

Abstract
To initiate infection, herpesviruses must navigate to and transport their genomes across the nuclear pore. VP1-2 is a large structural protein of the virion that is conserved in all herpesviruses and plays multiple essential roles in virus replication, including roles in early entry. VP1-2 contains an N-terminal basic motif which functions as an efficient nuclear localization signal (NLS). In this study, we constructed a mutant HSV strain, K.VP1-2ΔNLS, which contains a 7-residue deletion of the core NLS at position 475. This mutant fails to spread in normal cells but can be propagated in complementing cell lines. Electron microscopy (EM) analysis of infection in noncomplementing cells demonstrated capsid assembly, cytoplasmic envelopment, and the formation of extracellular enveloped virions. Furthermore, extracellular virions isolated from noncomplementing cells had similar profiles and abundances of structural proteins. Virions containing VP1-2ΔNLS were able to enter and be transported within cells. However, further progress of infection was prevented, with at least a 500- to 1,000-fold reduction in the efficiency of initiating gene expression compared to that in the revertant. Ultrastructural and immunofluorescence analyses revealed that the K.VP1-2ΔNLS mutant was blocked at the microtubule organizing center or immediately upstream of nuclear pore docking and prior to gene expression. These results indicate that the VP1-2 NLS is not required for the known assembly functions of the protein but is a key requirement for the early routing to the nuclear pore that is necessary for successful infection. Given its conservation, we propose that this motif may also be critical for entry of other classes of herpesviruses.
INTRODUCTION
The nuclear pore is the conduit for transport between the cytoplasm and the nucleus, and as such, it represents an obligatory pathway that must be navigated very early after infection by many classes of human viruses (9, 20, 21, 27, 36, 44, 50, 60, 62). For herpesviruses, capsid-tegument assemblies must be transported across the cytoplasm, be targeted to and interact with pores, and undergo structural rearrangements promoting genome exit and transport across the pore to the nucleus, where virus immediate early gene transcription ensues (14). A prime candidate within the virus-encoded proteins for a possible role at this stage of infection is the large tegument protein VP1-2, the product of the UL36 gene in herpes simplex virus (HSV). This protein is conserved across the herpesvirus family and is essential for virus replication (13, 15, 31, 34, 38). It is a complex, multifunctional protein playing crucial and distinct roles at various points in the virus life cycle, including entry, capsid transport, and virion assembly (7, 13, 15, 31, 34, 38, 52, 55). Consistent with a role at the earliest stages of infection, VP1-2 is among a subset of components classed as inner tegument proteins based on tight association with capsids during biochemical extraction and in vivo during entry observed by immunoelectron and confocal microscopy (19, 30, 37, 40, 45, 51, 61). Evidence for a key role for VP1-2 early after infection originated from studies of the temperature-sensitive (ts) mutant virus tsB7 where, at the restrictive temperature, full capsids accumulated at the nuclear pore and virus gene expression was profoundly blocked (5, 31). The defect in tsB7 was mapped specifically to a single amino acid at residue 1453 in VP1-2 (1). Supporting evidence for a role in nuclear entry was obtained from studies of a full deletion mutant of the UL36 gene (52). While this mutant does not assemble virions, the authors examined a potential role for VP1-2 by chemically fusing initially infected cells with surrounding cells and measuring infection in these secondary nuclei. The UL36 deletion mutant was completely unable to infect such nuclei in the fused polykaryocytes, while in parallel, a UL36-positive but UL37-negative virus was able to promote infection.
However, as indicated above, VP1-2 also clearly plays a pivotal role later in infection, in virion assembly. Thus, an additional late defect was identified in strains carrying ts VP1-2, resulting in defective cytoplasmic envelopment and a failure to produce infectious virus (1, 3, 4). Furthermore, analyses of VP1-2 deletion mutants have shown that while C capsids assemble relatively normally in the nucleus, cytoplasmic envelopment and production of infectious virus are completely blocked (13, 15, 52), indicating a critical role in assembly after capsid formation. Current work indicates, consistent with its definition as an inner tegument protein, that VP1-2 is among the first proteins, if not the first, to be recruited onto assembled capsids and is essential for the further recruitment of additional tegument proteins and subsequent envelopment (15, 30, 32, 52). VP1-2 has also been reported to have additional functions, including capsid motility on microtubules, a role that could also be connected to assembly (37, 38, 51, 55, 61). The protein also encompasses a unique class of ubiquitin-specific proteases embedded within its extreme N terminus (28, 54) which, while potentially dispensable for core functions of virus assembly in tissue culture, may play a role in aspects of replication in vivo (24, 35).
However, the essential nature, multiplicity of roles, and very large size of VP1-2 have made it difficult to mechanistically dissect the pathways in which it is involved. In particular, considering the dual roles in assembly and entry, deletion of the protein results in a failure to assemble virions and prevents further analysis of its role in entry. Progress in characterization of specific determinants and a mechanistic framework with which to understand the detailed mechanisms involved requires identification of variants which support virion assembly but remain defective for replication. Such variants represent candidates for identifying a role in virus entry. As part of our aim to understand both herpesvirus early entry mechanisms and the multiplicity of roles of VP1-2, we previously demonstrated that VP1-2 contains an efficient nuclear localization signal (NLS) adjacent to the N-terminal ubiquitin hydrolase domain. A similar motif in the pseudorabies virus (PRV) VP1-2 homologue has also been shown to be functional in nuclear localization (43). In the present study, we constructed and analyzed an HSV mutant containing a short deletion of this NLS. Our data indicate that the short NLS at position 475 in VP1-2, which exhibits strong positional and sequence conservation in the homologues in all herpesviruses (2), is essential for virus spread and, furthermore, that while it is dispensable for virion assembly per se, it is key to normal entry early in infection via routing to the nuclear pore.
MATERIALS AND METHODS
Cells, viruses, infections, and transfection.
Vero and HS30 were grown in Dulbecco's modified minimal essential medium (DMEM; Gibco) containing 10% newborn calf serum (NCS) and penicillin-streptomycin. HS30 cells are derived from Vero cells and contain the UL36 gene (13). Rabbit skin cells (RSC) and RSC-HAUL36 cells were grown in DMEM containing 10% fetal calf serum (FCS) supplemented with nonessential amino acids. RSC-HAUL36 cells are derived from RSC cells and contain the UL36 gene (52). HS30 and RSC-HAUL36 cells were propagated in 0.5 and 10 mg/ml Geneticin, respectively. For some experiments, we used HaCaT cells (human keratinocytes) grown in DMEM containing 10% FCS. Not only do these cells represent a relevant cell type for HSV replication, but in a comparative analysis, we also obtained a severalfold increase in extracellular virus yield for any strain analyzed, and therefore we used these cells to produce wild-type (wt) and mutant viruses. Routine characterization of virus infection was performed exactly as described previously (6).
Construction of a virus mutant expressing VP1-2ΔNLS.
A mutant virus expressing VP1-2 lacking the core NLS was constructed in the background of HSV-1 strain KOS, previously cloned into a bacterial artificial chromosome (BAC) (16). The bacmid was grown in Escherichia coli strain GS1783. The strategy involved recombinational insertion of a cassette for kanamycin resistance adjacent to the mutated sequence, selection for kanamycin resistance, and then recombination excision and counterselection, carried out exactly as described previously (6). PCR was carried out with the primers listed below, using pEPKan-S (57) as the template. Template DNA was digested by incubating the PCR mix with 20 U DpnI for 1 h at 37°C, and the PCR product was purified using a QIAQuick gel extraction kit (Qiagen) after separation in a 0.8% agarose gel. The primers used were as follows: COL414 (forward primer for UL36 NLS deletion), CGTCCCGGTTGGTCGGCACCGGGCACGCTACTCGGCCGGCACCTGGACTCCGCCTTCCAGAGGATGACGACGATAAGTAGGG; and COL417 (reverse primer for UL36 NLS deletion), CCGAAGTCAGGTCTTCGACGCTGGAAGGCGGAGTCCAGGTGCCGGCCGAGTAGCGTGCCCCAACCAATTAACCAATTCTGATTAG.
The PCR product (100 ng) was electroporated into bacteria, and bacteria were selected for resistance to chloramphenicol and kanamycin. Resistant colonies were screened by restriction digestion analysis. Appropriate colonies were grown and induced with I-SceI, and the culture was heat shocked at 42°C for 30 min. Candidate colonies that had lost the kanamycin resistance gene were identified by replica plating, and BAC DNA was isolated and examined by restriction digestion analysis. BAC DNA (100 μg) together with 1 μg pGS403, encoding Cre recombinase, was then transfected into HS30 cells to reconstitute HSV-1 and excise the BAC backbone from the viral genome. Plaques were isolated and multiply plaque purified in complementing RSC-HAUL36 cells. These cells were used for subsequent routine propagation because spontaneous revertants were not produced, while revertants were observed in complementing HS30 cells (though still at a low frequency). The mutant virus was designated K.VP1-2ΔNLS. The presence of a deletion covering 7 residues of the VP1-2 NLS substitution was confirmed by sequencing the first 1,353 nucleotides from purified DNA stocks of mutant or parental virus isolates. No other mutations were observed in this region, and this fragment was sufficient to rescue K.VP1-2ΔNLS by cotransfection with viral DNA and recombination. A revertant of K.VP1-2ΔNLS was generated by cotransfecting DNA from the K.VP1-2ΔNLS mutant virus together with the wt UL36 gene, followed by growth and plaque purification on noncomplementing RSC. The revertant virus was designated K.VP1-2ΔNLS.R. Restoration of the deleted residues was confirmed by sequencing of DNA from the isolated revertant virus. For purification of extracellular virions by density gradient centrifugation, RSC-HAUL36 cells were infected with K.VP1-2ΔNLS or the revertant at a multiplicity of infection (MOI) of 0.001, the medium was collected after complete cytopathic effect (CPE) and clarified by centrifugation (2,000 rpm for 10 min), and virus was pelleted from the supernatant by centrifugation at 19,000 rpm for 90 min. The pellet was then resuspended in phosphate-buffered saline (PBS) (overnight at 4°C) and loaded onto 5 to 15% Ficoll gradients made in PBS, and samples were centrifuged at 25,000 rpm for 2 h. The lower band, containing enveloped virions, was extracted by needle puncture and concentrated (25,000 rpm for 60 min), and the pellet was then resuspended in PBS and frozen.
