

Abstract
We report on a new anti-influenza virus agent, SA-19, a lipophilic glycopeptide derivative consisting of aglycoristocetin coupled to a phenylbenzyl-substituted cyclobutenedione. In Madin-Darby canine kidney cells infected with influenza A/H1N1, A/H3N2, or B virus, SA-19 displayed a 50% antivirally effective concentration of 0.60 μM and a selectivity index (ratio of cytotoxic versus antiviral concentration) of 112. SA-19 was 11-fold more potent than unsubstituted aglycoristocetin and was active in human and nonhuman cell lines. Virus yield at 72 h p.i. was reduced by 3.6 logs at 0.8 μM SA-19. In contrast to amantadine and oseltamivir, SA-19 did not select for resistance upon prolonged virus exposure. SA-19 was shown to inhibit an early postbinding step in virus replication. The compound had no effect on hemagglutinin (HA)-mediated membrane fusion in an HA-polykaryon assay and did not inhibit the low-pH-induced refolding of the HA in a tryptic digestion assay. However, a marked inhibitory effect on the transduction exerted by retroviral pseudoparticles carrying an HA or vesicular stomatitis virus glycoprotein (VSV-G) fusion protein was noted, suggesting that SA-19 targets a cellular factor with a role in influenza virus and VSV entry. Using confocal microscopy with antinucleoprotein staining, SA-19 was proven to completely prevent the influenza virus nuclear entry. This virus arrest was characterized by the formation of cytoplasmic aggregates. SA-19 appeared to disturb the endocytic uptake and trap the influenza virus in vesicles distinct from early, late, or recycling endosomes. The aglycoristocetin derivative SA-19 represents a new class of potent and broad-acting influenza virus inhibitors with potential clinical relevance.
INTRODUCTION
Influenza A and B viruses are highly contagious respiratory pathogens and the cause of a considerable medical and socioeconomical burden. The annual influenza epidemics and unpredictable yet potentially severe pandemics are the consequence of the continuing variation of these viruses, which is mainly related to the antigenic instability of the viral surface proteins hemagglutinin (HA) and neuraminidase (NA). Currently available drugs for prevention and treatment of influenza virus infections are the M2 ion channel blockers (amantadine and rimantadine) and the neuraminidase inhibitors (oseltamivir and zanamivir) (9). The potential usefulness of amantadine and rimantadine is limited, due to their lack of activity against influenza B virus, the global distribution of amantadine-resistant influenza A viruses, and the occurrence of neurological side effects (11). In clinical practice, the NA inhibitors are preferred, since they are effective against influenza A and B, are well tolerated, and have a higher barrier for drug resistance. Nevertheless, the isolation of oseltamivir-resistant mutants in A/H3N2- and A/H5N1-infected patients receiving this drug and the worldwide spread (during the period 2007 to 2009) of oseltamivir-resistant A/H1N1 strains, even among untreated patients, underline the urgent necessity for novel antiviral drugs to be adequately prepared for future influenza epidemics and pandemics (26, 35).
In humans, influenza presents as an acute infection, causing an abrupt and predominantly respiratory illness with a strong inflammatory component (15). Because of this acute onset, an intervention to block the initial virus entry into the host cell is an attractive antiviral strategy. Initiation of influenza virus infection requires the cooperative binding of multiple HA molecules to terminal sialic acid moieties on cell surface glycoproteins and glycolipids. This interaction may possibly be blocked by multivalent sialic acids, which have been described as potent inhibitors of adenovirus binding to sialic acids (22). A compound for use in an alternative strategy, enzymatic removal of the sialic acid moieties from the host cell receptors, is currently being explored in the form of Fludase (also known as DAS181) (4). After internalization of the bound influenza virions by endocytosis, the acidic pH inside the endosome triggers an extensive and irreversible rearrangement of the HA protein to adopt its fusogenic conformation, resulting in fusion of the viral and endosomal membranes. For most influenza virus strains, the optimal pH to trigger this fusion process is in the range of 5.0 to 6.0, and it is therefore assumed that the virus can be released from the endosomes only after their maturation from the early to the late form (6). However, influenza virus HA mutants which are able to fuse at a pH as high as 6.4 have been previously described (8). Several groups, including ours, have been able to identify small-molecule inhibitors of the acid-induced conformational change of HA. Unfortunately, clinical development of these influenza virus fusion inhibitors has been hampered by their low barrier for resistance selection (largely related to the rapid selection of HA mutants with increased fusion pH) and the group- or subtype-specific antiviral activity (46, 59). An exception is the broad-spectrum antiviral drug arbidol, which has been reported to inhibit HA-mediated fusion by preventing the HA conformational change (28), although its fusion-inhibiting effect may also be related to membrane perturbation (61). HA dependency may be circumvented by targeting the more conserved viral nucleoprotein (NP), as recently proposed by Kao et al. (24). That group identified a compound, named nucleozin, that was shown to cause NP aggregation, thereby preventing nuclear import of the viral ribonucleoproteins (vRNPs). Besides targeting viral factors, alternative approaches focus on inhibition of cellular proteins involved in influenza virus entry, such as PI3K/Akt signaling factors (13) or protein kinase C (PKC) (45).
We previously reported on the chemical synthesis (38, 54) and basic anti-influenza virus activity of a new series of aglycoristocetin derivatives (36). Aglycoristocetin is the aglycone form of the glycopeptide compound ristocetin, which was discovered many years ago (7) and belongs to the same class of antibiotics as vancomycin (Fig. 1). Their antibacterial activity is based on a specific binding interaction with the d-Ala-d-Ala termini of peptidoglycan precursors in the bacterial cell membrane. Although vancomycin is one of the few antibiotics with activity against the methicillin-resistant Staphylococcus aureus (MRSA) pathogen, the increasing incidence of vancomycin-resistant isolates has been a major reason for concern. Fortunately, these resistant isolates remain sensitive to a new class of hydrophobic side-chain-substituted glycopeptides (designated “lipoglycopeptides”) (12). The complex glycopeptide backbone structure, with its characteristic heptapeptide core, permits a variety of structural modifications to fine tune the pharmacological profile of new derivatives. Interestingly, a number of modified glycopeptides have been reported as active against various viruses, i.e., HIV, hepatitis C virus (HCV), and coronaviruses (1, 2, 37). Whereas the structure-activity relationships with respect to these unrelated viruses are clearly divergent, the addition of a hydrophobic substituent on the glycopeptide backbone appears to be a common prerequisite. Their versatility is further illustrated by the observation that the published lead compounds for HIV and coronaviruses were found to act during viral entry (1, 2), whereas a different glycopeptide compound with anti-HCV activity was shown to inhibit a postentry event (37). The study reported here is the first to elaborate on the anti-influenza virus potential of glycopeptide derivatives. We focused on a compound designated SA-19, which contains an aglycoristocetin moiety coupled to a phenylbenzyl-substituted cyclobutenedione and displays consistent activity against a wide range of influenza A and B viruses. Since SA-19 was found to interfere with a step in the virus entry process preceding its nuclear uptake, thorough mechanistic studies were performed to understand its precise mode of action. Our combined data indicate that SA-19 interacts with a cellular component, causing disturbance of the endocytic uptake of the virus.
Fig 1.

Chemical structures of SA-19 and related glycopeptide derivatives. SA-19 is a derivative of aglycoristocetin in which the primary amine group is coupled to a phenylbenzylgroup via a cyclobutenedione group. The identical lipophylic substitution is present in SA-48 (the teicoplanin analogue of SA-19); ERJ-147 (which lacks the chloro substituents on rings C and E of SA-48); and SA-26 (the vancomycin analogue of SA-19). The glycopeptide ristocetin (not shown) differs from aglycoristocetin in having three sugar substituents at the positions marked with an asterisk: d-mannose on ring A, l-ristosamine next to ring C, and the tetrasaccharide d-arabinose–d-mannose–d-glucose–l-rhamnose on ring D.
MATERIALS AND METHODS
Cells and virus strains.
Madin-Darby canine kidney (MDCK) cells and MDCK-SIAT1 cells (MDCK cells stably expressing human 2,6-sialyltransferase) (29) (a kind gift from M. Matrosovich, Marburg, Germany) were cultured as described previously (59). For MDCK-SIAT1 cells, the medium was supplemented with 1 mg per ml G418(Geneticin). Human lung carcinoma A549 cells, quail fibroblasts (QT6), and human embryonic kidney 293T cells (obtained from the American Type Culture Collection [ATCC]) were subcultivated as recommended by the supplier. African green monkey Vero cells were cultured in minimum essential medium (MEM), supplemented with 10% fetal calf serum (FCS) (Integro), 2 mM l-glutamine, and 0.11% sodium bicarbonate.
The human influenza virus strains A/PR/8/34 (A/H1N1) and B/HK/5/72 were obtained from ATCC. The strains A/X-31 (A/H3N2) [A/Aichi/2/68 (H3N2) × A/PR/8/34 (H1N1)] and A/HK/7/87 (A/H3N2) were a gift from J. Neyts (Leuven, Belgium), whereas the following strains were generously provided by R. Fouchier (Rotterdam, The Netherlands): A/FM/1/47 (A/H1N1); A/HK/2/68 and A/Victoria/3/75 (A/H3N2); and B/Lee/40. The A/Ishikawa/7/82 strain was a kind gift from M. Hosoya (Fukushima, Japan) (21). The A/X-31 strain (HA-D1122N; the suffix 1 or 2 denotes the location in the HA1 or HA2 subunit, respectively) was selected in the presence of the fusion inhibitor 4c (59). For these laboratory-adapted strains, virus stocks were prepared in 10-day-old embryonated hen eggs. In addition, two clinical isolates [A/Ned/378/05 (A/H1N1) and B/Ned/537/05; a generous gift from R. Fouchier], which underwent one passage in eggs, were used. The A/Virginia/ATCC3/2009 (A/H1N1) virus (purchased from ATCC) underwent passage once in MDCK cells.
To perform the immunocytochemistry and electron microscopy experiments, the A/X-31 virus was purified and concentrated. After clarification of the allantoic fluid by centrifugation (1,800 × g; 30 min; 4°C), the virus was pelleted by ultracentrifugation (35,000 × g; 3 h; 4°C) in a sucrose-NTE buffer (consisting of 30% sucrose, 0.1 M NaCl, 0.01 M Tris HCl, 0.001 M EDTA at pH 7.4) and resuspended in phosphate-buffered saline (PBS).
Test compounds and antiviral assays.
The chemical synthesis of the glycopeptide derivatives aglycoristocetin, SA-19, and SA-26 has been described before (36), whereas the synthesis of the compounds SA-48 and ERJ-147 is to be published elsewhere. The following reference compounds were included: ribavirin (Virazole from ICN Pharmaceuticals); rimantadine (from Sigma); oseltamivir carboxylate (GS-4071) (a kind gift from T. Cihlar, Gilead Sciences, Foster City, CA); chloroquine (from Sigma); bisindolylmaleimide I (45) (from Calbiochem); nucleozin (24) (from Sigma); 6-methyl-N-(2,8-dimethyl-3-oxo-1-thia-4-azaspiro[4.5]dec-4-yl)imidazo[2,1-b][1,3]thiazole-5-carboxamide (4c) (59) (synthesized by N. Cesur, Istanbul, Turkey); and NMSO3 (25) (a generous gift from G. Wright, Microbiotix, Worcester, MA).
