

Abstract
Human papillomaviruses (HPV) activate the ataxia telangiectasia mutated (ATM)-dependent DNA damage response to induce viral genome amplification upon epithelial differentiation. Our studies show that along with members of the ATM pathway, HPV proteins also localize factors involved in homologous DNA recombination to distinct nuclear foci that contain HPV genomes and cellular replication factors. These studies indicate that HPV activates the ATM pathway to recruit repair factors to viral genomes and allow for efficient replication.
TEXT
The life cycle of human papillomaviruses (HPV) is dependent on epithelial differentiation and is controlled through the action of viral as well as cellular factors (20). In undifferentiated cells, viral genomes are maintained as low-copy episomes that replicate once per cell cycle along with cellular DNA. Upon differentiation, viral genomes are amplified to thousands of copies per cell along with induction of late gene expression and the assembly of progeny virions (20). While normal epithelial differentiation results in exit from the cell cycle, expression of the E6 and E7 proteins pushes a subset of differentiating cells into S or G2/M phases to induce genome amplification (26). E6 and E7 also activate the ataxia telangiectasia mutated (ATM) DNA damage response that is necessary for the differentiation-dependent amplification of viral genomes (24).
The DNA damage response (DDR) plays a crucial role in the maintenance of genomic stability by coordinating cell cycle progression with DNA repair. The DDR is regulated by two main kinases, ATM and ATR (ATM and Rad3 related), that belong to the phosphoinositide-3-kinase-related protein kinase family (PIKKs) (12). ATM responds primarily to double-strand breaks (DSBs), while ATR is activated in response to single-stranded DNA (ssDNA) at stalled replication forks. ATM and ATR phosphorylate multiple substrates in response to DNA damage, including proteins involved in cell cycle checkpoints, DNA repair, and apoptosis (7, 21). The MRN complex, consisting of NBS1, Mre11, and Rad50, serves as the sensor for DSBs and recruits ATM to these sites, as well as promotes ATM activation through autophosphorylation (8, 17, 18). ATM activation leads to the phosphorylation of many substrates at sites of DNA damage, including Chk2, BRCA1, and NBS1, as well as the histone H2A variant H2AX (referred to as γH2AX) (5, 12).
Previous studies demonstrated that high-risk HPV31 induces an ATM-dependent DNA damage response in both undifferentiated and differentiating cells; however, this activity is required only for genome amplification and not stable maintenance replication (24). In HPV-positive cells, members of the ATM DNA damage pathway, including γH2AX, Chk2, BRCA1, and NBS1, are recruited in an ATM-dependent manner into distinct foci resembling those seen following DNA damage by ionizing radiation. HPV genomes are also replicated at specific nuclear loci (24, 38), but it was unclear if these regions also contained activated members of the ATM pathway or were localized at distinctly separate locations.
To determine if DNA repair factors colocalize with HPV genomes, we used immunofluorescence (IF) followed by fluorescence in situ hybridization (FISH) to screen for HPV DNA. For these assays, we utilized the CIN 612 cell line, which stably maintains HPV31 genomes and was previously shown to exhibit activation of the ATM-dependent DNA damage response (1, 24). CIN 612 cells were plated on glass coverslips and induced to differentiate in high-calcium medium at approximately 90% confluence or harvested as an undifferentiated sample (0 hours after end of log-phase growth [T0]). For the localization assays, cells were first extracted prior to fixation using a cytoskeleton buffer containing Triton X-100, as described previously (15). This method removes proteins that are not bound tightly to chromatin and provides a clearer view of protein localization. Following extraction, cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 in phosphate-buffered saline, and IF for DNA repair factors was performed. The samples were then cross-linked in methanol-acetic acid, and FISH for HPV DNA was performed using tyramide-enhanced fluorescence (Invitrogen), as described previously (15). IF/FISH was performed on detergent-extracted cells a minimum of two times for each DNA repair factor. The number of foci positive for both HPV DNA and each repair factor was quantified, with 25 to 40 FISH-positive cells being counted for each experiment. Although all cells contain HPV genomes, only a subset of cells amplify viral DNA upon differentiation and are FISH positive (24). Consistent with previous reports (23, 24), incubation in high-calcium medium induces epithelial differentiation, as evidenced by the expression of differentiation-specific markers, such as involucrin and keratin 10 (K10), as well as viral genome amplification (data not shown).
