

Abstract
In vivo cells receive simultaneous signals from multiple extracellular ligands and must integrate and interpret them to respond appropriately. Here we investigate the interplay between pathways downstream of two transforming growth factor β (TGF-β) superfamily members, bone morphogenetic protein (BMP) and TGF-β. We show that in multiple cell lines, TGF-β potently inhibits BMP-induced transcription at the level of both BMP-responsive reporter genes and endogenous BMP target genes. This inhibitory effect requires the TGF-β type I receptor ALK5 and is independent of new protein synthesis. Strikingly, we show that Smad3 is required for TGF-β's inhibitory effects, whereas Smad2 is not. We go on to demonstrate that TGF-β induces the formation of complexes comprising phosphorylated Smad1/5 and Smad3, which bind to BMP-responsive elements in vitro and in vivo and mediate TGF-β-induced transcriptional repression. Furthermore, loss of Smad3 confers on TGF-β the ability to induce transcription via BMP-responsive elements. Our results therefore suggest that not only is Smad3 important for mediating TGF-β's inhibitory effects on BMP signaling but it also plays a critical role in restricting the transcriptional output in response to TGF-β.
INTRODUCTION
The transforming growth factor β (TGF-β) superfamily comprises more than 30 ligands that play essential roles during early vertebrate development in the specification and subsequent patterning of the germ layers and in tissue homeostasis in adult organisms. Deregulation of signaling downstream of many of these ligands has been implicated in human diseases such as cancer and fibrosis, in wound healing disorders, and in several hereditary conditions (7, 37). The TGF-β superfamily ligands elicit their pleiotropic effects by signaling to the nucleus and inducing new programs of gene expression. The large number of ligands in this superfamily signal to the nucleus through a much smaller number of receptors and Smads, which act as intracellular signal transducers (14). Thus, different ligands utilize common pathway components. This raises important questions about how cells respond specifically to individual ligands and how cells integrate and interpret signals received from multiple ligands simultaneously.
TGF-β superfamily ligands signal by bringing together heteromeric complexes of type I and type II serine/threonine kinase receptors, which in turn phosphorylate and activate a subset of Smads known as the receptor-regulated Smads (R-Smads) at two serines at the extreme C terminus (46). The activated R-Smads form homomeric and heteromeric complexes with Smad4 that accumulate in the nucleus, where they directly activate or repress the transcription of target genes (46). The TGF-β superfamily pathways have traditionally been split into two branches. The TGF-βs, activins, and Nodals form one subfamily, which signal through the type I receptors ALK4, -5, and -7 and Smad2 and -3. The bone morphogenetic proteins (BMPs) and growth and differentiation factors (GDFs) form a second subfamily and signal through ALK1, -2, -3, and -6, and Smad1, -5, and -8 (14).
The DNA binding specificity of phosphorylated Smad1/5/8-Smad4 complexes formed in response to BMPs/GDFs is thought to be distinct from that of phosphorylated Smad2-Smad4 or phosphorylated Smad3-Smad4 complexes formed in response to TGF-β/activin/Nodal (42), and this has been assumed to account for the distinct sets of genes that are induced in response to the different classes of ligand. Indeed, ligands from the two different subfamilies play very different roles in vivo, as evidenced, for example, by the phenotypes of mice in which different ligands, receptors, and Smads have been knocked out (19, 52).
Antagonism between ligands of these two major subfamilies in a variety of biological contexts, such as vertebrate embryonic development, stem cell maintenance, and osteoblast differentiation, has been described (8, 35, 54, 55), and several different mechanisms have been proposed to be responsible for this antagonism. In early Xenopus embryos, heterodimerization between Nodal and BMPs has been demonstrated, accounting for some of the antagonism between these two classes of ligands (55). In the same system, competition for Smad4 has been reported to explain antagonism between BMP2/4 signaling and another Nodal-related ligand, Vg-1 (8). Finally, in mouse embryonic stem cells, induction of the negative Smad Smad7 in response to Nodal has been shown to inhibit BMP responses (15). Smad7 is induced by many TGF-β superfamily members and is thought to act by recruiting both E3 ubiquitin ligases Smurf1/2 and the phosphatase PP1 to both TGF-β and BMP receptor complexes to downregulate their activity (13, 27, 45).
Recent work has shown that the signaling pathways downstream of the two major subfamilies of TGF-β superfamily ligands are not as distinct as previously thought. In addition to inducing phosphorylation of Smad2 and Smad3, TGF-β strongly induces phosphorylation and activation of Smad1/5/8 in many different cell types, including endothelial cells, epithelial cells, fibroblasts, and epithelium-derived cancer cells (6, 9, 20, 33, 51). These observations raise critical questions of how the activation of Smad1/5/8 by TGF-β influences BMP responses and why TGF-β cannot induce BMP-like transcriptional responses.
To answer these questions, we have investigated how the TGF-β and BMP pathways influence each other. We demonstrate a potent direct inhibition of BMP-induced transcriptional responses by TGF-β. We show that this occurs downstream of receptor activation, is independent of new protein synthesis, and is mediated by novel complexes formed between phosphorylated Smad1/5 and phosphorylated Smad3 that are induced by TGF-β and are substantially increased when cells are induced with both TGF-β and BMPs. This work not only uncovers the mechanism by which these two TGF-β superfamily pathways antagonize each other but also explains why TGF-β cannot elicit a BMP-like transcriptional response.
MATERIALS AND METHODS
Cell culture, stable cell lines, and treatments.
All cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum. For overnight starvation, cells were washed twice with phosphate-buffered saline and incubated in Opti-MEM (Invitrogen) before induction. Stable MDA-MB-231 cell lines expressing either BMP-responsive element (BRE)–luciferase or CAGA12-luciferase together with thymidine kinase (TK)-Renilla (called MDA-MB-231 BRE and MDA-MB-231 CAGA, respectively) were generated by transfecting cells as described below with the appropriate plasmids and pCMV-Bsd (Invitrogen) for blasticidin resistance. The stable MDA-MB-231 cell line expressing FLAG-ALK2 was generated by cotransfecting the FLAG-ALK2 expression vector together with pSUPER-retro-puro (OligoEngine) for puromycin resistance. The stable HaCaT cell line expressing hemagglutinin (HA)-Smad3 was generated in the same way. TGF-β and BMP7 (Peprotech EC Ltd.) were used at 2 ng/ml and 100 ng/ml, respectively, unless otherwise stated. BMP2 (R&D Systems) was used at 20 ng/ml. SB-431542 (Tocris) was used at 10 μM, and emetine (Sigma) was used at 400 μM.
Antibodies and plasmids.