The medium used for virus infections consisted of UltraMDCK medium (Lonza), supplemented with 0.0225% sodium bicarbonate, 2 mM l-glutamine, and 2 μg/ml TPCK (tosylphenylalanylchloromethylketon)-treated trypsin (from Sigma). Anti-influenza virus activity was determined in MDCK cells using a multicycle cytopathic effect (CPE) reduction assay (59). At day −1, MDCK cells were seeded into 96-well plates at 7,500 cells per well. Serial dilutions of the compounds were added together with the influenza virus (multiplicity of infection [MOI], 50 50% cell culture infective doses [CCID50s] per well, corresponding to 0.0004 PFU per cell). After 3 days of incubation at 35°C, the CPE was scored by microscopy and antiviral activity was expressed as the compound concentration producing 50% inhibition of virus-induced CPE (EC50). In addition, the minimum cytotoxic concentration (MCC) of a test compound was defined as the concentration causing minimal alterations in cell morphology. Then, the formazan-based MTS cell viability assay was performed (CellTiter 96 AQueous One Solution Cell Proliferation assay from Promega) and the data expressed as the optical density at 490 nm (OD490) were used to calculate the 50% effective concentration (EC50) and 50% cytotoxic concentration (CC50).
Separate plates containing uninfected MDCK cells were used to determine the compound cytostatic activity. After 72 h of incubation with serial dilutions of the compounds, the cells were counted with a Z1 Coulter Counter apparatus (Beckman Coulter). Data were expressed as the concentration causing 50% inhibition of cell proliferation (IC50).
The amount of virus released in the supernatant was determined in virus yield assays based on titration of infectious virus. MDCK cells were incubated with virus and compounds as described above, and at day 3 postinfection (p.i.), the supernatants were collected. These samples were serially (5-fold) diluted and added to 96-well plates containing 7,500 MDCK cells per well. After 3 days of incubation at 35°C, microscopy was performed to score the viral CPE and the virus titer was calculated by the CCID50 method (42).
Inhibition of A/X-31 replication in different cell lines.
MDCK, MDCK-SIAT1, A549, Vero, and QT6 cells were seeded into 24-well dishes at 125,000 cells per well. After 16 h of incubation at 35°C, the compounds plus virus (A/X-31 at an MOI of 0.0004 PFU per cell) were added. At 8 h p.i., the supernatant was removed and the cells were subjected to RNA extraction with an RNeasy minikit (Qiagen). In order to quantify the negative-sense virus RNA (vRNA), the samples were analyzed by two-step real-time reverse transcriptase PCR (RT-PCR) (59). cDNA synthesis was performed on 0.5 μg of total RNA using Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen) and 500 nM M1-FOR primer. Then, real-time PCR was performed, using M1-FOR and M1-REV primers, the M1 probe and standard, and quantitative PCR (qPCR) MasterMix (Eurogentec) (59). All samples were analyzed in duplicate.
Time-of-addition experiment.
MDCK cells were seeded into 24-well dishes at 125,000 cells per well. After 16 h of incubation at 35°C, influenza virus A/X-31 was added at an MOI of 0.0004 PFU per cell, and the compounds were added at −30 min, 0 h, 30 min, 1 h, 3 h, 5 h, or 8 h p.i. At 10 h p.i., the medium was discarded and total cellular RNA extracts were prepared to quantify the negative (−) vRNA, using the two-step real-time RT-PCR assay as described above.
Virus binding assay.
MDCK cells were seeded into 24-well plates at 250,000 cells per well and incubated for 16 h at 35°C. After 2 h of incubation at 35°C with a compound (SA-19, SA-26, or aglycoristocetin [final concentration, 10 μM] or NMSO3 [25] [final concentration, 200 μM]), the cells were cooled on ice during 15 min and infected with precooled A/X-31 virus at an MOI of 0.04 PFU per cell. Then, the cells were incubated on ice for 1 h, followed by three careful washings with ice-cold UltraMDCK infection medium. Total cellular RNA extraction and two-step real-time RT-PCR quantification of (−) vRNA were performed as described above.
Fusion assays.
A detailed description of the trypsin susceptibility assay can be found elsewhere (59). Briefly, influenza virus A/X-31 (allantoic stock; diluted 1:10 in PBS) was incubated with compound at 37°C for 15 min. The pH was lowered to pH 5.0, and after 10 min of incubation at 37°C, the mixture was neutralized to pH 7.2. Then, samples were incubated with 10 mg per ml trypsin (from Sigma) at 37°C for 1 h, and the reaction was terminated by incubation (20 min at 37°C) with 5 mg per ml soybean trypsin inhibitor (from Sigma). Subsequently, the samples were analyzed by Western blotting using anti-HA1 mouse monoclonal antibody clone InA246 (from Hytest) and a horseradish peroxidase-linked goat anti-mouse immunoglobulin polyclonal antibody (from Dako).
To perform the polykaryon assay (see reference 59 for all details), HeLa cells seeded in 12-well plates were transfected with the pCAGEN-HA (A/X-31) plasmid (derived from Addgene plasmid 11160 [30]) using Lipofectamine 2000 reagent and Opti-MEM I (both from Invitrogen). Two days later, the HA was first cleaved by incubation with TPCK-treated trypsin during 15 min at 37°C. After preincubation with test compound during 15 min at 37°C, the cells were incubated with a pH 5.0 buffer containing the corresponding concentration of test compound. After exactly 15 min of incubation at 37°C, the cells were rinsed, and medium containing 10% FCS was added, followed by 3 h of incubation at 37°C. When indicated, test compound was further added during this syncytium formation stage. Finally, the cells were fixed with 96% ethanol, stained with Giemsa, and examined by microscopy at ×200 magnification.
Confocal microscopy.
MDCK cells were plated on 8-well glass Lab-Tek chamber slides (from Nunc) at 55,000 cells per well and incubated for 16 h at 35°C. The compounds were added to the cells, followed by addition of the ultracentrifugated A/X-31 virus at an MOI of 4 PFU per cell. After 1 h of incubation at 35°C, cells were washed three times with PBS and fixed with 4% formaldehyde (Formaldehyde Ultra Pure; from Polysciences)–PBS for 15 min at room temperature. After two washes and permeabilization with 0.01% Triton X-100 during 10 min at room temperature, the cells were blocked with 10% goat serum (diluted in PBS). Then, the primary antibody (diluted in 10% goat serum–PBS) was incubated with the cells for 1 h at room temperature. After two washes, the secondary antibody, diluted in PBS with 10% goat serum, was added for 1 h at room temperature. After two washes with PBS, the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) during 5 min. The primary antibodies were monoclonal mouse anti-NP IgG2 (from Abcam), rabbit polyclonal anti-early endosome autoantigen 1 (EEA1) (from Abcam), and monoclonal mouse anti-lysobisphosphatidic acid (anti-LBPA) IgG1 (Z-PLBPA; from Echelon). The secondary antibodies used were Alexa Fluor 488- or Alexa Fluor 568-labeled goat anti-mouse IgG2 immunoglobulin G, Alexa Fluor 568-labeled goat anti-rabbit IgG, Alexa Fluor 568-labeled goat anti-mouse IgG1, and Alexa Fluor 633-labeled goat anti-mouse IgG antibody (all from Invitrogen). In double-labeling experiments, the cells were first incubated with a mixture of the two relevant primary antibodies and then with a mix of the two corresponding secondary antibodies.
After mounting of the cells with Prolong Gold Antifade reagent (Invitrogen), the preparations were analyzed on a Leica TCS SP5 confocal microscope (Leica Microsystems) equipped with an acousto-optical beam splitter (AOBS), using an HCX PL APO 63.0× oil immersion lens (numerical aperture [NA], 1.40). Different fluorochromes were detected sequentially using excitation lines of 405 nm (DAPI), 488 nm (AlexaFluor 488), and 561 nm (AlexaFluor 568) or an excitation line of 633 nm (AlexaFluor633), and emission was detected between 410 and 480 nm, between 493 and 565 nm, and between 566 and 670 or between 650 and 720 nm, respectively. For presentation, images were processed by applying a 3-by-3 median filter and mild contrast adjustment.
To achieve transient expression of green fluorescent protein (GFP)-coupled Rab4, Rab5, Rab7, and Rab11 or of yellow fluorescent protein (YFP)-coupled LAMP-1, expression plasmids were used (Rab4, Rab5, Rab7, and Rab11 plasmids were a kind gift from P. Courtoy, Louvain-La-Neuve, Belgium; the LAMP-1 plasmid was plasmid 1816 from Addgene [49]). MDCK cells were seeded in chamber slides as described above. Transfection was carried out with 73 ng plasmid DNA per well and Lipofectamine 2000, following the manufacturer's instructions. After 23 h of incubation at 35°C, virus was added at an MOI of 16 PFU per cell, and at 1 h p.i., fixation, permeabilization, and NP staining were performed, as described above.
To perform the acridine orange staining, the MDCK cells were seeded in 35-mm-diameter glass-bottom culture dishes (MatTek Corporation) at 650,000 cells per dish and incubated for 16 h at 35°C. After treatment of the cells with SA-19, bafilomycin A1, or chloroquine, during 1 h at 35°C, acridine orange was added (at a final concentration of 4 μg per ml) and the cells were analyzed by confocal microscopy. Acridine orange fluorescence was detected after 488-nm excitation in two emission windows: 493 to 560 nm and 590 to 720 nm, respectively.
Experiments with pseudotyped retroviral particles.
GFP-expressing pseudotyped retroviral MMLV particles carrying influenza virus HA and NA, or the vesicular stomatitis virus glycoprotein (VSV-G), were generated as previously published (5), with some modifications. The expression plasmids for A/PR/8/34 HA (pVP-HA) and NA (pVP-NA), which are part of the eight-plasmid reverse genetics system, were generated and kindly donated by M. Kim (Daejeon, South Korea). Human embryonic kidney 293T cells were seeded in 60-mm-diameter dishes (at 700,000 cells per dish), in Dulbecco's modified Eagle's medium (DMEM) with 10% FCS, and grown overnight at 37°C. Cells were transfected using calcium phosphate, with 10 μg pQCXIP-AcGFP (derived from pQCXIP; Clontech) and 8 μg pCGGagPol (56) per dish and either 1 μg pVP-HA plus 1 μg pVP-NA or 2 μg pCF-VSVG (56). After overnight incubation at 37°C, the medium was replaced by 2 ml DMEM. On the next day, the supernatant containing the pseudotyped particles was harvested. The HA was activated by addition of 30 μg/ml TPCK-treated trypsin and 20 min of incubation at 37°C. Finally, the harvests were clarified by centrifugation (10 min at 1,500 × g) and stored at −80°C.
To determine the inhibitory effect of the compounds on pseudovirus transduction, MDCK cells were seeded in 96-well plates at 2,500 cells per well and grown overnight. After removal of the medium and a single wash step with PBS, UltraMDCK infection medium containing the test compound was added to the cells, followed by addition of the pseudotyped virus. After 72 h of incubation, the cells were trypsinized and fixed with paraformaldehyde (final concentration, 3%) and transduction efficiency was finally examined by flow cytometric analysis on a FACSCalibur instrument (BD Biosciences) equipped with a 488-nm argon-ion laser and a 530/30-nm bandpass filter (FL1; detector of GFP).
RESULTS
Activity of glycopeptide derivatives against human influenza A/H1N1, A/H3N2, and B viruses.