HPV DNA foci colocalize with markers of double-stranded DNA breaks.
Previously, we found that HPV induces the formation of nuclear foci containing γH2AX, a target of ATM that is frequently used as a marker of DNA breaks. p53 binding protein 1 (53BP1) is also an ATM target that is used to identify sites of DNA damage. H2AX represents 2 to 20% of histone 2A and when phosphorylated marks the chromatin around DSBs (9, 14). 53BP1 is subsequently recruited to sites of DNA damage in response to histone ubiquitylation (2). Importantly, 53BP1 is necessary for the intra-S and G2/M checkpoints (22, 35, 39). Western blot analysis of cell extracts indicated that both γH2AX and 53BP1 were present at increased levels in both undifferentiated and differentiated HPV-positive cells compared to HPV-negative human foreskin keratinocytes (HFKs) (Fig. 1A). To determine if γH2AX and 53BP1 localize to HPV DNA foci, CIN 612 cells were induced to differentiate in high-calcium medium and examined by IF to detect γH2AX and 53BP1 and FISH to detect HPV DNA. As shown in Fig. 1B, γH2AX and 53BP1 (Fig. 1C) were found localized to HPV DNA foci in both undifferentiated and differentiating cells. γH2AX was localized to 94 to 100% of cells containing HPV DNA foci in both undifferentiated and differentiated cells. Interestingly, 53BP1 was localized to 47 to 50% of undifferentiated cells that were FISH positive, and this increased to 88 to 93% upon differentiation. Consistent with viral genome amplification, the size of the foci containing γH2AX and 53BP1 increased in differentiating cells. Interestingly, upon differentiation, we observed FISH-positive cells containing either one or two large HPV DNA foci or multiple smaller foci. As shown in Fig. 1B, γH2AX localized to both focus types in detergent-extracted cells, and we observed similar results for 53BP1 as well as for the other repair factors tested (data not shown). This increase in focus size coincides with increased replication of viral episomes as determined by Southern blot analyses (24). To ensure that episomal copies of the virus were not lost upon detergent extraction, resulting in only the detection of integrated HPV foci (15), we performed the IF/FISH assay without the preextraction step. In the absence of preextraction, we observed a similar distribution of cells containing large HPV DNA foci and multiple foci. In addition, the localization of γH2AX to HPV DNA foci was not affected (Fig. 1B). These results indicate that DNA repair factors are recruited to episomal copies of HPV. It is possible that these large foci are the result of the coalescence of replication compartments, as has been shown for herpes simplex virus (HSV) (6, 30, 31). In contrast to HPV-positive cells, only faint focal staining of γH2AX and 53BP1 was observed in normal HFKs. Since both γH2AX and 53BP1 are important for the localization of cellular factors involved in damage signaling and repair, these findings indicate that HPV promotes the recruitment of host repair factors to HPV genomes.
Fig 1.

Proteins associated with damaged DNA colocalize with HPV DNA foci. (A) Whole-cell extracts were harvested from CIN 612 cells or normal HFKs at T0 or after 48 and 96 h of differentiation in high-calcium medium, as described previously (23). Immunoblotting was performed using antibodies to γH2AX (Cell Signaling) and 53BP1 (Abcam). GAPDH served as a loading control. Ca, calcium. Protein levels were quantified by ImageJ and normalized to levels observed in uninfected keratinocytes. Shown is a representative experiment. (B) IF for γH2AX (Millipore) was performed, followed by FISH for HPV DNA on detergent-extracted CIN 612 cells harvested at T0 or after 72 h of calcium-induced differentiation. IF for γH2AX was performed on detergent-extracted normal HFKs at T0 or 72 h postexposure to high-calcium medium. Cellular DNA was counterstained with DAPI (4′,6-diamidino-2-phenylindole). Omission of the preextraction step did not alter the localization of γH2AX to HPV DNA foci (no preextraction). Images were collected on a Zeiss 710 confocal laser-scanning microscope and were processed using the Zeiss Zen software. (C) IF for 53BP1 (Abcam) was performed on preextracted CIN 612 cells, followed by FISH for HPV DNA. IF for 53BP1 was also performed on detergent-extracted normal HFKs at T0 or after 72 h of differentiation in high-calcium medium.
Phosphorylated ATM and Chk2 localize to HPV DNA foci.