The antibodies used were obtained from Santa Cruz Biotechnology (anti-Smad4 [B-8], anti-Smad3 [38-Q], anti-MCM7 [141.2], anti-MCM6 [C-20], and anti-Alk5 [V-22]), Sigma (anti-FLAG antibody [M2]), Life Technologies (anti-Smad1 [catalog no. 385400] and anti-Smad2 [catalog no. 511300]), Cell Signaling Technology (anti-Smad5 [catalog no. 9517], anti-phospho-Smad2 [catalog no. 3108], anti-phospho-Smad3 [catalog no. 9520S], and anti-phospho-Smad1/5/8 [catalog no. 9516]), Abcam (chromatin immunoprecipitation [ChIP] grade anti-Smad3 [ab28379] for immunoprecipitation [IP] and ChIP), Covance (RNA polymerase II [PolII; 8WG16]), and BD Biosciences (Smad2/3 [catalog no. 610843]). Secondary antibodies (Dako) were peroxidase-conjugated secondary goat anti-mouse or goat anti-rabbit antibodies. The following plasmids were as previously described: CAGA12-luciferase, BRE-luciferase, TK-Renilla, FLAG-ALK2, FLAG-Smad4, HA-Smad3, and ID2-luciferase (9, 22, 38).
Transfections, siRNA, and reporter assays.
Plasmid transfections were performed using FUGENE HD (Promega) according to the manufacturer's protocol. Luciferase reporter assays were done as previously described (32), with 8-h ligand inductions. Small interfering RNA (siRNA) transfections into MDA-MB-231 and MDA-MB-231 BRE cells were performed using INTERFERin (Polyplus Transfection) and a final concentration of 10 nM siRNA. Cells were seeded 1 day prior to transfection and analyzed 72 h after knockdown. The siRNA oligonucleotides (Thermo Scientific [Dharmacon RNAi Technologies]) used were Smad2 (D-003561-01, D-003561-02, and D-003561-04), Smad3 (D-020067-01 and D-020067-04), Smad4 (D-003902-05 and D003902-07), Smad7 (D-020068-01 and D-020068-04), Smad1 (M-012723-00), and Smad5 (M-015791-00), and ALK5 (D-003929-05 and D-003929-08). The control siRNA was ON-TARGETplus nontargeting siRNA no. 1 (D-001810-01-20).
Protein extraction, recombinant proteins, and immunoprecipitation.
Whole-cell extracts and nuclear extracts for Western blotting were prepared as previously described (18, 50), and IP with endogenous proteins was performed as described previously (9). Western blot assays were visualized using Immobilon Western detection reagents (Millipore) either on film or using an ImageQuant LAS4000 mini (GE Healthcare) when the bands were quantitated using ImageQuant TL. Recombinant phosphorylated Smad1 and phosphorylated Smad3 were prepared by coexpressing Smad1 and Smad3 with the kinase domains of ALK2 and ALK5, respectively, in insect cells using baculovirus. Details of the purification will be published elsewhere. For the IP, 1.25 μg of each protein diluted in hypotonic buffer (50) containing 140 mM NaCl and 1 mg/ml bovine serum albumin was used.
DNA pulldown (DNAP), quantitative PCR (qPCR), and ChIP assays.
DNAP assays were performed as described previously (32), except that nuclear extracts were prepared as described in reference 44. The oligonucleotides used were Mutant (top) (5′-biotin-GGAGGTGCGCGGAGTCAGGCATATATATATATACAGCATGCATGCATGGTCGGCA) and the unbiotinylated reverse complement Mutant (bottom), BRE (top) (5′-biotin-CCGCGCGGCGCCAGCCTGACAGCCCGTCCTGGCGTCTAACGGTCTGAGCCG) and the unbiotinylated reverse complement BRE (bottom), ID2 −2979/−2920 (probe A top): (5′-biotin-CGCGGTTGCCATGGCAGCCGCCTGAGCGGCGCCGCGAGGACAAGGCTGCAGGGCGGCGTG) and the unbiotinylated reverse complement ID2 −2979/−2920 (probe A bottom), ID2 −2919/−2860 (probe B top) (5′-biotin-AATGGGCGGCGTCACGCGCCTGGCGCCAGAGAGTCTGCTCCGGGGCTCCGGCTCCGGCCC) and the unbiotinylated reverse complement ID2 −2919/−2860 (probe B bottom), and ID1 −1070/1025 (top) (5′-TGAATGGGTGACGTCACGGGCCTGGCGTCTAACGGTCTGAGCCGCTTG-3′) and the 3′-biotinylated reverse complement ID1 −1070/1025 (bottom).
For qPCR, RNA was prepared using TRIzol according to manufacturer's instructions and further cleaned up using GenElute (Sigma). cDNA was prepared using the cDNA construction kit (Agilent Technologies) and analyzed using the qPCR ABI 7500 Fast system (Applied Biosystems) with SYBR green Master Mix (Applied Biosystems). The qPCR primers used were as follows: ID2 (fwd), CGCCGCTGACCACCCTCAAC; ID2 (rev), AGCCACACAGTGCTTTGCTGTCA; ID3 (fwd), GGCCCCCACCTTCCCATCC; ID3 (rev), GCCAGCACCTGCGTTCTGGAG; ATOH8 (fwd), CCTGAGGATCGCCTGTAACT; ATOH8 (rev), TGGTCGGCACTGTAGTCAAG; ZNF114 (fwd), GAGTGGACCCTGCTGGAC; ZNF114 (rev), TTTTACATGGAGTTGCCCAAT; DUSP10 (fwd), AAGAGGCTTTTGAGTTCATTGAG; DUSP10 (rev), CAAGTAAGCGATGACGATGG; BIRC3 (fwd), TTGAACAGCTGCTATCCACATC; BIRC3 (rev), TCCAGGTTCAAAATGGATAATTG; Smad7 (fwd), CTTAGCCGACTCTGCGAACT; Smad7 (rev), CCAGGCTCCAGAAGAAGTTG; Smad3 (fwd), GGTCAAGAGCCTGGTCAAGA; Smad3 (rev), TTGAAGGCGAACTCACACAG; ALK5 (fwd), GCCGTTTGTATGTGCACCCTCTTC; ALK5 (rev), GCTGCCAGTTCCACAGGACCAA; PAI-1 (fwd), TGATGGCTCAGACCAACAAG; PAI-1 (rev), GTTGGTGAGGGCAGAGAGAG; GAPDH (fwd), CTTCAACAGCGACACCCACT; GAPDH (rev), GTGGTCCAGGGGTCTTACTC.
ChIPs were performed as described previously (1). The regions of PAI-1 amplified were the Smad binding region (SBR; −724/−542 using 5′-CAGCCAGACAAGGTTGTTGACACA-3′ and 5′-CCAGCCACGTGATTGTCTAGGTTT-3′) and the transcription start site (TSS; −156/−17 using 5′-ACACACACACACACACATGCCTCA-3′ and 5′-CCAGATGTGGGCAGGAAATAGATG-3′). The regions of ID2 amplified were the BRE (−2941/−2840 using 5′-CAGGGATCACTCGCGGGGTC-3′ and 5′-CTCCCTGCACATCTGGCGCAA-3′) and the TSS (−459/−399 using 5′-GCCCGTGTAGCTGTGATTTTAGA-3′ and 5′-CAGACCAAGCCCTACACACCTT-3′).
Microarray analysis.
