

Abstract
Human immunodeficiency virus type 1 (HIV-1), hepatitis B virus (HBV), and herpes simplex virus (HSV) have been incurable to date because effective antiviral therapies target only replicating viruses and do not eradicate latently integrated or nonreplicating episomal viral genomes. Endonucleases that can target and cleave critical regions within latent viral genomes are currently in development. These enzymes are being engineered with high specificity such that off-target binding of cellular DNA will be absent or minimal. Imprecise nonhomologous-end-joining (NHEJ) DNA repair following repeated cleavage at the same critical site may permanently disrupt translation of essential viral proteins. We discuss the benefits and drawbacks of three types of DNA cleavage enzymes (zinc finger endonucleases, transcription activator-like [TAL] effector nucleases [TALENs], and homing endonucleases [also called meganucleases]), the development of delivery vectors for these enzymes, and potential obstacles for successful treatment of chronic viral infections. We then review issues regarding persistence of HIV-1, HBV, and HSV that are relevant to eradication with genome-altering approaches.
INTRODUCTION
Chronic viral infections cause enormous suffering among infected individuals, highlighting the need for curative therapies. Thirty-three million people worldwide are infected with human immunodeficiency virus type 1 (HIV-1), and the annual global incidence is approximately 2.6 million infections (186). With the advent of highly active antiretroviral therapy (HAART), HIV is now managed as a chronic rather than a terminal disease (108, 120) and treated patients have a normal life span (6). However, only 36% of those in need of antiretroviral regimens in low- and middle-income countries have access to therapy (186). Without treatment, the median time for progression to AIDS is 9 to 10 years and the average life expectancy upon developing AIDS is 9 months (113). Moreover, HAART does not cure the infected individual or reverse all disease manifestations, and therapy is lifelong. Other sobering facts include possible development of drug resistance and long-term adverse effects of current drugs, as well as the enormous financial burden of lifelong treatment in populations in which HIV is endemic (120, 183).
Hepatitis B virus (HBV) is another infection of enormous public health importance. Though an effective vaccine is available, global uptake is low (51, 100). Over 350 million people are chronically HBV carriers, and more than 50% of people within certain regions of Asia and Africa have a history of HBV exposure. Cirrhosis, liver failure, and hepatocellular carcinoma (HCC) related to HBV claim 500,000 to 1.2 million lives annually (36, 94). Antiviral therapy is effective for preventing these outcomes but does not eliminate all reservoirs of virus. Only a small fraction of the infected population has access to therapy, which typically must be given over years (209). Liver transplantation, an option for infected persons with end-stage disease, is unavailable for the majority of those in need.
Herpes simplex virus (HSV) is also a cause of significant morbidity. HSV-2 is the leading cause of genital ulcers worldwide, and 16% of Americans are seropositive (208). HSV-1 prevalence exceeds 50% in the United States: this serotype can cause both oral and genital ulceration and is the most common etiology of infectious blindness (keratitis) and viral encephalitis (158). Both varieties of HSV cause severe infections in newborns and immunocompromised hosts (17). Importantly, HSV-2 is a key risk factor for HIV-1 acquisition and transmission (163, 196). While antiviral therapy decreases the severity of primary infection and recurrent ulcer formation and also decreases the frequency of asymptomatic viral shedding and recurrences, it is imperfect for each of these indications (38). Several candidate vaccines failed to demonstrate efficacy (37).
Effective antiviral therapies exist for treatment of each of these infections, and development of these agents represents one of the major successes in medicine during the previous decades. Accordingly, the search for new antiviral medications continues to be a major focus within virology. Unfortunately, antiviral therapies may have limited room for improvement. While existing treatments for HIV, HBV, and HSV inhibit replication and cellular entry of the virus extremely potently (164), they do not target latent viral stores which exist in a reversible nonreplicating state of infection. HIV-1, HBV, and HSV establish long-lived reservoirs from which newly synthesized viruses can continually arise. When antiviral agents are stopped, robust viral replication often resumes, and symptomatic manifestations of disease typically follow.
Because antiviral therapy is safe and mostly effective for these infections, relatively little attention has been paid to approaches that might rid the body of latent virus. However, a deeper understanding of the molecular nature of latent viral genomes and their cellular and anatomic sites has raised the possibility that new therapies may directly attack the Achilles' heel of chronic viral infections. Recently, new technologies that may allow specific disruption of latent viral genomes have been developed. In this review, we outline why highly specific, DNA-cleaving enzymes, an exciting technology that was recently recognized as the “Method of the Year” (110), may enhance the likelihood of a cure. We review the major classes of such enzymes and consider specific issues for their use in addressing the unique latent reservoirs for HIV-1, hepatitis B, and HSV-2.
SPECIFIC TYPES OF CLEAVAGE ENZYMES
For a DNA-targeting enzyme to be useful for gene therapy, it must possess adequate specificity for the target sequence such that it will not recognize off-target genomic sites. Therefore, enzymes need to target DNA stretches of sufficient length. Assuming homogeneous mixing of nucleotide sequences across the 2,900,000,000-bp human genome, the probability of a complementary sequence to a viral target site can be estimated according to the formula 1 − [1 − (1/4x)]2,900,000,000, where x is the number of base pairs in the enzyme binding site. This suggests that the use of cleavage enzymes that target 17 or more nucleotides will minimize the likelihood of off-site binding (Fig. 1). (There are 417 or 1.72 × 1010 possible 17-nucleotide sequences for an enzyme that targets a DNA sequence with overall specificity equivalent to that of an invariant 17-nucleotide region.) Factors such as sequence homology due to incorporation of ancient retroviral sequences into the human genome may enhance the probability of equivalent binding sites, while chromatin may impede access to complementary sequences within noncoding portions of the genome. Therefore, the extent of off-target binding is difficult to estimate precisely. Nevertheless, these considerations suggest that a DNA-binding enzyme of sufficient specificity might successfully target viral DNA while leaving the host genome intact.
Fig 1.
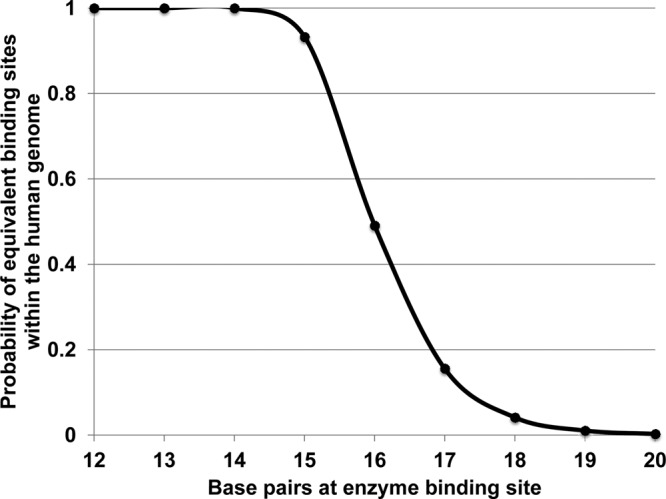
Probability of cDNA base pairs within the human genome according to the base pair length of the cleavage enzyme. The calculation assumes random ordering of nucleotides within the human genome.
Currently, 3 classes of DNA-recognizing/modifying enzymes with the required high degree of specificity are known: zinc finger nucleases (ZFNs), transcription activator-like (TAL) effector nucleases (TALENs), and homing endonucleases (HEs). Each class of proteins has unique features that are potentially advantageous and disadvantageous for eradication of chronic viral infection (Fig. 2).
Fig 2.
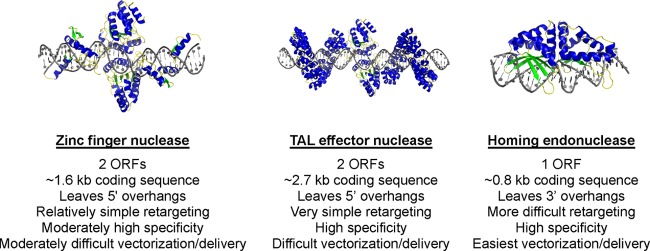
Structures of cleavage enzymes.
Zinc finger nucleases.
The zinc finger nucleases (ZFNs) are the most highly developed of the site-specific nucleases. DNA specificity results from the specific binding of tandem arrays of zinc fingers, each of which recognizes either a unique or a slightly degenerate DNA triplet within the context of the longer cognate target site (190). To achieve DNA cleavage activity, an array of zinc fingers is covalently tethered to the nonspecific nuclease domain from the R.FokI (FokI) restriction endonuclease. Since the FokI nuclease is active only as a dimer, two separately encoded, appropriately spaced zinc finger arrays must be designed to bind opposing half-sites of a desired DNA target, thereby facilitating cleavage at the desired site. In most systems, three or four zinc fingers are incorporated in each protein subunit. The outcome of ZFN activity is a DNA double-strand break with 5′ overhangs, located between the zinc finger-defined DNA-binding motifs.
The ZFNs are attractive gene-targeting constructs since the DNA triplets recognized by many individual zinc fingers have already been defined. However, ZFN technology also has certain limitations (162). Zinc fingers targeting certain DNA triplets are not yet available, and thus, only a limited number of DNA sequences can be accessed. ZFN specificity can be influenced by context dependence, in which neighboring zinc fingers can alter either the recognition specificity or the affinity of a given zinc finger, further limiting possible targets. Zinc fingers can also have substantial binding affinity for sequences that are similar but not identical to their intended targets. When coupled with the nonspecific DNA cleavage activity of the FokI nuclease, this can result in substantial off-target DNA cleavage and toxicity by some ZFN designs (68). Off-target cleavage has been reduced by the development of FokI variants that can function only as heterodimers, thus preventing cleavage by undesired homodimers (176), but questions remain regarding the ultimately achievable specificity of ZFNs. Despite these potential limitations, ZFNs have achieved major successes, having been used extensively in the study of knockout of the HIV coreceptor CCR5 (75, 137, 204) and more recently to directly target HBV covalently closed circular DNA (cccDNA) in infected hepatocytes (39).
TALENs.
The transcription activator-like (TAL) proteins were originally identified in certain plant-pathogenic species of Xanthomonas (reviewed in reference 13). The TAL proteins contain highly modular architectures that include an N-terminal region which interacts with the bacterial type III secretion system, a central region containing multiple tandem copies of a unique DNA-recognizing repeat domain, and C-terminal nuclear localization signal (NLS) and transcriptional activation regions. After being delivered from bacteria into plant cells and nuclei, these proteins specifically bind DNA targets within promoter regions of the plant genome, after which the transcriptional activation domain induces gene overexpression. The role that this process plays in bacterial pathogenesis in plants is currently an area of intense investigation.
The DNA specificity of the TAL proteins is mediated by a variable number (often 15 to 20) of tandem repeats of a 34-amino-acid repeat sequence. The repeats are nearly identical, with the exception of two variable amino acids at position 12 and 13, which comprise the repeat variable diresidue (RVD) motif (12, 114). The RVD motif specifies the DNA base recognized by each repeat domain, and the “code” of RVDs specifying each of the 4 DNA bases has been determined (12, 114). By combining the DNA recognition domains of the TAL proteins with the FokI nuclease, specifically targeted DNA cleavage enzymes (TAL effector nucleases [TALENs]) have been generated (28, 97).
The TALENs have certain advantages that make them particularly attractive constructs for specific DNA targeting. As noted above, each repeat of the DNA recognition domain mediates the interaction with one nucleotide. Furthermore, there is very little apparent context dependence between individual repeats. Thus, many desired DNA sequences can be targeted, and the resulting TALENs typically display DNA specificity and cleavage activity (24, 98).
The major drawback to the use of the TALENs is their large size. Each repeat of the DNA-binding domain (which recognizes a single nucleotide) is bulkier than an individual zinc finger (which recognizes a DNA triplet). The extremely large nature of these proteins may raise significant difficulties in vectorization and delivery, especially for in vivo use. Moreover, similar to zinc finger nucleases, DNA cleavage requires expression or delivery of two protein chains that subsequently dimerize the FokI nuclease domain at the site of cleavage.
Homing endonucleases.
Homing endonucleases are an especially promising class of DNA-cleaving enzymes (173). In nature, these proteins are encoded by homing endonuclease genes, which are selfish genetic elements found within introns in certain microbial organisms, phages, and viruses. Homing endonucleases introduce DNA double-strand breaks within homologous alleles that lack the corresponding intron. These breaks are then repaired via homologous recombination using the allele containing the homing endonuclease gene, thus driving the selfish genetic element into the susceptible target.
