

Abstract
Influenza virus has evolved replication strategies that hijack host cell pathways. To uncover interactions between viral macromolecules and host proteins, we applied a phage display strategy. A library of human cDNA expression products displayed on filamentous phages was submitted to affinity selection for influenza viral ribonucleoproteins (vRNPs). High-mobility-group box (HMGB) proteins were found to bind to the nucleoprotein (NP) component of vRNPs. HMGB1 and HMGB2 bind directly to the purified NP in the absence of viral RNA, and the HMG box A domain is sufficient to bind the NP. We show that HMGB1 associates with the viral NP in the nuclei of infected cells, promotes viral growth, and enhances the activity of the viral polymerase. The presence of a functional HMGB1 DNA-binding site is required to enhance influenza virus replication. Glycyrrhizin, which reduces HMGB1 binding to DNA, inhibits influenza virus polymerase activity. Our data show that the HMGB1 protein can play a significant role in intranuclear replication of influenza viruses, thus extending previous findings on the bornavirus and on a number of DNA viruses.
INTRODUCTION
Influenza viruses represent a major public health concern. They have the potential to cause devastating pandemics, and even during typical epidemic years approximately 250,000 to 500,000 people worldwide die as a result of severe complications. The genome of influenza A viruses consists of eight molecules of single-stranded RNA of negative polarity, present in the virions as ribonucleoproteins (RNPs). Each viral RNA (vRNA) is encapsidated by multiple nucleoproteins (NP) and associated with a copy of the viral polymerase, which is a heterotrimeric complex of the PB1, PB2, and PA proteins (64). Upon virus internalization by endocytosis, the viral RNPs (vRNPs) are released in the cytoplasm and transported to the nucleus, where transcription and replication of the viral genome take place.
The polymerase subunits and NP are the minimum set of viral proteins required for the transcription and replication process (59), but there is increasing evidence that cellular factors are required for efficient transcription and/or replication (83) and affect the host range of influenza viruses (10a, 55). Nuclear import and assembly of vRNP components rely on molecular interactions with cellular importins (24, 29, 30, 65, 82) and with the Hsp90 chaperone (19, 57). In the nucleus, vRNPs are found mainly in the insoluble fraction, also known as the nuclear matrix (36, 46, 76), and they bind to nucleosomes (31). Interactions were reported between vRNPs and the PARP-1, DBB1, and RCC1 chromatin-associated factors (20, 51) and between the viral polymerase and the CHD6 chromatin remodeler (34, 47). The viral polymerase strongly associates with the actively transcribing form of the large subunit of cellular RNA polymerase II (Pol II) (26). This association probably facilitates its access to capped RNAs that are used as primers to initiate viral transcription (60) and mediates inhibition of Pol II transcription in infected cells (18, 81). The synthesis and processing of viral messenger RNAs also depends on cellular mRNA splicing and polyadeniling (44, 70) and on export machineries (62). A number of cellular factors, such as UAP56 (54), tat-SF1 (56), and the MCM helicase (40), are reported to stimulate vRNA synthesis in vitro. Although yeast two-hybrid (69, 75a) and proteomic (12, 37, 51) screens have extended the list of host proteins possibly interacting with vRNPs or vRNP components, our understanding of how vRNPs rely on the host cell machinery for viral gene expression and replication is still limited.
Based on an approach not previously used to screen for cellular partners of influenza vRNPs, we expressed a library of human cDNAs at the surface of filamentous phages and subjected it to affinity selection cycles against purified influenza vRNPs. This led to the identification of high-mobility-group box (HMGB) proteins as vRNP-binding proteins. The HMGB protein family includes the HMGB1, HMGB2, and HMGB3 chromosomal proteins (reviewed in reference 8), and the recently described HMGB4 protein (16). HMGB1 and HMGB2 share 79% amino acid sequence identity and a common organization, characterized by two tandem HMG boxes A and B followed by an acidic C-terminal peptide (see Fig. 1C). HMG boxes are DNA-binding domains with an L-shaped fold of three alpha-helices (2, 74). They bind to the minor groove of DNA, causing a local distortion of the double helix (2, 77). Although HMGB1 and HMGB2 are highly homologous, they are not redundant as indicated by differences in their tissue-specific expression pattern and in the phenotype of knockout mice. HMGB1 is an abundant and ubiquitous protein, whereas HMGB2 is expressed mostly in lymphoid organs and testis. HMGB1−/− mice die shortly after birth due to hypoglycemia, whereas HMGB2−/− mice survive and show defects in spermatogenesis (14, 66).
Fig 1.

Selection of influenza virus RNP-binding proteins by phage display. (A) Phage display system. The phages contain a single-stranded phagemid DNA packaged into a filamentous particle that contains three to five copies of the minor coat protein pIII at one of its tips. The three domains of pIII (D1, D2, and D3) are represented schematically. The pIII proteins derived from the KM13 helper phage contain a protease-cleavable linker between domains D2 and D3 (indicated by an arrow). The pIII proteins encoded by the phagemid are fused at their N-terminal ends to the Jun peptide. The Fos peptide is at the N terminus of a glycine/serine-rich linker (G3S)7SG2 which is fused to the human cDNA expression product encoded by the phagemid. The Fos and Jun peptides are bound by two disulfide bonds (black lines). (B) Schematic of the serial affinity selection cycles and monoclonal phage-protein screen. (C) Human cDNA sequences identified upon phage display. Two human HMGB2-derived sequences selected by affinity for viral RNPs (HMGB2 C1 and C2) and the truncated human HMGB2-del sequence are shown, together with the reference amino acid sequences of the human HMGB2 (gene ID 3148) and HMGB1 (gene ID 3146) proteins. HMGB2 and HMGB1 share two DNA-binding domains named HMG box A (highlighted in red) and HMG box B (highlighted in blue), as well as an acidic tail (highlighted in green). The residues indicated in italic derive from the 5′ noncoding region of the cDNA inserts, in the context of the Fos peptide-linker-cDNA expression product fusion.
From the functional point of view, the HMGB1 protein is most documented. As a chromatin architectural factor, it regulates the interactions between DNA-binding proteins and their binding site within chromatin (reviewed in reference 8). In addition to its nuclear function, HMGB1 can be actively secreted from immune cells or passively released from necrotic cells and can act as an extracellular inflammatory cytokine (reviewed in reference 9). HMGB1, along with HMGB2 and HMGB3, was also proposed to contribute to DNA- or RNA-mediated activation of innate immune responses (85).
There is accumulating evidence for HMGB1 being involved during the course of viral infections. An interaction between HMGB1 and the phosphoprotein of the Borna disease virus was shown to repress p53-mediated transcription (87). HMGB1 was found to stimulate the DNA cleavage activity of the adeno-associated virus protein Rep (21), to promote the assembly of an enhanceosome on the Epstein-Barr virus promoter BHLF1 (52), and to facilitate RTA-mediated viral gene expression in gamma-2 herpesviruses (72). Moreover, HMGB1 was shown to be released from cells infected with a range of viruses including dengue virus, hepatitis C virus, and human immunodeficiency virus (HIV) (6, 38, 39). Interestingly, HMGB1 seems to have antagonistic effects on HIV infection. On one hand, it may reduce viral replication in acute infection by downregulating the long terminal repeat (LTR)-directed transcription and by increasing the release of inhibitors of HIV entry such as RANTES or macrophage inflammatory protein 1α; on the other hand, it may trigger viral replication in latently infected dendritic cells (for a review, see reference 33).
The involvement of HMGB1 in influenza virus infection, and its contribution to the excessive proinflammatory cytokine response associated with severe influenza cases, remains largely unknown. Increased levels of HMGB1 were detected in sera from patients with concurrent influenza virus infection and bacterial pneumonia (41) and in sera from H1N1pdm09-infected children with severe pneumonia (35). In a mouse model of severe influenza virus infection, elevated HMGB1 levels were observed in bronchoalveolar lavage fluids, and the HMGB1-receptor RAGE (receptor for advanced glycation end products) was shown to be detrimental (78). An increase in plasma HMGB1 levels was also observed in the serum of influenza virus-infected mice but did not correlate with death, suggesting that active secretion of HMGB1 is not a marker for the severity of the disease (3).
We show here that HMGB1 binds to the NP component of influenza vRNPs independently of the presence of viral RNA in vitro and associates with the viral NP in infected cells. Using HMGB1-depleted cells, we demonstrate that HMGB1 promotes viral growth and enhances the transcription/replication activity of the viral polymerase. Finally, we show that HMGB1 binding to DNA is required for the enhancement of influenza virus replication.
MATERIALS AND METHODS
Phagemids.
The pJLE phagemid (13) was modified by the insertion of two SfiI sites in order to allow the subsequent subcloning of human cDNAs in the three reading frames. The resulting pJLESfiI, pJLESfi2, and pJLESfi3 phagemids were further converted into pJLESfi1L, pJLESfi2L, and pJLESfi3L phagemids by inserting a sequence encoding a glycine and serine-rich linker (G3S)7SG2 between the fos gene and the cDNA insert. The sequences of the oligodeoxynucleotides used for pairwise hybridization and insertion between the BglII and KpnI sites of pJLE are oligodeoxynucleotides 1 and 2, 3 and 4, and 5 and 6 for pJLESfiI, pJLESfi2, and pJLESfi3, respectively (see Table S1 in the supplemental material). Linker insertion at the BglII site of the pJLESfiI, pJLESfiI2, and pJLESfiI3 plasmids was done with oligodeoxynucleotides 7 and 8. The cDNAs encoding the full-length human HMGB1 (gene identification number [ID] 3146) and HMGB2 (gene ID 3148) were amplified from a human spleen cDNA library (Clontech) and subcloned at the SfiI site of the pJLESfi1L phagemid. All constructs were verified by sequencing positive clones using a BigDye terminator sequencing kit and an automated sequencer (Perkin-Elmer).