Total RNA was prepared from MDA-MB-231 cells induced with TGF-β and/or BMP7 in duplicate for 1 h or 6 h. Labeled cDNA was prepared and analyzed on Affymetrix GeneChip Human Exon 1.0 ST arrays at the Paterson Institute Microarray Service. The raw microarray data were analyzed using Bioconductor (17). First the data were normalized using the RMA procedure (24), and then the log n-fold changes and their standard errors were calculated using the limma package (48). The standard errors were moderated using an empirical Bayes factor (49). The linear models required by limma were designed to use all of the data across the experimental conditions (two TGF-β statuses, two BMP7 statuses, and three time points), and then individual comparisons were calculated as contrasts within this model. Genes were chosen using a false discovery rate threshold of <0.05 to compensate for multiple testing and an absolute n-fold change of >2.
Invasion.
One hundred microliters of collagen gel (1.5 mg/ml) in serum-free DMEM supplemented with 10 mM HEPES (pH 7.5) and 1 mM NaHCO3 was added to 8-μm-pore-size transwells (Corning) and left to solidify at 37°C for 1 h. MDA-MB-231 cells were allowed to adhere to the underside of the chamber for 4 h. Serum-free medium was added to the lower chamber, and complete medium was added to the upper chamber, with the ligands as appropriate. Cells were left to invade the collagen matrix for 48 h. After fixation, the cells were stained with fluorescein isothiocyanate (FITC)-phalloidin (Invitrogen) and z sections were taken by confocal microscopy every 10 μm. Five stacks were analyzed per condition. Fluorescence quantification was performed using ImageJ software. Levels of fluorescence in each section were normalized such that that in the first section was 100.
RESULTS
TGF-β inhibits BMP-induced transcriptional responses in an ALK5-dependent manner.
To investigate interactions between BMP and TGF-β signaling pathways, we used MDA-MB-231 cells stably expressing either BRE-luciferase or CAGA12-luciferase together with an internal control, TK-Renilla (53). The BRE-luciferase reporter is driven by two copies of two BREs derived from the ID1 promoter (29), while the CAGA12-luciferase reporter is driven by Smad3-Smad4 binding sites derived from the highly TGF-β-inducible PAI-1 promoter (11). As expected, BMP7 and TGF-β robustly induced the BRE-luciferase and CAGA12-luciferase reporters, respectively (Fig. 1A). TGF-β alone could not induce the BRE-luciferase reporter, but strikingly, stimulation of cells with TGF-β substantially inhibited BMP7-induced BRE-luciferase activity. In contrast, BMP7 had no effect on TGF-β-induced CAGA12-luciferase activity (Fig. 1A). The inhibitory effect of TGF-β was not confined to BMP7 signaling, as we could also demonstrate that TGF-β inhibited BMP2-induced transcription, measured in the same assay (Fig. 1B). Furthermore, TGF-β inhibited BMP-induced luciferase induction in transient-transfection assays in a number of different cell lines and also inhibited BMP7-induced activation of a luciferase reporter driven by the promoter region of ID2, a well-known BMP-responsive gene (38) (Fig. 1C and D; data not shown).
Fig 1.

TGF-β inhibits BMP-induced transcriptional responses in an ALK5-dependent manner. (A, B, and E to G) MDA-MB-231 BRE cells and MDA-MB-231 CAGA cells were induced with the ligands indicated and assayed for luciferase and Renilla activities after 8 h. (C) C2C12 and HaCaT cells were transiently transfected with BRE-luciferase and TK-Renilla. After 24 h, the C2C12 cells were induced with the ligands shown and luciferase/Renilla assays were performed. At 24 h after transfection, HaCaT cells were starved overnight in Opti-MEM before ligand induction. (D) MDA-MB-231 cells were transfected with ID2-luciferase and TK-Renilla for 24 h prior to ligand induction. In panel F, SB-431542 was added 15 min before the ligands shown, and in panel G, either dimethyl sulfoxide (DMSO) or SB-431542 was added 15, 30, or 60 min after ligand addition, as shown. All experiments were performed in triplicate. The data are presented as luciferase activity normalized to Renilla activity and are the mean and standard deviation from a single representative experiment. In panel E (right side), in parallel with the luciferase assay, MDA-MB-231 BRE cells were treated with ligands as indicated for 1 h. Whole-cell extracts were Western blotted using antibodies against PSmad1/5, PSmad2, and PSmad3, as well as total Smad1, -2, and -3. MCM7 is a loading control. The asterisk indicates a nonspecific band.
A TGF-β titration experiment revealed that inhibition of BMP7-induced transcriptional responses was dose dependent; 2 ng/ml and 0.2 ng/ml TGF-β had similar inhibitory effects, while 0.02 ng/ml TGF-β had only a weak inhibitory effect and 0.002 ng/ml had no effect (Fig. 1E, left side). The doses of TGF-β that inhibited BMP signaling were those that induced robust levels of phosphorylated Smad1/5 (PSmad1/5), PSmad2, and PSmad3 (Fig. 1E, right side). We also demonstrated that inhibition of BMP7-induced transcription by TGF-β requires ALK5 activity, as preincubation of the cells with the ALK5 inhibitor SB-431542 (23) abolished TGF-β's inhibitory effect (Fig. 1F). To establish the minimal duration of TGF-β signaling required, we added SB-431542 at different times after ligand addition to inhibit TGF-β signaling, harvesting all of the samples 8 h after the initial TGF-β and BMP7 stimulation. This experiment revealed that 1 h of TGF-β signaling was sufficient to inhibit BMP7-induced transcription (Fig. 1G).
TGF-β inhibits BMP induction of cell invasion and transcription of endogenous genes.
To demonstrate physiological relevance for BMP7 stimulation of MDA-MB-231 cells and for the observed TGF-β-dependent inhibition of BMP signaling, we asked whether TGF-β could inhibit BMP7-induced cell invasion, which has previously been reported in MDA-MB-231 cells (3). Cells stimulated with BMP7 invaded the collagen matrix much more effectively than unstimulated cells, and this was substantially inhibited by costimulation with TGF-β (Fig. 2A). We then sought to identify BMP7 target genes in MDA-MB-231 cells whose induction was inhibited by TGF-β by performing an Affymetrix microarray. We found 19 genes in total that were induced at least 2-fold by BMP7 at either the 1-h or the 6-h time point (see Table S1 in the supplemental material). This is consistent with other microarray analyses that have revealed many fewer BMP-responsive genes than TGF-β-responsive genes in tissue culture cells (2, 40). For nine of these genes, costimulation with TGF-β substantially decreased the BMP7 responsiveness, and this was confirmed for six of these induced genes by qPCR (Fig. 2B).
Fig 2.

TGF-β inhibits BMP induction of cell invasion and transcription of endogenous genes. (A) MDA-MB-231 cells that were either left untreated or stimulated with the ligands shown were allowed to invade a collagen gel for 48 h. Cells were stained with FITC-phalloidin, and their depth of invasion was determined by measuring fluorescence in a series of z stacks using a Zeiss LSM 780 confocal microscope. Fluorescence was normalized such that that in the first stack was 100. Five stacks were analyzed per condition. Means and standard deviations are plotted. (B) MDA-MB-231 cells were incubated overnight in Opti-MEM and then induced with BMP7 and/or TGF-β for the times indicated. Total RNA was prepared, and the levels of the genes indicated, relative to that of the gene for glyceraldehyde-3-phosphate dehydrogenase, were measured by qPCR. The qPCRs were performed in triplicate, and means and standard deviations of a representative experiment are shown.