Homing endonucleases are attractive as DNA-targeting enzymes for two main reasons. First, unlike the ZFNs or TALENs, DNA-cleaving activity is inherent to the same protein domains also responsible for DNA binding (note that the cleavage mechanism of HEs results in 3′ DNA overhangs, in contrast to the 5′ overhangs resulting from the FokI nuclease used in ZFNs and TALENs). HEs are therefore significantly smaller (generally around 250 amino acids) than ZFNs or TALENs, thus facilitating vectorization and delivery. Second, HEs typically display extremely high specificity for their DNA targets (a property that presumably has resulted from long-term selective pressure to avoid fitness costs on their biologic hosts) and therefore theoretically will induce less off-target cleavage than ZFNs. Thus, many HEs show little if any toxicity in cells, even when expressed at very high levels for extended periods (8).
The major obstacle to widespread use of HEs has been the technical difficulties that are inherent in the process of redirecting their cleavage specificity to desired DNA targets. The DNA contact surfaces and residues of HEs do not possess the modular structure of either zinc fingers or the TAL proteins. Thus, their protein-DNA interactions and corresponding specificity tend to display significant context dependence that is strongly influenced by neighboring amino acids and DNA contacts. Furthermore, the association of DNA binding and cleavage activity within the same protein domain means that alterations in protein structure to facilitate DNA binding can have deleterious effects on the cleavage activity of the HE. Despite these obstacles, there have recently been several impressive achievements in redirecting HEs toward useful targets (7, 63, 185), and technical advances in HE redesign and targeting are occurring continually (173). Thus, the 3 classes of proteins (ZFNs, HEs, and TALENs) each have unique advantages and disadvantages, and the ideal protein may vary depending on the particular application envisioned.
EFFECTS OF DNA TARGETING AND GENE KNOCKOUT
The ability to induce DNA double-strand breaks at defined sites using ZFNs, TALENs, or HEs has important implications for gene therapy. DNA double-strand breaks are incompatible with continued cell viability, and thus, powerful DNA repair mechanisms are triggered. In mammalian cells, the presence of DNA double-strand breaks increases the frequency of homologous recombination by orders of magnitude (155, 171). This raises the possibility of targeted gene correction by the simultaneous delivery of a corrected DNA repair template along with the DNA-cleaving enzyme. This targeted repair would avoid dangers inherent in random insertional gene therapy and also ensures that the corrected sequence remains under the proper endogenous expression control elements (reviewed in reference 140). Such targeted repair has been used in a variety of mammalian cell types (reviewed in reference 143).
However, in mammalian cells, the predominant repair pathway for a DNA double-strand break is not homologous recombination but, rather, nonhomologous end joining (NHEJ). During NHEJ, the ends of the cleaved DNA are directly religated, and in most cases, repair is precise, resulting in restoration of the original DNA sequence (Fig. 3). However, in the presence of a targeted DNA endonuclease, precise repair restores the recognition sequence for the enzyme, leading to repeated rounds of cleavage and repair. The cycle repeats until an imprecise repair event, such as an insertion/deletion (indel) or more complex mutation, occurs. Once the recognition site for the enzyme is altered, the enzyme no longer targets the site. Cleavage enzyme-induced deletions and frame shifts within a DNA target can eliminate the synthesis of functional protein, making this an attractive strategy for genetic therapy.
Fig 3.

Targeted gene knockout by DNA-editing enzymes. The target sequence (red) is bound by the enzyme, leading to a DNA double-strand break. HE cleavage leaves 3′ overhangs as depicted here; ZFNs and TALENs leave 5′ overhangs. Such DNA double-strand breaks are typically repaired by precise nonhomologous end joining (NHEJ), which restores the original sequence. However, in the continued presence of DNA-editing enzymes, repeated rounds of cleavage and repair ultimately lead to mutagenic NHEJ pathways. In the example shown here, there is a deletion of 16 bp, resulting in a loss of amino acids and a frameshift mutation in the encoded protein. Small deletions such as this are common, although larger deletions and more-complex insertions/deletions can also be observed.
As noted above, targeted DNA endonucleases have already been applied successfully for the knockout of the HIV coreceptor CCR5 (75, 137). We believe that these same endonucleases might also be used to directly target latent or persistent viral genomes, conceptually offering the possibility of a cure for otherwise lifelong infections (8, 39). We will consider the differing biologies of three such infections (HIV, HBV, and HSV) and the implications that their biology has on the potential for curative therapy using targeted DNA endonucleases.
GENERAL CONSIDERATIONS IN THE USE OF DNA-TARGETING ENZYMES FOR VIRAL INFECTIONS
If DNA cleavage enzymes can successfully enter infected cells and eliminate production of functional viral proteins, then a critical remaining challenge will be to determine whether these enzymes can be vectorized and incorporated within therapeutic regimens that can completely eliminate large reservoirs of latently infected cells. The challenge of repeatedly delivering nucleases to target cells cannot be overstated. The discovery process will need to include cell culture experiments to obtain quantitative measures of latent viral inactivation, followed by dose escalation studies in animal models of infection to test for efficacy. As with antiviral therapy, dynamical mathematical modeling of enzyme delivery and latent pool kinetics will be essential at each step (135) to precisely define barriers to eradication. Potential hurdles may include inadequate enzyme delivery to infected cells located within anatomically sequestered sites and reexpansion of the pool of latently infected cells between cleavage enzyme doses.
Pharmacokinetic (PK) and pharmacodynamic (PD) properties of DNA cleavage enzymes are conceptually different from those of antiviral medications. Unlike antiviral agents, which exert an effect only when present at adequate concentrations, cleavage enzymes will have a permanent effect on latent genomes within infected cells. On the other hand, cells with inactivated latent viruses that have been cleaved may quickly become susceptible to reinfection. Therefore, residual viremia in the case of HIV or HBV or persistent viral reactivations in the case of HSV may rapidly reseed the latent pool of cells. Suppressive antiviral therapy may be necessary prior to and during therapy with cleavage enzymes. In addition, repeated cleavage enzyme dosing at narrow time intervals may be needed to prevent reaccumulation of the latent pool of infected cells.
As with antiviral therapy, drug resistance to specific enzymes is a potential problem. While in some cells, an enzyme may fail to cut all the available recognition sites, a more worrisome situation would occur if a genetic mutation destroys the enzyme recognition site but induces a deletion of only 3 or 6 nucleotides, resulting in the loss of 1 or 2 nonessential amino acids. In certain circumstances, this event might leave a functional viral protein and a latently infected cell which is immune to any further activity from the same enzyme. A similar scenario can be imagined for a virus that gains a mutation within the enzyme recognition sequence that prevents binding or cleavage without affecting viral fitness. Such escape is analogous to the development of HIV resistance to antiviral agents. In clinical practice, drug resistance to HIV-1, hepatitis B, and hepatitis C is minimized by concurrent use of multiple agents. A comparable approach will probably be necessary for cleavage enzyme-based curative therapy.
One of the most important challenges to the use of DNA cleavage enzymes in treating viral infections is the method of delivery used to target an enzyme to the site of latent infection. As with all genetics-based therapies, the target cell types for latent viral infections vary from one virus to another. Latently infected cells exhibit little to no viral gene expression, and vectors will need to bind and enter a given cell type whether it is infected or uninfected. While successful gene therapy has been established with adeno-associated virus (AAV) and retroviral vectors for human metabolic disorders (3, 119), it is unknown whether certain sequestered tissues, such as the nervous system, may serve as sanctuaries from successful delivery. Furthermore, the target cell types can even vary between patients with the same infection. For example, a latent HSV infection in one patient might reside in the dorsal root ganglia, while in another, it might reside in trigeminal ganglia (TG). While these latent reservoirs are in similar cell types, the challenges of enzyme delivery to each site are different and make the method of delivery important.
A number of factors should be taken into consideration when choosing which of the many available gene delivery vehicles would best suit a given therapy. Is there a nonviral or viral gene delivery system that can efficiently target the cell types harboring latent infection? Does the chosen delivery system need to be administered locally, or can a systemic administration route be used? Is the chosen gene delivery system toxic to either the target cells or other organs and cell types exposed upon administration? Will the therapy require transient or persistent expression of the DNA-editing enzyme within the target cell population? Will delivery of high levels of the DNA-editing enzyme result in target cell toxicity? All of these factors should be considered individually before choosing which gene transfer vector suits a given therapy best.
HUMAN IMMUNODEFICIENCY VIRUS
Anatomic sites of HIV-1 latency for cleavage enzyme delivery.
While HIV represents the most urgent target for DNA cleavage enzymes, the nature of HIV-1 latency poses tremendous challenges (Table 1). Latency is formally defined as a state of reversibly nonproductive infection of individual cells (170). A subtype of cells harboring latent genomes is termed a reservoir. A phylogenetic definition of reservoirs which contain latent viruses has been established: because reservoirs are continually seeded during active viral replication and because HIV-1 mutates considerably during the lifetime of the human host, reservoirs harbor considerable genetic diversity, though on average, strains diverge less dramatically from a common ancestor than do nonreservoir strains. This occurs because reservoirs include sequences from throughout the phylogenetic tree, including those archived soon after primary infection (122). A compartment is a site, which may be defined anatomically, in which independent evolution, which is substantially divergent from the common ancestor, continues to occur. While both compartments and reservoirs may also serve as sanctuaries where drug delivery is poor (123), the reservoir is not targeted by current antiviral therapies and, as such, is the critical target for endonuclease therapies. A major current research goal is to define reservoir sites and characteristics.
TABLE 1.
Considerations for cleavage enzyme therapy for HIV, HBV, and HSV infections
| Consideration | Information for infection with: |
||
|---|---|---|---|
| HIV | HBV | HSV | |
| Potential target sites | |||
| Genome size (kb) | 9.7 | 3.2 | 154 |
| No. of transcriptional ORFs | 9 | 4 | >74 |
| No. of essential viral proteins or transcriptional ORFs | 11 essential viral proteins | 4 essential transcriptional ORFs | 31 essential transcriptional ORFs |
| Target site mutability | |||
| Mutability | High | Low to intermediate | Low |
| Polymerase mutation rate | 2 × 10−5 mutations/site/cycle | 1.4 × 10−5-3.2 × 10−5 mutations/site/year | 3 ×10−8 mutations/site/cycle |
| Size/composition of latent reservoir | 107 latently infected cells; diverse, poorly characterized reservoir | >99% hepatocytes; ∼2 × 1011 hepatocytes; evidence of diffuse infection in multiple organs and PBMCs (unclear significance) | 2-10% of ganglionic neurons (HSV-1); ≤20,000 neurons; presumably only neuronal tissue |
| Burden of infection per cell | PBMCs, 1 genome/cell; spleen, ≤5 genomes/cell | 1-50 genomic copies/hepatocyte | 2-50 genomic copies/neuron |
| Maintenance of latency | Homeostatic proliferation of latent pool and/or reseeding due to low-level ongoing replication | High burden of infection in liver due to nonlytic viral replication; turnover of hepatocytes with cccDNA | Permanent infection of neurons during primary infection vs reseeding of neurons |
| Localization of latent genome | Proviral HIV-1 DNA incorporated into host genome | Extrachromosomal episomal cccDNA | Extrachromosomal episomal viral DNA forms |
| Key relevant molecular features of latency | Steric considerations due to repressive chromatin at the LTR, including loss of activating histone modifications, presence of repressive modifications, and presence of DNA methylation | Steric considerations due to chromatin surrounding the cccDNA minichromosome | Posttranslational histone modification of LAT; terminally differentiated neurons |
The most important HIV-1 reservoir consists of memory CD4+ T cells, as these cells harbor integrated HIV DNA but do not permit viral transcription (31, 213). Presumably, these cells are formed when a small subset of actively infected cells naturally reverts to a memory state. Replication may resume within a memory cell when the cell transforms back to an effector state upon contact with a cognate antigen or cytokine (14, 57, 116). However, many memory cells never leave a resting state and never express host transcription factors that stimulate viral reactivation, thereby explaining lifelong persistence of latency.
Other possible anatomic and cellular reservoirs of HIV-1 latency in the human host remain relatively uncharacterized (172). Studies of suppressive HIV-1 therapy reveal at least three phases of viral decline (134). The second stage of viral decay initiates after rapid first-phase decay and is considerably slower than primary decay, irrespective of the antiviral agent (66). While primary decay represents death of actively infected CD4+ T cells, the source of second-phase decay remains unknown (172). Third phase is characterized by very-low-level residual viremia and appears to last for the lifetime of the infected host. Residual viremia persists with treatment intensification and therefore is likely to represent the release of integrated virus from stable reservoirs of CD4+ memory T cells rather than ongoing replication (58). Sequence analysis of residual free virus in the plasma of patients undergoing HAART compared to provirus in resting CD4+ T cells or lymph nodes indicates that virus detected in plasma overlaps somewhat with the latent CD4+ T-cell reservoir but may also arise from another, as-yet-unidentified cellular source (9, 16). Proposed alternative reservoirs include dendritic cells, macrophages, astrocytes, and hematopoietic stem cells (22, 23, 83, 89, 194), though none of these sites have been firmly established to harbor replication-competent integrated HIV DNA for prolonged durations of time.