Plasmids.
The pcDNA and pPolI plasmids for reverse genetics of the A/WSN/33 (WSN) influenza virus (28) were kindly provided by G. Brownlee (Sir William Dunn School of Pathology, Oxford, United Kingdom). The pCAGGS plasmid allowing the expression of the bacteriophage T7 cytoplasmic RNA polymerase (45) was kindly provided by A. Billecocq (Institut Pasteur, Paris, France).
We generated a plasmid allowing the expression of a pseudoviral Renilla luciferase reporter influenza virus RNA in murine cells (pRF42-FluA-Renilla), or of pseudoviral CAT reporter influenza virus RNA (pT7-FluA-CAT-Rz) and pseudoviral CAT reporter vesicular stomatitis virus (VSV) RNA (pT7-VSV-CAT-Rz) under phage T7 RNA polymerase promoter. To generate these plasmids, the antisense Renilla open reading frame (ORF) or the antisense CAT ORF, both flanked by the 5′ and 3′ noncoding region of the A/WSN/33 NS segment, or the CAT ORF flanked by the antigenomic leader and trailer sequences of the VSV Indiana strain, were amplified using the pPR7-FluA-Renilla (5) or pPR7-FluA-CAT (23) plasmids as a template and oligonucleotides containing the flanking regions. The resulting amplicons were cloned into pRF42 vector (27) (kindly provided by Ramon Flick) or pT7-Rz plasmid, a derivative of the pPR7 plasmid (23) in which the Pol I promoter was replaced by the T7 promoter using BbsI or BsmBI restriction sites, respectively. The sequences encoding VSV-P, VSV-N, and VSV-L from the pBS plasmids (73) kindly provided by J. Rose (Yale University School of Medicine, New Haven, CT) were subcloned into the pCI vector (Promega) using XhoI and EcoRI (for L) or XhoI and NotI (for N and P) restriction sites. The Gateway-compatible pG-Luc1 and pG-Luc2 plasmids are described by Cassonnet et al. (15). Standard Gateway technology or standard PCR-based protocols were used to generate the indicated Luc1 or Luc2 derivative plasmids in a pDON-207 (Invitrogen) or a pCI plasmid (Promega), respectively. To generate the pCI-HMGB1 plasmid, the HMGB1 ORF was subcloned between XhoI and MluI into the pCI vector (Promega). The resulting plasmid was further modified by directed mutagenesis using QuikChange II multi-site-directed mutagenesis kit (Stratagene). The sequences of the oligonucleotides used for plasmids constructions can be obtained upon request.
Library construction.
Total mRNAs (13 μg) were isolated from 2 × 108 Ramos cells using an mRNA isolation kit (FastTrack; Invitrogen). The corresponding cDNAs were synthesized using a Creator Smart cDNA library construction kit (Clontech), and the 5′-ATTCTAGAGGCCGAGGCGGCCGACATGT30N′N-3′ (with “N′” representing A, G, or C and “N” representing A, G, C, or T) and 5′-AAGCAGTGGTATCAACGCAGAGTGGCCATTACGGCCGGG-3′ oligodeoxynucleotides as primers for the reverse transcription and second-strand synthesis, respectively. The cDNAs were cut with SfiI, size fractionated using Chroma spin 400 columns (Clontech), and ligated into the SfiI-digested vector pDNR-LIB (Clontech) prior to transformation in Escherichia coli. After large-scale plasmid purification and SfiI digestion, the cDNA inserts were subcloned into the pJLESfi1L, pJLESfi2L, and pJLESfi3L phagemids. Phage particles were prepared from E. coli cells transformed with the cDNA library phagemids by infection with the KM13 helper phage (42). The phage particles were purified from the culture supernatant by two polyethylene glycol 6000 induced precipitations prior to resuspension in phosphate-buffered saline (PBS; 25 mM sodium phosphate, 0.1 M NaCl [pH 7.4]).
Screening of monoclonal phages by ELISA.
Phagemid-containing E. coli clones were grown in 96-well plates in 2×YT medium supplemented with ampicillin, kanamycin, and 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 30°C for 16 h. Culture supernatants containing monoclonal phages were harvested, and the infectious titers were determined. Viral RNPs were purified from a concentrated suspension of a 6:2 (HA:NA) reassortant A/PR/8/34 × A/New Caledonia/20/99 influenza virus (kindly provided by Sanofi-Pasteur) as described previously (7). The expression and purification of the wild-type A/PR/8/34 NP and the corresponding R416A mutant NP were as described previously (11). Enzyme-linked immunosorbent assay (ELISA) plates were coated for 18 h at 4°C with 2 μg of purified vRNPs or NPs or with bovine serum albumin (BSA) as a control. After blocking with PBS–3% BSA for 2.5 h at 37°C, the plates were incubated for 2 h in the presence of the indicated amount of infective phage particles. Bound phage particles were detected using an horseradish peroxidase (HRP)-coupled anti-M13 antibody (Amersham) and the HRP substrate tetramethylbenzidine (Sigma).
Selection from phage-displayed protein libraries.
About 1012 infectious phage particles displaying expression products from the human cDNA library were added for 2 h at 20°C to tubes coated with purified influenza virus RNPs for 16 h at 4°C and then blocked at 37°C for 2 h with 2% milk in PBS as described earlier (79). After 10 washes with PBS containing 0.1% Tween 20 for the first round and 20 washes for the second round, phage particles that did not display a fusion protein were rendered noninfective by the addition of 10 mg of trypsin/liter for 10 min at 37°C prior to the addition of E. coli TG1 for infection. The E. coli population transformed with the selected cDNAs was then plated on ampicillin-containing petri dishes. To characterize the cDNA inserts, individual colonies were used for bacterial cell culture, prior to plasmidic DNA extraction using a Qiagen miniprep kit.
Cells.
Ramos cells (ATCC CRL-1596) were grown in RPMI 1640 supplemented with 2 mM glutamine, 10 mM HEPES, 1 mM sodium pyruvate, and 10% fetal bovine serum. A549, BSR, and 293T cells were grown in complete Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS). MDCK cells were grown in modified Eagle medium supplemented with 5% FCS. The wild-type (MEF-WT), HMGB1-deficient (MEF-HMGB1−/−), and HMGB2-deficient (MEF-HMGB2−/−) mouse embryonic fibroblasts were purchased from HMG Biotech (Milan, Italy) and grown in complete DMEM supplemented with 10% FCS and 1 mM sodium pyruvate. When indicated, glycyrrhizin (AcrosOrganics, Gells, Belgium) was added to the culture medium.
Viruses.
Wild-type A/WSN/33 influenza virus was produced by reverse genetics using a procedure adapted from Fodor et al. (28). VSV, Indiana strain, was kindly provided by O. Delmas (Institut Pasteur, Paris, France). VSV and WSN virus were amplified on BSR cells at a multiplicity of infection (MOI) of 10−3 PFU/cell and on MDCK cells at an MOI of 10−4 PFU/cell, respectively. They were titrated using a plaque assay adapted from Matrosovich et al. (49) on BSR and MDCK cells, respectively.
Protein complementation assay.
For transient protein complementation assay, the indicated pCI-Luc1 and pCI-Luc2 derived plasmids (100 and 200 ng, respectively) were cotransfected into 5 × 104 293T cells using Fugene-HD transfection reagent (Roche). At 24 h posttransfection, Gaussia luciferase activity was measured using the Renilla luciferase assay system kit (Promega) and a Tecan luminometer (Berthold). The normalized luminescence ratios (NLR) of Gaussia luciferase activity were calculated as described by Cassonnet et al. (15).
Viral infection assays.
For multicycle growth assays and single-cycle infection assays, confluent monolayers of MEF-WT, MEF-HMGB1−/−, and MEF-HMGB2−/− were infected at the indicated MOIs. After 1 h of adsorption, the cells were further incubated at 35°C in DMEM supplemented with 1% FCS. Virus titers in the supernatants collected at different time points were measured using a plaque assay as described above. Total cell lysates were prepared at different time points by direct lysis in Laemmli buffer.
Fluorescence cell-based PLA.
A549 cells grown on glass coverslips were fixed with PBS–4% paraformaldehyde for 20 min, permeabilized with PBS–0.1% Triton X-100 for 10 min, treated with 3% normal rabbit serum for 60 min, and then incubated with a mixture of mouse anti-NP monoclonal antibody (Argene) diluted 1:200 and anti-HMGB1 rabbit polyclonal antibody (ab18256; Abcam) diluted 1:650. Further, to detect in situ whether the epitopes are within 40-nm proximity (71), the cells were subjected to a proximity ligation assay (PLA) with a Duolink II fluorescence kit (Olink Biosciences) according to the manufacturer's protocol. Anti-mouse PLA Plus probe, anti-rabbit PLA Minus probe, and detection reagent orange were used. Fluorescein isothiocyanate (FITC)-conjugated anti-mouse antibody (Santa-Cruz Biotechnology) diluted 1:100 was present in the mixture of PLA Plus and PLA Minus probes. Microscopy was performed using a TCS SP2 AOBS confocal laser scanning microscope (Leica Microsystems, Germany) with a ×100 1.44 NA oil immersion objective lens. The excitation and emission were set at 488 nm and 500 to 550 nm for FITC, 405 nm and 420 to 470 nm for DAPI, 561 nm and 590 to 670 nm for Duolink II detection reagent orange (referred to as “PLA signal”). For all multicolor imaging, signals were acquired sequentially.
Western blots.
