

Abstract
We analyzed three human genes that were >200 kbp in length as they are switched on rapidly and synchronously by tumor necrosis factor alpha and obtained new insights into the transcription cycle that are difficult to obtain using continuously active, short, genes. First, a preexisting “whole-gene” loop in one gene disappears on stimulation; it is stabilized by CCCTC-binding factor and TFIIB and poises the gene for a prompt response. Second, “subgene” loops (detected using chromosome conformation capture) develop and enlarge, a result that is simply explained if elongating polymerases become immobilized in transcription factories, where they reel in their templates. Third, high-resolution localization confirms that relevant nascent transcripts (detected using RNA fluorescence in situ hybridization) lie close enough to be present on the surface of one factory. These dynamics underscore the complex transitions between the poised, initiating, and elongating transcriptional states.
INTRODUCTION
It is widely assumed that an RNA polymerase transcribes by first diffusing to a promoter wherever that promoter might be in the nucleus, binding, and then tracking down the template. However, an alternative sees the active form of the enzyme housed in a transcription factory; then, a promoter would diffuse to a factory, where it would bind a transiently immobilized polymerase, before that polymerase reeled in the template as it extruded the transcript (8, 32, 33). Nucleoplasmic factories are polymorphic (13, 14), and in a HeLa cell one is typically associated with ∼16 loops tethered through engaged polymerases and transcription factors to a ∼90-nm core (10). Chromosome conformation capture (3C) and fluorescence in situ hybridization (FISH) provide strong support for this alternative; sequences lying far apart on the genetic map lie together in three-dimensional (3D) nuclear space, and the contacting sequences are often transcribed and/or associated with bound transcription factors (2, 5, 7, 21, 24, 29, 33, 37, 47).
We present here a detailed analysis of the changing conformations of three human genes as they become active. Our approach depends on the use of a rapid and synchronous gene switch. Diploid human umbilical vein endothelial cells (HUVECs) are arrested in the G0 phase of the cell cycle by serum starvation, and tumor necrosis factor alpha (TNF-α) is added; this cytokine orchestrates the inflammatory response and induces a subset of genes to become active within minutes (44). We chose three rapidly responding genes of >200 kbp for analysis, since their great length provides ample temporal and spatial resolution. After switching them on, we used 3C to analyze their changing conformations over a period of 85 min. We found that “subgene” loops develop soon after initiation, and these then grow as pioneering polymerases elongate.
We also analyzed an exceptional “whole-gene” loop seen in one gene before stimulation which, in contrast to the subgene loops, disappears on stimulation. Whole-gene loops have been detected in various organisms, including mammals (1, 31, 35, 40–42, 50). Here, the presence of the whole-gene loop correlated with the binding of CCCTC-binding factor (CTCF) and a general transcription factor, TFIIB, to each end of the gene. Although TFIIB has been shown to associate with the 3′ ends of genes (17, 27, 45), and CTCF (26, 28) and TFIIB (40) have been implicated in stabilizing chromatin loops, this combination is observed here for the first time.
MATERIALS AND METHODS
Cell culture.
HUVECs from pooled donors (Lonza) were grown to 80 to 90% confluence in endothelial basal medium 2-MV with supplements (EBM; Lonza) and 5% fetal bovine serum (FBS), regrown (“starved”) for 16 to 18 h in EBM plus 0.5% FBS, treated with TNF-α (10 ng/ml; Peprotech), and harvested at different times after stimulation. In some cases, 50 μM 5,6-dichloro-1-β-d-ribofuranosyl-benzimidazole (DRB; Sigma-Aldrich) was added 25 min before harvesting.
Oligonucleotides.
PCR primers were designed using Primer3Plus (version 3.0; Whitehead Institute for Biomedical Research [http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi]) to have an optimal length of 20 to 22 nucleotides, to have a melting temperature of 62°C, and to yield amplimers of 125 to 225 bp. For quantitative PCR (qPCR), they were designed after activating qPCR settings. All primer sequences are available on request.
3C.