Latent reservoir heterogeneity may be of high importance when considering curative regimens: if cleavage enzymes are delivered to a high proportion of one latent reservoir but poorly access another, then this may impact the feasibility of eradication or, at a minimum, the number of doses needed to achieve eradication. The possibility of anatomically sequestered compartments, which also serve as viral reservoirs, must therefore be considered. It is postulated that gut-associated lymphoid tissues (GALT) (111), the central nervous system (CNS) (89), lungs (194), and genital tissues (21) may be sites of latency. Animal models of HIV latency in nonhuman primates under HAART have recapitulated many of the findings in infected humans, including early infection of the CNS (44). If these sites also serve as sanctuaries from vector delivery, then they may prohibit eradication and allow viral replication when ART is eventually stopped.
HIV-1 genome and cleavage enzyme targeting sites.
The HIV-1 genome is a challenging target due to its small size of ∼9.7 kb. However, the genome is gene-rich, containing nine open reading frames (ORFs) that produce 15 proteins, 11 of which are essential for viral replication. Furthermore, the final gene products from the gag, pol, and env genes are produced via proteolytic cleavage of a polyprotein (128), which means that a deletion or frameshift upstream may disrupt all downstream proteins within the polyprotein. Thus, mutation of a single target could knock out the function of several proteins at once.
Another issue that may hinder efforts to cure HIV-1 is the extremely high mutability that the virus exhibits during replication, presumably as a means of immune evasion. During the HIV infectious cycle, the viral RNA genome is reverse transcribed by the virally encoded reverse transcriptase (RT) to produce the DNA provirus. Reverse transcription has a high error rate of 1.4 × 10−5 to 4 × 10−5 mutations/base pair/replication cycle (1, 105), resulting in a virus population that is genetically diverse with a complex fitness and mutational landscape. Mathematical models predict that within a single HIV-infected patient, every possible nucleotide substitution is represented (202). Despite the fact that cleavage enzymes seek to eliminate virus from reservoirs in which viral replication is rare or absent altogether during antiretroviral therapy, nucleotide sequence diversity is likely to be high due to continual seeding of the reservoir throughout infection as the virus evolves in the context of immune pressure (122). Thus, use of a single DNA-editing enzyme may select for resistant viral variants, and successful therapy will likely require simultaneous use of multiple enzymes targeting different regions of the virus, as discussed in more detail below. On the other hand, the high mutation rate of HIV-1 comes at a significant fitness cost: many mutations render viral particles incapable of further infection and replication (73). Therefore, not all nucleotide changes at cleavage enzyme target sites will prove deleterious toward eradication efforts.
Latent HIV-1 DNA viral load.
Despite the challenges of reservoir diversity and genome mutability, the number of cells harboring integrated HIV DNA is estimated to be quite low (∼107 infected cells) (29), a fact which may increase the feasibility of a cure. Recent studies also suggest that most latently infected peripheral blood mononuclear cells (PBMCs) contain only one integrated HIV-1 provirus (80), again consistent with an overall low burden of infection. Yet PBMCs may not be representative of the most-critical anatomic regions of latency: in one study, infected splenocytes harbored a median of 5 proviral copies (81). Moreover, it is unknown whether the size of the latent reservoir varies considerably between infected persons and whether such variability impacts disease phenotype.
The establishment of a latent reservoir occurs with systemic spread of virus early during primary infection. Even patients who initiate HAART during the first weeks after viral acquisition are rendered incurable of persistent infection with currently available interventions (32, 174). Macaque challenge studies suggest that viral replication successfully bypasses mucosal immune control and disseminates widely throughout lymphatic tissue in the gut within 2 weeks after simian immunodeficiency virus (SIV) inoculation into the genital tract (192). Depletion of gastrointestinal CD4+ T cells is likely to coincide with establishment of reservoirs of latent infection, though within infected tissues, the proportion of target cells that are directly infected is small (29). If ART is not administered immediately after a high-risk exposure to HIV and infection takes hold, then the latent reservoir is irreversibly established within 10 days of primary infection (30).
During the 8 to 10 years of untreated infection that typically precede AIDS and death, plasma CD4 counts slowly decrease while the latent reservoir diversifies and slowly diverges from the founder strain. In patients who receive HAART, latently infected cells survive for decades despite the absence of high-level viremia and the immune cell depletion. Even successfully treated patients, who have no detectable HIV DNA in their peripheral CD4+ T cells or GALT and appear to have complete elimination of viral replication, experience a rebound of viremia within 50 days of cessation of therapy (32). This indicates that an undetectably small CD4+ T-cell or other reservoir that is either persistent or constantly replenishing is able to reconstitute infection (33) and will also have to be targeted with cleavage enzyme therapy.
The long-term mechanism that maintains the latent infection despite HAART is of importance when considering viral eradication strategies. One possibility is that resting CD4+ T cells are reseeded with low-level replication within the activated CD4+ T-cell compartments (33, 146). Proviral HIV-1 replication can be induced in latently infected CD4+ T cells (34). One study suggests that high multiplicity of infection within single cells renders HIV resistant to complete elimination (169), though this phenomenon has not been observed in vivo. This model of viral persistence assumes the presence of anatomic drug sanctuaries as a mechanism to support ongoing viral replication.
However, there are several findings to suggest that viral replication is nearly completely eliminated during successful HAART. Development of de novo drug resistance clones does not occur in circulating virions during fully suppressive HAART (84, 206). Moreover, viral blips persist at an unchanged rate following ultraintensification of regimens with four or more agents (48), and it is now believed that current ART regimens bind viral replication enzymes cooperatively and potently suppress nearly all replication (164, 165). Finally, recent mathematical models suggest that the latent pool of infected cells is not replenished due to low-level viral replication despite HAART and that the stability of the reservoir can be attributed to the long life span and occasional activation of latently infected cells (153, 161). The HIV reservoir includes T cells from two distinct populations: central memory T cells (TCM) and transitional memory cells (TTM). The long life of the TCM and the low levels of antigen-driven proliferation lead to the stability of this population, although it is slowly depleted over time (27). On the other hand, it has also been proposed that latently infected TTM persist via interleukin-7 (IL-7)-mediated homeostatic proliferation, which preserves not only the size of the reservoir but also its genetic variability. HIV-1-infected hematopoietic stem cells (HSCs) have been recently reported in vivo (22, 23), and these cells, with their capacity to self-renew over the lifetime of the individual, may possibly represent another component of the HIV reservoir. Yet more-recent reports identify no HIV DNA within CD34+ bone marrow progenitor cells (52, 79).
Molecular features of HIV-1 latency.
During latency, the integrated HIV provirus is transcriptionally repressed by a number of mechanisms which may impact success of the cleavage enzyme activity. When the provirus integrates into actively transcribed host genes, which occurs >90% of the time, transcriptional read-through from the upstream promoter can prevent formation of the initiation complex at the long terminal repeat (LTR) (72, 95, 160). In addition, resting cells lack the levels of host transcription factors necessary for robust viral gene expression (116, 181). The formation of repressive chromatin at the LTR—including loss of activating histone modifications, presence of repressive modifications, and presence of DNA methylation—has also been implicated in HIV latency (82, 132, 191). The accessibility of the DNA to recognition and cleavage by therapeutic enzymes may be compromised by the presence of heterochromatin and possibly by nucleosome positioning.
Gene delivery and genome-targeting efforts to date for HIV-1.
One novel approach to the problem of HIV latency is the use of gene editing to render T cells resistant to HIV. Initial experiments in mice have used a variety of approaches to downregulate CCR5, an entry receptor for HIV-1 strains (5, 47, 86, 91, 99, 166). Cells deficient for CCR5 expression would theoretically expand and become enriched due to the selective advantage conferred by resistance to HIV infection. If this therapy succeeds, then latent reservoirs would be unable to propagate widespread infection. This approach holds promise based on an apparently successful cure of HIV-1 achieved in a leukemia patient who was transplanted with CCR5Δ32/Δ32 stem cells (4, 78), although chemotherapy, radiation, antithymocyte globulin, and graft-versus-host disease may have all contributed to HIV eradication in this case.
Accordingly, there have been promising attempts to permanently knock down or knock out CCR5 expression with the use of engineered Zn finger nucleases (ZFN) that permanently disrupt the CCR5 gene in cells. Fifty percent of CCR5 alleles were disrupted using this technique: when treated human CD4+ cells were transplanted into a humanized mouse model, the mice exhibited lower viremia and slower decay of CD4+ cells after infection with HIV compared to mice transplanted with untreated cells (137). The same approach was used to decrease CCR5 expression in human hematopoietic stem cells (HSCs), although at a much lower frequency than in terminally differentiated cells: treated HSCs developed into multiple lineages of progeny in which CCR5 was nonfunctional. These engineered cells also successfully engrafted in a humanized mouse model, again allowing for lower viremia and less-severe CD4+ depletion after HIV-1 challenge (75).
Based on these encouraging results, Zn finger nucleases that cleave CCR5 have entered phase I clinical trials. Adverse effects due to the loss of CCR5 function are a concern. CCR5Δ32 has been implicated in more-severe disease during West Nile virus infection (61, 102), indicating an important role in certain protective immune pathways. In addition, all therapies targeting CCR5 can potentially select for the more pathogenic CXCR4-tropic HIV-1. However, ZFNs have also been used to target CXCR4 (204), which is not essential for normal T-cell function.
A second approach would modify the provirus within latent cells, thus preventing reactivation. This technique would not require adoptive transfer but would have challenges associated with delivery. One of the first attempts to target the latent provirus directly involved the use of a site-specific recombinase (SSR) to selectively excise integrated provirus. Recombinant HIV containing loxP sites in the U3 region of the long terminal repeat (LTR) was excised after integration by Cre recombinase (56). A Cre-derived SSR that recognized the HIV LTR successfully excised an integrated provirus containing the targeted LTR (157). However, the LTR sequence was chosen based on its similarity to the Cre recognition site and was atypical for HIV. LTR sequences from other HIV strains are much more divergent from the wild-type Cre recognition sequence. Directed evolution of these recombinases has been shown to change their sequence specificity, so it is possible that SSRs that recognize a variety of HIV LTRs may be generated (15, 19, 20).
Our lab recently published a more general demonstration of the ability of homing endonucleases to target and inactivate integrated virus (8) without excision of proviral DNA. We developed a lentivirus reporter bearing a recognition site for the HE Y2-AniI, a variant of I-AniI (178), inserted between the translational start site and the coding sequence for a short-half-life green fluorescent protein (GFP) reporter. Cleavage of the target site and subsequent repair by nonhomologous end joining caused small insertions and deletions at the site of cleavage, resulting in frame shifts, loss of the translational start site, or disruption of essential GFP-coding sequences. Thus, successful HE attack was monitored by loss of GFP expression. We found that Y2-AniI could efficiently target the integrated reporter lentivirus, resulting in mutagenesis of the target region in over 97% of Y2-AniI-expressing cells without evidence of detectable toxicity. These data suggest the possibility that incorporated proviral DNA can be selectively disabled without excision and without damage to the host cell.
As mentioned above, there are two potential strategies for a genome-targeting-based approach to HIV treatment. Strategies aimed at both the integrated provirus and cellular genes that permit HIV infection are promising but require enzyme delivery to specific but distinct cell types. For an approach targeting integrated HIV provirus, targeting enzymes to CD4+ cells showing active HIV replication in addition to CD4+ memory T cells that harbor reservoirs of latent HIV infection would be desirable. In contrast, the disruption of cellular genes that permit HIV infection would ideally be done in CD4− HSCs that would, if unaltered, give rise to CD4+ T cells permissive for HIV-1 infection (85). Although the disruption of cellular genes in CD4+ T cells can prevent HIV spread (137), HSC disruption would prevent reseeding of CD4+ T-cell reservoirs following therapy.
Efficient gene delivery to cells of hematopoietic origin, including CD4+ and CD34+ cells, can be achieved using a number of different approaches, and the methods available to investigators have been reviewed in detail elsewhere (117). In the context of DNA-editing enzyme delivery, the number of studies in CD4+ and CD34+ cells is small, although efficient enzyme delivery and genome targeting have been achieved. The nonviral Amaxa nucleofection gene delivery system has been used to efficiently deliver ZFNs targeting the IL-2R gamma gene or CCR5 to CD4+ T cells and CD34+ HSCs in ex vivo gene transfer protocols (189). Targeted CD34+ cells were able to engraft upon transplantation into immunodeficient mice, demonstrating the efficacy of this approach. Other groups have chosen viral vectors to target CD4+ and CD34+ cells. Adenovirus vectors that use CD46 as an entry receptor have been used to efficiently deliver ZFNs to CD4+ T cells that could engraft upon transplantation into immunodeficient mice (137, 204). Nonintegrating lentivirus vectors (also referred to as integrase-deficient lentiviruses [IDLVs]) have also successfully been used to deliver gene-targeted ZFNs to CD34+ cells (104). Although only a few delivery systems have been used to deliver DNA-editing enzymes to CD4+ and CD34+ cells, the existing data suggest that genome targeting could be a successful strategy for the elimination of HIV infection. However, delivery to every single infected cell may ultimately be necessary to achieve a cure.