Purification of viral DNA from K.VP1-2ΔNLS.
For purification of viral DNA, RSC-HAUL36 cells (10 175-cm2 flasks) were infected with virus (MOI of 0.001), and the virus was isolated from the medium after advanced CPE was observed. The medium was first clarified by centrifugation (2,000 rpm for 20 min), and then the virus was pelleted from the supernatant (19,000 rpm for 90 min). The pelleted virus was resuspended gently in 5 ml of buffer (10 mM Tris-HCl [pH 7.6], 1 mM EDTA, 1% SDS) containing 50 μg/ml proteinase K and incubated at 50°C for 4 h, and DNA was extracted by successive rounds of phenol and phenol-chloroform extraction. The final aqueous phase was precipitated by addition of 0.1 volume of 3 M sodium acetate (pH 5.3) and 2 volumes of ethanol, gentle mixing, and incubation at −20°C for 30 min. Finally, the DNA was pelleted (3,500 rpm for 30 min) at 4°C, washed with 70% ethanol, air dried, and then resuspended in 100 to 200 μl of H2O. The DNA was stored at −80°C.
Immunofluorescence studies.
Immunofluorescence analysis was performed exactly as described previously (3), using the following antibodies: ICP4 (1:400) (Virusys), ICP8 (1:2,000) (a kind gift from Roger Everett), pUL37 (1:800) (a kind gift from Prashant Desai), VP16 (LP1 antibody; 1:400) (a kind gift from Tony Minson), VP5 (1:500) (East Coast Bio), polyclonal VP1-2 (anti-VP1-2NT1r; 1:250) (3), monoclonal VP1-2 (CB4; 1:2), and anti-PCM1 (1:50) (Santa Cruz Technologies) antibodies. PCM1 antibody was used for detection of the region around the microtubule organizing center (MTOC) because it gave better staining under conditions for capsid detection than the other markers tested, such as γ-tubulin. Samples were collected at the indicated times, washed with PBS, fixed with methanol (5 min at −20°C), and blocked with PBSB (PBS containing 10% NCS) supplemented for anti-VP1-2NT1r with 0.5 mg/ml human IgG (Sigma) for 1 h at room temperature. After processing, images were collected using Zeiss ×10, ×40 LD, and ×100 (Plan-Apochromat, 1.4-numerical-aperture) lenses. Images for each channel were captured sequentially with a Retiga 2000R camera using Image Pro Plus software. Composite illustrations were prepared using Adobe software. Example images shown are representative of numerous images gathered for each virus and condition. For studies of the presence and subcellular distribution of HSV virions, infections were performed at an MOI of 100. The cells were prechilled at 4°C prior to infection. The monolayers were washed once with cold medium, and the inoculum was added for further incubation at 4°C for 1 h. The inoculum was then removed, prewarmed medium containing 2% NCS was added, and the cells were incubated for different periods at 37°C. The samples were then washed three times with cold PBS, fixed with 3.7% paraformaldehyde (PFA) in PBS for 20 min at room temperature, and permeabilized with 0.1% Triton X-100 prior to incubation with anti-VP5, pUL37, VP1-2 (CB4), or anti-PCM1 antibody. Controls included parallel analysis of infected cells incubated at 4°C without shifting to 37°C. Images for capsid localization were collected with a Zeiss LSM510 confocal microscope with ×40 and ×63 (Plan-Apochromat, 1.4-numerical-aperture) lenses. For studies of viral entry, so as not to be selective, images were collected throughout the cell in multiple z stacks and compiled, and the total images are shown. To demonstrate capsid congregation around the MTOC, e.g., in Fig. 7, single confocal slices were taken using a pinhole size of 2. Channels were captured sequentially using Axiovision acquisition software, and composite illustrations were prepared using Adobe software. For quantitation of capsid localization, a region of interest (ROI) comprising a 2-μm-diameter circle centered around the MTOC (identified by PCM1 staining) was defined for each cell in a field. In an unbiased way, i.e., including all cells with definable MTOCs, the ROIs were then applied to the green channel (VP5 localization), and the pixel intensity was calculated for every ROI after background subtraction. This analysis was performed for approximately 100 cells in several independent fields for the mutant or revertant virus. The results were plotted as the pixel intensity within each MTOC-associated ROI in each cell for each virus.
Fig 7.

Immunofluorescence analysis of K.VP1-2ΔNLSNC virus entry. Cells (RSC) were infected with extracellular virus of either the revertant ΔNLS.R virus or the mutant ΔNLS virus produced from noncomplementing cells. Infection was done at an MOI of 100 for the revertant and with an equivalent amount of the mutant, based on standardization of VP5 in the extracellular purified virus. To help to synchronize infection, cells were washed with cold medium and incubated at 4°C after addition of the inoculum. After 1 h, warmed medium was added, and the cells were further incubated for 2 or 4 h at 37°C prior to fixation and permeabilization. Control samples maintained at 4°C were also examined as controls. Samples were stained for the major capsid protein VP5 and counterstained with DAPI to detect nuclei. The panels show typical examples of many fields examined (using a ×40 objective to capture multiple cells in each field) and present both channels for each virus at the indicated time. The VP5 channel only is shown in panels IV and VIII, highlighting the contrast between the revertant and mutant viruses. Bars, 50 μm. (b) (I) High-resolution confocal section of cells infected with the ΔNLS virus and stained with anti-VP5 (green), anti-PCM1 (red), and DAPI. Bar, 10 μm. The inset (bar, 2 μm) shows the pericentriolar PCM1 accumulations with congregating capsids in the same location, though the capsids do not necessarily directly overlap the PCM1-specific material. (II and III) Higher-magnification images of capsids (VP5) and tegument proteins (pUL37 and VP1-2) colocalizing at the MTOC perinuclear accumulations during ΔNLS virus infection. Bar, 5 μm. (c) Graph showing quantification of MTOC accumulation for revertant and mutant capsids. Defined and unbiased areas of interest were applied around the PCM1 pericentriolar region, and the accumulated density of pixels in the green channel for VP5 in the same areas was calculated. Each vertical bar represents an individual cell. Approximately 130 cells were evaluated for each virus.
Electron microscopy (EM) studies.
For analysis of assembly (see Fig. 5), RSC or Vero cells were infected with wt KOS or K.VP1-2ΔNLS at an MOI of 5 and processed 16 h after infection. For early entry studies (see Fig. 8 and 9), cells were infected at an MOI of 500. Cells were washed with ice-cold PBS, fixed for 30 min at room temperature in 0.5% glutaraldehyde in 200 mM sodium cacodylate buffer, washed in buffer, processed in Epon, and sectioned. Samples were viewed by using an FEI Tecnai G2 electron microscope (FEI, Eindhoven, The Netherlands) with a Soft Imaging System Megaview III charge-coupled device camera. Images were collected at 1,376 by 1,032 by 16 pixels by using analysis version Docu software (Olympus Soft Imaging Solutions, Münster, Germany). For staining of virus particles, suspensions of extracellular pelleted virus were absorbed onto 400-mesh, Formvar-carbon-coated, glow-discharged grids and negatively stained with 1% uranyl acetate.
Fig 5.

Ultrastructural analysis of capsid and virion formation in K.VP1-2ΔNLS-infected cells. RSC were infected in parallel with KOS or the mutant virus K.VP1-2ΔNLS at an MOI of 5, fixed at 16 h postinfection, and processed for thin-section electron microscopy as described in Materials and Methods. (a to c) K.VP1-2ΔNLS; (d to f) KOS. Capsid formation was readily apparent for both the mutant and wt viruses (a and d; small white arrows indicate nucleocapsids, and the large white arrowhead indicates a particle in primary envelopment). Cytoplasmic envelopment was also observed for the mutant and wt viruses (b and e; black arrows indicate wrapping during secondary envelopment). Finally, panels c and f show the presence of assembled virions for the mutant and the revertant, respectively. (g and h) Further examples of assembled extracellular virions, which were readily observed for the mutant virus. In general, little difference in the main ultrastructural features of infection were observed for the wt versus the mutant strain, and all qualitative features of capsid and virion assembly could be observed. Similar results were obtained with the K.VP1-2ΔNLS revertant virus and with different cell lines (Vero and HaCaT cells). Bars, 500 nm.
Fig 8.

EM analysis showing nuclear pore interaction during entry of the ΔNLS revertant virus. Vero cells were infected with the revertant virus K.VP1-2ΔNLS.R at an MOI of 500 PFU/cell. The cells were washed with cold medium and incubated at 4°C after addition of the inoculum. After 1 h, warmed medium was added, and the cells were incubated for 4 h at 37°C and then fixed and processed for thin-section (70 nm) ultrastructural analysis as described in Materials and Methods. N, nucleus; C, cytoplasm. An example section is shown, with the inset illustrating the presence of empty capsids in close proximity to the nuclear pore. Additional insets are shown for additional independent sections illustrating the same features of nuclear pore proximity and empty capsids. Bars, 500 nm in the main image and 250 nm in the insets. Like the case in thin-section analysis, capsids immediately adjacent to the NE represented only a subset of infecting capsids but were readily observed for the wt virus. Total numbers of capsids within 100 nm of the NE are quantitated in Fig. 9c.