Cell lysates were analyzed by electrophoresis on a denaturing NuPAGE 4 to 12% Bis-Tris gel (Invitrogen), followed by Western blotting with polyvinylidene difluoride membranes (GE Healthcare). The membranes were incubated overnight at 4°C with rabbit polyclonal antibodies directed against A/PR/8/34 virions recognizing in particular NP and M1 proteins (80), HMGB1 (ab18256; Abcam), histone 3 (ab1791; Abcam), mouse monoclonal antibodies against VSV-G (V5507; Sigma), bacteriophage T7 RNA polymerase (catalog no. 70566; Merck), and α-tubulin (T6199; Sigma). Membranes were then incubated for 1 h at room temperature with peroxidase-conjugated secondary antibodies (GE Healthcare) and with the ECL Plus substrate (GE Healthcare). The membranes were scanned, and the chemiluminescence was acquired using a G-Box and GeneSnap software (SynGene) and then quantified using GeneTools software (SynGene).
Transcription/replication assay.
A total of 104 MEFs were transfected with the pCMV-Firefly (20 ng) and pRF42-FluA-Renilla (25 ng) plasmids, together with pCDNA-WSN-PB1, pCDNA-WSN-PB2, pCDNA-WSN-PA (50 ng), and pCDNA-WSN-NP (100 ng) using the Jet Prime reagent (PolyPlus transfection). Alternatively, MEFs were transfected with the pCMV-firefly (20 ng) and pRF42-FluA-Renilla (25 ng) plasmids, and 24 h later they were infected with the WSN virus at an MOI of 5, 50, 150, or 200 PFU/cell. At 24 h posttransfection or 24 h postinfection (hpi), respectively, the Renilla and firefly luciferase activities were measured using the Renilla luciferase and firefly luciferase assay system kits (Promega) and a Tecan luminometer (Berthold).
A total of 4 × 105 293T cells were transfected with a mixture of plasmids pcDNA-WSN-PB1, pcDNA-WSN-PB2, pcDNA-WSN-PA, and pcDNA-WSN-NP (0.25, 0.25, 0.25, and 1 μg), pT7-FluA-CAT (0.75 μg), and pCAGGS-T7-polymerase (0.5 μg) or with a mixture of plasmids pCI-VSV-P, pCI-VSV-L, and pCI-VSV-N (0.5, 0.5, and 1 μg), pT7-VSV-CAT (0.5 μg), and pCAGGS-T7-polymerase (0.5 μg), using the Fugene-HD reagent (Roche). At 48 h posttransfection, the chloramphenicol acetyltransferase (CAT) levels and total protein concentrations were measured by using a CAT ELISA kit (Roche) and a microBCA kit (Pierce), respectively.
Flow cytometry.
A total of 4 × 105 293T cells were transfected with the pCI-HMGB1, pCI-mut-HMGB1, or pCI plasmid (1 μg). At 24 h posttransfection, the cells were further infected with the WSN virus at a MOI of 0.3 PFU/cell. At 6 hpi, they were fixed with PBS–4% paraformaldehyde and coimmunostained with anti-NP monoclonal antibody (Argene) and anti-HMGB1 antibody (ab18256; Abcam). The cells were sorted using a FACSCalibur cytometer (BD Sciences) and analyzed with the CellQuest Pro software (Becton Dickinson).
RESULTS
Construction of a library of human cDNA expression products displayed on phages.
A library of more than 1.5 × 106 human cDNAs (>750 bp in length) was subcloned into phagemid vectors in all three reading frames. The cDNA expression products displayed on the surface of the filamentous phages were fusions with the Fos peptide bound by two disulfide bridges to the Jun peptide, itself fused to the minor coat protein pIII at the tip of the phage particle, as described previously (22) except for two modifications (Fig. 1A). First, domain 3 of protein pIII was replaced by the full-length protein pIII to make use of a selection background reduction strategy (42). Second, the cDNA expression product was linked to the Fos peptide through a long glycine- and serine-rich linker (G3S)7SG2 to facilitate its independent folding. After electroporation in E. coli, 3 × 108 colonies were recovered. After infection by the helper phage, the library of cDNA expression products displayed on filamentous phages was recovered.
Selection by phage display and characterization of influenza virus RNP-binding proteins.
Phage particles displaying the human cDNA products at the surfaces of their capsids were purified from the bacterial supernatant and subjected to two serial affinity selection cycles against vRNPs purified from influenza A/PR/8/34 virions (Fig. 1B). Two cycles were found to be sufficient to significantly reduce library diversity, in agreement with the high enrichment factors found in previous experiments (58, 79). A total of 3 × 105 of 1012 infective phages were recovered after the first selection round, whereas 106 of 1012 infective phages were recovered after the second round (Fig. 1B). Ninety-six clones derived from the second selection cycle were randomly picked; the corresponding monoclonal phages and protein were screened for vRNP binding by ELISA, and 27 clones were detected as positive. The 27 corresponding cDNAs, which were all in frame with the fos sequence, contained at least a part of the hmgb2 ORF. Between the fos sequence and the initiating codon of hmgb2, eight distinct lengths of the hmgb2 5′ noncoding region were represented, ranging between 0 and 107 nucleotides depending on the cDNA clone. Two sequences occurred more than three times among the 27 cDNAs (Fig. 1C, HMGB2-C1 and HMGB2-C2). The same strategy for the isolation of binding proteins, when used in parallel with vRNPs or NPs purified from Rift Valley fever virus or from measles virus, led to the selection of sets of cDNAs that encoded proteins distinct from HMGB2 or any other protein of the HMGB family, thus arguing for the specificity of the HMGB2 hit.
We then sought to determine whether the HMGB1 protein could also bind to vRNPs because HMGB1 and HMGB2 show 79% amino acid identity overall (reference sequences, Fig. 1C). We reasoned that the hmgb1 ORF might have been under-represented in the phagemid library, given the fact that cDNA reverse transcription was initiated with an oligo(dT) primer and that human HMGB1 mRNAs have a very long 3′ untranslated region compared to HMGB2 mRNAs (2,671 and 704 nucleotides, respectively). Thus, the cDNAs encoding HMGB1 and HMGB2 were subcloned into a phagemid in order to express the full-length proteins on the surfaces of phage particles. HMGB1- and HMGB2-expressing phages bound to purified vRNPs with an at least 5-fold signal/background ratio, as measured from the optical densities. Control phages did not bind (Fig. 2A). Parallel experiments were performed in the presence of purified RNA-free viral NP instead of vRNPs. Two variants of the A/PR/8/34-derived NP were used: the wild-type NP, which is known to oligomerize spontaneously (67), and the R416A mutant NP, which remains monomeric (11, 86). The HMGB1- and HMGB2-expressing phages, but not the control phages, bound both to the wild-type NP and to the mutant NP (Fig. 2B and C, respectively). Interestingly, a phage displaying a cDNA product corresponding to the first 63 amino acids of HMGB2 (HMGB2-del in Fig. 1C) was also found to bind the wild-type NP proteins (>5-fold signal to background ratio), thereby indicating that the N-terminal domain of HMGB2 is sufficient for NP binding.
Fig 2.
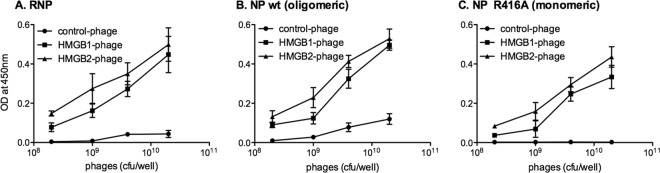
In vitro binding of HMGB1- and HMGB2-displaying phages to purified influenza virus RNPs and NP proteins as determined by ELISA. ELISA plates were coated with purified influenza virus RNPs (A), wild-type viral NPs proteins in an oligomeric state (B), or R416A mutant NPs in a monomeric state (C). Suspensions of monoclonal phage particles displaying the full-length HMGB1 or HMGB2 proteins were added (2 × 108 to 2 × 1010 CFU per well). Monoclonal phage particles expressed from a phagemid having no cDNA insert were used as a control. Bound phage particles were detected by an anti-M13-HRP antibody. Vertical bars represent ± the standard deviations (SD) and are representative of triplicates.
Association of HMGB1 and HMGB2 with influenza virus NPs in cells.
We assessed the association of HMGB proteins with influenza virus NP in cultured cells. A protein complementation assay that allows the detection of weak and/or transient interactions was used. The proteins of interest were fused to the Luc1 or Luc2 complementary fragments of the Gaussia princeps luciferase (63), and protein-protein interactions were monitored by measuring the luciferase activities in cells transiently coexpressing fusion proteins.
The PB1 and PB2 subunits of the influenza virus polymerase being well documented (32, 61, 75), coexpression of PB1-Luc1 and Luc2-PB2 gave a strong luminescence activity which was used as a positive control and as a reference (Fig. 3A, thin-hatched bar). The NP from the A/WSN/33 (WSN) strain and from the pandemic H1N1 isolate A/Paris/2590/2009 were tested in parallel. When NP-Luc1 proteins were coexpressed with the homologous Luc2-PB2, significant luminescence activities representing 18% ± 7% and 17% ± 4% of the above-mentioned reference were measured (Fig. 3A, thick-hatched bars), in agreement with published data showing an interaction between PB2 and NP (10, 43). Similarly, when the NP-Luc1 proteins were coexpressed with Luc2-HMGB1 or Luc2-HMGB2, the luminescence activities ranged between 25% ± 1% and 32% ± 12% (Fig. 3A, white bars) and were >20-fold higher than the background (Fig. 3A, horizontal dotted line). Very similar data were obtained when the Luc1 fragment was fused at the N-terminal instead of the C-terminal end of the NP (Fig. 3B, white bars).
Fig 3.