HEPATITIS B VIRUS
Anatomic sites of chronic HBV infection for cleavage enzyme delivery.
HBV is primarily an infection of the liver, an organ that is highly vascularized and receives approximately 25% of systemic blood flow, with 33% and 67% contributions from arterial and portal circulation, respectively. Within the liver, specialized, fenestrated capillaries, called sinusoids, allow hepatocytes to be continually bathed in blood. Moreover, enzyme entry into hepatocytes occurs prior to first-pass hepatic metabolism. For these reasons, cleavage enzyme delivery to the liver should occur at high levels.
A potential challenge is that HBV infection disseminates widely beyond the liver: various investigators have detected viral DNA and surface antigen in lymph nodes, spleen, bone marrow, kidney, skin, gastrointestinal tract, pancreas, testes, and periadrenal ganglia. A broad range of cell lines, including endothelial cells, epithelial cells, neurons, macrophages, peripheral blood mononuclear cells, and polymorphic nuclear leukocytes are permissive for HBV replication, and extrahepatic manifestations of disease include medium- and small-vessel vasculitis, glomerulonephritis, aplastic anemia, myocarditis, and polyarthritis (45, 107, 139, 142, 205, 211). It is unknown whether these extrahepatic sites could serve as viable reservoirs for reseeding of the liver following isolated eradication of cccDNA genomes from liver cells.
HBV genome and cleavage enzyme target sites.
Several features make HBV an attractive candidate for eradication with cleavage enzymes (Table 1). Though HBV has a very small genome (3.2 kb), viral survival is highly dependent on a small number of viral proteins for replication. The HBV genome comprises four open reading frames (envelope, nucleocapsid, polymerase, and X protein) which are translated into only seven proteins. The two nucleocapsid products are hepatitis B virus c and e antigens (HBcAg and HBeAg, respectively). HBcAg is involved in viral packaging, while HBeAg plays a possible role in immunosuppression (112). A cellular immune response to these antigens is thought to be important for viral clearance (133). The envelope comprises three polypeptides, designated the large, medium, and small surface antigens, which are all heavily glycosylated (18). The X protein, which is absolutely essential for viral replication, can modulate host and viral gene expression as well as affect host-cell signal transduction (217). Specifically targeting any one of these proteins would likely be sufficient to greatly reduce or eliminate viral replication.
The polymerase protein includes reverse transcriptase, DNA polymerase, and RNase H domains and is essential for encapsidation and replication of the viral genome through the reverse transcription process. After arriving in the nucleus, the HBV genome is converted from a partially double-stranded relaxed closed DNA (rcDNA) into a covalently closed circular form (cccDNA) (121). cccDNA is the template for all viral protein synthesis and viral replication through a DNA-to-RNA-to-DNA mechanism that requires viral reverse transcriptase. Integration of the genome is not required for replication (118), and cccDNA exists in an episomal state.
Two mechanisms likely contribute to cccDNA persistence and are of interest for eradication strategies via cleavage enzymes. First, the cccDNA pool is expanded and maintained by a mechanism in which newly produced rcDNA, whose principle destination is in nascent budding viral particles, instead is recycled back into the nucleus and converted into additional cccDNA copies (88, 184). In addition, hepatocyte division can result in asymmetric distribution of HBV in progeny cells, which can be one way that HBV is eliminated (212). The kinetics of these two processes will impact the number and frequency of doses required for eradication of cccDNA stores.
The HBV reverse transcriptase enzyme induces mutations at a considerably lower rate (1.4 × 10−5 to 3.2 × 10−5 mutations/base pair/year) than HIV (2 × 10−5 mutations/base pair/day) (103). It is estimated that in a typical patient during a 24-hour period, 1012 HBV particles are produced and cleared: based on the mutation rate and genome length, 9 × 1010 and 4.5 × 109 single- and double-base pair mutants, respectively, are also produced, thus exceeding the total possible numbers of single- and double-base pair mutants within the HBV genome by factors of 10 million and 100, respectively (136). As a consequence, current antiviral therapies can cause resistance mutations, leading to a poorer overall prognosis. A successful strategy for eliminating cccDNA from hepatocytes may need to account for an abundance of diverse circulating particles. To this end, cleavage enzymes may not be effective if suppressive antiviral therapy is not initiated first to lower the burden of replicating and mutating HBV.
On the other hand, there are several lines of evidence suggesting that cccDNA may exhibit less sequence diversity within a host. First, HBV is intolerant to many mutations. Drug-resistant HBV tends to emerge months to years after initiating treatment rather than immediately, as with hepatitis C and HIV-1 treatment failure (152, 180). This suggests that HBV resistance is not the primary cause of viral escape from antiviral therapy (42). Second, HBV reverse transcriptase can remove newly incorporated nucleotides during replication, allowing for ongoing proofreading of the HBV genome (188). Moreover, work in a duck model of infection suggests that superinfection of preinfected hepatocytes with a second drug-resistant strain cannot occur, even if the second virus possesses a replicative advantage (197).
The HBV genome is also not exceedingly diverse globally: there are 8 known HBV genotypes, each of which has less than 4% within-clade sequence divergence, while divergence exceeding 8% and sometimes approaching 16% can be observed between different clades (141). HBV therefore appears to survive within a fairly limited evolutionary space. Importantly, several regions, including the spacer region of the polymerase ORF, are conserved across all genotypes (Z. Chen, personal communication). Successful targeting and cleavage of a sequence within this region could feasibly cause mutations and/or frameshift insertions/deletions that render the polymerase gene product nonfunctional. It will be crucial to target such highly conserved target sites. Sequence diversity within potential target sites may also be overcome by targeting multiple sites concurrently.
Latent HBV viral load.
A major hurdle to eradicating HBV will be the massive pool of infected cells compared to those of HIV-1 and HSV. Due to the nonlytic nature of HBV replication, infected cells turn over only slightly more rapidly than uninfected cells according to the intensity of the host T-cell response. HBV therefore has sufficient time to spread efficiently between hepatocytes. The use of PCR in situ hybridization technology has revealed that in persons with chronic active hepatitis B and wide ranges of plasma viral loads ranging from 103 to 109 HBV DNA copies/ml, virtually all hepatocytes harbor HBV DNA. On the other hand, HBV RNA and surface antigen, which are markers of replication, are present only in certain hepatic regions (127). HBV DNA was also detected in a mean of 5% of hepatocytes in persons with occult hepatitis B (positive low-level plasma HBV DNA with negative HBV surface antigen); moreover, 50% of subjects had detectable levels of HBV in circulating PBMCs (150). Hence, during all forms of chronic infection, HBV is widely disseminated throughout the liver, and in active disease, nearly all of the total 2 × 1011 hepatocytes are infected.
If uninfected hepatocytes emerge during infection as a result of asymmetric cccDNA homeostatic proliferation (212), these cells are presumably at high risk of rapid infection due to high levels of surrounding HBV virions. Mathematical models suggest that under certain circumstances, regenerating hepatocytes may be refractory to infection based on the surrounding cytokine milieu: this is a key feature of the proposed mechanism for viral clearance, which occurs in approximately 90% of acutely infected adults (35). It is estimated from animal models that the entire supply of hepatocytes turns over 1 to 3 times during the first year following acute infection (67, 115, 175). Similar rates of infected cell turnover are probable during chronic active infection. Given that cleavage enzyme therapies are unlikely to enhance host immunological responses, replenished cells will likely remain largely susceptible to HBV infection, potentially adding kindling to the fire. The regeneration of target cells will present a challenge to curative strategies and may necessitate prolonged courses of therapy.
Treatment strategies that attempt to target the entire pool of hepatocytes containing viral genomes will need to account for the total genomic burden of infection rather than just the number of infected cells. Fortunately, the genome persists at relatively low levels within each infected cell. Individual nuclei from duck liver cells infected with duck HBV were isolated and analyzed with nested PCR: a significant fraction of all nuclei (13%) contained exactly one copy of cccDNA, and few nuclei contained more than 50 cccDNA copies (212). These data correlate with viral load measurements from human livers that were standardized to 106 total hepatocytes: in chronically infected and untreated patients, the total genomic burden of cccDNA exceeded the number of infected cells by only a maximum of one log. On the other hand, cccDNA represented only 0.7 to 22.0% (median, 5.9%) of total HBV DNA within the liver, highlighting the need for concurrent use of antiviral therapy with cleavage enzyme therapies (93).
The quantity of total liver cccDNA varies enormously between chronically infected persons (93), likely as a function of the strength of the immunological response. Indeed, a patient's outcome after acute HBV infection appears to hinge on their immune status rather than a specific latency-inducing mechanism contained in the virus. Viral expansion during acute infection is limited via noncytolytic reduction of viral proteins by cytokines produced by activated T lymphocytes, while clearance of plasma viremia correlates with an intense cytolytic T-cell response (59, 65, 148). Accordingly, chronic infection results from an inadequate initial lymphocyte response to viral antigens (130, 148), though other components of the immune response may be lacking as well (64). The ability to contain infection is age dependent: 1 to 5% of adults and 90% of neonates infected with HBV fail to develop a sufficient immune response to clear the virus and develop chronic, persistent infection (207).
Despite the importance of immunological control in determining clinical outcomes, nonreplicating viral forms partially avoid cytolytic effects. Even after apparent clinical clearance of viral surface antigen from plasma and conversion to a positive antibody status during occult infection, a high burden of cccDNA remains within infected hepatocytes (150). HBV e antigen (HBeAg) is commonly used as a marker of viral replication and infectivity. In untreated chronic HBV patients positive for HBeAg, cccDNA copy numbers are higher than they are in untreated HBeAg-negative patients (3.0 median copies/cell and 0.31 median copies/cell, respectively) (93). Despite this lower viral genome load, intrahepatic cccDNA maintains replicative capacity even in asymptomatic, HBeAg-negative carriers. This accounts for the high probability of viral reactivation in severely immunocompromised hosts who have definitive evidence of previous clearance of virus (71). In addition, asymptomatic carriers who are positive for HBsAg and negative for HBeAg and who maintain normal liver function yet are unable to clear the virus through adequate immune system activation are at risk for developing hepatocellular carcinoma, indicating incomplete viral control (210).
cccDNA remains in the nucleus for the lifetime of the infected hepatocyte, which may span months to years (2), and is the probable source for partial viral rebound that occurs in approximately 34% of patients during antiviral therapy, even in the absence of drug resistance (76). Accordingly, although total clearance of HBV DNA from plasma occurs 90% of the time with optimal treatment regimens (25, 92), this does not correlate with cccDNA eradication and relapse is common after therapy is stopped, particularly in the eAg-negative disease state (168). Successfully treated patients achieve only a median 0.8-log reduction in cccDNA levels compared to untreated patients (203). Moreover, their decrease in cccDNA levels does not coincide with a reduction in the percentage of hepatocytes displaying markers of HBV infection.
Molecular features of HBV latency.
cccDNA exists as a minichromosome in the form of a nucleosome similar to host cell chromatin (121). Minichromosome structure has important regulatory effects on the transcription of viral genes and may also influence the accessibility of target sequences to DNA-recognizing enzymes.
Gene delivery and HBV genome-targeting efforts to date.
There are few examples of gene therapy approaches targeting HBV. One possible explanation is the lack of an adequate cell culture model for HBV infection (43). RNA interference (RNAi)-based approaches have been investigated (43, 167) but are limited because they do not directly eliminate episomal viral cccDNA and, rather, only slightly reduce the cccDNA pool via an effect on the recycling of viral DNA back into the nucleus (187). Zinc finger proteins (ZFPs) and zinc finger nucleases (ZFNs) have been used to target viral DNA. A specifically designed ZFP avidly bound to duck hepatitis B virus DNA after delivery to the nucleus via attachment to an SV40 nuclear localization signal. While ZFPs do not cleave DNA due to their lack of endonuclease activity, this approach resulted in substantial reductions in viral RNA, proteins, and viral progeny in cell culture (215). ZFNs can cleave both episomal and proviral DNA, followed by repair through highly error-prone nonhomologous end joining. In cell culture experiments, an HBV-specific ZFN showed site-specific cleavage leading to a reduction in pregenomic RNA levels without a loss in cell viability (39).