The basic anti-influenza virus activity of the glycopeptide derivatives (see Fig. 1 for chemical structures) was evaluated in a CPE reduction assay with two virus strains, i.e., A/HK/7/87 (A/H3N2) and B/HK/5/72. Analysis of the antiviral EC50 data (Table 1) enabled us to further define the structure-activity relationship, initially described by Naesens et al. (36). The most potent compound, SA-19, which consists of aglycoristocetin coupled to a cyclobutenedione and substituted with a phenylbenzyl group (Fig. 1), showed an average EC50 of 0.39 μM. Its concentration causing minimal cytotoxicity (MCC; as determined by microscopy) was 20 μM, yielding a selectivity index (ratio of MCC to EC50) of 51. To determine the inhibitory effect of SA-19 on cell proliferation, a cell-counting assay was performed, and from the results (Table 1) the selectivity index of SA-19 (i.e., ratio of IC50 to EC50) was calculated as 172. Intermediate antiviral activity was observed for unsubstituted aglycoristocetin, which showed an 11-fold-higher EC50 than SA-19. Underivatized ristocetin (containing three sugar substituents; see legend to Fig. 1) was inactive (highest concentration tested, 500 μM). Interestingly, no activity was noted for SA-26, the aglycovancomycin analogue of SA-19, and SA-48, the aglycoteicoplanin analogue. These data indicate that both the aglycoristocetin moiety and the lipophilic substituent are crucial for anti-influenza virus activity. Aglycoristocetin and aglycoteicoplanin contain similar central heptapeptide cores, the structural differences being that (i) aglycoristocetin carries a β-hydroxy on the tyrosine of the E unit, (ii) aglycoristocetin lacks the chloro substituents on the C and E rings present in aglycoteicoplanin, and (iii) the F unit of aglycoristocetin consists of a 3,5-hydroxy-4-methyl phenylglycine, whereas the 4-methyl substituent is missing in aglycoteicoplanin. Of these structural differences, the chloro substituents appear to have the highest negative impact on anti-influenza virus activity, since removal of these chlorines to obtain compound ERJ-147 (Fig. 1) yields a favorable antiviral activity similar to that of SA-19 (EC50 of ERJ-147, 0.61 μM for the A/HK/7/87 virus).
TABLE 1.
Activity in influenza virus-infected MDCK cellsa
| Compoundb | Antiviral EC50c (μM) |
Cytotoxicityd |
|||||
|---|---|---|---|---|---|---|---|
| A/HK/7/87 |
B/HK/5/72 |
MCC (μM) | CC50 (μM) | IC50 (μM) | |||
| CPE | MTS | CPE | MTS | ||||
| Ristocetin | >500 | >500 | >500 | >500 | >500 | >500 | |
| Aglycoristocetin | 4.6 ± 1.4 | 2.5 ± 1.0 | 5.1 ± 1.8 | 5.4 ± 2.1 | 81 ± 16 | 44 ± 6 | |
| SA-19 | 0.36 ± 0.15 | 0.58 ± 0.06 | 0.30 ± 0.10 | 0.33 ± 0.08 | 20 ± 0 | 39 ± 7 | 67 ± 11 |
| ERJ-147 | 0.52 ± 0.13 | 0.70 ± 0.16 | >100 | >100 | 16 ± 0 | 20 ± 6 | |
| SA-48 | >50 | >50 | >50 | >50 | 0.80 ± 0.00 | 6.6 ± 0.1 | |
| SA-26 | >50 | >50 | >50 | >50 | >50 | >50 | |
| Oseltamivir carboxylate | 1.4 ± 0.8 | 2.7 ± 1.5 | 5.8 ± 2.2 | 4.9 ± 1.6 | >100 | >100 | 80 ± 7 |
| Ribavirin | 8.2 ± 1.1 | 6.4 ± 0.9 | 8.8 ± 0.5 | 7.2 ± 0.8 | 100 ± 0 | >100 | 16 ± 2 |
MDCK, Madin-Darby canine kidney cells. Data shown represent the means ± SEM of the results of 2 to 5 independent tests.
See Fig. 1 for chemical structures.
Antiviral activity was expressed as the EC50, defined as the compound concentration producing 50% inhibition of virus replication, as estimated by microscopic scoring of the cytopathic effect (CPE) or by measuring cell viability in the formazan-based MTS assay.
Cytotoxicity was expressed as the minimum cytotoxic concentration (MCC; compound concentration producing minimal changes in cell morphology, as estimated by microscopy); the 50% cytotoxic concentration (CC50; estimated by the MTS cell viability assay); or the concentration causing 50% inhibition of cell proliferation (IC50; determined by cell counting).
Next, the lead compound SA-19 was evaluated against a broad panel of human influenza viruses, including laboratory-adapted strains as well as clinical isolates of A/H1N1, A/H3N2, and B viruses (Table 2). SA-19 was shown to have very consistent activity against all virus strains tested, with an average EC50 of 0.60 μM. The compound was fully active against two amantadine-resistant strains, i.e., the 2009 pandemic A/H1N1 virus (strain A/Virginia/ATCC3/2009), which carries an S31N mutation in M2, and the A/PR/8/34 strain, that has two (V27T and S31N) amantadine resistance mutations in the M2 protein. SA-19 was equally effective against wild-type A/X-31 virus and an HA-D1122N A/X-31 mutant. The latter mutation results in an increased pH of HA-mediated fusion and renders the A/X-31 virus resistant to the fusion inhibitor 4c (59).
TABLE 2.
Antiviral activity in influenza A/H1N1, A/H3N2, and B virus-infected MDCK cellsa
| Strain | CPE reduction assay: antiviral EC50b (μM) |
Virus yield reduction assay: EC99c (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| SA-19 |
Ribavirin |
Oseltamivir carboxylate |
SA-19 | Ribavirin | Oseltamivir carboxylate | ||||
| CPE | MTS | CPE | MTS | CPE | MTS | ||||
| A/H1N1 | |||||||||
| A/PR/8/34 | 0.12 ± 0.03 | ≤0.069 | 10 ± 0 | 13 ± 3 | 8.7 ± 4.2 | 7.3 ± 4.6 | |||
| A/FM/1/47 | 0.30 ± 0.12 | 0.53 ± 0.47 | 11 ± 1 | 13 ± 0 | 2.4 ± 1.2 | ≤2.4 | |||
| A/Ned/378/05 | 0.22 ± 0.10 | <0.051 | 9.5 ± 2.6 | 7.8 ± 3.3 | 0.18 ± 0.04 | <0.20 | |||
| A/Virginia/ATCC3/2009 | 0.087 ± 0.019 | 0.067 ± 0.020 | 8.9 ± 0.0 | 9.4 ± 0.8 | ≥1.8 | ≥1.9 | |||
| A/H3N2 | |||||||||
| A/X-31 | 0.32 ± 0.08 | 0.67 ± 0.30 | 9.3 ± 0.7 | 11 ± 2 | <0.10 | <0.10 | 0.55 ± 0.03 | 10 ± 2 | 0.12 ± 0.06 |
| A/X-31 (HA2-D112N)d | 0.41 | 1.1 | 16 | 25 | 0.056 | 0.082 | |||
| A/HK/2/68 | 1.5 ± 0.5 | 1.9 ± 0.5 | 10 ± 0 | 10 ± 1 | 1.2 ± 0.6 | 5.9 ± 2.8 | |||
| A/Victoria/3/75 | 0.86 ± 0.28 | 0.88 ± 0.26 | 8.3 ± 1.4 | 8.3 ± 2.0 | ≤0.20 | <0.20 | |||
| A/HK/7/87 | 0.36 ± 0.15 | 0.58 ± 0.06 | 8.2 ± 1.1 | 6.4 ± 0.9 | 1.4 ± 0.8 | 2.7 ± 1.5 | |||
| A/Ishikawa/7/82 | 0.77 ± 0.22 | 0.73 ± 0.08 | 9.6 ± 0.6 | 9.5 ± 0.9 | 2.6 ± 1.4 | 1.1 ± 0.4 | |||
| B | |||||||||
| B/Lee/40 | 1.4 ± 0.8 | 0.91 ± 0.49 | 14 ± 7 | 16 ± 4 | ≥1.3 | 1.4 ± 0.9 | |||
| B/HK/5/72 | 0.30 ± 0.10 | 0.33 ± 0.08 | 8.8 ± 0.5 | 7.2 ± 0.8 | 5.8 ± 2.2 | 4.9 ± 1.6 | |||
| B/Ned/537/05 | 1.1 ± 0.6 | 0.74 ± 0.29 | 7.8 ± 2.3 | 6.8 ± 2.5 | 2.5 ± 1.2 | ≤0.34 | |||
MDCK, Madin-Darby canine kidney cells. Data shown represent the means ± SEM of the results of 3 to 5 independent tests.
Antiviral activity was expressed as the EC50, defined as the compound concentration producing 50% inhibition of virus replication, as estimated by microscopic scoring of the cytopathic effect (CPE) or by measuring cell viability in the formazan-based MTS assay.
The EC99 value represents the compound concentration producing a 2-log10 reduction in virus yield. Virus released in the supernatant at 72 h p.i. was quantified by determining the infectious virus titer, using the CCID50 method.
Data are from one individual experiment.
The strong activity of SA-19 was confirmed in virus yield experiments. SA-19 (at a concentration of 0.8 μM) produced a 3.6-log10 reduction in virus titer in the supernatant of A/X-31-infected MDCK cells at 72 h p.i., based on titration of infectious virus by the CCID50 method. The EC99 values (i.e., the compound concentrations producing a 99% reduction in virus titer) were 0.55 ± 0.03 μM for SA-19, 0.12 ± 0.06 μM for oseltamivir, and 10 ± 2 μM for ribavirin (Table 2). These values were very similar to the antiviral EC50s for the A/X-31 virus strain determined by the CPE reduction assay.
SA-19 inhibits influenza virus replication in various human and nonhuman cell lines.
A one-cycle assay was performed to determine the anti-influenza virus activity of SA-19 in A549 (human lung carcinoma) cells, Vero (African green monkey kidney) cells, QT6 (quail fibroblast) cells, MDCK cells, and MDCK-SIAT1 cells (an MDCK transfectant expressing 2-fold-larger amounts of 6-linked sialic acids and 2-fold-smaller amounts of 3-linked sialic acids, compared to parent MDCK cells) (29). All five cell lines were found to efficiently support A/X-31 vRNA synthesis. Fluorescence-activated cell sorter (FACS) analysis of the expression levels of the NeuAcα(2,3)-Gal [NeuAcα(2,3)-galactosidase]-type and NeuAcα (2,6)-Gal-type sialylated receptors (using the fluorescein isothiocyanate [FITC]-labeled Maackia amurensis lectin and Sambucus nigra-I lectin, respectively) demonstrated that all cell lines expressed both types of receptors. Compared to the expression seen with MDCK cells, the expression level of the α (2, 6) receptor type (which is preferred by the A/X-31 virus [53]), was 3-fold lower in QT6 cells, 2-fold lower in Vero cells, and 2-fold higher in MDCK-SIAT1 cells, whereas A549 cells showed the same α (2, 6) expression level as MDCK cells. The A/X-31 virus was able to replicate in all cell lines used, since the increase in vRNA copies between 0 and 8 h p.i. was at least 50-fold in all cases (Fig. 2). In the presence of 10 μM SA-19, viral RNA replication was decreased by 97% to 99% (≤2-log10 reduction) in the four mammalian cell lines tested. SA-19 also proved effective in avian QT6 cells, although in this case the inhibition was less pronounced (90% or a 1-log10 reduction in vRNA).