Since γH2AX and 53BP1 were localized to HPV DNA foci, we reasoned that ATM and Chk2 should also localize to these sites. pATM and pChk2 are activated at higher levels in HPV-positive cells than in normal HFKs, and this is maintained upon differentiation (Fig. 2A). To examine the possibility that these factors localize to HPV DNA foci, we performed IF/FISH on detergent-extracted CIN 612 cells. We observed that pATM (55 to 60% of FISH-positive cells) (Fig. 2B) and Chk2 (83 to 90% of FISH-positive cells) (Fig. 2C) colocalized at sites containing HPV DNA in undifferentiated cells. Upon differentiation, the intensity of the signal and size of the foci increased, indicating that activated ATM and Chk2 are recruited to newly synthesized viral genomes. In differentiating cells, pATM and Chk2 were localized to HPV DNA foci in 64 to 74% and 82 to 86% of FISH-positive cells, respectively. Though we observed some Chk2 foci in the absence of HPV FISH foci (Fig. 2C), we believe this reflects HPV's ability to induce a general DNA damage response as well as the limited sensitivity of the FISH assay (24). Normal HKFs exhibited only background staining of pATM and Chk2 (data not shown), as demonstrated previously (24).
Fig 2.

pATM and Chk2 localize to HPV DNA foci. (A) Whole-cell extracts were harvested from CIN 612 cells and normal HFKs that were undifferentiated (T0) or induced to differentiate in high-calcium medium for 48 and 96 h, as described previously (23). Western blot analysis was performed using an antibody to ATM phosphorylated on Ser1981 (Epitomics), total ATM (Calbiochem), Chk2 phosphorylated on Thr68 (Cell Signaling), and total Chk2 (Santa Cruz), followed by horseradish peroxidase (HRP)-linked secondary antibodies (Cell Signaling). GAPDH served as a loading control. Protein levels were quantified by ImageJ and normalized to levels observed in uninfected keratinocytes. Ca, calcium. Shown is a representative experiment. (B, C) Immunofluorescence (IF) for pATM Ser1981 (Epitomics) (B) and Chk2 (Abcam) (C) was followed by FISH for HPV DNA on detergent-extracted CIN 612 cells that were undifferentiated (T0) or differentiated in high-calcium medium for 72 h. Cellular DNA was counterstained using DAPI.
HPV DNA foci represent compartments containing actively replicating genomes.
To ensure that the HPV foci we observed by FISH represented active sites of viral replication, we next examined the levels and localization of the cellular replication proteins PCNA and replication protein A subunit 32 (RPA32) in relation to HPV DNA. As shown in Fig. 3A, HPV-positive cells exhibited higher levels of PCNA than normal HFKs, and this was maintained upon differentiation in high-calcium medium, while levels of the RPA32 were similar between the two sets of cells. When examined by IF/FISH, we found that both PCNA and RPA32 also formed foci that colocalized with HPV genomes in undifferentiated cells, and these foci increased in size upon differentiation, indicating that these are indeed active viral replication centers (Fig. 3B). PCNA and RPA were localized to 81% and 77% of FISH-positive undifferentiated cells, respectively, and 80% and 85%, respectively, of differentiating cells. Although we could detect PCNA and RPA at replication foci in undifferentiated cells, the intensity of the foci was very faint.
Fig 3.

Cellular replication factors localize to HPV replication foci. (A) Immunoblotting was performed using antibodies to PCNA (Abcam), RPA32 (Bethyl Laboratories), and pRPA32 S33 (Bethyl Laboratories) on whole-cell lysates extracted from CIN 612 cells and HFKs that were undifferentiated or differentiated in high-calcium medium for 48 and 96 h. GAPDH served as a loading control. Protein levels were quantified by ImageJ and normalized to levels observed in uninfected keratinocytes. Shown is a representative experiment. (B, C) IF for PCNA (Abcam) (B), RPA32 (Bethyl Laboratories) (B), and pRPA32 S33 (Bethyl Laboratories) (C) was performed, followed by FISH for HPV DNA on detergent-extracted CIN 612 cells that were undifferentiated or differentiated in high-calcium medium for 72 h. DAPI was used to counterstain cellular DNA. (C) IF was performed on detergent-extracted HFKs at T0 or after 72 h differentiation in high-calcium medium using an antibody to pRPA32 S33. DAPI was used to counterstain cellular DNA.