Fig 9.

EM analysis of the block in K.VP1-2ΔNLSNC virus entry. In parallel with the analysis shown in Fig. 8, cells were infected with the mutant virus K.VP1-2ΔNLSNC, fixed, and processed. N, nucleus; C, cytoplasm. We readily detected cytoplasmic capsids, frequently in close proximity to the centrioles of the MTOC, but capsids close to the NE were rare. (a and b) Representative field (a) indicating the position within the cell of a higher-magnification inset (b) showing full capsids of the infecting mutant. The single large arrowhead indicates the position of one of the centrioles, with small arrows pointing to 6 capsids accumulated around the MTOC area. (c) Quantification of full and empty capsids from K.VP1-2ΔNLS- and revertant-infected cells. The total perimeter of the NE in 100 cells was scanned for capsids in close proximity to the NE (within 100 nm of the NE), which were scored as empty or DNA-containing capsids. We found only rare examples of capsids close to the NE, and these were not adjacent to pores and contained DNA. Bars, 500 nm.
SDS-PAGE and Western blotting.
Mock-infected or infected cell monolayers were harvested in standard SDS sample buffer. Samples were lysed either by use of a sonicator water bath or by needle shearing using a 25G needle and were boiled for 5 min prior to electrophoresis. Equal cell samples were analyzed in 3 to 8% gradient Tris-acetate gels or 10% Tris-glycine gels and transferred to nitrocellulose membranes. Membranes were blocked in PBS containing 0.5× blocking solution (Li-Cor Biosciences). Primary and secondary antibodies for immunodetection were diluted in PBST (PBS plus 0.1% Tween 20 containing 5% dried milk). Target proteins were visualized using DyLight-conjugated secondary antibodies (1/10,000; Pierce) and developed using a Li-Cor Biosciences Odyssey infrared imaging system. Odyssey v3.0 software was used for quantification, with linearity of measurement being confirmed using a standardization bioassay with serial dilutions of sample inputs. Values for specific viral protein intensities were normalized against actin values. Primary antibodies included anti-ICP4 (1/1,000) (Virusys), anti-ICP8 (1/2,000), anti-VP5 (1/3,000) (East Coast Bio), anti-hemagglutinin (anti-HA; 1/10,000) (Invitrogen), anti-pUL37 (1/10,000) (a gift of P. Desai), anti-pUL25 (1/1,000) (a gift of V. Preston), LP1, anti-VP16 (1/4,000), anti-VP1-2 (anti-NT1; 1/2,000), anti-gB (1/10,000), anti-gD (1/10,000) (a gift of G. Cohen), anti-pUL6 (1/1,000) (a gift from Arvind Patel), and monoclonal anti-actin (1/500) (Sigma) antibodies.
RESULTS
Growth characteristics of K.VP1-2ΔNLS.
We previously identified a short, highly conserved basic motif in VP1-2, embedded within an otherwise poorly conserved region at the N terminus of the protein (Fig. 1a). We demonstrated that this motif could function as an efficient nuclear localization signal (2). Further complementation assays with a mutant VP1-2 protein lacking the NLS indicated that the NLS was important for virus growth. However, definitive elucidation of its role requires the analysis of a virus lacking the motif. Therefore, we constructed a mutant HSV KOS strain containing a short deletion of the 7 amino acids which provided the core NLS function (see Materials and Methods). Anticipating that this determinant would be important for virus growth, we isolated and plaque purified the mutant virus on complementing cell lines (either HS30 or RSC-HAUL36) containing the wt gene for VP1-2. The mutant replicated on the complementing cells, purified virus stocks were obtained, and the deletion was confirmed by sequencing across the UL36 gene. This virus, designated K.VP1-2ΔNLS, replicated only in complementing cell lines, with plaque formation roughly comparable to that of the KOS wt parental virus (Fig. 1b and c). In striking contrast, in parallel assays with noncomplementing Vero cells, absolutely no plaque formation was observed, and plaque-forming efficiency was decreased by at least 7 orders of magnitude compared to that on complementing cells (Fig. 1b and c). Identical results were obtained by comparing plaque formation in complementing RSC-HAUL36 cells to that in RSC (data not shown). Basically, no plaques of any size were observed in Vero cells, RSC, Hep2 cells, or any other cell type tested, though CPE was observed with high levels of virus in the inoculum. We next compared replication by use of multistep growth curves for complementing versus noncomplementing cell lines, assaying yields on complementing cells. Consistent with the lack of plaque formation, we observed a virtually complete absence of production of infectious virus (Fig. 1d).
Fig 1.

Deletion of VP1-2 abolishes plaque formation. (a) Schematic summary of VP1-2, indicating the N-terminal USP domain, the highly basic NLS motif contained within a poorly conserved region, the large central region (gray), and a C-terminal conserved region (blue). Regions relevant to interactions with VP16, pUL37, and pUL25 are also indicated (10, 15, 30, 34, 41, 47, 59). (b) Serial dilutions of K.VP1-2ΔNLS were examined for plaque formation in complementing HS30 cells versus normal Vero cells. (c) Quantitative analysis of plaque-forming efficiency of the mutant versus wt KOS in Vero and HS30 cells. Plaque formation was diminished by at least 7 logs and was essentially undetected in Vero cells compared to HS30 cells. (d) Comparison of the wt and mutant strains by multistep growth curves after low-MOI infection (0.01 PFU/cell, based on titers on complementing cells) of Vero (solid symbols) or HS30 (open symbols) cells. Yields were titrated on HS30 cells. (e) Comparison of wt and mutant strain replication by single-step growth curves after high-MOI infection (5 PFU/cell, based on titers on complementing cells) of Vero or HS30 cells.
We also tested growth after high-multiplicity infection of both Vero versus HS30 cells and RSC versus RSC-HAUL36 cells, with virus yields titrated on the complementing cells. The results were similar in both situations and are shown in Fig. 1e for Vero versus HS30 cells. Growth of the KOS parental virus was approximately the same in both cell types. For K.VP1-2ΔNLS, while replication kinetics and yields were similar to those of the parental virus in HS30 cells, yields in the noncomplementing Vero cells were substantially delayed and reduced by an order of 3 to 4 logs (Fig. 1e). Notwithstanding this reduction, it was clear that after the eclipse phase, infectious virus was produced from noncomplementing cells after a high-MOI infection and could infect and form plaques in complementing cells but not noncomplementing cells. Similar results were obtained by comparing RSC and RSC-HAUL36 cells (data not shown).
To ensure that the profound phenotype of the mutant was due to the NLS deletion, we rescued K.VP1-2ΔNLS with wt UL36, isolated a revertant termed K.VP1-2ΔNLS.R, and confirmed the restoration of the 7 basic amino acids by sequencing of purified viral DNA. The revertant could now propagate and form plaques on any noncomplementing cell type. A comparison of growth on noncomplementing RSC, demonstrating complete restoration of plaque formation, is shown in Fig. S1a and b in the supplemental material, with identical plaque-forming efficiencies on noncomplementing and complementing cells. Plaque size for the K.VP1-2ΔNLS.R revertant was also comparable with that for the parental wt KOS strain (see Fig. S1c). Protein expression, exemplified by ICP4 and ICP8 expression, was restored in the revertant, demonstrating progressive accumulation after low-MOI multistep replication, unlike the virtually complete block for the mutant (see Fig. S1d). Taken together, the observation of complementation in cells containing UL36 and the reversion of phenotype by restoration of the basic motif confirm that this 7-amino-acid deletion of the NLS was the basis of the profound defect in replication and spread.
K.VP1-2ΔNLS amplified on complementing cells (which we term K.VP1-2ΔNLSC for ease of future reference) clearly entered the complementing cells and spread. We therefore wished to examine whether although K.VP1-2ΔNLSC could not form plaques at all, it could enter noncomplementing cell types, with the defect in plaque formation being in some later aspect of propagation. To examine this, we used serial dilutions on Vero cells similar to those used in the plaque assays (Fig. 1), but we assayed the cells by immunofluorescence to detect virus protein expression at the cellular level. We observed that after 2 to 3 days, when normal plaques of wt KOS had spread and contained hundreds of infected cells, single antigen-positive cells were observed for K.VP1-2ΔNLSC (Fig. 2a, arrows), but these cells showed no progression of infection to surrounding cells. Identical results showing bright antigen-positive single cells were obtained for a number of virus proteins, demonstrating firstly their expression and, at the same time, a lack of virus spread. Identical results showing initial single infections and no spread were obtained on a number of standard cell types, including RPE, HFF, and HaCaT cells (data not shown).
Fig 2.

Initial infection of single cells by K.VP1-2ΔNLS and failure of the virus to spread. (a) Vero cells were infected with K.VP1-2ΔNLS or wt KOS at an MOI calculated to infect 1/500 cells. Monolayers were examined for virus proteins after 3 days by immunofluorescence. Green, ICP4; red, pUL37; blue, nuclear counterstain. For the wt virus, plaques had obviously grown and contained numerous antigen-positive cells. In contrast, for the mutant, single antigen-positive cells were observed (both antigens were detected, though the red channel obscures the green in this merged example), but infection was never detected and did not progress in surrounding cells. (b) To examine the induction of expression of wt VP1-2 during propagation in complementing cells, RSC-HAUL36 cells were infected with K.VP1-2ΔNLS at an MOI of 5 (based on titers on HS30 cells), and samples were harvested at the indicated times and examined for expression of endogenous wt VP1-2 by virtue of the HA tag, of total VP1-2 by using anti-VP1-2, or of the major capsid protein VP5. Extracellular virus (also see Fig. 5) from K.VP1-2ΔNLS and K.VP1-2ΔNLS.R (Rev), amplified in complementing RSC-HAUL36 cells and purified by Ficoll density gradient centrifugation, was analyzed to assess the incorporation of endogenous VP1-2 into the virion particles produced in these cells in relation to that of VP5 (lanes 10 and 11).