Fig 2.
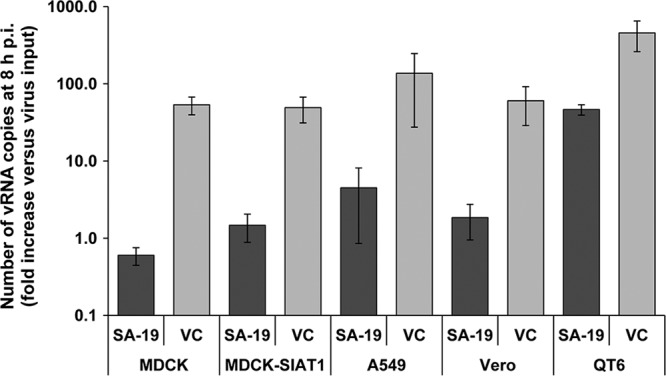
SA-19 inhibits influenza virus replication in different cell lines. Cells were infected with A/X-31 virus, at an MOI of 0.0004 PFU per cell, in the presence of 10 μM SA-19. At 8 h p.i., total cellular RNA was extracted and the number of vRNA copies was determined by real-time RT-PCR. Virus replication is shown as the fold increase in vRNA copy number relative to the virus input at time zero. Data shown represent the means ± standard errors of the means (SEM) of the results of 3 to 5 independent experiments. VC: virus control receiving no compound.
SA-19 acts at an early stage in the replication cycle.
In a one-cycle time-of-addition assay, the antiviral compounds were added at times in a range from −30 min to 8 h p.i. The following reference compounds were included: 4c (an inhibitor of the acid-induced conformational change of the HA, which is crucial for fusion of the viral and endosomal membranes) (59); rimantadine (an M2 ion channel blocker); chloroquine (a weak base that increases the endosomal pH); bisindolylmaleimide I (a protein kinase C inhibitor that prevents nuclear entry of the vRNPs) (45); and ribavirin (which inhibits viral RNA synthesis). At 10 h p.i., vRNA copies in the cells were quantified by RT-PCR. In the absence of compounds, the increase in the number of vRNA copies, relative to the virus added at 0 h, was 150-fold (Fig. 3A). When SA-19 was added at −30 min p.i., vRNA synthesis was strongly inhibited, while the compound lost its antiviral effect when added at 1 h p.i. or later. Partial inhibition was observed under the conditions that included addition of SA-19 at 0 or 30 min p.i. A similar time dependency was observed for 4c, rimantadine, and chloroquine. The curve was slightly shifted to the right for the nuclear entry blocker bisindolylmaleimide I. With ribavirin, ongoing RNA synthesis between 1 h and 8 h p.i. was reflected in a gradual increase in vRNA synthesis as a function of compound addition time.
Fig 3.

SA-19 inhibits virus entry yet has no effect on virus binding or on low-pH-induced HA refolding. (A) Inhibitory effect of SA-19 on vRNA synthesis as a function of the time of compound addition. MDCK cells were infected with A/X-31 influenza virus at an MOI of 0.0004 PFU per cell, and compounds were added at −30 min, 0 h, 30 min, 1 h, 3 h, 5 h, or 8 h p.i. At 10 h p.i., the cells were subjected to RNA extraction for subsequent vRNA analysis by real-time RT-PCR. The vRNA synthesis is presented as the fold increase in the number of vRNA copies at 10 h p.i. relative to the amount of virus particles added at time zero. Data shown represent the means ± SEM (numbers of experiments are shown in parentheses). (B) Effect of SA-19 and related derivatives on influenza virus binding to MDCK cells. After 2 h of incubation with compounds at 35°C, MDCK cells were cooled and allowed to bind the A/X-31 virus (MOI: 0.04 PFU per cell) for 1 h on ice in the further presence of compound. In order to quantify the virus bound to the cells, cellular RNA extracts were prepared and two-step real-time RT-PCR was performed. Virus binding is shown as the vRNA copy number, relative to that in the virus control receiving no compound. Compound concentrations were 10 μM for SA-19, SA-26, and aglycoristocetin and 200 μM for NMSO3. Data shown represent the means ± SEM of 3–6 independent determinations. (C) SA-19 does not inhibit the conformational change of HA at low pH, as demonstrated by trypsin digestion assay. A/X-31 virus was incubated at 37°C for 15 min in the presence of various concentrations of SA-19 or 4c, and the pH was lowered to 5.0. After neutralization, the mixtures were treated with trypsin. The lysates were subjected to Western blot analysis under reducing conditions and using an anti-HA1 antibody. The low-pH-induced conformational change renders the HA sensitive to trypsin digestion, causing disappearance of the HA1 band. (D) SA-19 has no effect on low-pH-induced polykaryon formation in HA-transfected HeLa cells. HeLa cells expressing A/X-31 HA were treated with trypsin to cleave HA0, washed, and incubated during 15 min in the presence of compound. Then, the pH was lowered to 5.0 and the cells were incubated for 15 min at 37°C in the presence of compound. Following syncytium formation for 3 h at 37°C in the presence of compound, the cells were fixed, stained with Giemsa, and examined by microscopy (original magnification, ×200). Representative fields are shown.
SA-19 has no effect on influenza virus binding.
We next performed a binding inhibition assay with influenza A/X-31 virus on MDCK cells, preincubated with compound during 2 h at 35°C, followed by cooling on ice for 10 min. After addition of the virus, the cells were further incubated during 1 h at 4°C, and, after removal of unbound virus, the attached virus was quantified by RT-PCR. Under the conditions in which no compound was added, the total number of bound viral copies was found to be ∼2 × 107 copies per well (corresponding to 70 virus particles per cell), which was 100-fold above the detection limit of our RT-PCR assay. In the presence of NMSO3, a sulfated sialyl lipid (25), influenza virus binding to the cells was reduced by 98% (Fig. 3B). In the case of SA-19, a moderate binding inhibition of 51% was observed. However, a similarly modest inhibition of virus binding was observed with SA-26, the aglycovancomycin analogue which was antivirally inactive in the CPE assay, and aglycoristocetin, which is 11-fold less active than SA-19. The finding that the effect of these compounds on binding was rather limited and uncorrelated to their antiviral activity in the multicycle CPE assay suggests that inhibition of virus binding does not account for the potent antiviral effect of SA-19. Instead, it is likely related to the tendency of these lipophilic glycopeptides to exhibit aspecific protein binding (57), causing some interference with the virus-receptor interaction.
Next, the potential effect of SA-19 on the acid-induced conformational change of HA and subsequent membrane fusion was addressed in an HA trypsin susceptibility assay and a polykaryon assay in HA-expressing HeLa cells. As a control compound, we included the fusion inhibitor 4c, which binds to prefusion HA and prevents its conformational change (59). The trypsin susceptibility assay is based on the exposure of trypsin cleavage sites in the HA1 polypeptide upon acid-induced refolding of the HA protein. Virus that was preincubated with 4c prior to acidification and subsequent trypsin treatment was clearly protected against trypsin digestion, since its HA1 was shown to be intact (Fig. 3C). In contrast, the HA1 band was absent in the presence of SA-19, indicating that SA-19 does not inhibit HA refolding at pH 5. Likewise, SA-19 was found to be inactive in the polykaryon assay (Fig. 3D). HA-expressing HeLa cells were preincubated with compound and then briefly exposed to pH 5 to trigger refolding of HA, resulting in fusion of plasma membranes and syncytium formation. Whereas polykaryon formation was fully inhibited in cells exposed to 4c (Fig. 3D), no inhibition was seen with SA-19, whether added during the preincubation stage or the acid pulse and/or syncytium formation phase. The absence of any inhibitory effect under the latter conditions argues against an aspecific effect of SA-19 on membrane fluidity.
Virus sensitivity to SA-19 remains unchanged after repeated virus passages.
To determine whether influenza virus acquires resistance to SA-19 upon prolonged exposure to the compound, we performed a resistance selection study, using amantadine and oseltamivir as the reference compounds (in a procedure similar to that described in reference 59). These experiments were done with the A/Ned/378/05 (A/H1N1) virus strain, which has a wild-type (i.e., amantadine-sensitive) M2 protein. Its HA sequence was shown to be >99% identical to that of the contemporary A/Rheinland-Pfalz/58/2005 isolate (3a). The A/Ned/378/05 virus underwent passage in MDCK cells in the continuous presence of SA-19, amantadine, or oseltamivir. A control (no-compound) experiment was included to detect any potential changes associated with cell culture adaptation. With each virus passage, the supernatants from selected wells showing 5% to 100% CPE at day 3 p.i. were collected and used to infect fresh MDCK cells, with further addition of compound at gradually increasing concentrations. In the presence of SA-19, no sign of resistance to the compound was observed after 15 passages with SA-19 at up to 10 μM. The resulting virus stock was subjected to plaque purification, and three clones (designated 15-cl1, -cl2, and -cl3) were randomly selected for antiviral testing. Their antiviral EC50s for SA-19, amantadine, and oseltamivir (Table 3) were similar to the values obtained for the A/Ned/378/05 starting virus (Table 2) or the control clones obtained after 15 passages in the absence of compound (Table 3). In the presence of amantadine, virus breakthrough was observed after only 5 passages. Two amantadine-resistant virus clones were isolated, and these viruses displayed a 32-fold increase in antiviral EC50 for amantadine compared to the control clones. In the case of oseltamivir, virus clones collected after 7 passages showed a 324-fold increase in sensitivity to the compound. Neither of the clones showed cross-resistance among the three test compounds. Sequencing revealed that both amantadine-resistant clones that we selected contained a mutation in HA (D181Y or H252Q; numbering according to reference 16), whereas their M2 protein was unchanged. Since both amino acid substitutions produce a charge change, they likely result in an increased fusion pH, as previously described for other HA mutations associated with amantadine resistance (8). Somewhat unexpectedly, the clones selected after 7 passages with oseltamivir contained no mutations in the neuraminidase but possessed two changes in HA: R1561G and G1891R. This agrees with previous reports that in vitro passages with oseltamivir can select for mutations in HA which render the virus less dependent on NA and, hence, confer cross-resistance to oseltamivir and zanamivir (as also observed with our mutant virus) (18). In the three virus clones isolated after 15 passages in the presence of SA-19 (none of which showed phenotypic resistance to any of the test compounds), an E692G change in HA was detected. The control virus that underwent passage in the absence of compound contained no mutations in HA.
TABLE 3.
