBsAg interacts in a highly specific manner with host cellular factors involved in the clathrin-mediated transport pathway. The process of HBV infection through clathrin-mediated endocytosis may ensure fast and specific virion entry. More of the early steps of HBV entry in vivo need to be clarified. A more complete understanding of the life cycle of HBV will allow a better understanding of the pathology caused by HBV in human liver diseases. This study suggests new strategies for manipulating HBV infection in order to treat disorders associated with liver injury and cirrhosis.
ACKNOWLEDGMENTS
We thank Kunitada Shimotohno (Kyoto University, Japan) for providing the HuS-E/2 cells. We thank Ming-Fu Chang (National Taiwan University, Taiwan) for helpful comments on this article.
This work was supported by research grants NSC 99-2321-B-002-015-MY3, NSC 99-2321-B-077-001-MY3, and NSC 100-2313-B-134-001-MY3 from the National Science Council and NRICM100-DBCM-10 and NRICM101-DBCM-09 from the National Research Institute of Chinese Medicine of the Republic of China.
Footnotes
Published ahead of print 27 June 2012
REFERENCES
- 1. Aly HH, et al. 2007. Serum-derived hepatitis C virus infectivity in interferon regulatory factor-7-suppressed human primary hepatocytes. J. Hepatol. 46:26–36 [DOI] [PubMed] [Google Scholar]
- 2. Beasley RP. 1988. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer 61:1942–1956 [DOI] [PubMed] [Google Scholar]
- 3. Bremer CM, Bung C, Kott N, Hardt M, Glebe D. 2009. Hepatitis B virus infection is dependent on cholesterol in the viral envelope. Cell. Microbiol. 11:249–260 [DOI] [PubMed] [Google Scholar]
- 4. Chai N, et al. 2007. Assembly of hepatitis B virus envelope proteins onto a lentivirus pseudotype that infects primary human hepatocytes. J. Virol. 81:10897–10904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen CC, et al. 2007. Long-term inhibition of hepatitis B virus in transgenic mice by double-stranded adeno-associated virus 8-delivered short hairpin RNA. Gene Ther. 14:11–19 [DOI] [PubMed] [Google Scholar]
- 6. Chou YC, et al. 2005. Evaluation of transcriptional efficiency of hepatitis B virus covalently closed circular DNA by reverse transcription-PCR combined with the restriction enzyme digestion method. J. Virol. 79:1813–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chouteau P, et al. 2001. A short N-proximal region in the large envelope protein harbors a determinant that contributes to the species specificity of human hepatitis B virus. J. Virol. 75:11565–11572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Falco S, et al. 2001. Cloning and expression of a novel hepatitis B virus-binding protein from HepG2 cells. J. Biol. Chem. 276:36613–36623 [DOI] [PubMed] [Google Scholar]
- 9. Dorobantu C, et al. 2011. Cholesterol depletion of hepatoma cells impairs hepatitis B virus envelopment by altering the topology of the large envelope protein. J. Virol. 85:13373–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ehrlich M, et al. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 118:591–605 [DOI] [PubMed] [Google Scholar]
- 11. Galibert F, Mandart E, Fitoussi F, Tiollais P, Charnay P. 1979. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 281:646–650 [DOI] [PubMed] [Google Scholar]
- 12. Ganem D, Prince AM. 2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350:1118–1129 [DOI] [PubMed] [Google Scholar]
- 13. Ghaziani T, et al. 2007. Serum measures of iron status and HFE gene mutations in patients with hepatitis B virus infection. Hepatol. Res. 37:172–178 [DOI] [PubMed] [Google Scholar]
- 14. Gillespie JR, Uversky VN. 2000. Structure and function of alpha-fetoprotein: a biophysical overview. Biochim. Biophys. Acta 1480:41–56 [DOI] [PubMed] [Google Scholar]
- 15. Glebe D, Urban S. 2007. Viral and cellular determinants involved in hepadnaviral entry. World J. Gastroenterol. 13:22–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gong ZJ, et al. 1999. Transfection of a rat hepatoma cell line with a construct expressing human liver annexin V confers susceptibility to hepatitis B virus infection. Hepatology 29:576–584 [DOI] [PubMed] [Google Scholar]
- 17. Gripon P, et al. 1988. Hepatitis B virus infection of adult human hepatocytes cultured in the presence of dimethyl sulfoxide. J. Virol. 62:4136–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gripon P, et al. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 99:15655–15660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gudima S, et al. 2008. Primary human hepatocytes are susceptible to infection by hepatitis delta virus assembled with envelope proteins of woodchuck hepatitis virus. J. Virol. 82:7276–7283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang C, Chang SC, Yu IC, Tsay YG, Chang MF. 2007. Large hepatitis delta antigen is a novel clathrin adaptor-like protein. J. Virol. 81:5985–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hwang SB, Lai MM. 1993. Isoprenylation mediates direct protein-protein interactions between hepatitis large delta antigen and hepatitis B virus surface antigen. J. Virol. 67:7659–7662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ilan E, et al. 1999. The hepatitis B virus-trimera mouse: a model for human HBV infection and evaluation of anti-HBV therapeutic agents. Hepatology 29:553–562 [DOI] [PubMed] [Google Scholar]
- 23. Inoue J, et al. 2011. Enhanced replication of hepatitis B virus with frameshift in the precore region found in fulminant hepatitis patients. J. Infect. Dis. 204:1017–1025 [DOI] [PubMed] [Google Scholar]