Considering the known recruitment of VP1-2 to virions, the reasonable interpretation of these results is that when K.VP1-2ΔNLSC is propagated in complementing cells, the wt VP1-2 protein from the cell line is recruited into virions, which can then infect noncomplementing cells, but in which further progression is profoundly blocked at one or more stages (also see below). To confirm the recruitment of wt VP1-2, we analyzed the expression of wt VP1-2 in the complementing cells, in this case RSC-HAUL36 cells, during infection with K.VP1-2ΔNLSC. This was made possible because the wt VP1-2 protein in this line contains an HA epitope tag enabling selective detection (52). The results show that while mock-infected RSC-HAUL36 cells expressed no detectable VP1-2.HA, infection with K.VP1-2ΔNLSC induced the endogenous protein, resulting in detectable levels from 12 h onwards (Fig. 2b). We further analyzed gradient-purified extracellular virus particles of K.VP1-2ΔNLSC (see below), which demonstrated the presence of HA-tagged wt VP1-2 in the virions (Fig. 2b, lanes 10 and 11). These data confirm that, as expected, complementation is due to the incorporation of wt VP1-2 during propagation of K.VP1-2ΔNLSC. It is possible that in complementing cells, mutant VP1-2 is recruited to virions in addition to the wt protein, but we cannot discriminate between the two proteins. The ΔNLS virus propagates in complementing cells, while there is no spread at all in noncomplementing cells. In the following series of experiments, we addressed the key issues of whether particles were assembled in noncomplementing cells and whether they contained the mutant VP1-2 protein (which in noncomplementing cells could only be mutant VP1-2), and if these events occurred, what was the nature of the profound block to virus spread.
Analysis of protein expression in K.VP1-2ΔNLS-infected cells.
To that end, we first examined various parameters of infection in noncomplementing cells, comparing K.VP1-2ΔNLSC with the revertant virus K.VP1-2ΔNLS.R during high-MOI infection (based upon titers obtained on complementing cells). Examining virus protein synthesis at 2- to 4-h intervals for 24 h, we observed virtually normal kinetics and accumulation for representative immediate early, early, and late virus proteins, including in the latter class those proteins which have been shown to interact with VP1-2 (Fig. 3a). No major differences were observed for a number of additional representative proteins, including VP5, pUL37, pUL25, and the glycoprotein gD. For VP1-2 itself, we noted a modest decrease in levels compared to those for infection with the revertant, though this would not be likely of itself to explain the profound phenotype in replication (also see below). We also noted, in a more refined analysis of the early part of infection with sampling every hour to 12 h, that there appeared to be a slight delay in accumulation of ICP4, although again this did not appear to have a significant effect on, e.g., accumulation of ICP8 (Fig. 3b) or other late proteins, as indicated above.
Fig 3.
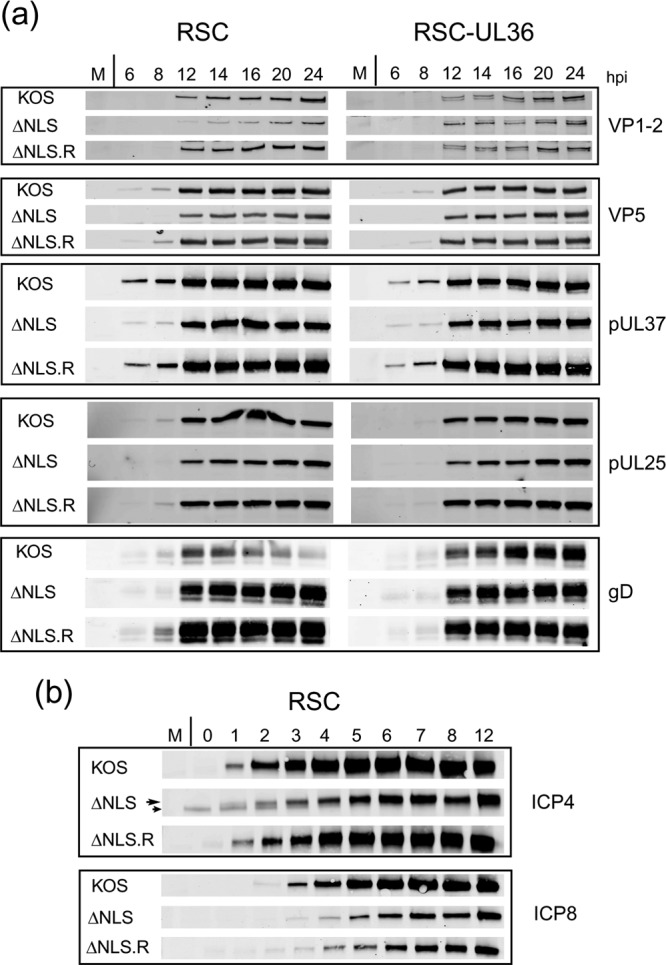
Comparison of protein expression during infection by K.VP1-2ΔNLS and its revertant. (a) RSC or RSC-HAUL36 cells were infected in parallel with the mutant virus K.VP1-2ΔNLS, the revertant virus K.VP1-2ΔNLS.R, or wt KOS virus at an MOI of 5. Samples were harvested at the indicated times, and expression of a series of delayed early or late proteins, i.e., VP5, pUL37, UL25, gD, and VP1-2 itself, was examined by Western blotting using fluorescent antibodies and quantitative detection as described in Materials and Methods. (b) Same as panel a, but examining immediate early (ICP4) and delayed early (ICP8) proteins every hour for 12 h. Some reduction in the abundance of ICP4 (long arrow) was observed, though there was little difference in the amount of ICP8. (A nonspecific band migrating just below ICP4 was occasionally observed, as indicated by the short arrow.) The parental KOS strain exhibited slightly increased expression of both proteins, estimated by quantitative analysis as a 2- to 4-fold increase over expression by the ΔNLS and ΔNLS.R viruses.
Normal localization of candidate virus proteins.
We also analyzed the localization of candidate proteins after infection of noncomplementing cells by using immunofluorescence analysis. Typical results obtained at 16 h postinfection are shown in Fig. 4. Generally, expression and localization of proteins, including VP5, VP16, and pUL37, could not be differentiated between K.VP1-2ΔNLSC and the revertant virus (Fig. 4a). VP5 and VP16 were observed to have nuclear patterns, with accumulation in replication compartments with additional cytoplasmic VP16, as previously reported. pUL37 was also observed in a cytoplasmic pattern that could not be differentiated between the mutant and revertant viruses. For VP1-2 itself, we previously reported on its localization in infected Vero cells, showing that although the protein contains an NLS, the majority of it is found in the cytoplasm, though with a discernible minor population in the nucleus (2). Interestingly, in the current work with RSC, we found little evidence for a population of nuclear VP1-2, detecting mainly a cytoplasmic pattern that was frequently perinuclear or aggregated, though the comparatively rapid rounding up of infected RSC made localization studies more difficult (Fig. 4b). Nevertheless, we found little difference in localization of VP1-2 by comparing infections with the mutant and revertant viruses. To further examine whether we could detect any difference in VP1-2, we also examined its localization in infected Vero cells (Fig. 4b). In this case, while the majority of the protein was in the cytoplasm, frequently in large perinuclear puncta, we could now detect a population of nuclear VP1-2 as previously reported. Indeed, a discernible difference was observed, with K.VP1-2ΔNLS exhibiting little detectable nuclear VP1-2. This result serves to illustrate a potential phenotype of the NLS deletion on de novo-synthesized VP1-2, at least in cells where nuclear VP1-2 can be detected. However, such a difference was not readily discernible in RSC or the other cell types analyzed, such as HaCaT cells (data not shown), in which the wt virus could clearly replicate normally but without significant levels of nuclear VP1-2. We do not know the reason for the cell type difference in VP1-2 localization, but possible explanations include differences in VP1-2 cleavage or nonspecific breakdown in different cells. A comprehensive analysis of the localization of all virus-encoded proteins, ultimately aimed at addressing virion formation in K.VP1-2ΔNLS, is beyond the scope of this work and of tractability. However, within the limits for these types of analyses, the results indicate normal expression levels and localization for a series of representative proteins. Moreover, such a comprehensive analysis is rendered unnecessary in the light of additional results described below.
Fig 4.