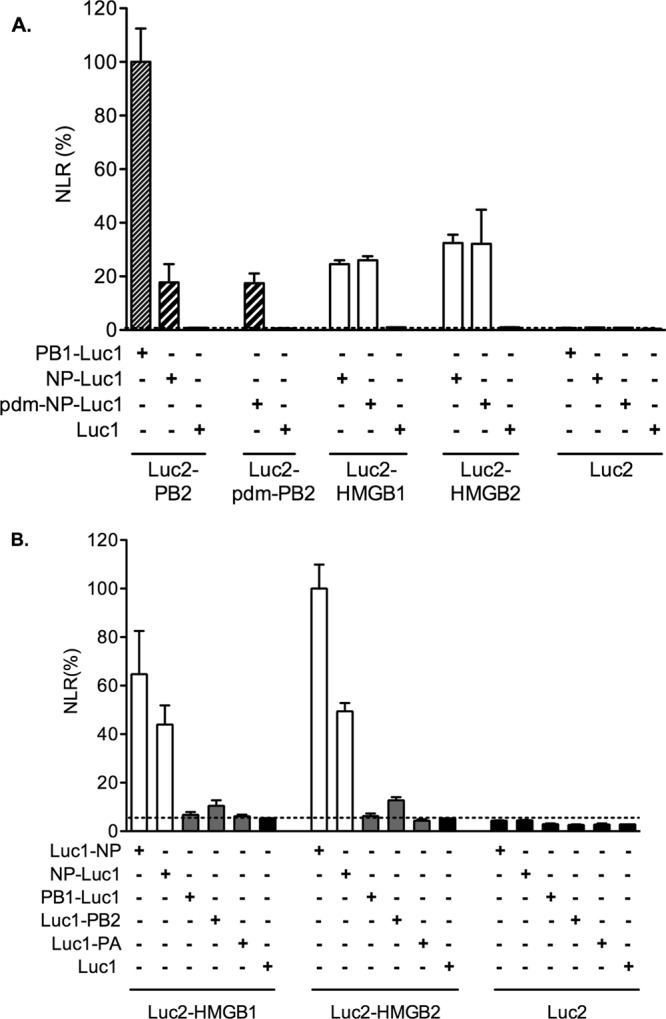
In cell association of HMGB1 and HMGB2 with influenza virus NP detected in a protein complementation assay. 293T cells were cotransfected with the indicated combinations of the Luc1-fusion or control Luc1 expression plasmid on one hand and the Luc2-fusion or control Luc2 plasmid on the other hand. The viral fusion proteins were derived from the A/WSN/33 strain, or from the pandemic (pdm) A/Paris/2590/2009 virus when indicated. Cell extracts were prepared at 24 h posttransfection and tested for Gaussia princeps luciferase activity. The normalized luminescence ratios (NLR) as defined by Cassonnet et al. (15) were expressed as percentages and as means ± the SD of triplicates. The dotted line represents the maximum NLR measured with the negative control plasmids.
The interaction of HMGB1 and HMGB2 with the other components of the viral RNPs was also examined. The coexpression of WSN-derived PB1-Luc1, Luc1-PB2, or Luc1-PA, together with Luc2-HMGB1 or Luc2-HMGB2, led to close to background-level luminescence activities (Fig. 3B, gray bars). Overall, our results indicated that HMGB1 and HMGB2 associate specifically with influenza virus NP when transiently expressed in cells.
Finally, the association of the endogenous HMGB1 protein with the viral NP was documented in the context of infection. To this end, the Duolink II cell-based fluorescence PLA was used, since it allows the detection of transient interactions occurring between native proteins (71). In PLA, a signal is generated only when two oligonucleotide-labeled secondary antibodies (“PLA probe Plus” and “PLA probe Minus”) are bound in close proximity (<40 nm). Each pair of PLA probes is visualized as a single fluorescent spot, as a result of ligation which yields a closed DNA circle, subsequent rolling-circle amplification by a DNA-dependent DNA polymerase, and detection of the amplification product with fluorescently labeled oligonucleotides. Influenza virus-infected A549 cells were incubated with a mixture of anti-HMGB1 and anti-NP antibodies, and then incubated with a mixture of the “PLA probe Minus” and “PLA probe Plus.” In the nuclei of infected cells fixed at 4 hpi, numerous PLA spots were observed, and the number of spots per nucleus correlated with the overall brightness of NP signal visualized by FITC-conjugated anti-mouse antibody in the same samples (Fig. 4A, upper panels, and Fig. 4B). Mock-infected cells showed very faint background signals (Fig. 4A, lower panels). When PLA was performed on HMGB1-deficient MEFs, no PLA signal was detected, although the NP was efficiently detected by immunostaining (not shown). Thus, PLA demonstrated that in the nuclei of infected cells, NP and endogenous HMGB1 are in close proximity (<40 nm) (71) and are thus indirectly or, most likely given our other data, directly interacting with each other.
Fig 4.

Association of endogenous HMGB1 with influenza virus NP in infected cells, detected in a Duolink II proximity ligation assay (PLA). A549 cells were infected with the WSN virus at a MOI of 3 PFU/cell (A, upper panels, and B) or mock infected (A, lower panels). At 4 hpi, the cells were subjected to Duolink II PLA as described in Materials and Methods, using primary anti-HMGB1 and anti-NP antibodies. In panel A, the PLA-specific fluorescent signal, the NP-immunostaining signal, and the DAPI nucleic staining were pseudo-colored red, green, and blue, respectively (scale bar, 20 μm). In panel B, orthogonal view of a three-dimensional reconstruction of an infected cell is shown, with PLA signal (red), NP immunostaining (green), and merge (yellow). Scale bar, 5 μm.
Mapping of the NP-interacting domain on HMGB1 and HMGB2.
In order to map the NP-interacting domain, we expressed deleted versions of HMGB1 and HMGB2 fused to Luc2 (schematically represented in Fig. 5). The Δ1-HMGB1 and Δ1-HMGB2 proteins, deleted of the C-terminal acidic tail, corresponded to the 185 N-terminal residues and encompassed the box A and box B domains. The Δ2-HMGB1and Δ2-HMGB2 proteins, further deleted from the box B domain, corresponded to the 89 N-terminal residues and encompassed the box A domain. Luciferase activities measured with the Δ1 and Δ2 proteins were ∼5- and ∼2-fold higher than with the corresponding full-length HMGB proteins (Fig. 5). Our data clearly indicate that the box A-box B domain alone, and to a lesser extent the box A domain alone, is able to bind the viral NP. The data also suggest that the presence of the C-terminal acidic tail reduces the binding to the NP.
Fig 5.
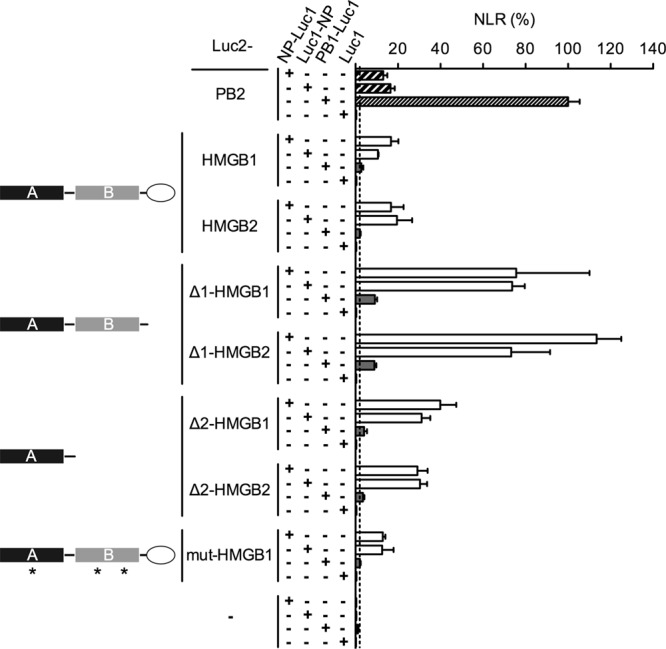
Mapping of the NP-binding domain on HMGB1 and HMGB2 proteins. 293T cells were cotransfected with the indicated combinations of the Luc1-fusion or control Luc1 plasmids on one hand and the Luc2-fusion or control Luc2 plasmids on the other hand. The deletion mutants Δ1-HMGB1/2 and Δ2-HMGB1/2 proteins correspond to the 185 and 89 N-terminal residues of the HMGB1/2 proteins, respectively. The mut-HMGB1 protein is a DNA-binding-defective triple mutant (F38A, F103A, and I122A, indicated by stars) (1). The NP, PB1, and PB2 fusion proteins were derived from the WSN strain. Cell extracts were prepared at 24 h posttransfection and tested for Gaussia princeps luciferase activity. The normalized luminescence ratios (NLR) as defined by Cassonnet et al. (15) were expressed as percentages and as means ± the SD of triplicates. The dotted line represents the maximum NLR measured with the negative control plasmids.
Finally, we sought to determine whether an HMGB1 protein whose DNA-binding activity was abolished was still able to bind the NP. When a DNA-binding-defective mutant of HMGB1 (F38A/F103A/I122A) (1) was fused to Luc2 and coexpressed with NP-Luc1, the luciferase activities were in the same range as with the wild-type HMGB1 protein (Fig. 5, mut-HMGB1), thus indicating that the HMGB1-NP interaction is independent of the presence of a functional DNA-binding site on HMGB1.
The production of infectious influenza virions is impaired in HMGB1-deficient cells.
To investigate the functional importance of HMGB proteins in the replication cycle of influenza viruses, we used MEFs derived from HMGB1- and HMGB2-deficient mice (MEF-HMGB1−/− and MEF-HMGB2−/−, respectively), in parallel with MEFs derived from wild-type mice (MEF-WT). The growth kinetics of the WSN influenza virus strain were compared for the three MEF types. The VSV Indiana strain, a mononegavirus that replicates in the cytoplasm, was used as a control of specificity. Because in a preliminary experiment VSV appeared to grow more efficiently than WSN virus on MEF-WT cells, MOIs of 10−4 and 10−3 PFU/cell were used for VSV and WSN virus, respectively. VSV replicated as efficiently on MEF-HMGB1−/− cells as on the MEF-WT and MEF-HMGB2−/− cells (Fig. 6A). The WSN virus also replicated efficiently on MEF-WT and MEF-HMGB2−/− cells, reaching titers of ∼107 PFU/ml at 96 hpi. However, it replicated at a slower rate and achieved 50-fold-lower titers on MEF-HMGB1−/− cells (2 × 105 PFU/ml at 96 hpi) (Fig. 6B).