TGF-β's inhibition of BMP-induced genes is independent of new protein synthesis.
The short duration of TGF-β signaling required to inhibit BMP-induced transcription suggested that the effect of TGF-β on BMP responses was direct and not dependent on transcription/translation of TGF-β target genes. Since ID2 and ID3 were the most strongly induced by BMP7 at the 1-h time point (Fig. 2B), we chose these genes to investigate this. Pretreatment of the cells with the translational inhibitor emetine had no effect on the TGF-β-mediated inhibition of BMP7-induced transcription of ID2 and ID3, indicating that it is independent of new protein synthesis (Fig. 3A). In a control experiment, we confirmed that emetine was highly effective for inhibiting translation in these cells (data not shown). A recent analysis of mouse embryonic stem cells showed that autocrine Nodal signaling has an inhibitory effect on BMP signaling through the synthesis of the inhibitory Smad Smad7 (15). Given the insensitivity of the TGF-β-mediated inhibition of BMP responses to de novo protein synthesis, we considered this mechanism unlikely to be responsible for our observations. Indeed, knocking down of Smad7 with two different siRNAs had no effect on TGF-β-mediated inhibition of BMP-responsive transcription (Fig. 3B). In contrast, as expected in light of the effect of SB-431542 (Fig. 1F), knocking down ALK5 abolished the effect and knocking down Smad4 inhibited BMP7-induced transcription (Fig. 3B).
Fig 3.
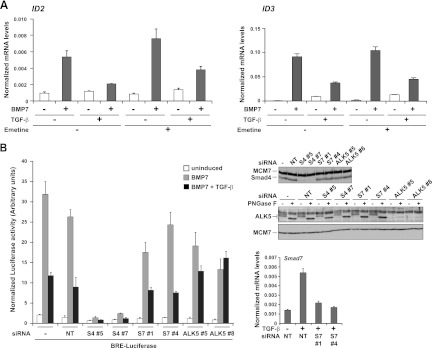
Inhibition of BMP7 responses by TGF-β does not require new protein synthesis and is independent of Smad7. (A) MDA-MB-231 cells were pretreated for 10 min with or without 400 μM emetine and then induced with the ligands indicated for 1 h. Total RNA was prepared, and levels of ID2 or ID3, relative to that of the gene for glyceraldehyde-3-phosphate dehydrogenase, were measured by qPCR. The qPCRs were performed in triplicate, and means and standard deviations are shown. (B) MDA-MB-231 BRE cells were transfected with the indicated siRNAs or a nontargeting (NT) control for 72 h before being treated with the ligands indicated for 8 h. Luciferase/Renilla assays were performed as described in the legend to Fig. 1. The knockdown efficiency of the Smad4 (S4) and ALK5 siRNAs is shown by Western blotting. For the ALK5 blot assay, extracts were treated with or without peptide N-glycosidase F (PNGase F) (12) to remove sugars from ALK5, enabling it to be more easily detected. The efficiency of the Smad7 (S7) knockdown was established by qPCR. Samples were treated for 1 h with TGF-β to induce levels of Smad7 in the control samples to be detectable by qPCR. Levels of Smad7, relative to that of the gene for glyceraldehyde-3-phosphate dehydrogenase, were calculated. Means and standard deviations of a representative experiment performed in triplicate are shown.
TGF-β has no effect on BMP-induced Smad1/5 phosphorylation and does not compete for BMP receptors.
To learn more about the mechanism whereby TGF-β inhibits BMP responses, we investigated whether TGF-β inhibited BMP-induced phosphorylation of Smad1/5 or accumulation of complexes containing PSmad1/5 in the nucleus. As expected, stimulation of cells with BMP7 or TGF-β led to the phosphorylation of Smad1/5 (9) (Fig. 4A and B). Costimulation of cells with both ligands actually resulted in a slightly increased level of PSmad1/5 in both whole-cell extracts and nuclear extracts compared with stimulation with BMP7 alone (Fig. 4A and B). These results rule out the simple hypothesis that TGF-β inhibits BMP-induced responses by inhibiting the ability of BMPs to induce Smad1/5 phosphorylation or accumulation of activated Smad complexes in the nucleus.
Fig 4.

TGF-β does not inhibit BMP's ability to induce phosphorylation of Smad1/5. (A) Whole-cell extracts were prepared from MDA-MB-231 cells treated with BMP7 and/or TGF-β for the times shown. Extracts were Western blotted using antibodies against phosphorylated Smad1/5 (PSmad1/5), Smad1, and MCM6 as a loading control. The blot assays were quantitated on an ImageQuant LAS4000 mini, and the levels of PSmad1/5 relative to Smad1 are plotted on the right. (B) Same as panel A, except that nuclear extracts were assayed. (C, left side) MDA-MB-231 cells or the same cells stably expressing FLAG-ALK2 were transiently transfected with BRE-luciferase and TK-Renilla. After 24 h, cells were induced with the ligands as indicated for 8 h. Luciferase/Renilla assays were performed as in the legend to Fig. 1. (C, right side) Whole-cell extracts were prepared from the same cell lines induced for 1 h with different concentrations of BMP7. The extracts were Western blotted using antibodies against PSmad1/5, Smad1, FLAG, and MCM6 as a loading control. WT, wild type.
We have previously shown that TGF-β induces phosphorylation of Smad1/5 via a heteromeric receptor complex comprising the TGF-β type II receptor with ALK5 and ALK2 or ALK3 (9). To eliminate the possibility that TGF-β inhibited BMP-induced responses by titrating out ALK2 or ALK3, which are BMP type I receptors (14), we generated a stable cell line overexpressing FLAG-tagged ALK2, which is the type I receptor preferentially recognized by BMP7 (34). This cell line responds more strongly to BMP7, indicating that the overexpressed ALK2 protein is active (Fig. 4C, right panel). However, no difference was detected between these ALK2-overexpressing cells and the parental cells in the ability of TGF-β (at two different concentrations) to inhibit BMP7-induced transcription (Fig. 4C, left panel), indicating that competition between TGF-β and BMP7 for the ALK2 receptor is not likely to be the mechanism responsible.
TGF-β stimulation results in a decrease in levels of PSmad1/5-Smad4 complexes formed in response to BMP7 and an increase in mixed R-Smad complexes.
We next used IPs to investigate whether TGF-β influenced the composition of the Smad complexes formed in response to BMP7. IP of Smad5 and blotting for Smad4 or IP of Smad4 and blotting for PSmad1/5 revealed that BMP7 induced PSmad1/5-Smad4 complexes (Fig. 5A). The presence of TGF-β significantly reduced the levels of these complexes (Fig. 5A). As previously reported, TGF-β induction resulted in the formation of so-called mixed R-Smad (PSmad2/3-PSmad1/5) complexes (9), which increased in cells treated with both TGF-β and BMP7 (Fig. 5A). TGF-β stimulation also induced the formation of PSmad2-Smad4 and PSmad3-Smad4 complexes, but the levels of these are not affected by the addition of BMP7 (Fig. 5B). To investigate the mixed R-Smad complexes more thoroughly, we used antibodies specific for Smad2 and Smad3 to immunoprecipitate these Smads. Both PSmad2-PSmad1/5 and PSmad3-PSmad1/5 complexes were formed in TGF-β-induced cells and in cells costimulated with TGF-β and BMP7 (Fig. 5C). These PSmad2-PSmad1/5 and PSmad3-PSmad1/5 complexes do not require Smad4 for their formation, as they are readily induced in cells in which Smad4 has been knocked down (Fig. 5C). To confirm that complex formation by PSmad3 and PSmad1 is direct and not dependent on bridging proteins, we performed IPs with recombinant PSmad1 and PSmad3. PSmad1-PSmad3 complexes were readily detected when either the PSmad1 or the PSmad3 component was immunoprecipitated (Fig. 5D).