RPA consists of three subunits, RPA70, RPA32, and RPA14, and is the major ssDNA binding protein in eukaryotic cells (3). RPA accumulates along stretches of ssDNA generated by stalled replication forks or DNA damage. In response to DNA damage, the N terminus of RPA32 is phosphorylated at multiple residues by DNA damage kinases (ATM, ATR, DNA-PK) (27), which has been suggested to redirect the function of RPA from DNA replication to repair DNA synthesis (27, 44). To determine if HPV alters the phosphorylation state of RPA, we examined phosphorylation of the RPA32 Ser33 residue (pRPA32 S33) by Western blot analysis and found that the levels of pRPA32 S33 were not significantly increased in HPV-positive cells upon differentiation compared to those in uninfected HFKs (Fig. 3A). However, using the IF/FISH assay, we detected pRPA32 S33 at HPV replication foci in 30 to 60% of undifferentiated FISH-positive cells (Fig. 3C). Interestingly, the number of foci containing pRPA that colocalized with HPV DNA increased to 83 to 85% in differentiated cells, with the size of the foci increasing in response to viral genome amplification. Since phosphorylated RPA localizes to nuclear foci where DNA repair is occurring following DNA damage (3, 45), HPV DNA synthesis may be dependent on repair replication.
Cellular factors involved in homologous recombination repair are localized to viral replication compartments.
Mammalian cells have two primary mechanisms by which they can repair double-stranded breaks: homologous recombination (HR) and nonhomologous end joining (NHEJ) (13, 28). HR is an accurate repair process that involves template-directed recombination and is restricted to the S and G2 phases of the cell cycle (25, 34). The MRN complex is the primary sensor that detects DSBs in the case of HR, and ssDNA generated by resection of the DSB is protected by RPA containing phosphorylated RPA32. NHEJ is an error-prone mechanism that can be carried out at any phase of the cell cycle, with the Ku70/Ku80 complex being the primary sensor of DSBs (19). Since we previously observed formation of foci containing MRN components in HPV-positive cells (24) and can also detect them at sites of viral replication (data not shown), we explored the potential linkage between HPV replication and cellular DNA repair through HR. We first examined the levels of the HR proteins Rad51 and BRCA1 by Western blot analysis. HPV-positive cells have higher levels of both Rad51 and BRCA1 than undifferentiated HFKs (Fig. 4A). Upon differentiation, levels of Rad51 remain high through 48 h postexposure to calcium, whereas Brca1 is detected at higher levels throughout the time course (Fig. 4A). To determine if the increase in Rad51 and BRCA1 coincided with the localization of these repair factors to HPV genomes, we performed IF for Rad51 and BRCA1 and FISH for HPV DNA on detergent-extracted CIN 612 cells, as well as on normal HFKs. In undifferentiated cells, the amount of Rad51 colocalizing with HPV genomes was very faint and detected in 50 to 58% of FISH-positive cells (Fig. 4B). Upon differentiation, Rad51 colocalized with 60 to 68% of cells that were HPV positive by FISH, and the staining intensity increased upon viral genome amplification. Similar results were observed for BRCA1 (Fig. 4C), with localization occurring at 60 to 78% of undifferentiated FISH-positive cells and 64 to 73% of differentiating FISH-positive cells. In contrast to the localization of HR proteins to HPV DNA foci, we did not observe significant localization of the NHEJ factor DNA-PK (phosphorylated on Thr 2069) to sites of HPV replication (data not shown). The observation that HR proteins, including phosphorylated RPA, Rad51, and BRCA1 localize to HPV replication foci suggests that the amplification of viral genomes during the productive phase may require HR machinery to ensure high-fidelity replication.
Fig 4.

HPV-positive cells exhibit increased levels of homologous recombination factors, which are localized to HPV replication foci. (A) Whole-cell extracts were harvested from CIN 612 cells and HFKs that were undifferentiated or differentiated in high-calcium medium for the indicated times. Western blot analysis was performed using antibodies to Rad51 (Santa Cruz) and BRCA1 (Calbiochem). GAPDH served as a loading control. Protein levels were quantified by ImageJ and normalized to levels observed in uninfected keratinocytes. Shown is a representative experiment. (B, C) IF for Rad51 (B) and BRCA1 (C) was performed, followed by FISH for HPV DNA on detergent-extracted CIN 612 cells harvested at T0 or after 72 h differentiation in high-calcium medium. IF was also performed on detergent-extracted HFKs that were undifferentiated or differentiated in high-calcium medium using antibodies to Rad51 (B) and BRCA1 (C). Cellular DNA was counterstained with DAPI.