- 24. Ishikawa T, Ganem D. 1995. The pre-S domain of the large viral envelope protein determines host range in avian hepatitis B viruses. Proc. Natl. Acad. Sci. U. S. A. 92:6259–6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Isom HC, Georgoff I. 1984. Quantitative assay for albumin-producing liver cells after simian virus 40 transformation of rat hepatocytes maintained in chemically defined medium. Proc. Natl. Acad. Sci. U. S. A. 81:6378–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Isom I, Georgoff I, Salditt-Georgieff M, Darnell JE., Jr 1987. Persistence of liver-specific messenger RNA in cultured hepatocytes: different regulatory events for different genes. J. Cell Biol. 105:2877–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kirchhausen T. 2000. Clathrin. Annu. Rev. Biochem. 69:699–727 [DOI] [PubMed] [Google Scholar]
- 28. Lee CH, Chang SC, Wu CH, Chang MF. 2001. A novel chromosome region maintenance 1-independent nuclear export signal of the large form of hepatitis delta antigen that is required for the viral assembly. J. Biol. Chem. 276:8142–8148 [DOI] [PubMed] [Google Scholar]
- 29. Le Seyec J, Chouteau P, Cannie I, Guguen-Guillouzo C, Gripon P. 1999. Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre-S1 domain. J. Virol. 73:2052–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Macovei A, et al. 2010. Hepatitis B virus requires intact caveolin-1 function for productive infection in HepaRG cells. J. Virol. 84:243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marsh M, Helenius A. 1989. Virus entry into animal cells. Adv. Virus Res. 36:107–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marsh M, Helenius A. 2006. Virus entry: open sesame. Cell 124:729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martinelli AL, et al. 2004. Liver iron deposits in hepatitis B patients: association with severity of liver disease but not with hemochromatosis gene mutations. J. Gastroenterol. Hepatol. 19:1036–1041 [DOI] [PubMed] [Google Scholar]
- 34. Mellman I. 1996. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 12:575–625 [DOI] [PubMed] [Google Scholar]
- 35. Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu. Rev. Biochem. 79:803–833 [DOI] [PubMed] [Google Scholar]
- 36. Pelkmans L, et al. 2005. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 436:78–86 [DOI] [PubMed] [Google Scholar]
- 37. Piguet V, Trono D. 1999. The Nef protein of primate lentiviruses. Rev. Med. Virol. 9:111–120 [DOI] [PubMed] [Google Scholar]
- 38. Pontisso P, Alberti A. 1991. The role of preS1 in the interaction of hepatitis B virus with human hepatocytes. Hepatology 14:405–406 [PubMed] [Google Scholar]
- 39. Prange R, Werr M, Loffler-Mary H. 1999. Chaperones involved in hepatitis B virus morphogenesis. Biol. Chem. 380:305–314 [DOI] [PubMed] [Google Scholar]
- 40. Price JE. 1986. Clonogenicity and experimental metastatic potential of spontaneous mouse mammary neoplasms. J. Natl. Cancer Inst. 77:529–535 [PubMed] [Google Scholar]
- 41. Puertollano R. 2004. Clathrin-mediated transport: assembly required. Workshop on Molecular Mechanisms of Vesicle Selectivity. EMBO Rep. 5:942–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Puri V, et al. 2001. Clathrin-dependent and -independent internalization of plasma membrane sphingolipids initiates two Golgi targeting pathways. J. Cell Biol. 154:535–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robinson WS, Clayton DA, Greenman RL. 1974. DNA of a human hepatitis B virus candidate. J. Virol. 14:384–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rust MJ, Lakadamyali M, Zhang F, Zhuang X. 2004. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 11:567–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schulze A, Gripon P, Urban S. 2007. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 46:1759–1768 [DOI] [PubMed] [Google Scholar]
- 46. Schulze A, Schieck A, Ni Y, Mier W, Urban S. 2010. Fine mapping of pre-S sequence requirements for hepatitis B virus large envelope protein-mediated receptor interaction. J. Virol. 84:1989–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sells MA, Chen ML, Acs G. 1987. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. U. S. A. 84:1005–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stefas I, et al. 2001. Hepatitis B virus Dane particles bind to human plasma apolipoprotein H. Hepatology 33:207–217 [DOI] [PubMed] [Google Scholar]
- 49. Tatematsu K, Tanaka Y, Sugiyama M, Sudoh M, Mizokami M. 2011. Host sphingolipid biosynthesis is a promising therapeutic target for the inhibition of hepatitis B virus replication. J. Med. Virol. 83:587–593 [DOI] [PubMed] [Google Scholar]
- 50. Traub LM, Ostrom JA, Kornfeld S. 1993. Biochemical dissection of AP-1 recruitment onto Golgi membranes. J. Cell Biol. 123:561–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tsai CW, Chang SC, Chang MF. 1999. A 12-amino acid stretch in the hypervariable region of the spike protein S1 subunit is critical for cell fusion activity of mouse hepatitis virus. J. Biol. Chem. 274:26085–26090 [DOI] [PubMed] [Google Scholar]
- 52. Weiss TS, et al. 2008. Hepatic progenitor cells from adult human livers for cell transplantation. Gut 57:1129–1138 [DOI] [PubMed] [Google Scholar]
- 53. Wu PC, et al. 1996. Classification of hepatocellular carcinoma according to hepatocellular and biliary differentiation markers. Clinical and biological implications. Am. J. Pathol. 149:1167–1175 [PMC free article] [PubMed] [Google Scholar]
- 54. Wu SC, Chang SC, Wu HY, Liao PJ, Chang MF. 2008. Hepatitis C virus NS5A protein down-regulates the expression of spindle gene Aspm through PKR-p38 signaling pathway. J. Biol. Chem. 283:29396–29404 [DOI] [PMC free article] [PubMed] [Google Scholar]