Fig 5.

TGF-β induction leads to a loss of BMP-induced Smad1/5-Smad4 complexes and an increase in Smad1/5-Smad2/3 complexes. (A to C) MDA-MB-231 cells that were either untransfected (A and B) or transfected with nontargeting (NT) siRNA or an siRNA against Smad4 (C) were treated for 1 h with the ligands indicated. Whole-cell extracts were subjected to IP followed by Western blot assays with the antibodies shown. The lane marked “beads” correspond to an IP using beads alone. The inputs correspond to lysates prior to IP. (D) A mixture of purified recombinant PSmad1 and PSmad3 was immunoprecipitated with the antibodies shown or beads alone and then Western blotted as indicated.
Thus, we conclude that, compared to BMP7 induction alone, costimulation of cells with TGF-β and BMP7 alters the type of the activated Smad complexes formed, resulting in a decrease in levels of PSmad1/5-Smad4 complexes and an increase in PSmad3-PSmad1/5 complexes and PSmad2/3-Smad4 complexes.
TGF-β inhibits BMP responses by specifically inducing the formation of PSmad3-PSmad1/5 complexes.
Three possible mechanisms for TGF-β's inhibitory effects on BMP responses can be envisioned from these results. First, TGF-β stimulation might divert Smad4 from PSmad1/5-Smad4 complexes, which are crucial for BMP-induced transcription (29), to PSmad2/3-Smad4 complexes. Second, formation of PSmad2/3-PSmad1/5 complexes in response to TGF-β could titrate out PSmad1/5 from BMP-induced PSmad1/5-Smad4 complexes. Third, either PSmad2-PSmad1/5 or PSmad3-PSmad1/5 could have an active inhibitory effect on BMP-induced transcription.
To test the first possibility, we investigated whether Smad4 is limiting by overexpressing FLAG-tagged Smad4. We proved that the tagged Smad4 protein was active, as it restored BMP7-induced BRE-luciferase activity in a Smad4-null cell line, MDA-MB-468 (Fig. 6). However, increasing levels of Smad4 had no effect on the ability of TGF-β to inhibit BMP7-induced transcription in MDA-MB-231 cells, indicating that Smad4 is not limiting in these assays (Fig. 6).
Fig 6.
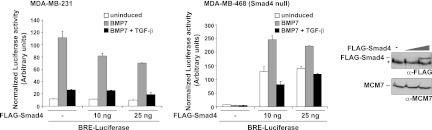
Smad4 is not responsible for TGF-β's inhibitory effects on BMP responses. MDA-MB-231 or MDA-MB-468 cells were transiently transfected with BRE-luciferase and TK-Renilla with or without increasing amounts of FLAG-Smad4 expression plasmid. After 24 h, cells were induced with the ligands shown and luciferase/Renilla assays were performed as described in the legend to Fig. 1. A Western blot assay showing the expression of FLAG-Smad4 in the MDA-MB-231 cells is shown. The asterisk denotes a nonspecific band.
To test the second and third possibilities, we dissected the roles of Smad2 and Smad3 in TGF-β's ability to inhibit BMP responses. Using MDA-MB-231 BRE cells, we found that knockdown of Smad3, but not knockdown of Smad2, abolished TGF-β-mediated inhibition (Fig. 7A and B). The fact that knockdown of Smad2 had no effect rules out a simple titration of PSmad1/5 from BMP-induced PSmad1/5-Smad4 complexes as the mechanism underlying TGF-β's inhibitory effects. If this were the mechanism, knockdown of Smad2 and knockdown of Smad3 would both be expected to abolish TGF-β-mediated inhibition, and moreover, knockdown of Smad2 should have a stronger effect, as Smad2 is more abundant than Smad3 in these cells (Fig. 7B). These results instead suggest that TGF-β-induced formation of PSmad3-PSmad1/5 complexes has a direct inhibitory effect on BMP-induced transcription.
Fig 7.

The inhibitory effect of TGF-β on BMP transcriptional responses depends on Smad3 but not on Smad2. (A to D) MDA-MB-231 BRE cells were transfected with nontargeting (NT) siRNA or siRNA duplexes against Smad2 (S2), Smad3 (S3), or ALK5, as indicated. After 72 h cells were stimulated with the ligands shown and luciferase/Renilla assays were performed as in the legend to Fig. 1 (A and C) or whole-cell extracts were prepared and blotted for Smad2/3, Smad4, or ALK5 as indicated (B and D). In the case of ALK5, samples were treated with or without peptide N-glycosidase F (PNGase F) as described in the legend to Fig. 3. (E) MDA-MB-231 BRE cells were treated with NT or Smad3 siRNA for 72 h as described above and then induced with ligands for 1 h. Total RNA was prepared, and levels of ID2, relative to that of the gene for glyceraldehyde-3-phosphate dehydrogenase, were measured by qPCR. The qPCRs were performed in triplicate, and means and standard deviations are shown. Controls for knockdown are those shown in panel D.
We have previously shown that TGF-β alone cannot induce the BRE-luciferase reporter (Fig. 1A) (9), and we hypothesized that this was due to the formation of mixed R-Smad complexes of phosphorylated Smad2/3 and Smad1/5 (9). The observation that Smad3 is required for TGF-β's inhibitory effect on BMP-induced transcription led us to investigate whether PSmad3-PSmad1/5 complexes were responsible for preventing TGF-β from inducing the BRE-luciferase reporter. Strikingly, knockdown of Smad3 with two different siRNAs conferred on TGF-β the ability to induce BRE-luciferase activity (Fig. 7C and D). Furthermore, knockdown of Smad3 also allowed TGF-β to induce endogenous ID2 expression, confirming that this observation was not restricted to a BMP-responsive reporter (Fig. 7E).
Taking these observations together, we conclude that PSmad1/5-PSmad3 complexes induced in response to TGF-β have a direct inhibitory effect on BMP-responsive elements. This explains the inability of TGF-β to induce BMP-responsive promoters when added alone and its ability to inhibit BMP-induced transcription when added with BMP7.
PSmad1/5-PSmad3 complexes bind to BMP-responsive elements in response to TGF-β in vitro and in vivo.