γH2AX associates with viral genomes.
The studies described above demonstrate that many components of the ATM DNA damage pathway colocalize with HPV genomes in foci that also contain cellular replication and homologous recombination repair factors. It is possible that these factors localize to these regions but are not bound to viral genomes. We therefore investigated if repair factors actually bind to HPV genomes. For our initial analyses, we focused on γH2AX and examined binding to viral DNA by chromatin immunoprecipitation (ChIP) assays. We screened for binding of γH2AX to sequences around the HPV origin of replication in the upstream regulatory region (URR) in undifferentiated as well as differentiated cells (Fig. 5). An increase of approximately 7-fold in γH2AX binding to the origin was detected in undifferentiated cells over the IgG control, and this increased upon differentiation to over 20-fold. This indicates that DNA repair enzymes are recruited to viral genomes during productive replication in differentiated cells.
Fig 5.
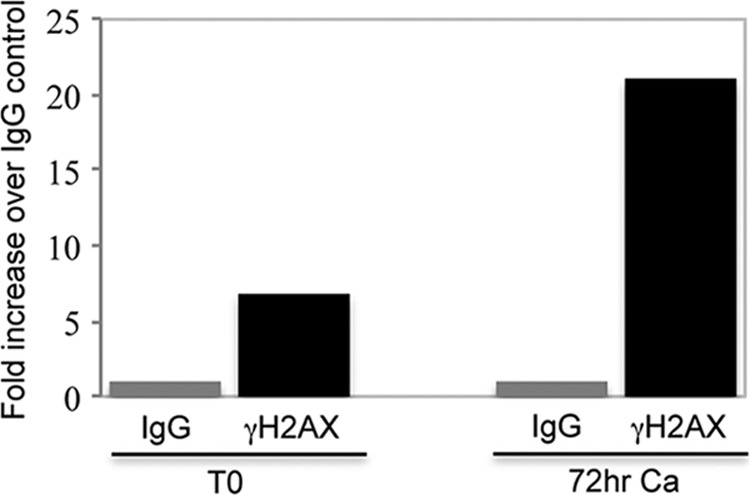
γH2AX associates with HPV genomes. Chromatin immunoprecipitation (ChIP) assays were performed on CIN 612 cells that were harvested at T0 or after 72 h of differentiation in high-calcium medium, as previously described by Wong et al. (42). Immunoprecipitations were performed using an antibody to phospho-H2AX Ser139 (γH2AX) (Millipore). Quantitative PCR was done using a Roche Lightcycler 480 Sybr green master kit (Roche Applied Sciences). The primers used were specific to the upstream regulatory region (URR) of HPV31. Forward, 5′ AAC TGC CAA GGT TGT GTC ATG C 3′; reverse, 5′ TGG CGT CTG TAG GTT TGC AC 3′.The data are expressed as fold activation over the control mouse IgG (Santa Cruz), where the fold level was set at 1 to allow for comparisons. Ca, calcium. Shown is a representative of two experiments.
Our studies demonstrate that members of the ATM DNA damage pathway are recruited to HPV replication foci in a manner resembling the effects seen following induction of DNA damage. We believe that the localization of the host cellular replication factors, PCNA and RPA, to sites of HPV DNA indicates that these centers contain viral genomes undergoing replication. The ATM DNA damage pathway can induce cell cycle arrest at either G1/S or G2/M, depending on which arm of the pathway is activated. ATM signaling through p53 induces G1/S arrest, while ATM signaling to Chk2 arrests cells in G2/M (7). HPV has been reported to amplify its DNA in cells that are in the G2 phase of the cell cycle, after cellular DNA has replicated (40). While HPV-induced ATM activation may contribute to productive replication via this mechanism, our studies suggest that the DDR factors also play an active role in HPV replication since they are recruited to HPV replication compartments.