Antiviral susceptibility of influenza virus after serial passages with SA-19, amantadine, or oseltamivir
| Selecting compound (concn)a | Passage no.-virus cloneb | Mutation(s)c | Antiviral EC50d (μM) of: |
||||
|---|---|---|---|---|---|---|---|
| SA-19 | Amantadine | Oseltamivir carboxylate | Zanamivir | Ribavirin | |||
| SA-19 (10 μM) | 15-cl1 | HA: E692G | 0.29 ± 0.11 | 11 ± 8 | 0.16 ± 0.10 | 0.16 | 7.1 ± 3.0 |
| 15-cl2 | HA: E692G | 0.36 ± 0.02 | 6.5 ± 3.7 | 0.58 ± 0.39 | 0.40 | 9.2 ± 0.3 | |
| 15-cl3 | HA: E692G | 0.45 ± 0.19 | 12 ± 9 | 0.16 ± 0.00 | 0.20 | 9.7 ± 1.1 | |
| Amantadine (500 μM) | 5-cl1 | M2, none; HA, D181Y | 0.39 ± 0.05 | >500 | 0.47 ± 0.04 | 0.47 | 9.8 ± 0.3 |
| 5-cl2 | M2, none; HA, H252Q | 0.37 ± 0.02 | >500 | 0.45 ± 0.19 | 0.26 | 9.9 ± 0.2 | |
| Oseltamivir carboxylate (100 μM) | 7-cl1 | NA, none; HA, R1561G; G1891R | 0.27 ± 0.04 | 189 ± 33 | >100 | >100 | 10 ± 0 |
| 7-cl2 | NA, none; HA, R1561G; G1891R | 0.29 ± 0.06 | 105 ± 24 | >100 | >100 | 12 ± 2 | |
| 7-cl3 | NA, none; HA, R1561G; G1891R | 0.42 ± 0.06 | 198 ± 98 | >100 | >100 | 11 ± 0 | |
| Control (no compound) | 15-cl1 | HA, M2, NA: none | 0.25 ± 0.10 | 11 ± 8 | 0.21 ± 0.05 | 0.10 | 9.1 ± 0.5 |
| 15-cl2 | HA, M2, NA: none | 0.21 ± 0.00 | 30 ± 17 | 0.46 ± 0.05 | 0.40 | 9.9 ± 0.2 | |
| 15-cl3 | HA, M2, NA: none | 0.25 ± 0.10 | 5.4 ± 2.2 | 0.26 ± 0.06 | 0.51 | 10 ± 0 | |
The highest compound concentration used during the selection procedure is shown in parentheses.
The influenza virus isolate A/Ned/378/05 (A/H1N1) was subjected to serial passage in MDCK cells in the presence of increasing concentrations of SA-19, amantadine, or oseltamivir carboxylate. After the indicated number of passages, virus clones were isolated by plaque purification and tested for susceptibility against the five test compounds.
Only the indicated genes supposedly involved in the antiviral mode of action were sequenced. HA numbering is according to reference 16; the suffix 1 or 2 denotes the location in the HA1 or HA2 subunit, respectively.
The EC50 represents the 50% effective concentration, or compound concentration producing 50% inhibition of virus-induced cytopathic effect in MDCK cells. Values represent the means ± SEM of the results of two to three tests (except for the zanamivir data, which are from one experiment).
SA-19 causes intracellular trapping of the virus and prevents its nuclear entry.
To unravel the precise mode of action of SA-19, MDCK cells were infected with A/X-31 influenza virus in the presence of SA-19 or several reference compounds, and, at 1 h p.i., virus was localized by confocal microscopy based on NP staining. In the absence of compounds, the vRNPs released from the endosomes efficiently entered the nucleus within 1 h p.i. (Fig. 4, panels E1 and E2). SA-19 (added at the time of infection at a concentration of 10 μM) produced a virtually complete inhibition of the nuclear entry of vRNP (panels A1 and A2). SA-19-treated cells contained large aggregates which showed an intense NP fluorescence staining. This indicates that the intracellular uptake of influenza virus was not reduced by SA-19. The pattern of NP staining in the presence of SA-19 was different from that obtained with the reference compounds. The recently described nucleozin (panels D1 and D2) triggers aggregation of the viral NP and inhibits its nuclear entry, leading to the formation of a halo of NP surrounding the perinuclear region (24). In the infected cells treated with bisindolylmaleimide I (panels C1 and C2), the NP staining showed a speckled pattern that was more diffuse throughout the cytoplasm. The fusion inhibitor 4c (panels B1 and B2) caused a punctate pattern, mostly unipolar to the nucleus, in what is assumed to be a late endosomal compartment. This NP localization is consistent with the mode of action of 4c and its activity in the fusion assays. Compared to the reference compounds, the trapping by SA-19 was characterized by the formation of larger and bright aggregates. A correlation between this virus-trapping effect and the inhibitory effect on virus replication in the multicycle CPE reduction assay was demonstrated by evaluating the structural analogues of SA-19. While the inactive aglycovancomycin analogue, SA-26 (Fig. 4, panels I1 and I2), and underivatized ristocetin (panels F1 and F2) did not inhibit the nuclear entry of the vRNP, we observed a moderate inhibition with aglycoristocetin (panels G1 and G2). With SA-48 (panels H1 and H2), the aglycoteicoplanin analogue of SA-19, a complete inhibition was observed, but only at cytotoxic concentrations.
Fig 4.

Localization of influenza virus NP in infected MDCK cells treated with (panels A to D) SA-19 or reference compounds or (panels F to I) analogues of SA-19. MDCK cells were infected with A/X-31 influenza virus (MOI: 4 PFU per cell) in the presence of compound and incubated for 1 h at 35°C. After fixation and permeabilization, the cells were stained with anti-NP antibody and Alexa Fluor 488-labeled secondary antibody (green). The nuclei were visualized with DAPI (blue). Bar, 25 μm. See Fig. 1 for the chemical structures of (aglyco)ristocetin, SA-19, SA-48, and SA-26.
Virus trapping by SA-19 is reversed by washing.
MDCK cells were incubated with A/X-31 virus and compound during 1 h at 35°C and then washed three times and incubated for another hour at 35°C. In the cells treated with SA-19, followed by washing, the virus was fully able to resume its nuclear entry process, resulting in exclusively nuclear staining for NP (Fig. 5, panels C1 to C3). The observation that these cells showed a less intense nuclear NP staining compared to the no-compound condition (Fig. 5, panels A1 to A3) is explained by the bulk of NP synthesis between 1 and 2 h p.i. in the cells that received no SA-19.
Fig 5.

Virus trapping by SA-19 is reversed by washing. MDCK cells were infected with A/X-31 influenza virus (at 4 PFU per cell) in the presence of 10 μM SA-19. After 1 h of incubation at 35°C, the cells were washed three times (if indicated) and incubated for an additional hour at 35°C. Then, the cells were fixed, permeabilized, and stained. Influenza virus was detected with anti-NP antibody and Alexa Fluor 488-labeled secondary antibody (green). The nuclei were visualized with DAPI (blue). Bar, 25 μm.
Colocalization studies with markers of early and late endosomes.
Two well-established endosomal markers, i.e., EEA1 and LBPA, were used to identify the early and late endosomes, respectively. Incubation with virus and compounds was performed as described above, and dual labeling was performed for the viral NP (green labeling; Fig. 6) and the early or late endosome markers (red labeling). The EEA1- and LBPA-positive endosomes showed a punctate distribution near the nucleus. The virus captured by the 4c fusion inhibitor was constrained at a similar juxtanuclear location, although colocalization between the anti-NP antibody (green) and the EEA1 or LPBA (red) markers was not apparent. In contrast, SA-19 caused the virus to be trapped at a location that was clearly distinct from that of the EEA-1- or LBPA-positive endosomes. In mock-infected cells, staining for EEA1 and LBPA was unchanged by SA-19 or 4c (data not shown), indicating that neither of the two compounds exerts an aspecific effect on the formation of early and late endosomes.
Fig 6.

Dual staining for influenza virus NP and the endosomal markers EEA1 and LBPA. MDCK cells were infected with A/X-31 virus (at 4 PFU per cell) in the presence of 10 μM SA-19 or 100 μM 4c and incubated at 35°C for 1 h. After fixation and permeabilization, the cells were double labeled. Influenza virus was visualized with anti-NP antibody and Alexa Fluor 488-labeled secondary antibody (green). Analysis was performed with anti-EEA1 and LBPA primary antibodies as early and late endosome markers and Alexa Fluor 568-labeled secondary antibodies (red). The nuclei were visualized with DAPI (blue). Bar, 25 μm.
Next, we transiently expressed GFP-coupled Rab proteins in MDCK cells and then performed virus infection followed by NP staining at 1 h p.i. Rab GTPases are key regulators of the sequential transport steps along the endocytic-recycling pathway, and their detection enables discrimination of the different endosomal stages. Rab5 mediates the formation of early endosomes; Rab4 and Rab11 regulate fast and slow endocytic recycling, respectively; and Rab7 acts upon the degradative (lysosomal) traffic. In addition, we included an expression plasmid for the lysosomal marker lysosome-associated membrane protein-1 (LAMP-1). To avoid the possibility that overexpression of the Rab proteins or LAMP-1 could modify their localization, the amount of Rab or LAMP-1 plasmid DNA used for transfection was kept as small as possible (32). Upon confocal microscopy, the green color representing GFP-coupled Rab4, Rab5, Rab7, or Rab11 or YFP-coupled LAMP-1 (Fig. 7) showed a punctate pattern, indicating that the bulk of the expressed proteins was indeed associated with endosomal vesicles. Virus infection of these transfected cells in the absence or presence of SA-19, followed by NP staining at 1 h p.i. (Fig. 7, red color), again demonstrated that SA-19 inhibits nuclear entry of the virus by causing the formation of intracellular aggregates. This means that overexpression of the endosomal-lysosomal proteins Rab4, Rab5, Rab7, and Rab11 or of LAMP-1 did not change the antiviral effect of SA-19. Strikingly, at least under our experimental conditions, none of the endosomal or lysosomal markers appeared to colocalize with the virus trapped by SA-19.
Fig 7.

Colocalization between influenza virus NP and Rab or LAMP-1 markers of endosomal-lysosomal compartments. MDCK cells were transfected with expression plasmids for GFP-coupled Rab4, Rab5, Rab7, or Rab11 or for YFP-coupled LAMP-1 (green), incubated for 23 h at 35°C, and subsequently infected with A/X-31 virus (at 16 PFU per cell) in the presence or absence of SA-19. After 1 h of incubation at 35°C, the cells were fixed, permeabilized, and stained with anti-NP and Alexa Fluor 633-labeled secondary antibody (red). The nuclei were visualized with DAPI (blue). Bar, 10 μm.
Finally, to exclude the possibility that SA-19 could influence the endosomal pH, staining with acridine orange was performed. Upon entry of acridine orange into acidic endosomes, this dye becomes protonated and sequestered and fluoresces red. This red fluorescence was not observed when endosomal acidification was prevented by the proton pump inhibitor bafilomycin A1 (66) or the weak lysosomotropic base chloroquine (50). In contrast, SA-19 was clearly shown to have no effect on the endosomal pH (Fig. 8).
Fig 8.

SA-19 has no effect on acidification of the endosomes. After treatment of MDCK cells with compound during 1 h at 35°C, acridine orange was added and the cells were subsequently analyzed by confocal microscopy. Acridine orange is an acidotropic dye. Its fluorescence is green at low concentrations, which changes to orange-red at high concentrations (as obtained in the lysosomes in which acridine orange is protonated and sequestered). Acridine orange also intercalates in DNA, resulting in green fluorescence in the nucleus. Upon treatment with the V-ATPase inhibitor bafilomycin A1, lysosomal acidification is completely inhibited, and, hence, the dye's accumulation in the lysosomes is prevented. Chloroquine induces lysosomal swelling and a decrease in lysosomal pH, giving a shift to yellow fluorescence. In the presence of SA-19, the acridine orange staining is identical to that observed in untreated cells. Bar, 25 μm.