The proof that costimulation with TGF-β and BMP results in inhibitory complexes comprising phosphorylated Smad1/5 and Smad3 binding to BMP-responsive promoters would be the direct detection of such complexes on BREs. We first investigated this in vitro by using DNAP assays of HaCaT cells. In these assays, oligonucleotides of interest are immobilized on beads and incubated with nuclear extract and bound proteins are detected by Western blotting. In response to BMP7, we could readily detect PSmad1/5 and Smad4 binding to an oligonucleotide corresponding to a single copy of the two BRE sequences that drive the BRE-luciferase reporter (Fig. 8A, BRE). In response to TGF-β and to costimulation with TGF-β and BMP7, we additionally detected Smad3 binding to this sequence (Fig. 8A; BRE). A similar pattern of binding was seen when we used the region of the ID1 promoter (−1070/−1025) that contains one of the two BREs present in the BRE-luciferase reporter (29) (Fig. 8A, ID1 −1070/−1025). We also used oligonucleotides corresponding to the two recently described BREs derived from the ID2 promoter (Fig. 8B) (38). PSmad3 and PSmad1/5 were detected on both of these elements in response to TGF-β and BMP7 (Fig. 8B). Because Smad4 was also found to bind these ID2 probes in the presence of BMP7 with or without TGF-β, it was important to distinguish between PSmad3 recruited by Smad4 and that recruited as part of PSmad3-PSmad1/5 complexes. In BxPC3 cells, which are Smad4 null (39), we found that PSmad3 was still recruited to both ID2 probes (data not shown), indicating that Smad4 is not required. Furthermore, when Smad1/5 was knocked down in MDA-MB-231 cells, very little Smad3 was found to bind to probe A (data not shown). Binding of Smad3 was still detected on probe B, however, but the residual PSmad1/5 in these knockdown cells evidently recruited it.
Fig 8.

Smad3 and PSmad1/5 bind BREs in vitro in response to costimulation with TGF-β and BMP7. (A and B) Nuclear extracts were prepared from HaCaT cells that had been starved overnight in Opti-MEM and then treated for 1 h with the ligands shown. They were analyzed either directly by Western blotting using antibodies against Smad2/3, PSmad1/5, PSmad3, and Smad4 as a control (Inputs) or by DNAP using an oligonucleotide containing mutated Smad binding sites (32) (mutant), one corresponding to a single copy of the BRE sequences that drive the BRE-luciferase reporter (BRE) and one corresponding to the −1070/−1025 region of the ID1 promoter (A) or the mutant oligonucleotide, oligonucleotides corresponding to the −2979/−2920 (probe A) and −2919/−2860 (probe B) regions of the ID2 promoter (B).
Finally we investigated whether TGF-β induced the recruitment of Smad3 and PSmad1/5 to BREs in vivo by using ChIP analysis. We focused on the ID2 gene, as this is an endogenous gene for which we observe a strong TGF-β-dependent inhibition of BMP7-responsive transcription. We analyzed two regions of the human ID2 gene: the BRE between −2980 and −2880 upstream of the start of transcription and the TSS itself (38). As a control for Smad3 recruitment in response to TGF-β, we used the PAI-1 gene, which is induced only by TGF-β in a Smad3-dependent manner (11), and analyzed the previously described SBR and the TSS (1). Antibodies against Smad3, PSmad1/5, and RNA PolII were used.
We could readily detect an increase in Smad3 occupation of the ID2 BRE in response to 1 h of stimulation with BMP7 or TGF-β, and strikingly, this was further increased when both ligands were added together (Fig. 9A). Levels of PSmad1/5 were significantly increased on the ID2 BRE in response to BMP7, as expected, and to a slightly lesser extent when TGF-β and BMP7 were added together but not when cells were induced with TGF-β alone. A substantial level of PolII was observed at the ID2 TSS in unstimulated cells, which was increased upon BMP7 stimulation, but not in response to TGF-β, and addition of TGF-β inhibited the BMP7-induced enrichment of PolII at the TSS (Fig. 9A). Remarkably, the enrichment of PolII at the ID2 TSS in response to these combinations of ligands faithfully reflected the levels of ID2 mRNA measured by qPCR produced after 1 h of stimulation with the same ligands (Fig. 9B). As a positive control for the ChIPs, we found that Smad3 was enriched on the PAI-1 SBR in response to TGF-β, and this was not affected by costimulation with BMP7. PSmad1/5 binding was not detected in the PAI-1 regulatory regions (Fig. 9A). Again we noted that the pattern of enrichment of PolII at the PAI-1 TSS reflected the levels of PAI-1 mRNA, measured by qPCR, produced after stimulation with the same ligands (Fig. 9B).
Fig 9.

Smad3 and PSmad1/5 accumulate on BMP-responsive elements in response to costimulation with TGF-β and BMP7 in vivo. (A) ChIP analysis of the BRE and TSS regions of the ID2 gene and the SBR and TSS of the PAI-1 gene using antibodies against Smad3, PSmad1/5, and RNA PolII. Cells were starved overnight in Opti-MEM and then stimulated with the ligands indicated. (B) Cells were treated with the ligands shown for 1 h and analyzed by qPCR for levels of ID2 and PAI-1 mRNAs. In panels A and B, the data are means and standard deviations of qPCRs performed in triplicate in a representative experiment. To increase the sensitivity of these assays, we used a HaCaT cell line that stably expresses low levels of HA-Smad3 (data not shown).
Thus, we conclude that costimulation of cells with BMP7 and TGF-β leads to an accumulation of Smad3 and phosphorylated Smad1/5 on the ID2 BRE.
DISCUSSION
A novel role for Smad3 in mediating TGF-β-induced transcriptional repression.
We and others have previously shown that, in addition to inducing the phosphorylation of Smad2 and Smad3, TGF-β robustly induces the phosphorylation and activation of Smad1/5/8, which had hitherto been thought to be unique to BMPs and GDFs (6, 9, 20, 33, 51). In fact, the class of R-Smads activated in response to a different subset of ligands had been thought to explain the particular repertoires of target genes induced by these ligands (43). The observation that TGF-β could induce so-called “BMP R-Smads” raised the important question of why TGF-β does not normally induce BMP-responsive genes. The work presented here now provides the molecular explanation. We show that TGF-β induces the formation of mixed R-Smad complexes containing PSmad1/5 and either PSmad2 or PSmad3. Importantly, knockdown of Smad3 abolishes TGF-β's ability to inhibit BMP-induced transcription. We show that the complexes formed by PSmad1/5 and PSmad3 are recruited to BREs in vivo and in vitro, where they act to repress transcription. This has two important consequences. First, it means that TGF-β cannot activate transcription via BREs unless Smad3 is absent. Second, it explains the antagonistic action of TGF-β on BMP-induced transcription. We do not know exactly how these PSmad3-PSmad1/5 complexes inhibit BMP-induced transcription. They could either compete for binding with activating PSmad1/5-Smad4 complexes at BREs or, alternatively, could bind in addition to the PSmad1/5-Smad4 complexes and confer repression, possibly by recruiting transcriptional corepressors.