As the liver is readily accessible to a number of gene delivery vectors, any novel anti-HBV therapy has an inherent advantage, since almost the entire target cell population can be treated. Furthermore, several vectors that efficiently transduce the liver are readily available, have been well characterized, and have even been used in clinical trials. The most-common gene delivery systems for hepatic gene transfer are recombinant adenovirus (Ad) and adeno-associated virus (AAV) vectors which can efficiently transduce the liver without the need for an invasive delivery procedure. Although other methods of hepatic gene transfer are available (74), the high efficiency of gene transfer seen with Ad and AAV vectors makes them ideal candidates for the delivery of anti-HBV therapeutics. Upon intravenous delivery of a relatively moderate dose, Ad vectors can transduce 100% of hepatocytes with minimal toxicity (96). Consequently, Ad vectors have been used to deliver anti-HBV RNAi sequences to the livers of HBV transgenic mice with considerable success (40, 147). AAV vectors are also able to efficiently transduce the liver after delivery of moderate vector doses that cause minimal toxicity (216). They have also been used to deliver anti-HBV RNAi sequences to the livers of HBV transgenic mice with significant success (26, 60).
HERPES SIMPLEX VIRUS
Anatomic sites of HSV latency for cleavage enzyme delivery.
The major issue unique to HSV latency is that it occurs exclusively in anatomic sanctuaries (the trigeminal ganglia, dorsal root, and spinal cord) (129) (Table 1). HSV establishes latency during primary infection. After replicating in epidermal cells in oral (HSV-1) or genital (HSV-2) skin and mucosas, HSV invades the peripheral nervous system and spreads from neuronal endings via axons to cell bodies in the ganglia (151). HSV infection in epidermal cells is rapid and lytic, resulting in characteristic oral (usually HSV-1) and genital (usually HSV-2) ulcers with dense inflammatory infiltrates (41, 214), while infection in neurons leads to limited tissue damage and lytic viral replication only during the first week following infection (69). Recent evidence suggests that while latency may be tightly established at the single-cell level, the ganglia leak virus nearly constantly toward the genital tract (159). Mouse models show early and late transcripts in small numbers of unperturbed ganglionic neurons even in the absence of reactivation (54). Therefore, low-level focal replication is likely to occur even while most neurons within the ganglia remain quiescent.
HSV genome and cleavage enzyme targeting sites.
Several features of HSV latency may facilitate the design of cleavage enzymes that target key sites within the latent genome. First, the HSV genome is large and contains numerous essential replication genes. HSV-1 and -2 are enveloped viruses with linear double-stranded DNA genomes that are 152 kb and 154 kb in length, respectively (49, 109). HSV-1 contains approximately 94 transcription units (ORFs), of which 84 encode a protein (145). For HSV-2, 74 ORFs have been confirmed and matched to HSV-1. Out of the 84 HSV-1 proteins with a described function, 31 are essential for replication in culture (151). Introduction of mutations within the DNA reading frame for many viral protein sites may permanently disable HSV genomes, and in silico analyses suggest that >100 potentially useful targets may exist (201).
Second, target sites are likely to be relatively well conserved among isolates. HSV DNA genomes are more stable than those of RNA viruses due to an overall low mutation rate (3.5 × 10−8 mutations/site/year) (156) and high accuracy of replication associated with HSV DNA polymerase 3′-to-5′ exonuclease proofreading activity (10). Yet different isolates that have evolved for prolonged periods of time within distinct ecologic niches may vary substantially from one another. The genome sequences of one clinical and one laboratory HSV-1 isolate had 1% sequence divergence compared to the only other sequenced HSV-1 genome (from laboratory strain 17) (177). This level of variability between the three isolates may be an issue for targeted mutagenesis. However, two of the isolates compared were laboratory strains that had artificially undergone multiple passages in culture. In fact, in a study examining the sequence variation of a segment comprising 3.5% of the genome of HSV-2 isolates from different region of the globe, there was high similarity (99.6%) between the two most distant HSV-2 isolates (125). A phylogenetic analysis of clinical HSV-1 isolates determined the sequence diversity on a region representing 2.3% of the HSV-1 genome. Depending on the gene of the sequenced region, sequence differences ranging from 0.6% to 3.1% were observed (124). HSV-1 and HSV-2 are closely related at the nucleotide level, with 83% nucleotide identity when HSV-1 laboratory strain 17 was aligned with HSV-2 laboratory strain HG52 (49). Therefore, target sites found in one type have a reasonably high likelihood of being conserved in the other type.
Another concern is that viral escape mutants may emerge and persist within a person over time, as has been observed occasionally with antiviral therapy. Several reports identified that recombinant viruses are shed asymptomatically in some infected individuals, suggesting coinfection with more than one virus (101, 179). However, it has not been demonstrated that individual neurons are coinfected with genetically distinct genomes.
Latent HSV DNA viral load.
In nonimmunocompromised individuals, at a single point in time, HSV latency is established in only a small percentage of sensory neurons, ranging from 2.0 to 10.5% for HSV-1 (198). There are approximately 10,000 to 20,000 neuronal cell bodies per ganglion, and approximately 10 ganglia are at risk for infection, though the spinal cord may serve as another reservoir of HSV DNA. The number of HSV genome copies in each latently infected neuron is also modest (2 to 50 copies/cell for HSV-1) in most infected cells (198, 199). This suggests an overall infectious burden of ∼105 genomes. In mice, viral burden, defined by copy number and the number of infected cells, appears to be linked to recurrent disease frequency/rate (77). Therefore, it may not be necessary to eliminate all copies of the latent genome within every infected neuron to limit disease severity or completely eliminate viral shedding.
On the other hand, HSV-2 reactivations defined by high-level viral replication in genital skin near sensory nerve endings occur approximately weekly in humans (195). Moreover, human shedding patterns remain mostly unchanged over decades (138), meaning either that a pool of latently infected neurons is permanently established during primary infection or that steady state is achieved via a balance between elimination and replenishment of HSV within neurons. There is indeed some indirect evidence that latency may not be a static process at the cellular level. In human cadaver studies, dense CD8+ and CD4+ T-cell infiltrates congregate around neurons containing HSV DNA and only some infected neurons produce latency-associated transcripts (193). In mice, CD8+ lymphocytes with a cytolytic phenotype inactivate lytic HSV replication, not by killing infected neurons but, rather, by inactivating the immediate early protein ICP4 (87). These data suggest that regional HSV DNA levels in ganglia may possibly fluctuate over time due to local immune pressure. If neurites are seeded with HSV during each mucosal reactivation and the pool of latently infected ganglionic neurons is periodically replenished, then a more comprehensive dosing strategy may be necessary to achieve elimination of latently infected neurons.
Molecular features of HSV latency.
If enzyme delivery to an adequate number of infected ganglionic cells is accomplished, then several molecular features of HSV latency are likely to impact whether the latent genome is amenable for targeted mutagenesis. On one hand, latent HSV DNA is extrachromosomal and predominantly maintained in circular or concatenated forms referred to as episomal “endless DNA” (53, 149). This is advantageous because it is perhaps less likely that the DNA breaks and repair of the episomal DNA will lead to cellular chromosomal mutation, as might occur with integrated virus.
However, viral episomes are associated with the nucleosome in a chromatin structure, which may present challenges for endonuclease binding (46). During its latent state, HSV usually does not produce viral progeny and expresses only one transcript, called the latency-associated transcript (LAT). Viral transcription during latency is regulated not by DNA methylation but by posttranslational histone modifications (50, 90). During latency, the LAT region is associated with acetylated histones or active chromatin, whereas lytic-gene promoters are associated with heterochromatin (repressive) forms of histones (90). The presence of acetylated histones may influence how accessible a viral target sequence(s) is to cleavage enzymes. Histones may also present steric impediments to delivery of cellular repair machinery: fortunately, nonhomologous end joining is active throughout the cell cycle while homologous recombination (which would not induce lethal mutations in key viral genomic segments) is preferentially active in the S and G2 phases of the cell cycle (154). Because neurons are likely to be in cellular arrest phase, lethal-mutation-inducing nonhomologous end joining is likely to be favored.
Despite being terminally differentiated and postmitotic, ganglionic neurons are highly active cells. The high metabolic and transcriptional activity within neurons increases the potential for genomic DNA damage, necessitating a robust DNA damage response. While all eukaryotic DNA repair systems operate in neurons, DNA repair activity is slower relative to dividing cells and, therefore, genomic errors accumulate more rapidly. Despite the fact that DNA repair is attenuated at the global genome level, it is maintained in expressed genes which are, in turn, less prone to mutation (126). In addition, the homologous-recombination repair pathway may not be essential due to the postmitotic state of the neurons (11). This may also favor nonhomologous-end-joining repair of cleaved viral targets, something which is a prerequisite for effective eradication therapies.
Gene delivery and genome-targeting efforts to date.
To date, only one report describes HSV genome targeting by HEs. In cultured nonneuronal cells transiently expressing HSV-specific HEs, 2.8 to 16% of HSV-1 genomes harbored a mutation at the HE target site. In addition, viral replication and infection of cultured cells was substantially diminished when HEs were introduced prior to HSV infection at various multiplicities of infection. This study demonstrated that HSV genomes can be mutated following HE exposure, although cellular toxicity was evident with certain engineered HEs (63).
A few considerations will need to be addressed with all potential delivery systems. While expression of transgenes carried by such vectors has been demonstrated, expression is not sustained over an extended period of time. To resolve this issue, the LAT promoter could be used to drive expression of cleavage enzymes. However, when examined in a cross section, only one-third of infected neurons express LAT, a finding which appears to be due to histone modifications (198). The expression of the transgene under a cytomegalovirus (CMV) promoter or a similar expression promoter may be sustained long enough for genome targeting to occur. An advantage of such a promoter is its self-limiting phenotype, which may limit the potential side effects of long-term expression of homing endonucleases or other cleavage enzymes.
Delivery of the HE to the neurons where HSV is latent could be achieved by using replication-incompetent HSV-based delivery vectors (200), which have been developed for gene therapy of diseases of the nervous system (62). In animal models, HSV-1 and -2 display different tropisms for sensory neurons, with HSV-1 more likely to be detected during latency in neurons displaying the surface marker A5 and HSV-2 more likely to be detected during latency in those displaying KH10 (106). Therefore, the use of HSV-based delivery systems may allow matching of the delivery vectors to the HSV serotype targeted.
Adenovirus vectors have been employed in primary cultures of latently infected trigeminal ganglia (TG) (70). AAV and lentivirus vectors have been considered for gene delivery to dorsal root ganglion neurons and have achieved efficient and sustained transduction within human sensory neurons in dissociated cultures, demonstrating the potential of these vectors for gene therapy applications in the peripheral nervous system (55, 70).
A particular challenge of cleavage enzyme delivery to the ganglia is the blood brain barrier, a structure that isolates and protects the CNS from harmful macromolecules. This is an area of great interest for research fields dedicated to the treatment of brain tumors, Alzheimer's disease, and Parkinson's disease. Various strategies are being investigated to avoid the need for direct intracranial drug delivery. Noninvasive strategies include drug manipulation, carrier-mediated drug delivery, receptor/vector-mediated delivery, and intranasal drug delivery (131, 144), and some of these approaches may prove useful in gene therapy efforts. Nanotechnology applications include the delivery of drugs and other small molecules, such as genes and oligonucleotides, across the barrier (182).
CONCLUSIONS
There is a new focus on strategies that directly target latent viral genomes with the goal of eradicating chronic viral infection. If cleavage enzymes can be successfully targeted to latently infected cells and can subsequently incapacitate key viral proteins, then the possibility of a viral cure exists. Several promising types of cleavage enzymes that target key regions within the HIV-1, HBV, and HSV genomes are in development. When in vivo delivery becomes a goal, considerations will need to include the unique pharmacokinetic/pharmacodynamic features of cleavage enzymes, the challenges with specific sites of latency for these infections, the development of viral resistance due to high viral mutability, the density of latently infected cells, and the molecular and steric constraints to enzyme delivery. These challenges call for the concurrent use of cell culture systems, animal models, mathematical models, and strategic clinic trial design in order to maximize the likelihood of success.
ACKNOWLEDGMENTS
We thank Barry Stoddard, Tae-Wook Chun, Lorne Tyrrell, and Todd Margolis for their helpful comments on the manuscript.
Portions of the work discussed here were supported by grant 107772-47-RGNT from the American Foundation for AIDS Research, Grand Challenges Explorations (GCE) Phase I grant 51763 and GCE Phase II grant OPP1018811 from the Bill and Melinda Gates Foundation, pilot awards from the Northwest Genome Engineering Consortium and the Vaccine and Infectious Disease Division of the Fred Hutchinson Cancer Research Center to K.R.J., and NIH U19 AI96111 to K.R.J. and Hans-Peter Kiem.
Biographies

Joshua T. Schiffer is a clinician specializing in infectious diseases with particular interests in the management of HIV-infected patients and other immunocompromised hosts. The aim of his research program is to gain a better understanding of the quantitative features of human pathogens and immune responses. In close collaboration with several colleagues at the Fred Hutchinson Cancer Research Center and the University of Washington, he designs mathematical models that capture growth and decay kinetics of infectious organisms. These models attempt to replicate detailed empirical datasets and, in turn, are used to inform subsequent human studies and laboratory experiments. Model results inform treatment strategies and attempt to identify hypotheses that may ultimately inform certain treatment and prevention strategies.