Fig 6.
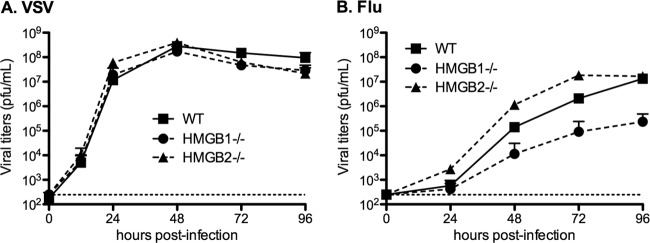
Viral growth curves on wild-type, HMGB1−/−, or HMGB2−/− MEFs. MEF-WT, MEF-HMGB1−/−, and MEF-HMGB2−/− cell monolayers were infected with the VSV (Indiana) (A) or the WSN influenza virus (B) at an MOI of 10−4 or 10−3 PFU/cell, respectively. At the indicated time points, the supernatants were harvested, and infectious virus titers were determined by plaque assays on BSR (A) or MDCK cells (B). The horizontal dotted lines represent the limit of detection of the plaque assays. The data are expressed as means ± the SD (n = 3).
Influenza virus protein synthesis is impaired in HMGB1-deficient cells.
We next examined whether the production of viral proteins was impaired in HMGB1-deficient cells. The MEF-WT, MEF-HMGB1−/−, and MEF-HMGB2−/− cells were infected in parallel at an MOI of 5 PFU/cell with the WSN virus or VSV. The accumulation of viral proteins was evaluated by Western blot analysis of total cell lysates at various times postinfection (Fig. 7A). Upon quantification, the normalized steady-state levels of the VSV-G protein were slightly higher in MEF-HMGB1−/− cells compared to MEF-WT and MEF-HMGB2−/− cells (Fig. 7B). In contrast, the normalized steady-state levels of the WSN-NP and M1 proteins showed a >40% reduction in MEF-HMGB1−/− cells compared to MEF-WT and MEF-HMGB2−/− cells (Fig. 7C and D). These data indicated that an early step of the influenza virus replication cycle was specifically impaired in HMGB1−/− cells but not in HMGB2−/− cells. We thus focused on HMGB1 in the following experiments.
Fig 7.
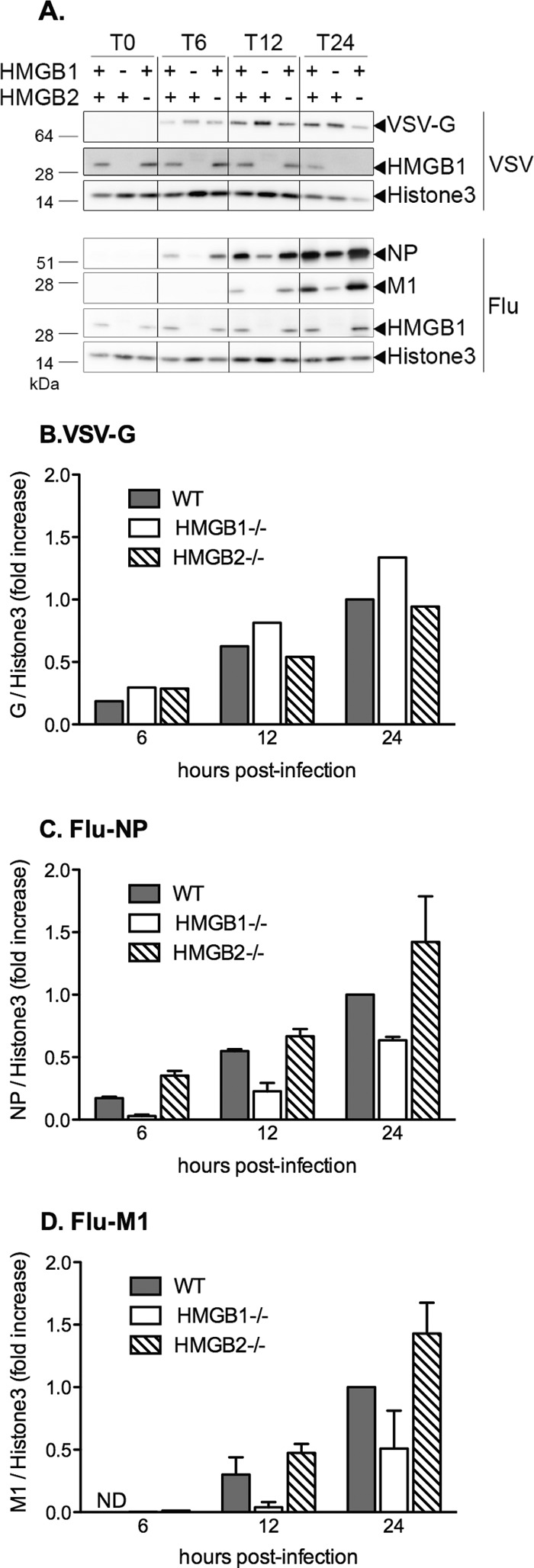
Steady-state levels of viral proteins after infection of wild-type versus HMGB1−/− or HMGB2−/− MEFs. (A) MEF-WT, MEF-HMGB1−/−, and MEF-HMGB2−/− cell monolayers were infected with VSV (upper panel) or WSN virus (lower panel) at an MOI of 5 PFU/cell. Whole-cell lysates were prepared at the indicated time points and analyzed by Western blotting as described in Materials and Methods, using antibodies recognizing the VSV-G glycoprotein (upper panel), the influenza virus NP and M1 proteins (lower panel), and the HMGB1 or the histone 3 proteins (upper and lower panels). One representative experiment of two with similar results is shown. (B to D) Graphic representation of the Western blot quantification. After the membranes were scanned with a G-Box (SynGene), the VSV-G, NP, M1, and histone 3 signals were quantified using GeneTools software (SynGene). The VSV-G (B), NP (C), and M1 (D) signals were normalized with respect to the histone 3 signal. The data are expressed as ratios and as means ± the SD of one (B) or two (C and D) independent experiments. ND, not detected.
HMGB1 promotes influenza virus polymerase transcription/replication activity.
We examined whether the activity of the WSN influenza virus polymerase was affected by the presence or absence of HMGB1. A luciferase reporter pseudo-influenza virus RNA was transiently expressed in MEF-WT and MEF-HMGB1−/− cells, which were subsequently infected with WSN virus at various MOIs ranging from 5 to 200 PFU/cell. The efficiency of the early steps of the viral cycle was monitored by measuring the luciferase activities in cell extracts. The normalized luciferase activities increased, together with the MOIs, in both cell types, but they remained 5- to 10-fold lower in MEF-HMGB1−/− cells than in MEF-WT cells (Fig. 8A).
Fig 8.

Effect of HMGB1 depletion or inhibition on the transcription/replication of an influenza virus-like minigenome. (A) Transcription/replication of an influenza virus-like minigenome in infected wild-type or HMGB1−/− MEFs. A plasmid allowing the expression of an influenza virus-like Renilla luciferase reporter RNA was cotransfected with a firefly luciferase expression plasmid in MEF-WT cells (gray bars) or in MEF-HMGB1−/− cells (white bars). At 24 h posttransfection, the cells were either mock infected (−) or infected with WSN virus at increasing MOIs (5 to 200 PFU/cell, black triangle). The Renilla luciferase activities measured in cell extracts at 24 hpi were normalized with respect to the firefly luciferase activities (RLU, relative light units). They are expressed as means ± the SD of triplicates and are representative of two independent experiments. (B) Transcription/replication of an influenza virus-like minigenome upon transient reconstitution of ribonucleoproteins in wild-type or HMGB1−/− MEFs. A plasmid allowing the expression of an influenza virus-like Renilla luciferase reporter influenza virus RNA was cotransfected with a firefly luciferase expression plasmid only (−) or together with PA, PB1, PB2, and NP expression plasmids (+) in MEF-WT cells (gray bars) or in MEF-HMGB1−/− cells (white bars). The Renilla luciferase activities measured in cell extracts at 24 h posttransfection were normalized with respect to the firefly luciferase activities (RLU, relative light units) and are expressed as means ± the SD of quadruplicates. (C and D) Transcription/replication of an influenza virus-like or a VSV-like minigenome in glycyrrhizin-treated 293T cells. 293T cells were cotransfected with plasmids, allowing the expression of a pseudoviral CAT reporter virus-specific RNA and the viral ribonucleoprotein components of VSV (opened squares) or WSN influenza virus (closed circles). At 8 h posttransfection, the culture medium was replaced with fresh medium supplemented with 1, 2, or 4 mM glycyrrhizin. (C) Cell extracts were prepared at 48 hpi and tested for CAT expression by ELISA. CAT levels were normalized with respect to the total protein concentration. The results are expressed as percentages and as means ± the SD of three independent experiments performed in triplicates (**, P = 0.0316, unpaired two-tailed Student t test [n = 3]). The horizontal dotted line represents the background signal. (D) Cell extracts were tested by Western blotting with antibodies against the NP, T7 polymerase (T7pol), HMGB1, and α-tubulin proteins. The results of one representative experiment from two with similar results are shown.