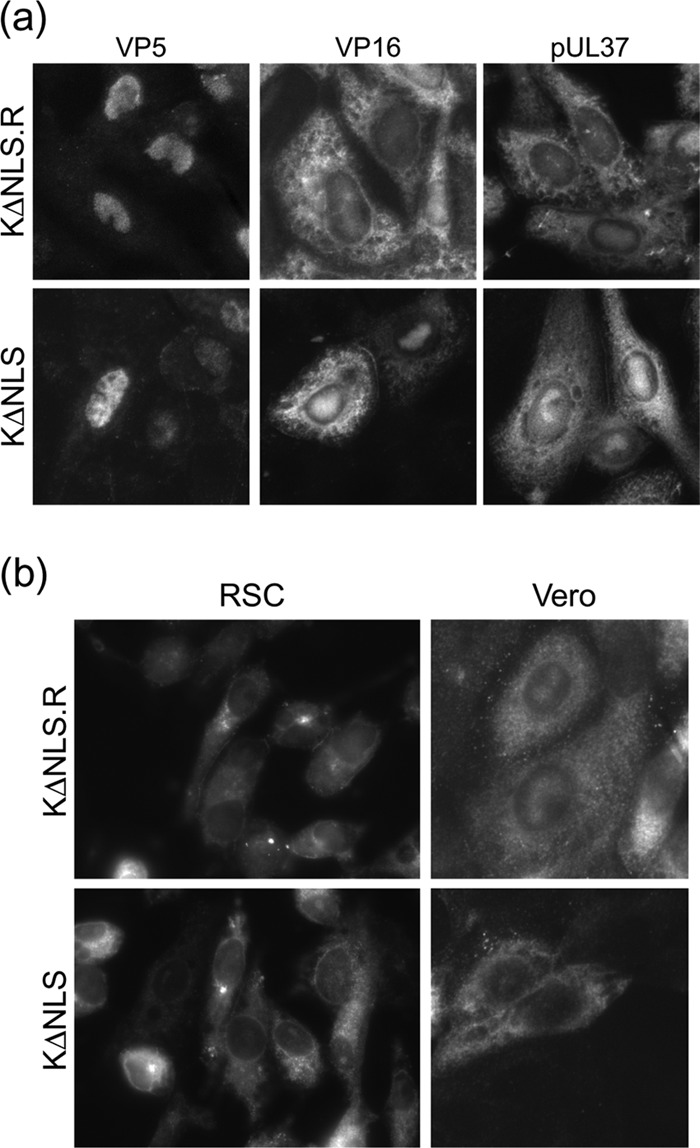
Comparison of protein localization during infection by K.VP1-2ΔNLS and wt KOS. (a) Vero cells were infected with KOS or the mutant virus K.VP1-2ΔNLS at an MOI of 5. Cultures were fixed after 16 h and processed for immunofluorescence as described in the text. Patterns of localization discussed in the text were similar for both the wt and mutant viruses. (b) RSC or Vero cells were infected and processed as described for panel a and were stained for VP1-2 localization.
Virion assembly during infection by K.VP1-2ΔNLS.
Based on the results described above, the profound defect in K.VP1-2ΔNLSC infecting noncomplementing cells clearly occurs at a stage after initial infection. Considering the observation of normal protein synthesis, at least for all candidates examined, including late proteins, the next key question was whether capsids and virions would be assembled at all during infection with the mutant, with the defect being at some stage downstream of this, or whether the deletion of the motif in VP1-2 prevented some late stage of assembly itself. To address this, we first performed EM analyses of noncomplementing cells infected with the mutant or with the wt virus. The results (Fig. 5) generally indicate little discernible difference between the mutant and wt viruses. Importantly, formation of full capsids was observed in the nuclei of cells infected with K.VP1-2ΔNLS (Fig. 5a, arrows) and the wt virus (Fig. 5d, arrows). Cytosolic capsids were also observed, either as free capsids or in membrane-wrapping intermediates, examples of which are shown (Fig. 5b and e, arrows). Finally, enveloped particles were also observed at the cell surface for both K.VP1-2ΔNLS and the wt virus (Fig. 5c and f). Additional examples showing readily detectable cell surface or extracellular enveloped virions for the K.VP1-2ΔNLS mutant are shown in Fig. 5g and h. Over the course of this work, we could detect no significant difference in capsid or enveloped particle formation in the nucleus and cytoplasm between the mutant and wt viruses (see additional data below). Clearly, these data indicate that the profound defect in ΔNLS propagation is not due to a defect in the known requirement for VP1-2 for virion assembly per se. They also demonstrate that the deletion did not affect the global folding and functioning of the protein, since VP1-2 is known to be essential for assembly and for multiple interactions necessary for this complex activity. Given that we could now observe virion formation and extracellular virus after infection by K.VP1-2ΔNLSC in noncomplementing (i.e., UL36 negative) cells, we termed such virions K.VP1-2ΔNLSNC.
Extracellular virion formation by K.VP1-2ΔNLS.
With the observations of a profound defect in spread after low-multiplicity infection, on the one hand, and of very similar levels of protein expression and formation of enveloped virions, on the other hand, the implication of our studies was that extracellular K.VP1-2ΔNLSNC virions were formed but were blocked at some stage of subsequent infection in secondary cells. We next performed a more quantitative analysis of virion production and infectivity. Since multiround virion production was impossible, we performed high-MOI infections in noncomplementing cells (in this case, HaCaT human keratinocytes, which in a comparative analysis generally produced increased yields of all HSV-1 strains). Cells were infected with the mutant or revertant virus at an MOI of 1 (based on titers on complementing cells), and the medium was harvested at 16 h postinfection. The medium was clarified, and extracellular virus was then pelleted and analyzed firstly by SDS gel electrophoresis and total protein staining. The results showed similar levels of extracellular virion production. When normalized for levels of major capsid protein by minor adjustment, little difference in qualitative or quantitative terms could be observed between the virion protein profiles (Fig. 6a). In particular, there was no difference in the relative abundance of VP1-2 itself, e.g., compared to that of the major capsid protein VP5. Further analysis by Western blotting of a number of structural proteins, including VP5, VP1-2, pUL37, VP16, pUL25, gB, and pUL6, also showed little difference between the mutant and revertant particles (Fig. 6b). Without a complete comprehensive analysis of all virus (and host) proteins and their potential modifications, it is formally impossible to state that there were no qualitative differences in the protein profiles of the mutant and revertant particles (with the implication that the deletion of the NLS of VP1-2 was responsible for any proposed differences). However, within the limitations of SDS-PAGE and Western blot analyses of extracellular particles, we could not detect significant differences between the mutant and revertant protein profiles. We further analyzed these extracellular particles by negative staining and observed at least as many particles from the K.VP1-2ΔNLS-infected medium. While inherent in such analysis, stain penetration is not homogeneous and a range of particles are observed, but no significant differences were detected between the mutant and the revertant. Sample images showing stained particles for the mutant and revertant samples are shown in Fig. 6c. Taken together with the thin-section EM analysis and expression data, these data demonstrate few defects in virion assembly and release of K.VP1-2ΔNLS.
Fig 6.

Characterization of extracellular K.VP1-2ΔNLSNC virus produced from noncomplementing cells. HaCaT cells were infected with K.VP1-2ΔNLSC or the revertant virus at an MOI of 5 (estimated from titration on complementing cells). We used HaCaT cells because we find that they help to increase overall yields of all herpesvirus strains. The medium was harvested 16 h after infection and clarified, and extracellular virus was pelleted by centrifugation at 19,000 rpm for 90 min. Samples of pelleted virus showed slightly increased amounts of virion proteins for the mutant. Samples were adjusted (based on the major capsid protein VP5) and analyzed by SDS-PAGE. (a) Mutant and revertant viruses exhibited similar qualitative and quantitative profiles of structural proteins, including VP1-2 and VP5. (b) Equalized samples were analyzed by Western blotting for the presence of a number of structural components, as indicated. (c) Samples of extracellular particles from revertant- or mutant-infected cell medium, analyzed by negative staining. Various degrees of stain penetration were observed for both the mutant and the revertant, and examples illustrating partially stained intact particles are shown for both. Bars, 200 nm. (d) Comparison of specific infectivities of K.VP1-2ΔNLSNC and revertant extracellular virus particles, equalized on the basis of the major capsid protein and titrated for plaque formation on complementing HS30 cells. On a capsid-standardized basis, K.VP1-2ΔNLSNC particles exhibited approximately 600-fold lower infectivity. (e) Comparison of infectivities at the single-cell level. Vero or HS30 cells were infected with serial dilutions of the revertant virus designed to give low-MOI infections within the range of 1:3 to 1:100 cells infected. Cells were infected in parallel with K.VP1-2ΔNLSNC particles over an equivalent range based on equalized capsid protein. Cells were fixed 6 h after infection, stained with antibody to detect ICP4, and counterstained with DAPI (4′,6-diamidino-2-phenylindole) to enumerate cells. Numerous cells were counted for each virus at each dilution. Typical fields are shown for Vero cells with the largest amount of input virus to emphasize the difference in infectivity between the revertant and the mutant, as quantified in panel f. (g) Based on the titer of the revertant virus, cells were infected at a high MOI (5 PFU/cell) with the revertant virus or an equivalent input of mutant virus, based on standardization of the capsid protein in extracellular virus particles. Cells were harvested for Western blotting at the indicated times, with analysis in this case of a typical delayed early protein (ICP8) or the late major capsid protein VP5.
In contrast, there was a very considerable difference in infectivity. Normalizing for the amount of major capsid protein in the extracellular virus preparations, infectious titers of the mutant virus (analyzed by titration on complementing HS30 cells) were of the order of 1,000-fold lower than those of the revertant (Fig. 6d). This result is consistent with the initial analytical-scale analysis (Fig. 1), where despite a complete block in spread after single-cell infection, some infectious virus was produced after high-MOI infection, but at an approximately 1,000-fold lower efficiency. The implications of the current EM and extracellular virus analyses are that this decrease is not due to a defect in the production of virus particles but rather that such particles are produced and are profoundly reduced in the ability to initiate infection in a new cell.
Block to infection with K.VP1-2ΔNLSNC.
To characterize the block to infection further, we next examined the mutant virus now made in noncomplementing cells, i.e., K.VP1-2ΔNLSNC, in low-multiplicity infections in noncomplementing cells, identifying successfully infected cells by immunofluorescence similar to that shown in Fig. 2. The implication of the results described in the previous section is that with standardized amounts of infecting particles, the number of cells initiating infection will be reduced considerably for the mutant compared to the wt virus. We infected Vero or HS30 cells with a series of dilutions of the extracellular virus preparations, standardized for the major capsid protein (which we generally found to be more consistent than particle counts) and designed to result in 1/1,000, 1/100, or 1/3 cells infected (based on titers of the revertant virus), and enumerated antigen-positive cells in multiple fields for the revertant and the NLS mutant. Typical fields are shown for Vero cells with the largest amount of input virus, emphasizing the difference in infectivity between the revertant and the mutant (Fig. 6e). With this input, and evaluating 1,392 cells in several fields, 398 cells were positive for ICP4 protein synthesis for the revertant virus. An approximately 2-fold higher rate of infection was observed in HS30 cells (Fig. 6f). In striking contrast, for the mutant, for which 1,685 cells were evaluated, no positive cells were detected in Vero cells, and of 1,442 HS30 cells, 8 were positive for ICP4 (Fig. 6f).