Martine Aubert obtained her Ph.D in bacterial genetics at the University of Nancy I (France) and did her postdoctoral training on herpesvirus biology with Dr. J. A. Blaho in the Department of Microbiology at Mount Sinai School of Medicine, New York, NY. She is currently a Senior Staff Scientist in the laboratory of Dr. Keith R. Jerome, in the Vaccine and Infectious Disease Division at the Fred Hutchinson Cancer Research Center, Seattle, WA. Her research efforts have focused on new therapeutic approaches to cure latent viral infection by targeted mutagenesis using designer rare cutting nucleases since the initiation of the project in 2008. These efforts have resulted in many new and exciting collaborative multidisciplinary projects spanning the fields of virology, immunology, structural biology, and cell biology. While the original work targeted the elimination of latent HIV infection, it is now expanded to other latent infectious agents, including herpes simplex virus, hepatitis B virus, and human papillomavirus.

Nicholas D. Weber obtained his Ph.D. at the Universidad Autónoma de Madrid in Madrid, Spain, where he carried out research on nanoparticle delivery vectors for small interfering RNA (siRNA) to HIV-infected lymphocytes in the Laboratory of Molecular Immunology of Gregorio Marañón Hospital. He has been awarded internships at the National Institute of Allergy and Infectious Diseases of the NIH in Hamilton, Montana, where he worked with interactions between exogenous and endogenous retroviruses, and at the University of Montana Division of Biological Sciences to work with intracellular signal receptor transactivation and sorting in endosomes. He began his scientific career doing research in rechargeable satellite fuel cell degradation at the Aerospace Corporation in Los Angeles, CA, where he also obtained his bachelor's degree in Chemistry at Occidental College. He currently holds a postdoctoral fellowship at Fred Hutchinson Cancer Research Center, where he works with DNA-targeting enzymes as a way of potentially addressing hepatitis B virus infection.

Esther Mintzer received her B.S. from the University of Idaho, where she investigated nuclear pore complex degradation during rhinovirus infection in the laboratory of Dr. Kurt Gustin. She then worked at Pacific Northwest National Laboratory, conducting research in copper electrochemistry for the MAJORANA neutrinoless double-beta decay experiment. She is currently a Ph.D. student in microbiology at the University of Washington, working in the laboratories of Dr. Keith R. Jerome and Dr. David Baker. Her doctoral research is focused on computational design of homing endonucleases for use as HIV therapeutics.

Daniel Stone obtained a B.Sc in Biochemistry in 1997 and a Ph.D. in gene therapy in 2001 from the University of Manchester, United Kingdom. Subsequently, he moved to the United States and held positions as a Senior Fellow and then an Instructor at the University of Washington, Seattle. Dr. Stone has held positions as an Associate Specialist at the University of California, Berkeley, and an Associate Scientist at California Pacific Medical Center, San Francisco. He is currently a Staff Scientist at the Fred Hutchinson Cancer Research Center. Dr. Stone has been developing adenovirus and adeno-associated virus vectors for over 10 years, and he recently joined the group of Dr. Keith R. Jerome to help develop delivery systems for DNA-editing enzymes targeting latent viral infections.

Keith R. Jerome is Head of the Virology Division in the University of Washington Department of Laboratory Medicine and leads the combined program in Infectious Disease Sciences/Virology at the Fred Hutchinson Cancer Research Center. He received his M.D. and Ph.D. degrees from Duke University, and subsequently did a fellowship in virology at the University of Washington under the mentorship of Dr. Lawrence Corey. Dr. Jerome's clinical focus is on the diagnosis of viral infections and the role the laboratory can play in improved patient care. The aim of his research program is to use gene editing techniques to develop new therapeutic approaches to previously incurable viral infections, including HIV, hepatitis B virus, human herpesvirus, and human papillomavirus infections.
Footnotes
Published ahead of print 20 June 2012
REFERENCES
- 1. Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. 2010. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 84:9864–9878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Addison WR, et al. 2002. Half-life of the duck hepatitis B virus covalently closed circular DNA pool in vivo following inhibition of viral replication. J. Virol. 76:6356–6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aiuti A, et al. 2009. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 360:447–458 [DOI] [PubMed] [Google Scholar]
- 4. Allers K, et al. 2011. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood 117:2791–2799 [DOI] [PubMed] [Google Scholar]
- 5. Anderson JS, Walker J, Nolta JA, Bauer G. 2009. Specific transduction of HIV-susceptible cells for CCR5 knockdown and resistance to HIV infection: a novel method for targeted gene therapy and intracellular immunization. J. Acquir. Immune Defic. Syndr. 52:152–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antiretroviral Therapy Cohort Collaboration 2008. Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies. Lancet 372:293–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ashworth J, et al. 2010. Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res. 38:5601–5608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aubert M, et al. 2011. Successful targeting and disruption of an integrated reporter lentivirus using the engineered homing endonuclease Y2 I-AniI. PLoS One 6:e16825 doi:10.1371/journal.pone.0016825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bailey JR, et al. 2006. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J. Virol. 80:6441–6457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baker RO, Hall JD. 1998. Impaired mismatch extension by a herpes simplex DNA polymerase mutant with an editing nuclease defect. J. Biol. Chem. 273:24075–24082 [DOI] [PubMed] [Google Scholar]
- 11. Barzilai A, Biton S, Shiloh Y. 2008. The role of the DNA damage response in neuronal development, organization and maintenance. DNA Repair (Amst.) 7:1010–1027 [DOI] [PubMed] [Google Scholar]
- 12. Boch J, et al. 2009. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512 [DOI] [PubMed] [Google Scholar]
- 13. Bogdanove AJ, Schornack S, Lahaye T. 2010. TAL effectors: finding plant genes for disease and defense. Curr. Opin. Plant Biol. 13:394–401 [DOI] [PubMed] [Google Scholar]
- 14. Bohnlein E, et al. 1988. The same inducible nuclear proteins regulates mitogen activation of both the interleukin-2 receptor-alpha gene and type 1 HIV. Cell 53:827–836 [DOI] [PubMed] [Google Scholar]
- 15. Bolusani S, et al. 2006. Evolution of variants of yeast site-specific recombinase Flp that utilize native genomic sequences as recombination target sites. Nucleic Acids Res. 34:5259–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brennan TP, et al. 2009. Analysis of human immunodeficiency virus type 1 viremia and provirus in resting CD4+ T cells reveals a novel source of residual viremia in patients on antiretroviral therapy. J. Virol. 83:8470–8481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brown EL, et al. 2007. Effect of maternal herpes simplex virus (HSV) serostatus and HSV type on risk of neonatal herpes. Acta Obstet. Gynecol. Scand. 86:523–529 [DOI] [PubMed] [Google Scholar]
- 18. Bruss V, Ganem D. 1991. The role of envelope proteins in hepatitis B virus assembly. Proc. Natl. Acad. Sci. U. S. A. 88:1059–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buchholz F. 2008. Molecular evolution of the tre recombinase. J. Vis. Exp. doi:10.3791/791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buchholz F, Hauber J. 2011. In vitro evolution and analysis of HIV-1 LTR-specific recombinases. Methods 53:102–109 [DOI] [PubMed] [Google Scholar]
- 21. Bull M, et al. 2009. Compartmentalization of HIV-1 within the female genital tract is due to monotypic and low-diversity variants not distinct viral populations. PLoS One 4:e7122 doi:10.1371/journal.pone.0007122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carter CC, et al. 2011. HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe 9:223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carter CC, et al. 2010. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat. Med. 16:446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cermak T, et al. 2011. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39:e82 doi:10.1093/nar/gkr218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang TT, et al. 2006. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 354:1001–1010 [DOI] [PubMed] [Google Scholar]
- 26. Chen CC, et al. 2009. Comparative study of anti-hepatitis B virus RNA interference by double-stranded adeno-associated virus serotypes 7, 8, and 9. Mol. Ther. 17:352–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chomont N, et al. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 15:893–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Christian M, et al. 2010. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186:757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chun TW, et al. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188 [DOI] [PubMed] [Google Scholar]
- 30. Chun TW, et al. 1998. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 95:8869–8873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chun TW, et al. 1995. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1:1284–1290 [DOI] [PubMed] [Google Scholar]
- 32. Chun TW, et al. 2010. Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of HIV reservoir: implications for eradication. AIDS 24:2803–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chun TW, et al. 2005. HIV-infected individuals receiving effective antiviral therapy for extended periods of time continually replenish their viral reservoir. J. Clin. Invest. 115:3250–3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chun TW, et al. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 94:13193–13197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ciupe SM, Ribeiro RM, Nelson PW, Dusheiko G, Perelson AS. 2007. The role of cells refractory to productive infection in acute hepatitis B viral dynamics. Proc. Natl. Acad. Sci. U. S. A. 104:5050–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cohen C, et al. 2008. Underestimation of chronic hepatitis B virus infection in the United States of America. J. Viral Hepat. 15:12–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cohen J. 2010. Immunology. Painful failure of promising genital herpes vaccine. Science 330:304. [DOI] [PubMed] [Google Scholar]
- 38. Corey L, et al. 2004. Once-daily valacyclovir to reduce the risk of transmission of genital herpes. N. Engl. J. Med. 350:11–20 [DOI] [PubMed] [Google Scholar]
- 39. Cradick TJ, Keck K, Bradshaw S, Jamieson AC, McCaffrey AP. 2010. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 18:947–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Crowther C, et al. 2008. Efficient inhibition of hepatitis B virus replication in vivo, using polyethylene glycol-modified adenovirus vectors. Hum. Gene Ther. 19:1325–1331 [DOI] [PubMed] [Google Scholar]
- 41. Cunningham AL, Turner RR, Miller AC, Para MF, Merigan TC. 1985. Evolution of recurrent herpes simplex lesions. An immunohistologic study. J. Clin. Invest. 75:226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dahari H, Shudo E, Ribeiro RM, Perelson AS. 2009. Modeling complex decay profiles of hepatitis B virus during antiviral therapy. Hepatology 49:32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dandri M, Petersen J. 2012. Chimeric mouse model of hepatitis B virus infection. J. Hepatol. 56:493–495 [DOI] [PubMed] [Google Scholar]
- 44. Deere JD, Schinazi RF, North TW. 2011. Simian immunodeficiency virus macaque models of HIV latency. Curr. Opin. HIV AIDS 6:57–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dejean A, Lugassy C, Zafrani S, Tiollais P, Brechot C. 1984. Detection of hepatitis B virus DNA in pancreas, kidney and skin of two human carriers of the virus. J. Gen. Virol. 65(Part 3):651–655 [DOI] [PubMed] [Google Scholar]
- 46. Deshmane SL, Fraser NW. 1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 63:943–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DiGiusto DL, et al. 2010. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl Med. 2:36ra43 doi:10.1126/scitranslmed.3000931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dinoso JB, et al. 2009. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 106:9403–9408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dolan A, Jamieson FE, Cunningham C, Barnett BC, McGeoch DJ. 1998. The genome sequence of herpes simplex virus type 2. J. Virol. 72:2010–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dressler GR, Rock DL, Fraser NW. 1987. Latent herpes simplex virus type 1 DNA is not extensively methylated in vivo. J. Gen. Virol. 68(Part 6):1761–1765 [DOI] [PubMed] [Google Scholar]
- 51. Dumolard L, Gacic-Dobo M, Shapiro CN, Wiersma S. 2008. Implementation of newborn hepatitis B vaccination—worldwide, 2006. MMWR Morb. Mortal. Wkly. Rep. 57:1249–1252 [PubMed] [Google Scholar]
- 52. Durand CM, et al. 2012. HIV-1 DNA is detected in bone marrow populations containing CD4+ T cells but is not found in purified CD34+ hematopoietic progenitor cells in most patients on antiretroviral therapy. J. Infect. Dis. 205:1014–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Efstathiou S, Minson AC, Field HJ, Anderson JR, Wildy P. 1986. Detection of herpes simplex virus-specific DNA sequences in latently infected mice and in humans. J. Virol. 57:446–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feldman LT, et al. 2002. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc. Natl. Acad. Sci. U. S. A. 99:978–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fleming J, et al. 2001. Adeno-associated virus and lentivirus vectors mediate efficient and sustained transduction of cultured mouse and human dorsal root ganglia sensory neurons. Hum. Gene Ther. 12:77–86 [DOI] [PubMed] [Google Scholar]