Given the fact that HMGB1 is primarily a nuclear protein, we hypothesized that it might facilitate the influenza virus polymerase transcription/replication activity, which takes place in the nucleus of infected cells, unlike in VSV infection. We thus used a transient vRNP reconstitution assay to specifically monitor the activity of the viral polymerase in MEF-WT and MEF-HMGB1−/− cells. Upon transient expression of a pseudo-influenza virus luciferase reporter RNA, together with the viral NP and the polymerase subunits (PB1, PB2, and PA), we observed 5-fold-lower polymerase activities in MEF-HMGB1−/− cells than in MEF-WT cells (Fig. 8B). To confirm that HMGB1 was facilitating the viral polymerase activity, the assay was repeated in human 293T cells and in the presence of glycyrrhizin, an inhibitor of HMGB1 (53). A control VSV minigenome assay was used in parallel. The VSV polymerase activity remained unchanged in the absence or in the presence of 1 to 4 mM glycyrrhizin (Fig. 8C, open squares). No significant difference in influenza virus polymerase activity was observed at 1 and 2 mM glycyrrhizin, but a reduction of around 50% (P = 0.0316, n = 3) was repeatedly observed in the presence of 4 mM glycyrrhizin in the culture medium (Fig. 8C, closed circles). The high concentration of glycyrrhizin needed to observe an intracellular effect was consistent with the fact that glycyrrhizin binds HMGB1 with a dissociation constant of 0.15 mM in vitro (53). The levels of endogenous (HMGB1 and α-tubulin) and recombinant (NP or T7 RNA polymerase) proteins were assessed by Western blotting to control the absence of drug toxicity and the transfection efficiency (Fig. 8D). Overall, our data indicate that HMGB1 promotes influenza virus polymerase transcription/replication activity.
HMGB1 binding to DNA is required for the enhancement of influenza virus replication.
Since glycyrrhizin was shown to bind the DNA-binding site of HMGB1 and to reduce HMGB1 binding to DNA (53), we further examined whether HMGB1 DNA binding is involved in promoting influenza virus replication. We used the F38A/F103A/I122A DNA-binding-defective mutant of HMGB1, shown above to bind the NP as efficiently as the wild-type HMGB1 (Fig. 5). 293T cells were transiently transfected with plasmids encoding the DNA-binding-defective mutant or the wild-type HMGB1 protein and were subsequently infected with the WSN influenza virus. The levels of expression of HMGB1 and viral NP were evaluated by indirect immunofluorescence and cell sorting flow cytometry. In cells which expressed endogenous levels of HMGB1, 38% ± 1% of the cells were NP positive (Fig. 9). In the cellular subpopulation that overexpressed the wild-type HMGB1, the proportion of NP-positive cells showed a clear increase (50% ± 5%) (Fig. 9), in agreement with our other data showing that HMGB1 facilitates viral replication. Interestingly, in the cellular subpopulation that overexpressed the DNA-binding-defective HMGB1, the proportion of NP-positive cells remained unchanged (40% ± 2%), indicating that HMGB1 binding to DNA is required for the enhancement of viral replication.
Fig 9.
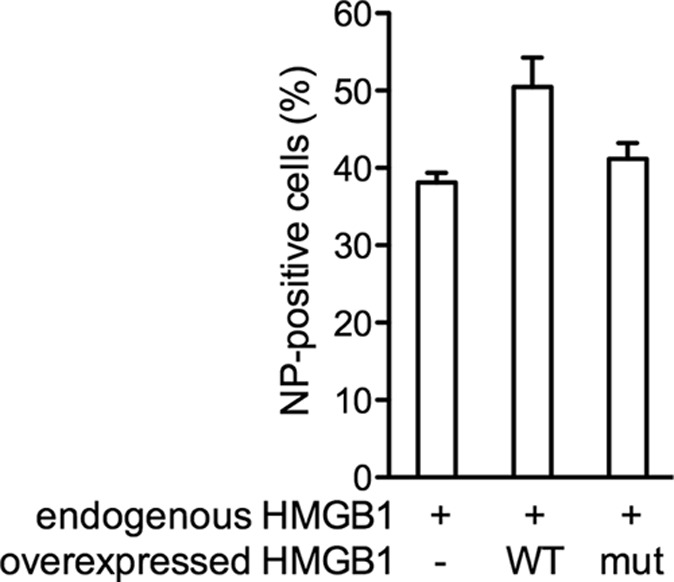
Effect of a DNA-binding-defective HMGB1 mutant on influenza virus replication. 293T cells were transiently transfected with a plasmid allowing the expression of the wild-type or a DNA-binding-defective HMGB1 or mock transfected. At 24 h posttransfection, the cells were infected at an MOI of 0.3 PFU/cell with WSN influenza virus. At 6 hpi, the HMGB1 and NP expression levels were evaluated using indirect immunofluorescent assay and flow cytometry. The percentages of NP-positive cells were evaluated in the cellular subpopulations expressing endogenous levels of HMGB1 only (−) and in subpopulations overexpressing the wild-type (WT) or mutant (mut) HMGB1 protein. The data are expressed as means ± the SD of duplicates and are representative of three independent experiments.
DISCUSSION
Understanding the relationship between pathogenic microorganisms and their human host necessitates the characterization of their interactions at the molecular and macromolecular levels. Successful approaches for the high-throughput detection of viral-host protein-protein interactions include the yeast two-hybrid screen, which does not require experimental handling of the proteins but only of the corresponding genes, and affinity purification of protein complexes followed by mass spectrometry analysis (17). However, these approaches cover only a fraction of the total number of protein-protein interactions within a cell, and they show a number of limitations (e.g., some proteins might be toxic to yeast in yeast two-hybrid screening, and some infectious cell lysates cannot be handled experimentally outside high-security laboratories for mass spectrometry analyses).
The strategy used here for the detection of human protein domains binding influenza virus ribonucleoproteins makes use of in vitro affinity selections of human protein domains for given targets. The proteins are displayed on the surfaces of filamentous phages for the straightforward recovery of the corresponding genes, which can then be further amplified and sequenced (48). The phagemid approach allows, at most, one copy of the cDNA expression product to be displayed on a phage particle, thereby avoiding biases during affinity selections due to avidity effects. However, it has one drawback, namely, that numerous phage particles do not display a cDNA expression product and contribute to background. A selection background reduction strategy using engineered phagemids and helper phages designed to render noninfective the phage particles which do not display proteins of interest (42) was therefore combined here with an adapted full-length cDNA expression system on the surfaces of filamentous phages deriving from the Jun and Fos peptides (22). The overall efficiency and selectivity of this combination of strategies is highlighted by the set of distinct cDNAs found to be in frame with the fos sequence, all of which contain the 5′ end of the hmgb2 ORF. To our knowledge, this report describes the first host-pathogen protein-protein interaction identified from a library of human cDNA expression products displayed on phages and validated using cell-based assays. We envision that this library, and more generally the strategy described here, will allow the identification of many more macromolecular interactions between pathogenic microorganisms and their hosts. The binding of HMGB proteins to influenza virus vRNPs has not been detected to date by affinity purification (12, 37, 51) or yeast two-hybrid assays (69, 75a), which underlines the complementarity of these methods and phage display.
Interestingly, the ELISA revealed that recombinant HMGB2 and HMGB1 phages could bind to purified RNA-free influenza virus NP, either in its wild-type oligomeric state or in a mutant monomeric state. Consistently, both HMGB proteins were found to bind to the NP but not to the polymerase subunits in cultured cells when a protein complementation assay was used. Overall, our data suggest that the cellular HMGB proteins interact with vRNPs through their NP component; they interact with the NP independently of the presence of RNA, and neither the NP-NP oligomerization domain nor the RNA-binding domain of the NP are involved in the interaction. In addition, our data show that the HMG box domains are sufficient for binding to the viral NP, whereas the presence of the C-terminal acidic tail reduces the binding to the NP. A likely hypothesis is that the C-terminal acidic tail, which can make intramolecular contacts with HMGB boxes (84), prevents the association of NP with the HMGB boxes by steric hindrance.
To investigate the functional importance of HMGB1 and HMGB2 proteins in influenza virus replication, we used MEF cells deficient in HMGB1 or HMGB2. These cells represented a relevant model, since murine and human HMGB2 proteins show 92% homology, and murine and human HMGB1 proteins are identical except for two glutamate/aspartate substitutions in the C-terminal acidic tail. We found that the absence of HMGB1, but not HMGB2, resulted in decreased production of infectious progeny in multicycle growth assays and in reduced expression of viral proteins in single-cycle infection assays. Sequence differences between the two proteins might contribute to the differential effect of HMGB1 and HMGB2 depletion. Indeed, despite their high degree of sequence homology, the two proteins clearly have distinct functions, as revealed by the distinct phenotypes of HMGB1 and HMGB2 knockout mice. The HMGB1 protein has a more essential and pleiotropic role than HMGB2 (14, 66). In addition, HMGB1 might be expressed at higher levels than HMGB2 in MEF cells, which would contribute to the preferential formation of HMGB1-NP complexes. In the lung in particular, the HMGB1 protein appears overexpressed compared to HMGB2 (68).
We investigated the mechanism through which HMGB1 exerts its positive effect on influenza virus replication. HMGB1 associates with the viral NP in the nucleus of infected cells, as demonstrated by an in situ fluorescence-based proximity ligation assay. The presence of a functional HMGB1 DNA-binding site is required for the enhancement of influenza virus replication, as shown with a DNA-binding-defective HMGB1 mutant. In minigenome assays, the presence of HMGB1 upregulates the transcription/replication activity of the viral polymerase; glycyrrhizin, which is known to reduce HMGB1 DNA-binding activity, inhibits influenza virus polymerase activity. Overall, these data are consistent with the facts that (i) viral transcription is critically dependent on the cellular transcription machinery (25), (ii) vRNPs bind nucleosomes (31) and chromatin-associated factors (20, 47, 51), and (iii) HMGB1 is a dynamic regulator of the accessibility of nucleosomally packaged DNA (8). We propose that binding of the viral NP to HMGB1 facilitates the recruitment of vRNPs at transcriptionally active sites of the chromatin, which in turn favors efficient viral transcription/replication. A recent study by Matsumoto et al. (50), which shows that HMGB1 is required to stabilize bornavirus RNPs on chromosomes and for efficient bornavirus RNA persistence in the nucleus, strengthens the likeliness of our hypothesis. However, further studies are required to elucidate the mechanism through which HMGB1 stimulates influenza virus replication.