The DSB markers γH2AX and 53BP1 were found to colocalize with replicating HPV DNA. Our studies suggest the possibility that DSBs play a role in HPV genome amplification. DSBs could be introduced into amplifying HPV DNA if productive replication is mediated through a rolling circle, which has been suggested as the mechanism of differentiation-dependent genome amplification (10). In addition, the dramatic increase in viral replication taking place in differentiating cells could lead to replication stress, resulting in stalled replication forks and the formation of DSBs. Activation of ATM signaling and the recruitment of cellular HR repair factors would provide an efficient mechanism for HPV to repair and synthesize viral DNA. The finding that γH2AX localizes to viral replication centers and is bound to viral chromatin suggests that γH2AX may recruit host repair proteins necessary to carry out viral DNA replication. Within HPV virions, viral genomes are associated with histones (37, 43), raising the possibility that H2AX is incorporated into newly replicated genomes and packaged into progeny viruses. This hypothesis will be investigated in more detail in future studies.
We previously found that HPV induces the activation of an ATM-dependent DNA damage response. While our current study indicates that an outcome of this response is the recruitment of repair factors to viral genomes, the mechanism by which HPV activates ATM signaling is unclear. One possibility is that expression of a viral protein elicits this response. In support of this, we and others have shown that expression of E7 is sufficient to activate ATM and its downstream targets (24, 29, 32). We have also found that E7 interacts with ATM (24), as well as the MRN complex (C. A. Moody, unpublished results). Recent studies indicate that the expression of the viral replication protein E1 can also activate ATM signaling (11, 15, 33). E7 and/or E1 may induce cellular DNA damage that consequently provides DNA repair/replication factors necessary for the synthesis of viral DNA. Alternatively, the interaction of HPV proteins with DNA repair factors could facilitate recruitment to viral DNA, which may be sufficient to nucleate a DNA damage response (36). It is also possible that viral replication results in the formation of intermediates that the cell recognizes as DNA damage. This may be sufficient to activate ATM signaling, resulting in the recruitment of repair factors necessary to allow viral genome amplification. Understanding how HPV triggers a DNA damage response will be the focus of future investigations.
Our findings that cellular factors involved in HR, including Rad51, BRCA1, and pRPA S33, are also recruited to HPV replication foci raises the intriguing possibility that homologous recombination is directly involved in HPV genome amplification. HSV, Epstein-Barr virus (EBV), and SV40 have all been shown to recruit Rad51 to replication compartments (4, 16, 41). For EBV, as well as SV40, knockdown of Rad51 diminishes viral genome synthesis, suggesting that HR is necessary for efficient replication (4, 16). The localization of HR factors to HPV DNA foci may indicate that viral replication compartments contain substrates, such as stalled replication forks or abnormal replication intermediates, that require HR activity to facilitate high-fidelity viral DNA synthesis upon differentiation. However, it is possible that HPV actively uses these factors to promote replication. In addition, if viral genome amplification does proceed through a rolling circle mechanism, HR may be required to circularize HPV genomes upon cleavage of viral concatemers to allow for packaging into virions. Overall, these studies show that members of the ATM DNA damage pathway along with factors involved in homologous recombination play active roles in the differentiation-dependent amplification of HPV genomes.
ACKNOWLEDGMENTS
We thank Dale Ramsden for generously providing the pDNA-PK Thr 2069 antibody.
L.A.L. was supported by grants from the NCI, and C.A.M was supported by a Pathway to Independence Award from the NCI.
Footnotes
Published ahead of print 27 June 2012
REFERENCES
- 1. Bedell MA, et al. 1991. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 65:2254–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bekker-Jensen S, Mailand N. 2010. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst.) 9:1219–1228 [DOI] [PubMed] [Google Scholar]
- 3. Binz SK, Sheehan AM, Wold MS. 2004. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst.) 3:1015–1024 [DOI] [PubMed] [Google Scholar]
- 4. Boichuk S, Hu L, Hein J, Gjoerup OV. 2010. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J. Virol. 84:8007–8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol. Cell 40:179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Bruyn Kops A, Knipe DM. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857–868 [DOI] [PubMed] [Google Scholar]
- 7. Derheimer FA, Kastan MB. 2010. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 584:3675–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Falck J, Coates J, Jackson SP. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434:605–611 [DOI] [PubMed] [Google Scholar]
- 9. Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. 2004. H2AX: the histone guardian of the genome. DNA Repair (Amst.) 3:959–967 [DOI] [PubMed] [Google Scholar]
- 10. Flores ER, Lambert PF. 1997. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 71:7167–7179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fradet-Turcotte A, et al. 2011. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 85:8996–9012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harper JW, Elledge SJ. 2007. The DNA damage response: ten years after. Mol. Cell 28:739–745 [DOI] [PubMed] [Google Scholar]