- 56. Flowers CC, Woffendin C, Petryniak J, Yang S, Nabel GJ. 1997. Inhibition of recombinant human immunodeficiency virus type 1 replication by a site-specific recombinase. J. Virol. 71:2685–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Folks T, et al. 1986. Induction of HTLV-III/LAV from a nonvirus-producing T-cell line: implications for latency. Science 231:600–602 [DOI] [PubMed] [Google Scholar]
- 58. Gandhi RT, et al. 2010. The effect of raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: a randomized controlled trial. PLoS Med. 7:e1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ganem D, Prince AM. 2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350:1118–1129 [DOI] [PubMed] [Google Scholar]
- 60. Giering JC, Grimm D, Storm TA, Kay MA. 2008. Expression of shRNA from a tissue-specific pol II promoter is an effective and safe RNAi therapeutic. Mol. Ther. 16:1630–1636 [DOI] [PubMed] [Google Scholar]
- 61. Glass WG, et al. 2006. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 203:35–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Glorioso JC, Fink DJ. 2004. Herpes vector-mediated gene transfer in treatment of diseases of the nervous system. Annu. Rev. Microbiol. 58:253–271 [DOI] [PubMed] [Google Scholar]
- 63. Grosse S, et al. 2011. Meganuclease-mediated inhibition of HSV1 infection in cultured cells. Mol. Ther. 19:694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guidotti LG, Matzke B, Schaller H, Chisari FV. 1995. High-level hepatitis B virus replication in transgenic mice. J. Virol. 69:6158–6169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guidotti LG, et al. 1999. Viral clearance without destruction of infected cells during acute HBV infection. Science 284:825–829 [DOI] [PubMed] [Google Scholar]
- 66. Gulick R, et al. 2006. Three- vs four-drug antiretroviral regimens for the initial treatment of HIV-1 infection: a randomized controlled trial. JAMA 296:769–781 [DOI] [PubMed] [Google Scholar]
- 67. Guo JT, et al. 2000. Apoptosis and regeneration of hepatocytes during recovery from transient hepadnavirus infections. J. Virol. 74:1495–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gupta A, Meng X, Zhu LJ, Lawson ND, Wolfe SA. 2011. Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res. 39:381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gussow AM, et al. 2006. Tissue-specific splicing of the herpes simplex virus type 1 latency-associated transcript (LAT) intron in LAT transgenic mice. J. Virol. 80:9414–9423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Halford WP, Kemp CD, Isler JA, Davido DJ, Schaffer PA. 2001. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J. Virol. 75:6143–6153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hammond SP, et al. 2009. Hepatitis B virus reactivation following allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 15:1049–1059 [DOI] [PubMed] [Google Scholar]
- 72. Han Y, et al. 2004. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J. Virol. 78:6122–6133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Harrison L., et al. 2010. The effect of transmitted HIV-1 drug resistance on pre-therapy viral load. AIDS 24:1917–1922 [DOI] [PubMed] [Google Scholar]
- 74. Herzog RW. 2005. Recent advances in hepatic gene transfer: more efficacy and less immunogenicity. Curr. Opin. Drug Discov. Devel. 8:199–206 [PubMed] [Google Scholar]
- 75. Holt N, et al. 2010. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 28:839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hongthanakorn C, et al. 2011. Virological breakthrough and resistance in patients with chronic hepatitis B receiving nucleos(t)ide analogues in clinical practice. Hepatology 53:1854–1863 [DOI] [PubMed] [Google Scholar]
- 77. Hoshino Y, Pesnicak L, Cohen J, Straus S. 2007. Rates of reactivation of latent herpes simplex virus from mouse trigeminal ganglia ex vivo correlate directly with viral load and inversely with number of infiltrating CD8+ T cells. J. Virol. 81:8157–8164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hutter G, et al. 2009. Long-term control of HIV by CCR5 delta32/delta32 stem-cell transplantation. N. Engl. J. Med. 360:692–698 [DOI] [PubMed] [Google Scholar]
- 79. Josefsson L, et al. 2012. Hematopoietic precursor cells isolated from patients on long term suppressive HIV therapy did not contain HIV-1 DNA. J. Infect. Dis. 206:28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Josefsson L, et al. 2011. Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc. Natl. Acad. Sci. U. S. A. 108:11199–11204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jung A, et al. 2002. Recombination: multiply infected spleen cells in HIV patients. Nature 418:144. [DOI] [PubMed] [Google Scholar]
- 82. Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. 2009. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 5:e1000495 doi:10.1371/journal.ppat.1000495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Keele BF, et al. 2008. Characterization of the follicular dendritic cell reservoir of human immunodeficiency virus type 1. J. Virol. 82:5548–5561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kieffer TL, et al. 2004. Genotypic analysis of HIV-1 drug resistance at the limit of detection: virus production without evolution in treated adults with undetectable HIV loads. J. Infect. Dis. 189:1452–1465 [DOI] [PubMed] [Google Scholar]
- 85. Kiem HP, Jerome KR, Deeks SG, McCune JM. 2012. Hematopoietic-stem-cell-based gene therapy for HIV disease. Cell Stem Cell 10:137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kim SS, et al. 2010. RNAi-mediated CCR5 silencing by LFA-1-targeted nanoparticles prevents HIV infection in BLT mice. Mol. Ther. 18:370–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Knickelbein JE, et al. 2008. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322:268–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kock J, et al. 2010. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 6:e1001082 doi:10.1371/journal.ppat.1001082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Koenig S, et al. 1986. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 233:1089–1093 [DOI] [PubMed] [Google Scholar]
- 90. Kubat NJ, Tran RK, McAnany P, Bloom DC. 2004. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 78:1139–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kumar P, et al. 2008. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell 134:577–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lai CL, et al. 2006. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 354:1011–1020 [DOI] [PubMed] [Google Scholar]
- 93. Laras A, Koskinas J, Dimou E, Kostamena A, Hadziyannis SJ. 2006. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology 44:694–702 [DOI] [PubMed] [Google Scholar]
- 94. Lavanchy D. 2004. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 11:97–107 [DOI] [PubMed] [Google Scholar]
- 95. Lenasi T, Contreras X, Peterlin BM. 2008. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe 4:123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Li Q, Kay MA, Finegold M, Stratford-Perricaudet LD, Woo SL. 1993. Assessment of recombinant adenoviral vectors for hepatic gene therapy. Hum. Gene Ther. 4:403–409 [DOI] [PubMed] [Google Scholar]
- 97. Li T, et al. 2011. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 39:359–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li T, et al. 2011. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 39:6315–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Liang M, et al. 2010. Inhibition of HIV-1 infection by a unique short hairpin RNA to chemokine receptor 5 delivered into macrophages through hematopoietic progenitor cell transduction. J. Gene Med. 12:255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Liaw YF, Chu CM. 2009. Hepatitis B virus infection. Lancet 373:582–592 [DOI] [PubMed] [Google Scholar]
- 101. Liljeqvist JA, Tunback P, Norberg P. 2009. Asymptomatically shed recombinant herpes simplex virus type 1 strains detected in saliva. J. Gen. Virol. 90:559–566 [DOI] [PubMed] [Google Scholar]
- 102. Lim JK, et al. 2010. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J. Infect. Dis. 201:178–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Locarnini S, Zoulim F. 2010. Molecular genetics of HBV infection. Antivir. Ther. 15(Suppl 3):3–14 [DOI] [PubMed] [Google Scholar]
- 104. Lombardo A, et al. 2007. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 25:1298–1306 [DOI] [PubMed] [Google Scholar]
- 105. Mansky LM, Temin HM. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Margolis TP, Imai Y, Yang L, Vallas V, Krause PR. 2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J. Virol. 81:1872–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mason A, Wick M, White H, Perrillo R. 1993. Hepatitis B virus replication in diverse cell types during chronic hepatitis B virus infection. Hepatology 18:781–789 [DOI] [PubMed] [Google Scholar]
- 108. May MT, et al. 2006. HIV treatment response and prognosis in Europe and North America in the first decade of highly active antiretroviral therapy: a collaborative analysis. Lancet 368:451–458 [DOI] [PubMed] [Google Scholar]
- 109. McGeoch DJ, et al. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69(Part 7):1531–1574 [DOI] [PubMed] [Google Scholar]
- 110. McMahon MA, Rahdar M, Porteus M. 2012. Gene editing: not just for translation anymore. Nat. Methods 9:28–31 [DOI] [PubMed] [Google Scholar]
- 111. Mehandru S, et al. 2004. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 200:761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Milich D, Liang TJ. 2003. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology 38:1075–1086 [DOI] [PubMed] [Google Scholar]
- 113. Morgan D, et al. 2002. HIV-1 infection in rural Africa: is there a difference in median time to AIDS and survival compared with that in industrialized countries? AIDS 16:597–603 [DOI] [PubMed] [Google Scholar]
- 114. Moscou MJ, Bogdanove AJ. 2009. A simple cipher governs DNA recognition by TAL effectors. Science 326:1501. [DOI] [PubMed] [Google Scholar]
- 115. Murray JM, Wieland SF, Purcell RH, Chisari FV. 2005. Dynamics of hepatitis B virus clearance in chimpanzees. Proc. Natl. Acad. Sci. U. S. A. 102:17780–17785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nabel G, Baltimore D. 1987. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326:711–713 [DOI] [PubMed] [Google Scholar]
- 117. Naldini L. 2011. Ex vivo gene transfer and correction for cell-based therapies. Nat. Rev. Genet. 12:301–315 [DOI] [PubMed] [Google Scholar]
- 118. Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res. 134:235–249 [DOI] [PubMed] [Google Scholar]
- 119. Nathwani AC, et al. 2011. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 365:2357–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Neuhaus J, et al. 2010. Risk of all-cause mortality associated with nonfatal AIDS and serious non-AIDS events among adults infected with HIV. AIDS 24:697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Newbold JE, et al. 1995. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 69:3350–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Nickle DC, et al. 2003. Evolutionary indicators of human immunodeficiency virus type 1 reservoirs and compartments. J. Virol. 77:5540–5546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Nickle DC, Shriner D, Mittler JE, Frenkel LM, Mullins JI. 2003. Importance and detection of virus reservoirs and compartments of HIV infection. Curr. Opin. Microbiol. 6:410–416 [DOI] [PubMed] [Google Scholar]
- 124. Norberg P, Bergstrom T, Rekabdar E, Lindh M, Liljeqvist JA. 2004. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J. Virol. 78:10755–10764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Norberg P, Kasubi MJ, Haarr L, Bergstrom T, Liljeqvist JA. 2007. Divergence and recombination of clinical herpes simplex virus type 2 isolates. J. Virol. 81:13158–13167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Nouspikel T, Hanawalt PC. 2000. Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Mol. Cell. Biol. 20:1562–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Nuriya H, et al. 2010. Detection of hepatitis B and C viruses in almost all hepatocytes by modified PCR-based in situ hybridization. J. Clin. Microbiol. 48:3843–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Nutt RF, et al. 1988. Chemical synthesis and enzymatic activity of a 99-residue peptide with a sequence proposed for the human immunodeficiency virus protease. Proc. Natl. Acad. Sci. U. S. A. 85:7129–7133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ohashi M, Bertke AS, Patel A, Krause PR. 2011. Spread of herpes simplex virus to the spinal cord is independent of spread to dorsal root ganglia. J. Virol. 85:3030–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Pasquetto V, Wieland S, Chisari FV. 2000. Intracellular hepatitis B virus nucleocapsids survive cytotoxic T-lymphocyte-induced apoptosis. J. Virol. 74:9792–9796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Pathan SA, et al. 2009. CNS drug delivery systems: novel approaches. Recent Pat. Drug Deliv. Formul. 3:71–89 [DOI] [PubMed] [Google Scholar]
- 132. Pearson R, et al. 2008. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 82:12291–12303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Penna A, et al. 1992. Hepatitis B virus (HBV)-specific cytotoxic T-cell (CTL) response in humans: characterization of HLA class II-restricted CTLs that recognize endogenously synthesized HBV envelope antigens. J. Virol. 66:1193–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Perelson A, et al. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188–191 [DOI] [PubMed] [Google Scholar]
- 135. Perelson A, Neumann A, Markowitz M, Leonard J, Ho D. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582–1586 [DOI] [PubMed] [Google Scholar]
- 136. Perelson AS, Ribeiro RM. 2004. Hepatitis B virus kinetics and mathematical modeling. Semin. Liver Dis. 24(Suppl 1):11–16 [DOI] [PubMed] [Google Scholar]
- 137. Perez EE, et al. 2008. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 26:808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Phipps W, et al. 2011. Persistent genital herpes simplex virus-2 shedding years following the first clinical episode. J. Infect. Dis. 203:180–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Pontisso P, Poon MC, Tiollais P, Brechot C. 1984. Detection of hepatitis B virus DNA in mononuclear blood cells. BMJ 288:1563–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Porteus MH, Carroll D. 2005. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 23:967–973 [DOI] [PubMed] [Google Scholar]