We also examined infection at nominally higher multiplicities and examined protein synthesis by Western blotting (Fig. 6g). We infected cells with virus purified from extracellular medium at an input of 5 PFU/cell (based on titers of the revertant) or with equal input for the mutant, again standardized based on VP5 capsid protein levels. The results demonstrated that while we could readily detect, e.g., ICP8 or VP5 protein synthesis for the revertant, we detected little protein synthesis for the VP1-2.ΔNLS mutant, with only minor amounts of these proteins observed very late after infection (Fig. 6g). Altogether, these data demonstrate a profound block to infection by K.VP1-2ΔNLSNC particles.
There may be several explanations for the block in K.VP1-2ΔNLSNC infection, including a block in virus entry into the cells or some block in virus gene expression per se. Such explanations would invoke a critical role of the NLS of VP1-2 in assembly of a particle that would fuse normally (which while formally possible seemed unlikely to us) or a role of the NLS in immediate early expression per se, which also seemed less likely than other explanations. Given the strong evidence from previous analyses for a role of VP1-2 in transport to and docking with the nuclear pore (1, 4, 31, 52), it seemed reasonable to propose that disruption in some aspect of the early intracellular pathway may underpin the defect in K.VP1-2ΔNLSNC.
To that end, we next examined localization of infecting capsids in cells infected with K.VP1-2ΔNLSNC or the revertant by immunofluorescence (Fig. 7) and by electron microscopy (Fig. 8 and 9). We wished to address the issue of whether K.VP1-2ΔNLSNC particles entered cells and, if so, to determine where the apparent block to infection may be. For these localization studies, multiple images were collected in the z dimension, and images were compiled to give an accurate representation throughout the cell. Cells were infected with extracellular virus as described above, in this case at an MOI of 100 for the revertant or with a standardized input of K.VP1-2ΔNLSNC. After infection at +4°C, the cells were washed, warmed to 37°C, and incubated for 2 or 4 h. At these times, the cultures were fixed, permeabilized, and stained with anti-VP5 antibody. In parallel control infections, cells were maintained at +4°C to prevent virus entry. This analysis showed a distinct difference in behavior of the mutant and revertant particles.
For the revertant virus after the shift to 37°C, particle staining at 2 h was distinct from that seen at +4°C, with numerous cytoplasmic particles reflecting particle internalization (Fig. 7a, panels I and II). By 4 h, virtually all of the cells also exhibited homogeneous nuclear staining, reflecting successful infection, gene expression, and de novo synthesis of VP5 (Fig. 7a, panels III and IV), with the latter being absent when analysis was performed in the presence of cycloheximide (data not shown). Considering that virtually all cells were successfully infected by the revertant and synthesized VP5 (Fig. 7a, panel IV), it was surprising that we could not see abundant nucleocapsid aggregation around the nuclear rim, though individual particle association could be observed. Nevertheless, a clear difference was observed with the ΔNLS mutant, though not that which we had originally anticipated. For the mutant virus, the pattern was clearly different at 37°C from that at +4°C, reflecting capsid internalization (Fig. 7a, panels V and VI). However, by 2 h, the pattern for the mutant could be distinguished from that for the revertant, and this became even more pronounced at 4 h. Thus, the ΔNLS mutant was seen congregating in distinct accumulations in a single perinuclear region, and though capsids were clearly internalized (see also the EM analysis described below), none of the cells exhibited the diffuse nuclear VP5 indicative of de novo synthesis. This pronounced difference in behavior between the revertant and mutant viruses is clear in Fig. 7a, panel IV versus panel VIII, which omits the nuclear counterstain. The perinuclear accumulation of the mutant was observed in many cells (Fig. 7a, panel VIII, arrows) and was reminiscent of congregation around the MTOC. The observation that the distinct localization of the ΔNLS capsids represents congregation around the MTOC is confirmed in Fig. 7b (panel I; also see Fig. S2 in the supplemental material), which shows a confocal section, this time costained with anti-PCM1, an antibody targeting one of a class of centriole-specific proteins which associates with cytoplasmic granules and concentrates around the MTOC and centriolar matrix (12). The extent of congregation was quantified for revertant and mutant capsids. Defined and unbiased areas of interest (2-μm-diameter circles) were applied around PCM1 foci, and the accumulated density of pixels in the green VP5 channel in the same areas was then calculated. We evaluated multiple fields and approximately 130 cells for each virus (Fig. 7c [each vertical bar represents a cell]). While a congregation pattern could be observed for the revertant virus in some cells, the distinction in MTOC association behavior between the two viruses was very clear. We found that a pixel density of approximately 2,000 by quantitative analysis corresponded to the classification of a cluster by visual inspection. Using this value as a threshold to aid in comparison, approximately 10% of the cells showed MTOC clustering for the revertant, compared to 75% for the mutant, though this represents a conservative estimate of the difference in behavior, which was highly significant (P < 0.0001).
The simplest explanation for our results is that the block in VP1-2ΔNLS capsid transport to the nucleus could be due to a requirement for the VP1-2 NLS for capsid interactions with cellular machinery. However, it is formally possible that the requirement for the NLS could reflect its involvement in retaining VP1-2 on the capsid during entry in order for it to carry out some other function. Considering that the NLS was not required for VP1-2 recruitment and virus assembly, this seems less likely in our view. However, such a scenario has been proposed for a C-terminal determinant of VP1-2 (53). We therefore examined the presence of VP1-2 during infection by K.VP1-2ΔNLS in confocal z sections stained with combinations of antibodies. Since our anti-VP5 and anti-VP1-2 combination was not suitable, we stained cells with either anti-VP5 and pUL37 or anti-VP1-2 and pUL37. The results show an almost quantitative association of pUL37 with VP5+ capsids and the same association for VP1-2 with pUL37+ capsids (Fig. 7b, panels II and III). These data indicate that VP1-2 remains on incoming capsids during infection with K.VP1-2ΔNLS and that the requirement for the NLS more likely reflects a direct involvement in capsid targeting.
To pursue the defect in K.VP1-2ΔNLS capsid entry, we performed a similar analysis by using electron microscopy of thin sections (70 nm) after high-MOI infection with the mutant and revertant viruses. For both the revertant and the mutant, we readily observed internalized capsids free of membranes in the cytosol of infected cells (Fig. 8 and 9). However, consistent with the immunofluorescence analysis, a key qualitative difference was observed between the two (quantified in Fig. 9c). The perimeters of the nuclear envelopes (NE) of 100 cells were scanned and scored for capsids in close proximity to the NE (within approximately 100 nm), which were also classed as empty or DNA-containing capsids. For the revertant virus, we could detect such capsids, typical examples of which are shown in Fig. 8, with approximately 85% of these being empty and the majority being adjacent to pore structures (Fig. 9c). In contrast, for K.VP1-2 ΔNLS, capsids in close proximity to the NE were rare, and the few that were observed retained DNA and were not associated with pores (Fig. 9c). Instead, we frequently observed capsids in close proximity to the centrioles within the microtubule organizing center and not wrapped in membranes, with all containing DNA. Figure 9a and b show an example field with multiple full capsids (small arrowheads) in close proximity to the centriole (large single arrowhead). Taken together, these data answer several questions. Firstly, they show that ΔNLS virions, which exhibit a profound block in infection, are able to enter cells. Moreover, although at the EM level it is difficult to be categorical, nevertheless we could not identify ΔNLS empty capsids in proximity to the nuclear pore. Altogether, with the distinct phenotype and more quantitative analysis of capsid localization by immunofluorescence and the clear failure to initiate gene expression, the most reasonable explanation for our data is that the ΔNLS mutant exhibits a major block in forward routing from the MTOC and/or docking with the nuclear pore, resulting in a profound block to the normal progression of infection. These data provide evidence that the short NLS at position 475 in VP1-2, which exhibits strong positional and sequence conservation in the homologues in all herpesviruses (2), is critical for virus spread and, furthermore, that while it is dispensable for virion assembly per se, this NLS is key to normal entry early in infection via routing to the nuclear pore.
DISCUSSION
Herpesvirus capsids must navigate the cytosolic environment of the cells which they infect, dock and engage with nuclear pores, and deposit their genomes across the pore for subsequent gene expression. Any mechanistic understanding of these early pathways requires the identification and dissection of the virus and host proteins involved. However, most of the potential virus candidates for an important role in early entry are essential proteins required for virus assembly. Thus, their complete deletion results in failure to assemble a particle and prevents any further detailed analysis of a specific role in entry and the mechanism involved. VP1-2 is conserved in all herpesviruses and is absolutely essential for virus assembly (13, 15, 34, 52). However, strong evidence for an additional role of VP1-2 in entry was provided from previous data on the retention of a subset of tegument proteins, including VP1-2, on infecting capsids (19, 23, 30, 37, 39, 40, 45, 51, 56, 61), on the failure of VP1-2-negative capsids to infect nuclei in polykaryocytes (52), and on a ts defect in entry mapping to VP1-2 (1, 5, 31). Nevertheless, any mechanistic understanding of the relevant pathways and roles of VP1-2 requires the identification of variants wherein virion assembly and secondary infection still occur yet virus propagation is defective. Here we constructed and characterized a variant in which infection and virus assembly proceed but virus propagation and cell-to-cell spread are profoundly blocked. A summary of the features of this virus mutant is illustrated schematically in Fig. 10. Taken together, our data provide strong support for the proposal that the basic motif of VP1-2 functions as an NLS to promote successful transport to and/or interaction with nuclear pore complexes and that this is a critical requirement for successful infection.