- 13. Hartlerode AJ, Scully R. 2009. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 423:157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huen MS, Chen J. 2008. The DNA damage response pathways: at the crossroad of protein modifications. Cell Res. 18:8–16 [DOI] [PubMed] [Google Scholar]
- 15. Kadaja M, Isok-Paas H, Laos T, Ustav E, Ustav M. 2009. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 5:e1000397 doi:10.1371/journal.ppat.1000397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kudoh A, et al. 2009. Homologous recombinational repair factors are recruited and loaded onto the viral DNA genome in Epstein-Barr virus replication compartments. J. Virol. 83:6641–6651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee JH, Paull TT. 2005. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308:551–554 [DOI] [PubMed] [Google Scholar]
- 18. Lee JH, Paull TT. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304:93–96 [DOI] [PubMed] [Google Scholar]
- 19. Lieber MR. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79:181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Longworth MS, Laimins LA. 2004. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 68:362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsuoka S, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316:1160–1166 [DOI] [PubMed] [Google Scholar]
- 22. Mochan TA, Venere M, DiTullio RA, Jr, Halazonetis TD. 2004. 53BP1, an activator of ATM in response to DNA damage. DNA Repair (Amst.) 3:945–952 [DOI] [PubMed] [Google Scholar]
- 23. Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. 2007. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc. Natl. Acad. Sci. U. S. A. 104:19541–19546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 5:e1000605 doi:10.1371/journal.ppat.1000605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moynahan ME, Jasin M. 2010. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11:196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Munger K, et al. 2004. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 78:11451–11460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nuss JE, et al. 2005. DNA damage induced hyperphosphorylation of replication protein A. 1. Identification of novel sites of phosphorylation in response to DNA damage. Biochemistry 44:8428–8437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pardo B, Gomez-Gonzalez B, Aguilera A. 2009. DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship. Cell. Mol. Life Sci. 66:1039–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pickering MT, Kowalik TF. 2006. Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene 25:746–755 [DOI] [PubMed] [Google Scholar]
- 30. Quinlan MP, Chen LB, Knipe DM. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857–868 [DOI] [PubMed] [Google Scholar]
- 31. Rixon FJ, Atkinson MA, Hay J. 1983. Intranuclear distribution of herpes simplex virus type 2 DNA synthesis: examination by light and electron microscopy. J. Gen. Virol. 64(Pt 9):2087–2092 [DOI] [PubMed] [Google Scholar]
- 32. Rogoff HA, et al. 2004. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol. Cell. Biol. 24:2968–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sakakibara N, Mitra R, McBride AA. 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 85:8981–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. San Filippo J, Sung P, Klein H. 2008. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77:229–257 [DOI] [PubMed] [Google Scholar]
- 35. Silverman J, Takai H, Buonomo SB, Eisenhaber F, de Lange T. 2004. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 18:2108–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Soutoglou E, Misteli T. 2008. Activation of the cellular DNA damage response in the absence of DNA lesions. Science 320:1507–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stunkel W, Bernard HU. 1999. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J. Virol. 73:1918–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swindle CS, et al. 1999. Human papillomavirus DNA replication compartments in a transient DNA replication system. J. Virol. 73:1001–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 2002. 53BP1, a mediator of the DNA damage checkpoint. Science 298:1435–1438 [DOI] [PubMed] [Google Scholar]
- 40. Wang HK, Duffy AA, Broker TR, Chow LT. 2009. Robust production and passaging of infectious HPV in squamous epithelium of primary human keratinocytes. Genes Dev. 23:181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wilkinson DE, Weller SK. 2004. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 78:4783–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wong PP, Pickard A, McCance DJ. 2010. p300 alters keratinocyte cell growth and differentiation through regulation of p21(Waf1/CIP1). PLoS One 5:e8369 doi:10.1371/journal.pone.0008369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wooldridge TR, Laimins LA. 2008. Regulation of human papillomavirus type 31 gene expression during the differentiation-dependent life cycle through histone modifications and transcription factor binding. Virology 374:371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zernik-Kobak M, Vasunia K, Connelly M, Anderson CW, Dixon K. 1997. Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. J. Biol. Chem. 272:23896–23904 [DOI] [PubMed] [Google Scholar]
- 45. Zou Y, Liu Y, Wu X, Shell SM. 2006. Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses. J. Cell. Physiol. 208:267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]