Activity of SA-19 against other viruses or pseudotyped lentiviral particles.
As previously reported (36), broad antiviral testing by CPE reduction assays revealed that SA-19 has no marked antiviral effect on other enveloped or nonenveloped RNA viruses, i.e., HIV, coronavirus, vesicular stomatitis virus (VSV), coxsackie B4 virus, respiratory syncytial virus, parainfluenza-3 virus, reovirus-1, Sindbis virus, and Punta Toro virus. When antiherpesvirus activity was evaluated, we noticed that SA-19 has a pronounced antiviral effect on varicella-zoster virus (VZV) (antiviral EC50 of 0.55 μM and selectivity index [ratio of cytotoxic concentration to antiviral concentration] of 180), whereas herpes simplex virus and cytomegalovirus were not markedly inhibited. Thus, a general antiviral effect of SA-19 on diverse viruses (whether naked or enveloped) can be excluded.
We next examined whether SA-19 inhibits the entry of GFP-expressing pseudotyped retroviral particles carrying the influenza virus HA and NA (to allow release of the particles from the producer cells) (62). As a reference, we included particles pseudotyped with the VSV-G fusion protein, which, like HA, requires an acidic pH to adopt a fusogenic conformation and cause fusion of the viral and endosomal membranes (44). MDCK cells were infected with these pseudotype particles in the presence of SA-19 or chloroquine (an inhibitor of endosome acidification), and flow cytometry was performed to quantify the transduction efficiency. Under our experimental conditions, the transduction efficiency of the HA-NA-pseudotyped particles was considerably lower than that of the VSV-G-containing particles, since the percentage GFP positivity in the control cells (receiving no compound) was 10% for the HA-NA- and 70% for the VSV-G-pseudotyped particles. Subset analysis of the fluorescent cell population was thus performed to estimate the inhibitory effect of the test compounds. Both SA-19 and chloroquine produced a marked and dose-dependent inhibition of the transduction by HA-containing pseudoparticles (Fig. 9). Interestingly, both compounds also inhibited the transduction of VSV-G-carrying particles. These data indicate that SA-19 inhibits a step in virus entry that is common to influenza virus and VSV. Furthermore, its effect appears to be directed toward the influenza virus spike protein (HA or NA) or the virus envelope, since the other influenza virion components (such as the M1 capsid protein) were not present in the influenza virus-pseudotyped retroviral particles. Finally, we noticed that the strong effect of SA-19 with respect to VSV entry, as observed in the VSV pseudoparticle assay (which is based on a single VSV-G-mediated entry event), was not detected in a multicycle and highly stringent CPE reduction test (36).
Fig 9.

Inhibition of transduction by GFP-expressing retroviral pseudoparticles carrying influenza virus HA and NA (upper panel) or VSV-G (lower panel). The test compounds (SA-19 at 1, 5, or 10 μM or chloroquine at 50 μM) were added to MDCK cells together with the pseudoparticles. After 72 h, the cells were subjected to flow cytometry for quantification of transduction efficiency. The bars represent the percentages of GFP-positive cells (relative to the untreated control) (data represent averages ± SEM of the results of three tests).
DISCUSSION
Despite the availability of influenza vaccines and two classes of anti-influenza virus drugs, influenza viruses remain the cause of significant morbidity and mortality, posing a serious health threat during seasonal outbreaks as well as periodic pandemics (34). The acute nature of influenza virus infection and the accompanying inflammatory disease make an intervention strategy blocking the early viral entry process particularly attractive (15). The only influenza virus entry inhibitors that are presently available, i.e., the M2 inhibitors amantadine and rimantadine, have a low barrier for resistance selection, which explains their limited value in current influenza virus therapy (9). In the present work, we report on the antiviral activity and mechanism of action of a new influenza virus entry inhibitor, the glycopeptide derivative SA-19. This molecule consists of a central aglycoristocetin moiety that is coupled to a phenylbenzyl-substituted cyclobutenedione. SA-19 shows robust activity against all influenza A and B virus strains tested, with an average EC50 of 0.60 μM, and a 50% cytostatic concentration of 67 μM, yielding a selectivity index of 112. From a comparison with closely related analogues, the following conclusions can be drawn regarding the essential structural elements in SA-19. First, the activity is confined to aglycoristocetin compounds, since the analogues derived from aglycovancomycin and aglycoteicoplanin were found to be inactive. For the latter compound, the lack of anti-influenza virus activity is clearly related to the two chloro functions in the heptapeptide core (cf. the inactive aglycoteicoplanin analogue SA-48 versus the active analogue ERJ-147, in which both chlorine atoms are absent). Second, compared to SA-19, unsubstituted aglycoristocetin displays 11-fold-lower antiviral activity, whereas the parent ristocetin compound is inactive. These data indicate that the anti-influenza virus activity requires removal of the sugar substituents, which is known to increase the lipophilicity of glycopeptide compounds (41). Likewise, the antiviral efficacy is considerably improved by addition of a lipophilic phenylbenzyl side chain, present in the optimized lead compound SA-19. As previously demonstrated in a more detailed SAR study, the neutral charge and steric bulkiness of this lipophilic group are important determinants for anti-influenza virus activity of these aglycoristocetin derivatives (36).
Although the lipophilic substituent of SA-19 causes a marked (11-fold) increase in antiviral potency, the aglycoristocetin moiety appears to be the essential part of the molecule, since unsubstituted aglycoristocetin is also active. Ristocetin (i.e., aglycoristocetin [Fig. 1] with three sugar substituents) is well known for its property of causing thrombocyte aggregation, due to stimulation of the interaction of the von Willebrand factor (vWF) with the platelet GP Ib-IX-V complex (23). This effect is related to the binding of ristocetin to proline-rich peptides containing proline-glycine loops (33). The rhamnose substituent is considered to play an essential role in the interaction of ristocetin with vWF, since removal of this rhamnose was found to eliminate the thrombocyte-aggregating activity (3, 55). Based on this, aglycoristocetin proved superior to ristocetin during clinical development of these antibiotics (7). Since SA-19 is a derivative of aglycoristocetin, and thus lacks this rhamnose moiety, there is no apparent parallel between the interaction of ristocetin with vWF and the anti-influenza virus activity of SA-19. The clinical implication of this is that SA-19 can be expected to have a minimal (if any) platelet-aggregating side effect. This is also in accordance with our studies on a similar series of aglycoristocetin derivatives carrying an hexose substituent at the R position (Fig. 1), since their anti-influenza virus activity was found to be uncorrelated to a platelet-aggregating effect (39).
The broad and robust antiviral activity against all influenza virus A and B strains tested, when examined in human or nonhuman (e.g., avian) cell lines, implies that SA-19 targets a conserved (possibly cellular) factor with a critical role in influenza virus replication. This is consistent with our observation that influenza virus remained fully sensitive to SA-19 after 15 sequential passages in MDCK cells in the presence of SA-19 (at concentrations up to 10 μM). In parallel, virus mutants with marked resistance to amantadine or oseltamivir appeared after only five or seven passages with amantadine or oseltamivir, respectively. The high resistance barrier for SA-19 may be considered an advantage in terms of its potential clinical application. The amantadine-resistant mutants that we selected contained mutations in HA (D181Y and H252Q, both located in the HA stem structure) which probably increase its fusion pH (8). The R1561G and G1891R mutations in the HA of our oseltamivir-resistant virus are located in the globular head of HA and most likely decrease the efficiency of binding of HA to its cell receptor, thereby reducing the neuraminidase dependency for virus release (18). An as-yet-unresolved yet intriguing observation is the E692G mutation in HA that was detected after 15 virus passages in the presence of SA-19. This virus manifested wild-type sensitivity to SA-19, amantadine, and oseltamivir and displayed no obvious replication defects. The Glu692 residue is located in the B-loop of HA that is relieved from the prefusion state in the early steps of fusion activation (65). Since the E692G mutant virus was equally sensitive to SA-19, we anticipate that its appearance may be the result of an accidental mutation. However, in order to definitely exclude a role for this Glu692 residue in the antiviral mode of action of SA-19, additional resistance selection experiments should be performed.
In time-of-addition experiments, SA-19 lost its anti-influenza virus activity when added at 1 h p.i., indicating that its target is situated in the viral entry process. Our confocal microscopy studies demonstrated that, under our experimental conditions, the influenza virus entered the nucleus of MDCK cells within 1 h p.i. The consecutive stages in influenza virus entry consist of (i) HA-mediated binding to sialylated glycan receptors; (ii) endocytosis of the virus particle, presumably by the clathrin-mediated pathway (although other clathrin-independent mechanisms have also been proposed) (51); (iii) virus uncoating (due to M2 proton channel activity) and low-pH-induced rearrangement of HA, resulting in fusion of the viral and endosomal membranes and release of the vRNP in the cytoplasm; and (iv) import of the vRNP into the nucleus (6). To pinpoint which of these events are inhibited by SA-19, appropriate mechanistic assays were performed. First, our virus binding experiments at 4°C provided evidence that SA-19 has only a minor inhibitory effect on receptor-mediated virus binding. The weak activity of SA-19 in both tests is in agreement with our confocal microscopy data, showing that, in the presence of SA-19, cellular uptake of the virus is not visibly inhibited, whereas nuclear entry of the vRNP is completely blocked. More specifically, SA-19-treated cells were found to contain NP-containing aggregates outside the nucleus. This intense intracellular NP staining demonstrates that SA-19 does not diminish the initial uptake of influenza virus at the site of the plasma membrane. A second possible mode of action for SA-19, i.e., prevention of the low-pH-induced conformational change of HA (which is required for fusion of the endosomal and viral membranes) is negated by the lack of any inhibitory effect in the tryptic digestion assay with virus particles or in the polykaryon assay in HA-expressing cells. Furthermore, an aspecific inhibitory effect on endosomal acidification was excluded by acridin orange staining. HA-mediated membrane fusion requires a low-pH-induced and extensive rearrangement of the coiled regions in the HA stem structure and extrusion of the conserved hydrophobic fusion peptide (6). The role of fatty acid moieties attached to the cytoplasmic HA domain is not entirely clear, although it is conceivable that these fatty acids might play a role at a later stage in the membrane fusion process by perturbing membrane lipid organization. Removal of the HA acylation sites has been shown to impair HA-mediated syncytium formation in some studies, whereas others have found contradicting data (60). Taken together, our negative results in the binding and fusion experiments contradict a direct interaction of SA-19 with either the receptor-binding site of HA or its regions involved in membrane fusion (i.e., the fusion peptide, stem structure, or fatty-acid-substituted intracellular domain). However, we do not rule out that SA-19 may interact with a part of HA that is exposed during endosome formation or trafficking. One argument in favor of this hypothesis is the appearance of the E692G mutation in HA after prolonged exposure to SA-19 (though not causing viral resistance to the compound).