Fig 10.

Summary of propagation of K.VP1-2ΔNLS in complementing and normal cell types. The three rows (a, b, and c) summarize the outcomes of K.VP1-2ΔNLS infection. (a) Complementing cells (shaded in green) contain the wt UL36 gene, which is induced after infection with the K.VP1-2ΔNLS mutant. The wt VP1-2 protein is recruited onto virions (green virions with green dots, indicating functional VP1-2). Note that a population of mutant VP1-2 (indicated in orange) could be recruited onto particles, and virions may have mixed populations and different distributions (II?). It may be that not all of the VP1-2 incorporated needs to be wt, though neither the distribution of wt versus mutant VP1-2 nor the amount of wt VP1-2 required for successful infection is known. The mutant can replicate, spread, and form plaques in complementing cells. (b) Upon infection of noncomplementing cells (shaded in gray) with the complemented virus, the virus can infect, undergo gene expression, and produce extracellular virus (orange virions), which now contains mutant VP1-2ΔNLS (orange dots; II). Our results indicate that this virus contains VP1-2ΔNLS, appears structurally similar to wt virus, with a similar cohort of proteins, and can infect cells but is blocked at the MTOC prior to successful nuclear engagement. (c) Producing K.VP1-2ΔNLS virus in noncomplementing cells results in the formation of extracellular noncomplemented virions. These virions have a profound block in infection at the MTOC prior to nuclear engagement.
There may be several non-mutually exclusive explanations to account for our observations, since distinct events in transport or docking or subsequent to nuclear pore engagement can all be involved in early nuclear entry (60). While VP1-2 has been shown to be required for intracellular microtubule-mediated capsid transport (38, 55, 61), our results indicate that a defect in the initial interaction and transport of the VP1-2ΔNLS mutant capsid, at least from the periphery of the cell, is not the most likely explanation for the block in infection. Although defects in early transport mechanisms, e.g., in peripheral interactions or in the kinetic efficiency of transport to the interior of the cell, are less likely explanations and are not supported by the current data, they will be addressed formally in future work when a fluorescent derivative of the VP1-2ΔNLS mutant can be constructed.
It may have been that capsids were transported to the pore without a requirement for the VP1-2 NLS (but with a requirement for other specific capsid/tegument determinants), with the NLS being involved in promoting tight engagement with the pore or a key event subsequent to pore interaction per se. For example, in adenovirus, capsid binding to nuclear pores has been reported to be independent of importin and, presumably, of any NLS motifs in capsid proteins and to be mediated instead by direct capsid binding to pore components, including Nup214 (58). However, our data support a role for VP1-2 NLS interactions specifically in routing to the pore, though there could be several non-mutually exclusive explanations regarding the detailed processes involved. For example, while the NLS is not required for peripheral transport, it is possible that it is required for promoting subsequent transport of capsids directly, i.e., with the NLS determinant interacting directly with the cytoskeleton-associated transport machinery, or indirectly, by binding some type of NLS receptor, e.g., an importin, and by virtue of this interaction promoting transport to the pore. It could also be that the NLS is involved in capsid detachment from microtubules after retrograde transport, and indeed, it is possible that these events are coupled (see below).
It has previously been reported that HSV virions stripped of their envelope by detergent extraction can bind to nuclear pores in vitro (46). Such in vitro binding could be inhibited to some degree either by a general broad antibody to multiple nucleoporins or by an antibody to importin β, and importin β was reported to be sufficient to promote capsid binding to isolated nuclei (46). No specific virus receptors were identified for such binding, but mild trypsin treatment of the capsids, which would remove several tegument proteins, including VP13/14, VP22, VP16, and VP1-2, was reported to inhibit capsid-nucleus interaction. Also, an antibody to VP1-2 itself which was scrape loaded onto cells resulted in a reduction in capsid accumulation at the nuclear pore (11). It is tempting to speculate that the pronounced requirement for the NLS of VP1-2 demonstrated here could reflect an involvement of importin β. However, it was also reported that importin α, which interacts directly with NLS motifs (17, 18), does not support capsid binding and that an NLS-peptide conjugate cannot compete for capsid-pore docking. This implies that the VP1-2 NLS does not bind importin α but rather has specificity for importin β. Moreover, an antibody to nucleoporins (46) also inhibited capsid binding in vitro, and small interfering RNA (siRNA) knockdown of nucleoporins reduced capsid accumulation at the nucleus in vivo (11). These effects, though modest, indicate that other interactions may be involved in capsid-pore interactions (see below).
The distinct congregation of the ΔNLS mutant capsids around the MTOC and our failure to detect any empty capsids around the pore indicate that the NLS somehow promotes the forward routing of capsids from the MTOC and/or their detachment from the MTOC or surrounding region. The most straightforward explanation is that without any necessary spatial restriction, the VP1-2 NLS interacts with a cellular component as it undergoes retrograde transport to the interior of the cell, retaining the component on the virus capsid. Some downstream event, possibly but not necessarily involving the NLS, then promotes release or alteration of the capsid and transport toward the pore dependent upon VP1-2 and its NLS-associated component(s). This explanation would not rule out additional downstream roles for the NLS once it is docked at the pore. The congregation of the ΔNLS mutant capsids around the MTOC would then be a result of the inefficient directed transport toward the pore and/or an actual retention of the ΔNLS capsids within and around the MTOC region.
It is also possible that the NLS functions or interacts with its potential receptor in a spatially restricted way once it arrives at the MTOC, a region known to have a particular structure and a matrix-like organization (12, 26, 33). One explanation for this could a regulated conformational exposure such as that thought to occur on the capsid of hepatitis B virus (HBV). HBV capsids (from bacterially expressed core protein) bind nuclear pores, at least in permeabilized cells, dependent upon phosphorylation at residues adjacent to an NLS motif. It is proposed that phosphorylation exposes the NLS for importin binding and pore docking (27). It could be that the disposition or activity of the VP1-2 NLS is regulated either to promote exposure and function in entry or to mask or inhibit its activity after de novo synthesis and incorporation into newly assembled capsids during exit. An example of an event which might promote function or presentation of the NLS is the proteolytic cleavage of VP1-2, which one report has indicated may be relevant at some stage of early infection (25).
Furthermore, it is intriguing that in certain circumstances, e.g., intra-axonal transport, certain importin species have been shown to be complexed with the retrograde motor dynein. Signaling pathways induce the rapid translation of spatially restricted importin β mRNA into a protein that is distant from the cell body and then complexes with importin α and dynein to enable nuclear translocation of cargo (22, 48). In this manner, it is possible that an NLS acts as a determinant for both microtubule-associated transport and nuclear pore interaction. While such a mechanism is possible, it is more plausible for the neurotropic herpesviruses. Other mechanisms not requiring spatially restricted NLS interactions are more likely to explain the requirement for the VP1-2 NLS in core replication pathways in culture. Thus, the NLS motif could be engaged at any of a number of points but still be critically important for forward transport from the MTOC area.
Clearly, other determinants in VP1-2 are also critical for virus replication. Insertional and deletion mutations in several regions abrogate VP1-2 function. Little is known about the basis of the VP1-2 disruption, though it is likely that such mutants affect the known essential roles of VP1-2 in recruiting additional tegument proteins to the assembly pathway. Indeed, certain mutants have been shown to specifically disrupt VP1-2 interactions with other tegument proteins required for assembly (7, 15, 29, 32, 34, 41, 42). One specific VP1-2 determinant, the extreme carboxy-terminal region, promotes interaction with the essential capsid protein pUL25 (8, 34). In pseudorabies virus, deletion of the extreme C-terminal 23 residues abrogates virus replication (34, 42), an effect which has been attributed to disruption of VP1-2 recruitment to the capsid, possibly by direct interaction with pUL25 (10, 47). Since pUL25 itself has also been implicated in early events during the initiation of infection (47, 49), such additional VP1-2 determinants clearly may play a role in entry events. Indeed, it has previously been shown that pUL25 expressed individually can bind nucleoporins, and thus it has been proposed that it may act on capsids as an interface with the nuclear pore (47). If this were the case, based on our results, it would be insufficient to promote nuclear pore engagement in the absence of the VP1-2 NLS. Nevertheless, it remains possible that VP1-2 plays a role in promoting pUL25 function after docking. It is possible, for example, that downstream of NLS-promoted routing to the pore, the NLS itself or additional determinants in VP1-2 and/or pUL25 engage with specific pore components such as Nup214 or Nup358. However, the role of the C terminus of VP1-2 may be more complicated than first anticipated, since recent results indicate that the C terminus is not required for VP1-2 function in virion assembly but rather is specifically required to retain the protein on capsids as it infects secondary cells (53). This apparently indicates that VP1-2 remains bound to newly formed capsids and promotes virion assembly without requiring the C-terminal pUL25-interacting region but detaches from capsids after they infect cells if this C-terminal region is not present. This determinant appears to act in a distinct fashion from the NLS identified here.
In conclusion, our results advance the understanding of the role of HSV VP1-2, specifically in the entry pathways of the virus. The characterization of the phenotype and the nature of the block to entry uncouple critical determinants in VP1-2 for early intracellular routing pathways. These results provide a framework for future detailed analysis of the host cell factors involved, since these can be linked to a physiologically relevant and critical determinant whose alteration has a profound phenotype. Furthermore, considering the conserved and essential nature of VP1-2 and the presence of the basic NLS-like motif at similar positions in all homologues (2), we believe that this motif may play a similar critical role in other herpesvirus species. Finally, in principle, targeting the function of the NLS could provide a useful therapeutic avenue, and the debilitated virus itself could prove useful in other settings, such as gene therapy, where entry is required but second-round spread is not desirable, or vaccine applications.