- 141. Purdy MA, Gonzales AC, Dimitrova Z, Khudyakov Y. 2008. Supragenotypic groups of the hepatitis B virus genome. J. Gen. Virol. 89:1179–1183 [DOI] [PubMed] [Google Scholar]
- 142. Pyrsopoulos NT, Reddy KR. 2001. Extrahepatic manifestations of chronic viral hepatitis. Curr. Gastroenterol. Rep. 3:71–78 [DOI] [PubMed] [Google Scholar]
- 143. Rahman SH, Maeder ML, Joung JK, Cathomen T. 2011. Zinc-finger nucleases for somatic gene therapy: the next frontier. Hum. Gene Ther. 22:925–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Rajadhyaksha M, Boyden T, Liras J, El-Kattan A, Brodfuehrer J. 2011. Current advances in delivery of biotherapeutics across the blood-brain barrier. Curr. Drug Discov. Technol. 8:87–101 [DOI] [PubMed] [Google Scholar]
- 145. Rajcani J, Andrea V, Ingeborg R. 2004. Peculiarities of herpes simplex virus (HSV) transcription: an overview. Virus Genes 28:293–310 [DOI] [PubMed] [Google Scholar]
- 146. Ramratnam B, et al. 2000. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 6:82–85 [DOI] [PubMed] [Google Scholar]
- 147. Rauschhuber C, Xu H, Salazar FH, Marion PL, Ehrhardt A. 2008. Exploring gene-deleted adenoviral vectors for delivery of short hairpin RNAs and reduction of hepatitis B virus infection in mice. J. Gene Med. 10:878–889 [DOI] [PubMed] [Google Scholar]
- 148. Rehermann B, Ferrari C, Pasquinelli C, Chisari FV. 1996. The hepatitis B virus persists for decades after patients' recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 2:1104–1108 [DOI] [PubMed] [Google Scholar]
- 149. Rock DL, Fraser NW. 1985. Latent herpes simplex virus type 1 DNA contains two copies of the virion DNA joint region. J. Virol. 55:849–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Rodriguez-Inigo E, et al. 2003. Distribution of hepatitis B virus in the liver of chronic hepatitis C patients with occult hepatitis B virus infection. J. Med. Virol. 70:571–580 [DOI] [PubMed] [Google Scholar]
- 151. Roizman B, KD 2007. Herpes simplex viruses and their replication. In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 152. Rong L, Dahari H, Ribeiro RM, Perelson AS. 2010. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci. Transl. Med. 2:30ra32 doi:10.1126/scitranslmed.3000544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Rong L, Perelson AS. 2009. Modeling latently infected cell activation: viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy. PLoS Comput. Biol. 5:e1000533 doi:10.1371/journal.pcbi.1000533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Rothkamm K, Kruger I, Thompson LH, Lobrich M. 2003. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 23:5706–5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Rouet P, Smih F, Jasin M. 1994. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 91:6064–6068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Sakaoka H, et al. 1994. Quantitative analysis of genomic polymorphism of herpes simplex virus type 1 strains from six countries: studies of molecular evolution and molecular epidemiology of the virus. J. Gen. Virol. 75(Part 3):513–527 [DOI] [PubMed] [Google Scholar]
- 157. Sarkar I, Hauber I, Hauber J, Buchholz F. 2007. HIV-1 proviral DNA excision using an evolved recombinase. Science 316:1912–1915 [DOI] [PubMed] [Google Scholar]
- 158. Schiffer JT, Corey L. 2009. New concepts in understanding genital herpes. Curr. Infect. Dis. Rep. 11:457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Schiffer JT, Wald A, Selke S, Corey L, Magaret A. 2011. The kinetics of mucosal herpes simplex virus-2 infection in humans: evidence for rapid viral-host interactions. J. Infect. Dis. 204:554–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Schroder AR, et al. 2002. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110:521–529 [DOI] [PubMed] [Google Scholar]
- 161. Sedaghat AR, Siliciano JD, Brennan TP, Wilke CO, Siliciano RF. 2007. Limits on replenishment of the resting CD4+ T cell reservoir for HIV in patients on HAART. PLoS Pathog. 3:e122 doi:10.1371/journal.ppat.0030122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Segal DJ, et al. 2003. Evaluation of a modular strategy for the construction of novel polydactyl zinc finger DNA-binding proteins. Biochemistry 42:2137–2148 [DOI] [PubMed] [Google Scholar]
- 163. Serwadda D, et al. 2003. Human immunodeficiency virus acquisition associated with genital ulcer disease and herpes simplex virus type 2 infection: a nested case-control study in Rakai, Uganda. J. Infect. Dis. 188:1492–1497 [DOI] [PubMed] [Google Scholar]
- 164. Shen L, et al. 2008. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 14:762–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Shen L, et al. 2011. A critical subset model provides a conceptual basis for the high antiviral activity of major HIV drugs. Sci. Transl. Med. 3:91ra63 doi:10.1126/scitranslmed.3002304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Shimizu S, et al. 2010. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood 115:1534–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Shlomai A, Lubelsky Y, Har-Noy O, Shaul Y. 2009. The “Trojan horse” model-delivery of anti-HBV small interfering RNAs by a recombinant HBV vector. Biochem. Biophys. Res. Commun. 390:619–623 [DOI] [PubMed] [Google Scholar]
- 168. Shouval D, et al. 2009. Relapse of hepatitis B in HBeAg-negative chronic hepatitis B patients who discontinued successful entecavir treatment: the case for continuous antiviral therapy. J. Hepatol. 50:289–295 [DOI] [PubMed] [Google Scholar]
- 169. Sigal A, et al. 2011. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477:95–98 [DOI] [PubMed] [Google Scholar]
- 170. Siliciano RF, Greene WC. 2011. HIV latency. Cold Spring Harb. Perspect. Med. 1:a007096 doi:10.1101/cshperspect.a007096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Smih F, Rouet P, Romanienko PJ, Jasin M. 1995. Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 23:5012–5019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Spivak AM, et al. 2011. Dynamic constraints on the second phase compartment of HIV-infected cells. AIDS Res. Hum. Retroviruses 27:759–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Stoddard BL. 2011. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure 19:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Strain MC, et al. 2005. Effect of treatment, during primary infection, on establishment and clearance of cellular reservoirs of HIV-1. J. Infect. Dis. 191:1410–1418 [DOI] [PubMed] [Google Scholar]
- 175. Summers J, et al. 2003. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. U. S. A. 100:11652–11659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Szczepek M, et al. 2007. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 25:786–793 [DOI] [PubMed] [Google Scholar]
- 177. Szpara ML, Parsons L, Enquist LW. 2010. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 84:5303–5313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Takeuchi R, Certo M, Caprara MG, Scharenberg AM, Stoddard BL. 2009. Optimization of in vivo activity of a bifunctional homing endonuclease and maturase reverses evolutionary degradation. Nucleic Acids Res. 37:877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Terasaki S. 1996. Latent multiple infections by herpes simplex virus type 1. Kurume Med. J. 43:127–136 [DOI] [PubMed] [Google Scholar]
- 180. Tipples GA, et al. 1996. Mutation in HBV RNA-dependent DNA polymerase confers resistance to lamivudine in vivo. Hepatology 24:714–717 [DOI] [PubMed] [Google Scholar]
- 181. Tong-Starksen SE, Luciw PA, Peterlin BM. 1987. Human immunodeficiency virus long terminal repeat responds to T-cell activation signals. Proc. Natl. Acad. Sci. U. S. A. 84:6845–6849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Tosi G, et al. 2011. Investigation on mechanisms of glycopeptide nanoparticles for drug delivery across the blood-brain barrier. Nanomedicine (Lond.) 6:423–436 [DOI] [PubMed] [Google Scholar]
- 183. Trono D, et al. 2010. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science 329:174–180 [DOI] [PubMed] [Google Scholar]
- 184. Tuttleman JS, Pourcel C, Summers J. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47:451–460 [DOI] [PubMed] [Google Scholar]
- 185. Ulge UY, Baker DA, Monnat RJ., Jr 2011. Comprehensive computational design of mCreI homing endonuclease cleavage specificity for genome engineering. Nucleic Acids Res. 39:4330–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.UNAIDS. Report on the global AIDS epidemic. 2010. http://www.unaids.org/globalreport/global_report.htm.
- 187. Uprichard SL, Boyd B, Althage A, Chisari FV. 2005. Clearance of hepatitis B virus from the liver of transgenic mice by short hairpin RNAs. Proc. Natl. Acad. Sci. U. S. A. 102:773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Urban S, Fischer KP, Tyrrell DL. 2001. Efficient pyrophosphorolysis by a hepatitis B virus polymerase may be a primer-unblocking mechanism. Proc. Natl. Acad. Sci. U. S. A. 98:4984–4989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Urnov FD, et al. 2005. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–651 [DOI] [PubMed] [Google Scholar]
- 190. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. 2010. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11:636–646 [DOI] [PubMed] [Google Scholar]
- 191. Van Lint C, Emiliani S, Ott M, Verdin E. 1996. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 15:1112–1120 [PMC free article] [PubMed] [Google Scholar]
- 192. Verhoeven D, Sankaran S, Silvey M, Dandekar S. 2008. Antiviral therapy during primary simian immunodeficiency virus infection fails to prevent acute loss of CD4+ T cells in gut mucosa but enhances their rapid restoration through central memory T cells. J. Virol. 82:4016–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Verjans GM, et al. 2007. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc. Natl. Acad. Sci. U. S. A. 104:3496–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Wagner TA, et al. 2009. Increased mutations in Env and Pol suggest greater HIV-1 replication in sputum-derived viruses compared with blood-derived viruses. AIDS 23:923–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Wald A, et al. 1997. Frequent genital herpes simplex virus 2 shedding in immunocompetent women. Effect of acyclovir treatment. J. Clin. Invest. 99:1092–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Wald A, Link K. 2002. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J. Infect. Dis. 185:45–52 [DOI] [PubMed] [Google Scholar]
- 197. Walters KA, Joyce MA, Addison WR, Fischer KP, Tyrrell DL. 2004. Superinfection exclusion in duck hepatitis B virus infection is mediated by the large surface antigen. J. Virol. 78:7925–7937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Wang K, Lau T, Morales M, Mont E, Straus S. 2005. Laser-capture microdissection: refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella-zoster virus DNA in human trigeminal ganglia at the single-cell level. J. Virol. 79:14079–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Wang K, et al. 2007. Diverse herpes simplex virus type 1 thymidine kinase mutants in individual human neurons and ganglia. J. Virol. 81:6817–6826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wayengera M. 2011. Identity of zinc finger nucleases with specificity to herpes simplex virus type II genomic DNA: novel HSV-2 vaccine/therapy precursors. Theor. Biol. Med. Model 8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Wayengera M, Kajumbula H, Byarugaba W. 2008. Identification of restriction endonuclease with potential ability to cleave the HSV-2 genome: inherent potential for biosynthetic versus live recombinant microbicides. Theor. Biol. Med. Model. 5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Wei X, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117–122 [DOI] [PubMed] [Google Scholar]
- 203. Werle-Lapostolle B, et al. 2004. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 126:1750–1758 [DOI] [PubMed] [Google Scholar]
- 204. Wilen CB, et al. 2011. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 7:e1002020 doi:10.1371/journal.ppat.1002020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Willson RA. 1997. Extrahepatic manifestations of chronic viral hepatitis. Am. J. Gastroenterol. 92:3–17 [PubMed] [Google Scholar]
- 206. Wong JK, et al. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291–1295 [DOI] [PubMed] [Google Scholar]
- 207. Wright TL, Lau JY. 1993. Clinical aspects of hepatitis B virus infection. Lancet 342:1340–1344 [DOI] [PubMed] [Google Scholar]
- 208. Xu F, et al. 2006. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296:964–973 [DOI] [PubMed] [Google Scholar]
- 209. Xu R, et al. 2003. Molecular therapeutics of HBV. Curr. Gene Ther. 3:341–355 [DOI] [PubMed] [Google Scholar]
- 210. Yang HI, et al. 2002. Hepatitis B e antigen and the risk of hepatocellular carcinoma. N. Engl. J. Med. 347:168–174 [DOI] [PubMed] [Google Scholar]
- 211. Yoffe B, Burns DK, Bhatt HS, Combes B. 1990. Extrahepatic hepatitis B virus DNA sequences in patients with acute hepatitis B infection. Hepatology 12:187–192 [DOI] [PubMed] [Google Scholar]
- 212. Zhang YY, et al. 2003. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. U. S. A. 100:12372–12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Zhou Y, Zhang H, Siliciano JD, Siliciano RF. 2005. Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J. Virol. 79:2199–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Zhu J, et al. 2009. Persistence of HIV-1 receptor-positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nat. Med. 15:886–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Zimmerman KA, Fischer KP, Joyce MA, Tyrrell DL. 2008. Zinc finger proteins designed to specifically target duck hepatitis B virus covalently closed circular DNA inhibit viral transcription in tissue culture. J. Virol. 82:8013–8021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. 2008. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 16:1073–1080 [DOI] [PubMed] [Google Scholar]
- 217. Zoulim F, Saputelli J, Seeger C. 1994. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 68:2026–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]