These results raise a fundamental question relating to the DNA-binding specificity of the Smads. Studies with Drosophila, mammalian cell culture, and in vitro experiments have suggested that Smad1 preferentially binds GC-rich sequences with the consensus GRCGNC, while Smad3 and Smad4 bind direct or inverted repeats of the sequence GTCT or its reverse/complement AGAC (called SBEs) (11, 16, 42, 47, 56). Until now, this Smad binding specificity seemed to adequately explain why BMP-activated Smads recognize certain composite elements while TGF-β-activated Smads recognize different elements. However, our observation that complexes comprising PSmad1/5 and PSmad3 bind to BREs now casts doubt on this view. The BRE-luciferase construct used in our present study and shown to bind PSmad1/5-PSmad3 complexes contains two separate BREs derived from the ID1 promoter. One contains the canonical Smad1-Smad4 binding site comprising a GC-rich element spaced 5 nucleotides from a GTCT, while the other contains a GC-rich element but no GTCT element (29). Moreover, the mapped BMP-responsive region of the ID2 promoter similarly contains both types of BRE (38) and we have shown by using DNAP assays that PSmad3 and PSmad1/5 bind to each of these BRE sequences. This suggests that PSmad1/5-PSmad3 complexes have a distinct binding specificity that we do not yet fully understand. Interestingly, a recent systematic binding study showed that, in fact, the Smad3 and Smad1 MH1 domains exhibit very similar affinities for GC-rich sequences and also for SBEs, indicating that the generally accepted Smad binding specificity is not entirely accurate (5). How the different active and repressive Smad complexes recognize promoter/enhancer elements therefore remains an unresolved question.
Mechanisms of TGF-β superfamily signaling antagonism.
In this report, we describe a novel mechanism of antagonism between TGF-β and BMP, mediated via complexes formed by PSmad1/5 and PSmad3. Previously, a mechanism underlying TGF-β-induced repression of ID1 was described (26) and it is important to distinguish that mechanism from what we describe here for ID2 and ID3. In the case of ID1, TGF-β-induced repression depends on new protein synthesis, as it is mediated by ATF3, which is itself induced by TGF-β (26). In contrast, the mechanism we describe here is independent of new protein synthesis. Furthermore, the binding site for ATF3 is not included in the BRE reporter derived from the ID1 promoter that we used in this study, nor is it present in any of the ID1 or ID2 probes used for the DNAP assays. Moreover, as shown in Table S1 in the supplemental material, TGF-β does not repress BMP7-induced ID1 transcription at early time points, in contrast to what we observed with ID2 and ID3.
Other studies have observed mutual antagonism between Nodal and BMP signaling but have ascribed different mechanisms, including competition for Smad4 (8), heterodimerization of ligands (55), and induction of the negative Smad Smad7 (15). We have ruled out all of these mechanisms for the direct antagonism we observed between TGF-β and BMP7, which occurs within 1 h of TGF-β stimulation. Our demonstration that ALK5 receptor activity is required rules out a ligand dimerization mechanism. In our system, overexpression of Smad4 failed to abolish TGF-β's inhibitory effect on BMP signaling, indicating that Smad4 is not a limiting component. Finally, we have shown that knockdown of Smad7 had no effect on TGF-β's antagonistic behavior. Thus, BMP-induced transcription can be inhibited by other TGF-β superfamily signaling pathways via a number of different mechanisms, depending on the biological context and the particular ligand. The mechanism we describe here requires TGF-β to induce the phosphorylation of Smad1/5. Crucially, it is not known whether this is also true of Nodal signaling, which is mediated via ALK4 and ALK7 (14), but it is important to discover whether this is the case.
Functional implications.
Our observation that Smad3 is essential for preventing TGF-β from inducing BMP-like transcriptional responses and also is responsible for TGF-β's antagonistic effects on BMP signaling has many functional implications. We predict that loss of Smad3 in cells that normally receive both TGF-β and BMP signals would result in enhanced BMP responses. Smad3-null mice die of defects in immune function and, in some cases, of colorectal cancer (reviewed in reference 19). They also show accelerated wound healing (4). It is not clear whether any of these defects result from increased BMP signaling. However, it has recently been reported that loss of Smad3 accelerates early bone fracture healing in mice, partly by promoting osteogenesis, which is a BMP-driven process (28, 41).
Our observation that loss of Smad3 confers on TGF-β the ability to induce ID2 could be relevant in cancer. The ID genes, which are normally inhibited by TGF-β (36), are potent tumor promoters, deregulating proliferation and conferring invasiveness, angiogenesis, and metastasis (21). Moreover, Smad3 is frequently downregulated in cancer and inactivating mutations have recently been discovered (10, 25, 30). In addition, tumor cells frequently secrete high levels of TGF-β (31) and we have previously shown that the ability of TGF-β to induce phosphorylation of Smad1/5 occurs at concentrations of TGF-β higher than that required for the phosphorylation of Smad2/3 (9). It will be exciting in the future to understand whether there is a link between downregulation or mutation of Smad3 and TGF-β-induced upregulation of ID genes in tumorigenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Peter ten Dijke and Yoshifumi Yokota for the BRE-luciferase and ID2-luciferase plasmids, respectively; Sean Harkin for generating the HA-Smad3 HaCaT cell line; Gavin Kelly for analysis of the microarray data; and the London Research Institute Equipment Park for technical support. We thank members of the Hill lab and Mike Howell for stimulating discussions and/or very useful comments on the manuscript.
This work was supported by Cancer Research UK, the European Commission Network of Excellence EpiGeneSys (HEALTH-F4-2010-257082), and by Marie Curie Intra-European Fellowships (PIEF-GA-2008-220135 to E.G. and PIEF-GA-2009-235995 to P.V.).