Beyond its role in facilitating influenza virus polymerase activity in the nucleus, the binding of NP to HMGB1 might modulate the cellular response to infection through various mechanisms. It could influence the chromatin structure and transcription pattern in infected cells, as shown in the case of Borna disease virus phosphoprotein-HMGB1 association (87). The NP-HMGB1 association could also restrict HMGB1 proinflammatory activity by reducing the active secretion of HMGB1 from immune cells or could interfere with the HMGB1-mediated induction of interferon (33).
In recent years, the potential of HMGB1 inhibition as an alternative strategy to reduce inflammation, in infectious as well as in noninfectious contexts, has become a major field of research (4). Our data suggest that targeting the binding of HMGB1 to influenza virus NP, in addition to its proinflammatory activity, could lead to the design of novel anti-influenza treatments.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge F. Rougeon for advice and suggestions during this work and thank B. Heyd (IP PTR 194 fellowship) for helper phage preparations and the final vRNP purification step. We thank G. Brownlee, A. Billecocq, J. Rose, and C. Chiang for providing plasmids. We thank O. Delmas for providing the VSV strain, and Sanofi-Pasteur for providing a concentrated influenza virus preparation. We thank the Plateforme de Séquençage at the Institut Pasteur for its expertise. N.N. and D.M. thank O. Delmas, D. Blondel, M. Schwemmle, G. Chase, C. Prehaud, and S. van der Werf for helpful discussions and C. Barbezange, S. Munier, and C. Chiang for critical reading of the manuscript.
X.G. was supported by a fellowship from the Chinese Academy of Sciences. S.V.A. was supported by an EIPOD fellowship (EMBL). This study was supported in part by the Institut Pasteur (PTR 194 in particular), the FLUINNATE (SP5B-CT-2006-044161) and the FLUPHARM (FP7-INFLUENZA-2010-259751) programs, and the ANR FLU INTERPOL contract (ANR-06-MIME-014-01).
Footnotes
Published ahead of print 13 June 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Agresti A, Scaffidi P, Riva A, Caiolfa VR, Bianchi ME. 2005. GR and HMGB1 interact only within chromatin and influence each other's residence time. Mol. Cell 18:109–121 [DOI] [PubMed] [Google Scholar]
- 2. Allain FH, et al. 1999. Solution structure of the HMG protein NHP6A and its interaction with DNA reveals the structural determinants for non-sequence-specific binding. EMBO J. 18:2563–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alleva LM, Budd AC, Clark IA. 2008. Systemic release of high mobility group box 1 protein during severe murine influenza. J. Immunol. 181:1454–1459 [DOI] [PubMed] [Google Scholar]
- 4. Andersson U, Tracey KJ. 2011. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29:139–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avilov SV, et al. 2012. Replication-competent influenza A virus that encodes a split-green fluorescent protein-tagged PB2 polymerase subunit allows live-cell imaging of the virus life cycle. J. Virol. 86:1433–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barqasho B, Nowak P, Abdurahman S, Walther-Jallow L, Sonnerborg A. 2010. Implications of the release of high-mobility group box 1 protein from dying cells during human immunodeficiency virus type 1 infection in vitro. J. Gen. Virol. 91:1800–1809 [DOI] [PubMed] [Google Scholar]
- 7. Baudin F, Bach C, Cusack S, Ruigrok RW. 1994. Structure of influenza virus RNP. I. Influenza virus nucleoprotein melts secondary structure in panhandle RNA and exposes the bases to the solvent. EMBO J. 13:3158–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bianchi ME, Agresti A. 2005. HMG proteins: dynamic players in gene regulation and differentiation. Curr. Opin. Genet. Dev. 15:496–506 [DOI] [PubMed] [Google Scholar]
- 9. Bianchi ME, Manfredi AA. 2007. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol. Rev. 220:35–46 [DOI] [PubMed] [Google Scholar]
- 10. Biswas SK, Boutz PL, Nayak DP. 1998. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J. Virol. 72:5493–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10a. Bortz E, et al. 2011. Host- and strain-specific regulation of influenza virus polymerase activity by interacting proteins. mBio 2(4):e00151-11 doi:10.1128/mBio.00151-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boulo S, et al. 2011. Human importin alpha and RNA do not compete for binding to influenza A virus nucleoprotein. Virology 409:84–90 [DOI] [PubMed] [Google Scholar]
- 12. Bradel-Tretheway BG, et al. 2011. Comprehensive proteomic analysis of influenza virus polymerase complex reveals a novel association with mitochondrial proteins and RNA polymerase accessory factors. J. Virol. 85:8569–8581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brunet E, Chauvin C, Choumet V, Jestin JL. 2002. A novel strategy for the functional cloning of enzymes using filamentous phage display: the case of nucleotidyltransferases. Nucleic Acids Res. 30:e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calogero S, et al. 1999. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 22:276–280 [DOI] [PubMed] [Google Scholar]
- 15. Cassonnet P, et al. 2011. Benchmarking a luciferase complementation assay for detecting protein complexes. Nat. Methods 8:990–992 [DOI] [PubMed] [Google Scholar]
- 16. Catena R, et al. 2009. HMGB4, a novel member of the HMGB family, is preferentially expressed in the mouse testis and localizes to the basal pole of elongating spermatids. Biol. Reprod. 80:358–366 [DOI] [PubMed] [Google Scholar]
- 17. Causier B. 2004. Studying the interactome with the yeast two-hybrid system and mass spectrometry. Mass Spectrom. Rev. 2004:350–367 [DOI] [PubMed] [Google Scholar]
- 18. Chan AY, Vreede FT, Smith M, Engelhardt OG, Fodor E. 2006. Influenza virus inhibits RNA polymerase II elongation. Virology 351:210–217 [DOI] [PubMed] [Google Scholar]
- 19. Chase G, et al. 2008. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 377:431–439 [DOI] [PubMed] [Google Scholar]
- 20. Chase GP, et al. 2011. Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PLoS Pathog. 7:e1002187 doi:10.1371/journal.ppat.1002187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Costello E, Saudan P, Winocour E, Pizer L, Beard P. 1997. High mobility group chromosomal protein 1 binds to the adeno-associated virus replication protein (Rep) and promotes Rep-mediated site-specific cleavage of DNA, ATPase activity and transcriptional repression. EMBO J. 16:5943–5954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crameri R, Suter M. 1993. Display of biologically active proteins on the surface of filamentous phages. Gene 137:69–75 [DOI] [PubMed] [Google Scholar]
- 23. Crescenzo-Chaigne B, Naffakh N, van der Werf S. 1999. Comparative analysis of the ability of the polymerase complexes of influenza viruses type A, B, and C to assemble into functional RNPs that allow expression and replication of heterotypic model RNA templates in vivo. Virology 265:342–353 [DOI] [PubMed] [Google Scholar]
- 24. Deng T, et al. 2006. Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J. Virol. 80:11911–11919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Engelhardt OG, Fodor E. 2006. Functional association between viral and cellular transcription during influenza virus infection. Rev. Med. Virol. 16:329–345 [DOI] [PubMed] [Google Scholar]
- 26. Engelhardt OG, Smith M, Fodor E. 2005. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J. Virol. 79:5812–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Flick R, Pettersson RF. 2001. Reverse genetics system for Uukuniemi virus (Bunyaviridae): RNA polymerase I-catalyzed expression of chimeric viral RNAs. J. Virol. 75:1643–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fodor E, et al. 1999. Rescue of influenza A virus from recombinant DNA. J. Virol. 73:9679–9682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gabriel G, Herwig A, Klenk HD. 2008. Interaction of polymerase subunit PB2 and NP with importin α1 is a determinant of host range of influenza A virus. PLoS Pathog. 4:e11 doi:10.1371/journal.ppat.0040011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gabriel G, et al. 2011. Differential use of importin-alpha isoforms governs cell tropism and host adaptation of influenza virus. Nat. Commun. 2:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garcia-Robles I, Akarsu H, Muller CW, Ruigrok RW, Baudin F. 2005. Interaction of influenza virus proteins with nucleosomes. Virology 332:329–336 [DOI] [PubMed] [Google Scholar]
- 32. Gonzalez S, Zurcher T, Ortin J. 1996. Identification of two separate domains in the influenza virus PB1 protein involved in the interaction with the PB2 and PA subunits: a model for the viral RNA polymerase structure. Nucleic Acids Res. 24:4456–4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gougeon ML, Melki MT, Saidi H. 2012. HMGB1, an alarmin promoting HIV dissemination and latency in dendritic cells. Cell Death Differ. 19:96–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huarte M, Sanz-Ezquerro JJ, Roncal F, Ortin J, Nieto A. 2001. PA subunit from influenza virus polymerase complex interacts with a cellular protein with homology to a family of transcriptional activators. J. Virol. 75:8597–8604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ito Y, et al. 2011. Increased levels of cytokines and high-mobility group box 1 are associated with the development of severe pneumonia, but not acute encephalopathy, in 2009 H1N1 influenza-infected children. Cytokine 56:180–187 [DOI] [PubMed] [Google Scholar]
- 36. Jackson DA, Caton AJ, McCready SJ, Cook PR. 1982. Influenza virus RNA is synthesized at fixed sites in the nucleus. Nature 296:366–368 [DOI] [PubMed] [Google Scholar]
- 37. Jorba N, et al. 2008. Analysis of the interaction of influenza virus polymerase complex with human cell factors. Proteomics 8:2077–2088 [DOI] [PubMed] [Google Scholar]