In the presence of SA-19, virus replication is arrested by the formation of NP-containing aggregates in the cytoplasm which exclude subsequent entry of the vRNPs into the nucleus. Interestingly, this virus-trapping effect by SA-19 was shown to be reversible, since the virus resumed its nuclear entry when SA-19 was washed away after an initial 1-h incubation with the compound. These data exclude the possibility that SA-19 could cause deviation of the entering virus to an end-stage degradative (lysosomal) compartment. Furthermore, irreversible (covalent) binding of SA-19 to a cellular or viral factor can be eliminated. Finally, the finding that SA-19 is readily removed from the cell by washing suggests that its membrane transport may occur via passive diffusion. The NP staining pattern in SA-19-treated cells (with the characteristic formation of aggregates) was clearly distinct from that seen with unrelated virus entry inhibitors, namely, the fusion inhibitor 4c, which prevents the conformational change of HA at low pH (59); bisindolylmaleimide I, a PKC inhibitor preventing nuclear entry of the vRNP (45); and nucleozin, which causes perinuclear accumulation of NP complexes (24). The NP-containing aggregates formed in the presence of SA-19 are not located in EEA1- or LBPA-positive endosomes. The EEA1 early endosome marker used in our study is specifically associated with the cytoplasmic face of the early endosome membrane (63), whereas the LBPA late endosome marker is present in the complex system of internal membranes within the lumen of the late endosomes (27). Since the virus trapped by SA-19 appears not to be located in early or late endosomes, it may be blocked at (or deviated to) an intermediate stage, such as recycling endosomes or multivesicular bodies (MVBs) (17). To address this issue, we performed virus infection in cells after transient overexpression of Rab4, Rab5, Rab7, Rab11, or the lysosomal marker LAMP-1. Rab GTPases are key regulators of endocytic vesicle formation and trafficking (43). Rab5 and Rab7, which regulate early and late endosome function, respectively, have been implicated in influenza virus endocytic trafficking (52). Strikingly, in virus-infected cells treated with SA-19, we did not observe colocalization between the NP-containing aggregates and any of the four Rab proteins or LAMP-1. It thus appears that SA-19 deviates the virus to a vesicular compartment which is separate from the dynamic endocytic sorting pathway (whether or not clathrin mediated) (47), which the virus depends on for its uptake, low-pH-induced fusion, and endosomal release. The most striking outcome of these colocalization experiments is the lack of costaining with Rab5, since virtually all endocytic pathways seem to merge at this early endosomal regulator (31). The most plausible interpretation is that SA-19 dysregulates the endocytic uptake of the virus at the site of the plasma membrane by capturing the entering virus in distinct vesicles. Support for this hypothesis is given by our studies in HA- or VSV-G-pseudotyped retroviral particles. Sieczkarski and Whittaker (52) demonstrated that, while influenza virus requires trafficking to the late endosomes to undergo its acid-induced fusion, VSV entry proceeds from the early endosomal compartment. This is consistent with the notion that the conformational rearrangement of the VSV-G fusion protein occurs at a higher pH than that of HA. The arrest of VSV-G pseudoparticle transduction by SA-19 should thus occur either before their uptake into or within the early endosomes. The last possibility seems contradicted by our observation that (at least for influenza virus) the arrest does not occur in Rab5- or EEA1-positive vesicles. Thus, a disturbance effect of SA-19 during early endocytic vesicle formation appears more plausible. In this regard, it is relevant that the HA and VSV-G proteins also govern the virus-receptor interaction, and, hence, our HA/NA or VSV-G-bearing retrovirus particles most likely follow the same receptor-mediated endocytic uptake mechanism as authentic influenza or VSV virions. Another virus that was found to be strongly inhibited by SA-19 is VZV (36). Although no mechanistic data for VZV are yet available, we tend to speculate that SA-19 may interfere with the receptor-mediated and low-pH-dependent endocytosis of VZV (14, 19). The inhibitory activity of SA-19 against three unrelated viruses (i.e., influenza virus, VSV, and VZV), combined with its high barrier for selection of resistant influenza virus, provides a strong argument that SA-19 interacts with a cellular component that is critically involved in the endocytic uptake of these heterogeneous viruses.
The chemical structure of SA-19 is reminiscent of that of the new antibiotic oritavancin, a lipoglycopeptide derivative of vancomycin carrying a p-chloro-phenylbenzyl side chain on the sugar moiety (12). Due to its marked amphipathic character, oritavancin is able to specifically interact with anionic phospholipids that are enriched in the bacterial membrane, causing membrane permeabilization. Likewise, in the case of SA-19, the potentiating effect of the lipophilic phenylbenzyl substituent could be related to a cell membrane binding effect. This effect, however, cannot be the principal antiviral mode of action, since the aglycovancomycin analogue carrying the same lipophilic substituent (SA-26) was shown to be antivirally inactive, whereas unsubstituted aglycoristocetin, lacking the lipophilic substituent, did show anti-influenza virus activity. In addition, the finding that SA-19 was devoid of any inhibitory effect in the polykaryon assay contradicts the notion that SA-19 could act as a membrane fluidity modulator at the level of the plasma membrane. To compare, the neutral glycolipid compound fattiviracin (FV-8) was shown to be markedly active in a cell-cell fusion assay in HIV-infected cells, and this inhibition was attributed to its membrane fluidity-modulating effect (20). On the other hand, an interaction of SA-19 with specific cell membrane lipids appears plausible, since it would be in accordance with the high selectivity index of SA-19 in influenza virus-infected cells. Tentative proof for this is provided by our observations in a pyrene excimer fluorescence-based fusion assay (10). When influenza virosomes were preincubated with SA-19, prior to their acidification to promote fusion with erythrocyte ghosts, premature membrane destabilization was seen, since the pyrene excimer fluorescence was found to be decreased before the acidification took place. We therefore hypothesize that the intracellular trapping of influenza virus by SA-19 may (at least in part) be related to a hydrophobic interaction between SA-19 and one or more cell membrane (phospho)lipids. To validate this hypothesis, specialized biochemical techniques with model membranes would be useful, provided that they sufficiently mimic the heterogeneous composition of the outer or inner leaflet of the plasma membrane or of the various endosomal membranes.
As an alternative, we performed transmission electron microscopy on SA-19-treated MDCK cells at 1 h p.i., in an attempt to characterize the NP-containing aggregates formed in the presence of SA-19. Under these conditions, we did not observe any signs of aberrant vacuolization or formation of virus particle aggregates. However, when SA-19 exposure was extended to 24 h, extensive accumulation of multivesicular bodies (MVBs) was seen, independent of whether the cells were virus infected or not (data not shown). Under physiological conditions, MVBs are endocytic sorting organelles intermediate between early and late endosomes which are characterized by the presence of intraluminal vesicles and have different functions related to protein sorting, recycling, transport, storage, and release (40). Several pharmacological agents (including the lipoglycopeptide oritavancin mentioned above) are known to induce accumulation of MVBs, which, in the case of oritavancin, appears to be related to a lipid storage disorder (58). Whether SA-19 has similar effects on lipid storage is presently unknown. In addition, it is not clear whether the MVB accumulation induced by SA-19 (after 24 h of exposure to the compound) is related to its anti-influenza virus activity, since its virus-trapping effect was obtained in a much shorter time, i.e., within 1 h after incubation of the cells with virus and compound.
During the last several years, diverse approaches to design inhibitors of influenza virus entry have been proposed in the literature, namely, interference with HA-mediated receptor binding (4); inhibition of HA-mediated membrane fusion (28, 46, 59); inactivation of the viral envelope (64); nucleoprotein aggregation (24); and inhibition of diverse cellular protein kinases (such as protein kinase C) (45, 48). By comparing SA-19 to several of these reference compounds in our mechanistic studies, we conclude that the antiviral mode of action of SA-19 is clearly distinct from any of these known concepts.
In summary, the lipophilic aglycoristocetin derivative SA-19 represents a broadly active inhibitor of influenza virus entry acting on a step subsequent to binding but preceding nuclear entry of the virus. SA-19 appears to reversibly disturb the endocytic uptake of the virus by the host cell, causing virus trapping in a distinct vesicular compartment that does not contain common markers of early, late, or recycling endosomes. Further exploration of SA-19 or related analogues for their potential use in influenza virus therapy is warranted.
ACKNOWLEDGMENTS
We dedicate this article to the memory of Ferenc Sztaricskai, who passed away during the preparation of the manuscript.
This study was supported by a Ph.D. grant from the KU Leuven (to E. Vanderlinden); the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO no. 9.0188.07); the Geconcerteerde Onderzoeksacties (GOA/10/014); the International Consortium for Anti-Virals (ICAV); and the Impulsfinanciering KU Leuven. The work was supported in part by the TÁMOP 4.2.1/B-09/1/KONV-2010-0007 project. The project is cofinanced by the European Union; the European Social Fund; and the Hungarian Research Fund (K 79126).
We appreciate the dedicated assistance of Leentje Persoons, Wim van Dam, Stijn Stevens, and Frieda De Meyer. We acknowledge the following colleagues for the kind donation of materials: R. Fouchier; M. Kim; M. Matrosovich; and W. Mothes. We are grateful to P. Courtoy for his useful advice and generous donation of the Rab expression plasmids and to J. Paeshuyse for valuable discussions.