Supplementary Material
ACKNOWLEDGMENTS
We thank Fraser Rixon and Valerie Preston for antibodies and cells and Prashant Desai for the complementing cell line HS30. We thank Tomi Kuyoro for help with virus titrations. We are grateful to David Leib for provision of the KOS bacmid and to Nick Osterrieder and Greg Smith for bacmid reagents. We thank Martin Spitaller for assistance with confocal microscopy.
C.M.C. was supported by a university research fellowship from the Royal Society. F.A. received a fellowship from the Spanish Ministerio de Educacion y Ciencia. This work was funded by Marie Curie Cancer Care.
Footnotes
Published ahead of print 20 June 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Abaitua F, Daikoku T, Crump CM, Bolstad M, O'Hare P. 2011. A single mutation responsible for temperature sensitive entry and assembly defects in the VP1-2 protein of HSV. J. Virol. 85:2024–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abaitua F, O'Hare P. 2008. Identification of a highly conserved, functional nuclear localization signal within the N-terminal region of herpes simplex virus type 1 VP1-2 tegument protein. J. Virol. 82:5234–5244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abaitua F, Souto RN, Browne H, Daikoku T, O'Hare P. 2009. Characterization of the herpes simplex virus (HSV)-1 tegument protein VP1-2 during infection with the HSV temperature-sensitive mutant tsB7. J. Gen. Virol. 90:2353–2363 [DOI] [PubMed] [Google Scholar]
- 4. Batterson W, Furlong D, Roizman B. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 45:397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Batterson W, Roizman B. 1983. Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J. Virol. 46:371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bolstad M, Abaitua F, Crump CM, O'Hare P. 2011. Autocatalytic activity of the ubiquitin-specific protease domain of HSV-1 VP1-2. J. Virol. 85:8738–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bottcher S, et al. 2007. Identification of functional domains within the essential large tegument protein pUL36 of pseudorabies virus. J. Virol. 81:13403–13411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bottcher S, et al. 2006. Identification of a 709-amino-acid internal nonessential region within the essential conserved tegument protein (p)UL36 of pseudorabies virus. J. Virol. 80:9910–9915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cohen S, Au S, Pante N. 2011. How viruses access the nucleus. Biochim. Biophys. Acta 1813:1634–1645 [DOI] [PubMed] [Google Scholar]
- 10. Coller KE, Lee JI, Ueda A, Smith GA. 2007. The capsid and tegument of the alpha herpesviruses are linked by an interaction between the UL25 and VP1-2 proteins. J. Virol. 81:11790–11797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Copeland AM, Newcomb WW, Brown JC. 2009. Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 83:1660–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dammermann A, Merdes A. 2002. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 159:255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608–11618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Flint SJ, Enquist LW, Krug RM, Racaniello VR, Skalka AM. 2009. Principles of virology. ASM Press, Washington, DC [Google Scholar]
- 15. Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 78:11879–11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gierasch WW, et al. 2006. Construction and characterisation of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J. Virol. Methods 135:197–206 [DOI] [PubMed] [Google Scholar]
- 17. Goldfarb DS, Corbett AH, Mason DA, Harreman MT, Adam SA. 2004. Importin alpha: a multipurpose nuclear-transport receptor. Trends Cell Biol. 14:505–514 [DOI] [PubMed] [Google Scholar]
- 18. Gorlich D, Kutay U. 1999. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15:607–660 [DOI] [PubMed] [Google Scholar]
- 19. Granzow H, Klupp BG, Mettenleiter TC. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 79:3200–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greber UF, Fassati A. 2003. Nuclear import of viral DNA genomes. Traffic 4:136–143 [DOI] [PubMed] [Google Scholar]
- 21. Greber UF, Fornerod M. 2005. Nuclear import in viral infections. Curr. Top. Microbiol. Immunol. 285:109–138 [DOI] [PubMed] [Google Scholar]
- 22. Hanz S, et al. 2003. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 40:1095–1104 [DOI] [PubMed] [Google Scholar]
- 23. Heine JW, Honess RW, Cassai E, Roizman B. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jarosinski K, Kattenhorn L, Kaufer B, Ploegh H, Osterrieder N. 2007. A herpesvirus ubiquitin-specific protease is critical for efficient T cell lymphoma formation. Proc. Natl. Acad. Sci. U. S. A. 104:20025–20030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jovasevic V, Liang L, Roizman B. 2008. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 82:3311–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kalt A, Schliwa M. 1993. Molecular components of the centrosome. Trends Cell Biol. 3:118–128 [DOI] [PubMed] [Google Scholar]
- 27. Kann M, Sodeik B, Vlachou A, Gerlich WH, Helenius A. 1999. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J. Cell Biol. 145:45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL. 2005. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 19:547–557 [DOI] [PubMed] [Google Scholar]
- 29. Kelly BJ, Diefenbach E, Fraefel C, Diefenbach RJ. 2012. Identification of host cell proteins which interact with herpes simplex virus type 1 tegument protein pUL37. Biochem. Biophys. Res. Commun. 417:961–965 [DOI] [PubMed] [Google Scholar]
- 30. Klupp BG, Fuchs W, Granzow H, Nixdorf R, Mettenleiter TC. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76:3065–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Knipe DM, Batterson W, Nosal C, Roizman B, Buchan A. 1981. Molecular genetics of herpes simplex virus. VI. Characterization of a temperature-sensitive mutant defective in the expression of all early viral gene products. J. Virol. 38:539–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ko DH, Cunningham AL, Diefenbach RJ. 2010. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J. Virol. 84:1397–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kollman JM, Merdes A, Mourey L, Agard DA. 2011. Microtubule nucleation by gamma-tubulin complexes. Nat. Rev. Mol. Cell Biol. 12:709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee JI, Luxton GW, Smith GA. 2006. Identification of an essential domain in the herpesvirus VP1/2 tegument protein: the carboxy terminus directs incorporation into capsid assemblons. J. Virol. 80:12086–12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee JI, et al. 2009. A herpesvirus encoded deubiquitinase is a novel neuroinvasive determinant. PLoS Pathog. 5:e1000387 doi:10.1371/journal.ppat.1000387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levin A, Loyter A, Bukrinsky M. 2011. Strategies to inhibit viral protein nuclear import: HIV-1 as a target. Biochim. Biophys. Acta 1813:1646–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luxton GW, et al. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 102:5832–5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mettenleiter T. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Michael K, Klupp BG, Mettenleiter TC, Karger A. 2006. Composition of pseudorabies virus particles lacking tegument protein US3, UL47, or UL49 or envelope glycoprotein E. J. Virol. 80:1332–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mijatov B, Cunningham AL, Diefenbach RJ. 2007. Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology 368:26–31 [DOI] [PubMed] [Google Scholar]
- 42. Mohl BS, et al. 2010. Random transposon-mediated mutagenesis of the essential large tegument protein pUL36 of pseudorabies virus. J. Virol. 84:8153–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mohl BS, et al. 2009. Intracellular localization of the pseudorabies virus large tegument protein pUL36. J. Virol. 83:9641–9651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Monette A, Pante N, Mouland AJ. 2011. HIV-1 remodels the nuclear pore complex. J. Cell Biol. 193:619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Newcomb WW, Brown JC. 2010. Structure and capsid association of the herpesvirus large tegument protein UL36. J. Virol. 84:9408–9414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 20:4922–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 83:6610–6623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Perry RB, Fainzilber M. 2009. Nuclear transport factors in neuronal function. Semin. Cell Dev. Biol. 20:600–606 [DOI] [PubMed] [Google Scholar]
- 49. Preston VG, Murray J, Preston CM, McDougall IM, Stow ND. 2008. The UL25 gene product of herpes simplex virus type 1 is involved in uncoating of the viral genome. J. Virol. 82:6654–6666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rabe B, et al. 2009. Nuclear entry of hepatitis B virus capsids involves disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog. 5:e1000563 doi:10.1371/journal.ppat.1000563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Radtke K, et al. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 6:e1000991 doi:10.1371/journal.ppat.1000991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roberts AP, et al. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1 (HSV-1). J. Virol. 83:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schipke J, et al. 2012. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 86:3682–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schlieker C, et al. 2007. Structure of a herpesvirus-encoded cysteine protease reveals a unique class of deubiquitinating enzymes. Mol. Cell 25:677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shanda SK, Wilson DW. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 82:7388–7394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smith GA, Enquist LW. 2002. Break ins and break outs: viral interactions with the cytoskeleton of mammalian cells. Annu. Rev. Cell Dev. Biol. 18:135–161 [DOI] [PubMed] [Google Scholar]
- 57. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 58. Trotman LC, Mosberger N, Fornerod M, Stidwill RP, Greber UF. 2001. Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat. Cell Biol. 3:1092–1100 [DOI] [PubMed] [Google Scholar]
- 59. Vittone V, et al. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566–9571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Whittaker GR, Kann M, Helenius A. 2000. Viral entry into the nucleus. Annu. Rev. Cell Dev. Biol. 16:627–651 [DOI] [PubMed] [Google Scholar]
- 61. Wolfstein A, et al. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227–237 [DOI] [PubMed] [Google Scholar]
- 62. Zhou L, et al. 2011. Transportin 3 promotes a nuclear maturation step required for efficient HIV-1 integration. PLoS Pathog. 7:e1002194 doi:10.1371/journal.ppat.1002194 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.