- 38. Jung JH, et al. 2011. Hepatitis C virus infection is blocked by HMGB1 released from virus-infected cells. J. Virol. 85:9359–9368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kamau E, et al. 2009. Dengue virus infection promotes translocation of high mobility group box 1 protein from the nucleus to the cytosol in dendritic cells, upregulates cytokine production and modulates virus replication. J. Gen. Virol. 90:1827–1835 [DOI] [PubMed] [Google Scholar]
- 40. Kawaguchi A, Nagata K. 2007. De novo replication of the influenza virus RNA genome is regulated by DNA replicative helicase, MCM. EMBO J. 26:4566–4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kosai K, et al. 2008. Elevated levels of high mobility group box chromosomal protein-1 (HMGB-1) in sera from patients with severe bacterial pneumonia coinfected with influenza virus. Scand. J. Infect. Dis. 40:338–342 [DOI] [PubMed] [Google Scholar]
- 42. Kristensen P, Winter G. 1998. Proteolytic selection for protein folding using filamentous bacteriophages. Fold Des. 3:321–328 [DOI] [PubMed] [Google Scholar]
- 43. Labadie K, Dos Santos Afonso E, Rameix-Welti MA, van der Werf S, Naffakh N. 2007. Host-range determinants on the PB2 protein of influenza A viruses control the interaction between the viral polymerase and nucleoprotein in human cells. Virology 362:271–282 [DOI] [PubMed] [Google Scholar]
- 44. Landeras-Bueno S, Jorba N, Perez-Cidoncha M, Ortin J. 2011. The splicing factor proline-glutamine rich (SFPQ/PSF) is involved in influenza virus transcription. PLoS Pathog. 7:e1002397 doi:10.1371/journal.ppat.1002397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee KJ, Perez M, Pinschewer DD, de la Torre JC. 2002. Identification of the lymphocytic choriomeningitis virus (LCMV) proteins required to rescue LCMV RNA analogs into LCMV-like particles. J. Virol. 76:6393–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lopez-Turiso JA, Martinez C, Tanaka T, Ortin J. 1990. The synthesis of influenza virus negative-strand RNA takes place in insoluble complexes present in the nuclear matrix fraction. Virus Res. 16:325–337 [DOI] [PubMed] [Google Scholar]
- 47. Lutz T, Stoger R, Nieto A. 2006. CHD6 is a DNA-dependent ATPase and localizes at nuclear sites of mRNA synthesis. FEBS Lett. 580:5851–5857 [DOI] [PubMed] [Google Scholar]
- 48. Marks JD, Hoogenboom HR, Griffiths AD, Winter G. 1992. Molecular evolution of proteins on filamentous phage. Mimicking the strategy of the immune system. J. Biol. Chem. 267:16007–16010 [PubMed] [Google Scholar]
- 49. Matrosovich M, Matrosovich T, Garten W, Klenk HD. 2006. New low-viscosity overlay medium for viral plaque assays. Virol. J. 3:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsumoto Y, et al. 2012. Bornavirus closely associates and segregates with host chromosomes to ensure persistent intranuclear infection. Cell Host Microbe 11:492–503 [DOI] [PubMed] [Google Scholar]
- 51. Mayer D, et al. 2007. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. J. Proteome Res. 6:672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mitsouras K, Wong B, Arayata C, Johnson RC, Carey M. 2002. The DNA architectural protein HMGB1 displays two distinct modes of action that promote enhanceosome assembly. Mol. Cell. Biol. 22:4390–4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mollica L, et al. 2007. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem. Biol. 14:431–441 [DOI] [PubMed] [Google Scholar]
- 54. Momose F, et al. 2001. Cellular splicing factor RAF-2p48/NPI-5/BAT1/UAP56 interacts with the influenza virus nucleoprotein and enhances viral RNA synthesis. J. Virol. 75:1899–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moncorge O, Mura M, Barclay WS. 2010. Evidence for avian and human host cell factors that affect the activity of influenza virus polymerase. J. Virol. 84:9978–9986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Naito T, et al. 2007. An influenza virus replicon system in yeast identified Tat-SF1 as a stimulatory host factor for viral RNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 104:18235–18240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Naito T, Momose F, Kawaguchi A, Nagata K. 2007. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J. Virol. 81:1339–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Orsi E, Jestin JL. 2003. Optimisation of in vitro enzyme selection. C. R. Chimie 6:501–506 [Google Scholar]
- 59. Pleschka S, et al. 1996. A plasmid-based reverse genetics system for influenza A virus. J. Virol. 70:4188–4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Plotch SJ, Bouloy M, Ulmanen I, Krug RM. 1981. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23:847–858 [DOI] [PubMed] [Google Scholar]
- 61. Poole EL, Medcalf L, Elton D, Digard P. 2007. Evidence that the C-terminal PB2-binding region of the influenza A virus PB1 protein is a discrete alpha-helical domain. FEBS Lett. 581:5300–5306 [DOI] [PubMed] [Google Scholar]
- 62. Read EK, Digard P. 2010. Individual influenza A virus mRNAs show differential dependence on cellular NXF1/TAP for their nuclear export. J. Gen. Virol. 91:1290–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Remy I, Michnick SW. 2006. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat. Methods 3:977–979 [DOI] [PubMed] [Google Scholar]
- 64. Resa-Infante P, Jorba N, Coloma R, Ortin J. 2011. The influenza virus RNA synthesis machine: advances in its structure and function. RNA Biol. 8:207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Resa-Infante P, et al. 2008. The host-dependent interaction of alpha-importins with influenza PB2 polymerase subunit is required for virus RNA replication. PLoS One 3:e3904 doi:10.1371/journal.pone.0003904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ronfani L, et al. 2001. Reduced fertility and spermatogenesis defects in mice lacking chromosomal protein Hmgb2. Development 128:1265–1273 [DOI] [PubMed] [Google Scholar]
- 67. Ruigrok RW, Baudin F. 1995. Structure of influenza virus ribonucleoprotein particles. II. Purified RNA-free influenza virus ribonucleoprotein forms structures that are indistinguishable from the intact influenza virus ribonucleoprotein particles. J. Gen. Virol. 76(Pt 4):1009–1014 [DOI] [PubMed] [Google Scholar]
- 68. Seyedin SM, Kistler WS. 1979. Levels of chromosomal protein high mobility group 2 parallel the proliferative activity of testis, skeletal muscle, and other organs. J. Biol. Chem. 254:11264–11271 [PubMed] [Google Scholar]
- 69. Shapira SD, et al. 2009. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139:1255–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shih SR, Krug RM. 1996. Novel exploitation of a nuclear function by influenza virus: the cellular SF2/ASF splicing factor controls the amount of the essential viral M2 ion channel protein in infected cells. EMBO J. 15:5415–5427 [PMC free article] [PubMed] [Google Scholar]
- 71. Soderberg O, et al. 2006. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3:995–1000 [DOI] [PubMed] [Google Scholar]
- 72. Song MJ, et al. 2004. The DNA architectural protein HMGB1 facilitates RTA-mediated viral gene expression in gamma-2 herpesviruses. J. Virol. 78:12940–12950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stillman EA, Rose JK, Whitt MA. 1995. Replication and amplification of novel vesicular stomatitis virus minigenomes encoding viral structural proteins. J. Virol. 69:2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stott K, Tang GS, Lee KB, Thomas JO. 2006. Structure of a complex of tandem HMG boxes and DNA. J. Mol. Biol. 360:90–104 [DOI] [PubMed] [Google Scholar]
- 75. Sugiyama K, et al. 2009. Structural insight into the essential PB1-PB2 subunit contact of the influenza virus RNA polymerase. EMBO J. 28:1803–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75a. Tafforeau L, et al. 2011. Generation and comprehensive analysis of an influenza virus polymerase cellular interaction network. J. Virol. 85:13010–13018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Takizawa N, Watanabe K, Nouno K, Kobayashi N, Nagata K. 2006. Association of functional influenza viral proteins and RNAs with nuclear chromatin and sub-chromatin structure. Microbes Infect. 8:823–833 [DOI] [PubMed] [Google Scholar]
- 77. Thomas JO, Travers AA. 2001. HMG1 and 2, and related ‘architectural’ DNA-binding proteins. Trends Biochem. Sci. 26:167–174 [DOI] [PubMed] [Google Scholar]
- 78. van Zoelen MA, et al. 2009. Receptor for advanced glycation end products is detrimental during influenza A virus pneumonia. Virology 391:265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vichier-Guerre S, Ferris S, Auberger N, Mahiddine K, Jestin JL. 2006. A population of thermostable reverse transcriptases evolved from Thermus aquaticus DNA polymerase I by phage display. Angew. Chem. Int. Ed. Engl. 45:6133–6137 [DOI] [PubMed] [Google Scholar]
- 80. Vignuzzi M, Gerbaud S, van der Werf S, Escriou N. 2001. Naked RNA immunization with replicons derived from poliovirus and Semliki Forest virus genomes for the generation of a cytotoxic T cell response against the influenza A virus nucleoprotein. J. Gen. Virol. 82:1737–1747 [DOI] [PubMed] [Google Scholar]
- 81. Vreede FT, Chan AY, Sharps J, Fodor E. 2010. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 396:125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang P, Palese P, O'Neill RE. 1997. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 71:1850–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Watanabe T, Watanabe S, Kawaoka Y. 2010. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7:427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Watson M, Stott K, Thomas JO. 2007. Mapping intramolecular interactions between domains in HMGB1 using a tail-truncation approach. J. Mol. Biol. 374:1286–1297 [DOI] [PubMed] [Google Scholar]
- 85. Yanai H, et al. 2009. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462:99–103 [DOI] [PubMed] [Google Scholar]
- 86. Ye Q, Krug RM, Tao YJ. 2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444:1078–1082 [DOI] [PubMed] [Google Scholar]
- 87. Zhang G, et al. 2003. Borna disease virus phosphoprotein represses p53-mediated transcriptional activity by interference with HMGB1. J. Virol. 77:12243–12251 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.