Footnotes
Published ahead of print 27 June 2012
REFERENCES
- 1. Balzarini J, et al. 2006. Inhibition of feline (FIPV) and human (SARS) coronavirus by semisynthetic derivatives of glycopeptide antibiotics. Antiviral Res. 72:20–33 doi:10.1016/j.antiviral.2006.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balzarini J, et al. 2003. Antiretroviral activity of semisynthetic derivatives of glycopeptide antibiotics. J. Med. Chem. 46:2755–2764 [DOI] [PubMed] [Google Scholar]
- 3. Bardsley B, Williams DH, Baglin TP. 1998. Cleavage of rhamnose from ristocetin A removes its ability to induce platelet aggregation. Blood Coagul. Fibrinolysis 9:241–244 [DOI] [PubMed] [Google Scholar]
- 3a. Bauer K, Richter M, Wutzler P, Schmidtke M. 2009. Different neuraminidase inhibitor susceptibilities of human H1N1, H1N2, and H3N2 influenza A viruses isolated in Germany from 2001 to 2005/2006. Antiviral Res. 82:34–41 [DOI] [PubMed] [Google Scholar]
- 4. Belser JA, et al. 2007. DAS181, a novel sialidase fusion protein, protects mice from lethal avian influenza H5N1 virus infection. J. Infect. Dis. 196:1493–1499 [DOI] [PubMed] [Google Scholar]
- 5. Bertaux C, et al. 2007. Entry of hepatitis C virus and human immunodeficiency virus is selectively inhibited by carbohydrate-binding agents but not by polyanions. Virology 366:40–50 doi:10.1016/j.virol.2007.04.008 [DOI] [PubMed] [Google Scholar]
- 6. Cross KJ, Burleigh LM, Steinhauer DA. 2001. Mechanisms of cell entry by influenza virus. Expert Rev. Mol. Med. 3:1–18 [DOI] [PubMed] [Google Scholar]
- 7. Crowley BM, Mori Y, McComas CC, Tang D, Boger DL. 2004. Total synthesis of the ristocetin aglycon. J. Am. Chem. Soc. 126:4310–4317 [DOI] [PubMed] [Google Scholar]
- 8. Daniels RS, et al. 1985. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40:431–439 doi:10.1016/0092-8674(85)90157-6 [DOI] [PubMed] [Google Scholar]
- 9. De Clercq E. 2006. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 5:1015–1025 doi:10.1038/nrd2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Jonge J, et al. 2007. Cellular gene transfer mediated by influenza virosomes with encapsulated plasmid DNA. Biochem. J. 405:41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deyde VM, et al. 2007. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J. Infect. Dis. 196:249–257 [DOI] [PubMed] [Google Scholar]
- 12. Domenech O, Dufrene YF, Van Bambeke F, Tukens PM, Mingeot-Leclercq MP. 2010. Interactions of oritavancin, a new semi-synthetic lipoglycopeptide, with lipids extracted from Staphylococcus aureus. Biochim. Biophys. Acta 1798:1876–1885 [DOI] [PubMed] [Google Scholar]
- 13. Ehrhardt C, Ludwig S. 2009. A new player in a deadly game: influenza viruses and the PI3K/Akt signalling pathway. Cell Microbiol. 11:863–871 doi:10.1111/j.1462-5822.2009.01309.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Finnen RL, et al. 2006. Postentry events are responsible for restriction of productive varicella-zoster virus infection in Chinese hamster ovary cells. J. Virol. 80:10325–10334 doi:10.1128/JVI.00939-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fukuyama S, Kawaoka Y. 2011. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr. Opin. Immunol. 23:481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gamblin SJ, et al. 2004. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303:1838–1842 doi:10.1126/science.1093155 [DOI] [PubMed] [Google Scholar]
- 17. Gruenberg J. 2001. The endocytic pathway: a mosaic of domains. Nat. Rev. Mol. Cell Biol. 2:721–730 [DOI] [PubMed] [Google Scholar]
- 18. Gubareva LV. 2004. Molecular mechanisms of influenza virus resistance to neuraminidase inhibitors. Virus Res. 103:199–203 doi:10.1016/j.virusres.2004.02.034 [DOI] [PubMed] [Google Scholar]
- 19. Hambleton S, Steinberg SP, Gershon MD, Gershon AA. 2007. Cholesterol dependence of varicella-zoster virion entry into target cells. J. Virol. 81:7548–7558 doi:10.1128/JVI.00486-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harada S, Yokomizo K, Monde K, Maeda Y, Yusa K. 2007. A broad antiviral neutral glycolipid, fattiviracin FV-8, is a membrane fluidity modulator. Cell Microbiol. 9:196–203 doi:10.1111/j.1462-5822.2006.00781.x [DOI] [PubMed] [Google Scholar]
- 21. Ikeda S, et al. 1994. In vitro and in vivo inhibition of ortho- and paramyxovirus infections by a new class of sulfonic acid polymers interacting with virus-cell binding and/or fusion. Antimicrob. Agents Chemother. 38:256–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johansson SM, et al. 2007. Multivalent sialic acid conjugates inhibit adenovirus type 37 from binding to and infecting human corneal epithelial cells. Antiviral Res. 73:92–100 doi:10.1016/j.antiviral.2006.08.004 [DOI] [PubMed] [Google Scholar]
- 23. Kang M, Wilson L, Kermode JC. 2008. Evidence from limited proteolysis of a ristocetin-induced conformational change in human von Willebrand factor that promotes its binding to platelet glycoprotein Ib-IX-V. Blood Cells Mol. Dis. 40:433–443 [DOI] [PubMed] [Google Scholar]
- 24. Kao RY, et al. 2010. Identification of influenza A nucleoprotein as an antiviral target. Nat. Biotechnol. 28:600–605 doi:10.1038/nbt.1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kimura K, et al. 2000. Antiviral activity of NMSO3 against respiratory syncytial virus infection in vitro and in vivo. Antiviral Res. 47:41–51 doi:10.1016/S0166-3542(00)00091-7 [DOI] [PubMed] [Google Scholar]
- 26. Kiso M, et al. 2004. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet 364:759–765 doi:10.1016/S0140-6736(04)16934-1 [DOI] [PubMed] [Google Scholar]
- 27. Kobayashi T, et al. 1999. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1:113–118 [DOI] [PubMed] [Google Scholar]
- 28. Leneva IA, Russell RJ, Boriskin YS, Hay AJ. 2009. Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol. Antiviral Res. 81:132–140 doi:10.1016/j.antiviral.2008.10.009 [DOI] [PubMed] [Google Scholar]
- 29. Matrosovich M, Matrosovich T, Carr J, Roberts NA, Klenk HD. 2003. Overexpression of the alpha-2,6-sialyltransferase in MDCK cells increases influenza virus sensitivity to neuraminidase inhibitors. J. Virol. 77:8418–8425 doi:10.1128/JVI.77.15.8418-8425.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsuda T, Cepko CL. 2004. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. U. S. A. 101:16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mayor S, Pagano RE. 2007. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 8:603–612 [DOI] [PubMed] [Google Scholar]
- 32. Medts T, et al. 2010. Acute ligand-independent Src activation mimics low EGF-induced EGFR surface signalling and redistribution into recycling endosomes. Exp. Cell Res. 316:3239–3253 doi:10.1016/j.yexcr.2010.09.001 [DOI] [PubMed] [Google Scholar]
- 33. Mohri H, et al. 1997. Synthetic peptide from the V3 loop consensus motif with a potent anti-HIV activity inhibits ristocetin-mediated vWF-GPIb interaction. Peptides 18:1289–1293 doi:10.1016/S0196-9781(97)00205-2 [DOI] [PubMed] [Google Scholar]
- 34. Moscona A. 2008. Medical management of influenza infection. Annu. Rev. Med. 59:397–413 [DOI] [PubMed] [Google Scholar]
- 35. Moscona A. 2009. Global transmission of oseltamivir-resistant influenza. N. Engl. J. Med. 360:953–956 [DOI] [PubMed] [Google Scholar]
- 36. Naesens L, et al. 2009. Anti-influenza virus activity and structure-activity relationship of aglycoristocetin derivatives with cyclobutenedione carrying hydrophobic chains. Antiviral Res. 82:89–94 doi:10.1016/j.antiviral.2009.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Obeid S, et al. 2011. Inhibition of hepatitis C virus replication by semi-synthetic derivatives of glycopeptide antibiotics. J. Antimicrob. Chemother. 66:1287–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pintér G, et al. 2009. Diazo transfer-click reaction route to new, lipophilic teicoplanin and ristocetin aglycon derivatives with high antibacterial and anti-influenza virus activity: an aggregation and receptor binding study. J. Med. Chem. 52:6053–6061 [DOI] [PubMed] [Google Scholar]
- 39. Pintér G, et al. 2010. Click reaction synthesis of carbohydrate derivatives from ristocetin aglycon with antibacterial and antiviral activity. Bioorg. Med. Chem. Lett. 20:2713–2717 [DOI] [PubMed] [Google Scholar]
- 40. Piper RC, Katzmann DJ. 2007. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 23:519–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pitkin DH, Mico BA, Sitrin RD, Nisbet LJ. 1986. Charge and lipophilicity govern the pharmacokinetics of glycopeptide antibiotics. Antimicrob. Agents Chemother. 29:440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 27:493–497 [Google Scholar]
- 43. Rink J, Ghigo E, Kalaidzidis Y, Zerial M. 2005. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122:735–749 doi:10.1016/j.cell.2005.06.043 [DOI] [PubMed] [Google Scholar]
- 44. Roche S, Albertini AA, Lepault J, Bressanelli S, Gaudin Y. 2008. Structures of vesicular stomatitis virus glycoprotein: membrane fusion revisited. Cell. Mol. Life Sci. 65:1716–1728 doi:10.1007/s00018-008-7534-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Root CN, Wills EG, McNair LL, Whittaker GR. 2000. Entry of influenza viruses into cells is inhibited by a highly specific protein kinase C inhibitor. J. Gen. Virol. 81:2697–2705 [DOI] [PubMed] [Google Scholar]
- 46. Russell RJ, et al. 2008. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 105:17736–17741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rust MJ, Lakadamyali M, Zhang F, Zhuang X. 2004. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 11:567–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmolke M, Viemann D, Roth J, Ludwig S. 2009. Essential impact of NF-kappaB signaling on the H5N1 influenza A virus-induced transcriptome. J. Immunol. 183:5180–5189 doi:10.4049/jimmunol.0804198 [DOI] [PubMed] [Google Scholar]
- 49. Sherer NM, et al. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic. 4:785–801 doi:10.1034/j.1600-0854.2003.00135.x [DOI] [PubMed] [Google Scholar]
- 50. Shichiri M, et al. 2012. A novel role for alpha-tocopherol transfer protein (alpha-TTP) in protecting against chloroquine toxicity. J. Biol. Chem. 287:2926–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sieczkarski SB, Whittaker GR. 2002. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 76:10455–10464 doi:10.1128/JVI.76.20.10455-10464.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sieczkarski SB, Whittaker GR. 2003. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 4:333–343 doi:10.1034/j.1600-0854.2003.00090.x [DOI] [PubMed] [Google Scholar]
- 53. Suzuki Y, Kato H, Naeve CW, Webster RG. 1989. Single-amino-acid substitution in an antigenic site of influenza virus hemagglutinin can alter the specificity of binding to cell membrane-associated gangliosides. J. Virol. 63:4298–4302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sztaricskai F, et al. 2006. A new series of glycopeptide antibiotics incorporating a squaric acid moiety. Synthesis, structural and antibacterial studies. J. Antibiot. (Tokyo) 59:564–582 [DOI] [PubMed] [Google Scholar]
- 55. Sztaricskai F, et al. 2007. N-glycosylthioureido aglyco-ristocetins without platelet aggregation activity. J. Antibiot. (Tokyo) 60:529–533 [DOI] [PubMed] [Google Scholar]
- 56. Ulm JW, Perron M, Sodroski J, Mulligan C. 2007. Complex determinants within the Moloney murine leukemia virus capsid modulate susceptibility of the virus to Fv1 and Ref1-mediated restriction. Virology 363:245–255 doi:10.1016/j.virol.2006.09.048 [DOI] [PubMed] [Google Scholar]
- 57. Van Bambeke F. 2004. Glycopeptides in clinical development: pharmacological profile and clinical perspectives. Curr. Opin. Pharmacol. 4:471–478 [DOI] [PubMed] [Google Scholar]
- 58. Van Bambeke F, Saffran J, Mingeot-Leclercq MP, Tulkens PM. 2005. Mixed-lipid storage disorder induced in macrophages and fibroblasts by oritavancin (LY333328), a new glycopeptide antibiotic with exceptional cellular accumulation. Antimicrob. Agents Chemother. 49:1695–1700 doi:10.1128/AAC.49.5.1695-1700.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vanderlinden E, et al. 2010. Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J. Virol. 84:4277–4288 doi:10.1128/JVI.02325-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Veit M, Thaa B. 25 July 2011, posting date Association of influenza virus proteins with membrane rafts. Adv. Virol. 2011:370606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Villalaín J. 2010. Membranotropic effects of arbidol, a broad anti-viral molecule, on phospholipid model membranes. J. Phys. Chem. B 114:8544–8554 [DOI] [PubMed] [Google Scholar]
- 62. Wang W, et al. 2010. A mutation in the receptor binding site enhances infectivity of 2009 H1N1 influenza hemagglutinin pseudotypes without changing antigenicity. Virology 407:374–380 doi:10.1016/j.virol.2010.08.027 [DOI] [PubMed] [Google Scholar]
- 63. Wilson JM, et al. 2000. EEA1, a tethering protein of the early sorting endosome, shows a polarized distribution in hippocampal neurons, epithelial cells, and fibroblasts. Mol. Biol. Cell 11:2657–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wolf MC, et al. 2010. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. U. S. A. 107:3157–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu R, Wilson IA. 2011. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol. 85:5172–5182 doi:10.1128/JVI.02430-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266:17707–17712 [PubMed] [Google Scholar]