Footnotes
Published ahead of print 21 May 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Agricola E, Randall RA, Gaarenstroom T, Dupont S, Hill CS. 2011. Recruitment of TIF1γ to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol. Cell 43:85–96 [DOI] [PubMed] [Google Scholar]
- 2. Alarcón C, et al. 2009. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-β pathways. Cell 139:757–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alarmo EL, et al. 2009. BMP7 influences proliferation, migration, and invasion of breast cancer cells. Cancer Lett. 275:35–43 [DOI] [PubMed] [Google Scholar]
- 4. Ashcroft GS, et al. 1999. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell Biol. 1:260–266 [DOI] [PubMed] [Google Scholar]
- 5. BabuRajendran N, et al. 2010. Structure of Smad1 MH1/DNA complex reveals distinctive rearrangements of BMP and TGF-β effectors. Nucleic Acids Res. 38:3477–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bharathy S, Xie W, Yingling JM, Reiss M. 2008. Cancer-associated transforming growth factor β type II receptor gene mutant causes activation of bone morphogenic protein-Smads and invasive phenotype. Cancer Res. 68:1656–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blobe GC, Schiemann WP, Lodish HF. 2000. Role of transforming growth factor β in human disease. N. Engl. J. Med. 342:1350–1358 [DOI] [PubMed] [Google Scholar]
- 8. Candia AF, et al. 1997. Cellular interpretation of multiple TGF-β signals: intracellular antagonism between activin/BVg1 and BMP-2/4 signaling mediated by Smads. Development 124:4467–4480 [DOI] [PubMed] [Google Scholar]
- 9. Daly AC, Randall RA, Hill CS. 2008. Transforming growth factor β-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell. Biol. 28:6889–6902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daly AC, Vizan P, Hill CS. 2010. Smad3 protein levels are modulated by Ras activity and during the cell cycle to dictate transforming growth factor-beta responses. J. Biol. Chem. 285:6489–6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dennler S, et al. 1998. Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17:3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dorey K, Hill CS. 2006. A novel Cripto-related protein reveals an essential role for EGF-CFCs in Nodal signalling in Xenopus embryos. Dev. Biol. 292:303–316 [DOI] [PubMed] [Google Scholar]
- 13. Ebisawa T, et al. 2001. Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 276:12477–12480 [DOI] [PubMed] [Google Scholar]
- 14. Feng XH, Derynck R. 2005. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21:659–693 [DOI] [PubMed] [Google Scholar]
- 15. Galvin KE, Travis ED, Yee D, Magnuson T, Vivian JL. 2010. Nodal signaling regulates the bone morphogenic protein pluripotency pathway in mouse embryonic stem cells. J. Biol. Chem. 285:19747–19756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao S, Laughon A. 2006. Decapentaplegic-responsive silencers contain overlapping mad-binding sites. J. Biol. Chem. 281:25781–25790 [DOI] [PubMed] [Google Scholar]
- 17. Gentleman RC, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Germain S, Howell M, Esslemont GM, Hill CS. 2000. Homeodomain and winged-helix transcription factors recruit activated Smads to distinct promoter elements via a common Smad interaction motif. Genes Dev. 14:435–451 [PMC free article] [PubMed] [Google Scholar]
- 19. Goumans MJ, Mummery C. 2000. Functional analysis of the TGFβ receptor/Smad pathway through gene ablation in mice. Int. J. Dev. Biol. 44:253–265 [PubMed] [Google Scholar]
- 20. Goumans MJ, et al. 2002. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 21:1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iavarone A, Lasorella A. 2006. ID proteins as targets in cancer and tools in neurobiology. Trends Mol. Med. 12:588–594 [DOI] [PubMed] [Google Scholar]
- 22. Inman GJ, Hill CS. 2002. Stoichiometry of active Smad-transcription factor complexes on DNA. J. Biol. Chem. 277:51008–51016 [DOI] [PubMed] [Google Scholar]
- 23. Inman GJ, et al. 2002. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 62:65–74 [DOI] [PubMed] [Google Scholar]
- 24. Irizarry RA, et al. 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones S, et al. 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321:1801–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang Y, Chen CR, Massagué J. 2003. A self-enabling TGFβ response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol. Cell 11:915–926 [DOI] [PubMed] [Google Scholar]
- 27. Kavsak P, et al. 2000. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF β receptor for degradation. Mol. Cell 6:1365–1375 [DOI] [PubMed] [Google Scholar]
- 28. Kawakatsu M, et al. 2011. Loss of Smad3 gives rise to poor soft callus formation and accelerates early fracture healing. Exp. Mol. Pathol. 90:107–115 [DOI] [PubMed] [Google Scholar]
- 29. Korchynskyi O, ten Dijke P. 2002. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 277:4883–4891 [DOI] [PubMed] [Google Scholar]
- 30. Leary RJ, et al. 2008. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proc. Natl. Acad. Sci. U. S. A. 105:16224–16229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Levy L, Hill CS. 2006. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 17:41–58 [DOI] [PubMed] [Google Scholar]
- 32. Levy L, et al. 2007. Arkadia activates Smad3/Smad4-dependent transcription by triggering signal-induced SnoN degradation. Mol. Cell. Biol. 27:6068–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu IM, et al. 2009. TGFβ-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFβ switch. EMBO J. 28:88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Macías-Silva M, Hoodless PA, Tang SJ, Buchwald M, Wrana JL. 1998. Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J. Biol. Chem. 273:25628–25636 [DOI] [PubMed] [Google Scholar]
- 35. Maeda S, Hayashi M, Komiya S, Imamura T, Miyazono K. 2004. Endogenous TGF-β signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J. 23:552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Massagué J. 2008. TGFβ in cancer. Cell 134:215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Massagué J, Blain SW, Lo RS. 2000. TGFβ signaling in growth control, cancer, and heritable disorders. Cell 103:295–309 [DOI] [PubMed] [Google Scholar]
- 38. Nakahiro T, Kurooka H, Mori K, Sano K, Yokota Y. 2010. Identification of BMP-responsive elements in the mouse Id2 gene. Biochem. Biophys. Res. Commun. 399:416–421 [DOI] [PubMed] [Google Scholar]
- 39. Nicolás FJ, Hill CS. 2003. Attenuation of the TGF-β-Smad signaling pathway in pancreatic tumor cells confers resistance to TGF-β-induced growth arrest. Oncogene 22:3698–3711 [DOI] [PubMed] [Google Scholar]
- 40. Ramel M-C, Hill CS. Spatial regulation of BMP activity. FEBS Lett., in press [DOI] [PubMed] [Google Scholar]
- 41. Rosen V. 2009. BMP2 signaling in bone development and repair. Cytokine Growth Factor Rev. 20:475–480 [DOI] [PubMed] [Google Scholar]
- 42. Ross S, Hill CS. 2008. How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 40:383–408 [DOI] [PubMed] [Google Scholar]
- 43. Schmierer B, Hill CS. 2007. TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8:970–982 [DOI] [PubMed] [Google Scholar]
- 44. Schreiber E, Matthias P, Muller MM, Schaffner W. 1989. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi W, et al. 2004. GADD34-PP1c recruited by Smad7 dephosphorylates TGFβ type I receptor. J. Cell Biol. 164:291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shi Y, Massagué J. 2003. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113:685–700 [DOI] [PubMed] [Google Scholar]
- 47. Shi Y, et al. 1998. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell 94:585–594 [DOI] [PubMed] [Google Scholar]
- 48. Smyth GK. 2005. Limma: linear models for microarray data, p 397–420 In Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W. (ed), Bioinformatics and computational biology solutions using R and Bioconductor. Springer, New York, NY [Google Scholar]
- 49. Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3:Article3 [DOI] [PubMed] [Google Scholar]
- 50. Wong C, et al. 1999. Smad3-Smad4 and AP-1 complexes synergize in transcriptional activation of the c-Jun promoter by transforming growth factor β. Mol. Cell. Biol. 19:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wrighton KH, Lin X, Yu PB, Feng XH. 2009. Transforming growth factor β can stimulate Smad1 phosphorylation independently of bone morphogenic protein receptors. J. Biol. Chem. 284:9755–9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu MY, Hill CS. 2009. TGF-β superfamily signaling in embryonic development and homeostasis. Dev. Cell 16:329–343 [DOI] [PubMed] [Google Scholar]
- 53. Wu MY, Ramel M-C, Howell M, Hill CS. 2011. SNW1 is a critical regulator of spatial BMP activity, neural plate border formation and neural crest specification in vertebrate embryos. PLoS Biol. 9:e1000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xiao L, Yuan X, Sharkis SJ. 2006. Activin A maintains self-renewal and regulates fibroblast growth factor, Wnt, and bone morphogenic protein pathways in human embryonic stem cells. Stem Cells 24:1476–1486 [DOI] [PubMed] [Google Scholar]
- 55. Yeo C, Whitman M. 2001. Nodal signals to Smads through Cripto-dependent and Cripto-independent mechanisms. Mol. Cell 7:949–957 [DOI] [PubMed] [Google Scholar]
- 56. Zawel L, et al. 1998. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1:611–617 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.