Chromosome conformation capture (3C) was performed as described previously (33). In brief, 107 cells were fixed (10 min at 20°C) in 1% paraformaldehyde (Electron Microscopy Sciences), aliquots of 106 cells in 0.125 M glycine in phosphate-buffered saline (PBS) were centrifuged, and the cells were resuspended in the appropriate restriction enzyme buffer and lysed (16 h at 37°C) in 0.3% sodium dodecyl sulfate (SDS). After sequestering SDS by adding 1.8% Triton X-100 (1.5 h at 37°C), the cells were treated overnight with HindIII or SacI (a total of 800 U added in four sequential steps; New England Biolabs), the enzyme was heat inactivated (25 min at 65°C), and the digestion efficiency was determined by qPCR. Samples with digestion efficiencies of >75% were ligated using T4 ligase (6,000 U [New England Biolabs]; DNA concentration, <0.5 ng/μl; 3 to 5 days at 4°C to minimize unwanted ligations), and cross-links were reversed (16 h at 65°C) in proteinase K (10 μg/ml; New England Biolabs) before the DNA was purified using an EZNA MicroElute DNA cleanup kit (Omega BioTek) and a PCR purification kit (Qiagen). To control for amplification of randomly ligated segments, nondigested/ligated and digested/nonligated 3C templates were also prepared. For 3C-PCRs, amplification efficiency controls using bacterial artificial chromosomes were as described previously (33). PCRs were conducted using 1.75 mM MgCl2, 1% dimethyl sulfoxide, 10 pmol of each primer, and GoTaq polymerase (Promega) per reaction (95°C for 2 min, followed by 31 cycles of 95°C for 55 s, 59°C for 45 s, and 72°C for 20 s, followed in turn by 1 cycle at 72°C for 2 min). Amplimers were resolved on 2.5% agarose gels, stained, and imaged on an FLA-5000 scanner (Fuji). The identities of 3C-PCR products were verified by sequencing (Geneservices, Oxford, United Kingdom) and/or restriction digestions. The results shown were reproduced using at least two independently obtained templates.
Chromatin immunoprecipitation (ChIP).
Approximately 107 HUVECs were cross-linked (10 min at 20°C) in 0.8% paraformaldehyde at the appropriate times after TNF-α induction. Chromatin was prepared, fragmented, washed, and eluted using a ChIP-It-Express kit according to the instructions for enzymatic shearing (Active Motif). Immunoprecipitations were performed using a rabbit polyclonal against the N terminus of the RNA polymerase II large subunit (sc-889X; Santa Cruz Biotechnology), TFIIB (sc-274X; Santa Cruz Biotechnology), and CTCF (ab70303; Abcam). DNA was purified using a MicroElute Cycle-Pure kit (Omega BioTek) prior to qPCR analysis. The region of GAPDH containing the TATA box was used as a positive control for RNA polymerase II binding, and a portion of the 3′ untranslated region of AFP served as a negative control. The results shown were reproduced using at least two independently obtained templates. For native ChIP (data not shown), the cross-linking step was omitted, and a rabbit polyclonal recognizing histone H3 (sc-10809X; Santa Cruz Biotechnology) was used. The ReChIP-It kit (Active Motif) was used for sequential ChIP (ReChIP).
Coimmunoprecipitation.
Approximately 5 × 107 HUVECs, grown and induced with TNF-α as described above, were lysed (20 min at 4°C) in 5 ml of lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5 mM MgCl2, 2 mM EDTA, 0.3 mM sucrose, 1% NP-40, 1 mM dithiothreitol) complemented with protein inhibitor cocktail (PIC; Roche) and spun at 10,000 × g (10 min; 4°C), and the supernatant was precleared (1 h; 4°C) with 5 μg of rabbit IgG (Upstate) in 50 μl of protein A-coupled agarose beads (Pierce). Protein complexes were then pulled down (16 h at 4°C) using anti-CTCF (2 μg) or anti-TFIIB (3 μg) in 100 μl of protein A-coupled agarose beads in 125 or 250 mM NaCl. The complexes were washed 10 times (for 10 min at 4°C each time) in wash buffer (50 mM Tris-HCl [pH 7.5], 0.02% Tween 20, 0.05% Triton X-100, 250 mM NaCl) plus PIC, eluted by boiling in 1× SDS loading buffer (50 μl/ml of lysate), separated in 10% acrylamide gels (Bio-Rad), and transferred onto nitrocellulose using an iBlot transfer system (Invitrogen), and CTCF or TFIIB was detected by immunoblotting (1:5,000 dilution) and visualized by chemiluminescence (SuperSignal West Pico; Pierce) with a ChemiDoc XRS+ imager (Bio-Rad).
qPCR.
For 3C and ChIP, quantitative real-time PCR (qPCR) was performed using a Rotor-Gene 3000 cycler (Corbett) and Platinum SYBR green qPCR SuperMix-UDG (Invitrogen). After incubation at 50°C for 2 min to activate the mix and treatment at 95°C for 5 min to denature the templates, reactions were carried out for 40 cycles at 95°C for 15 s and 60°C for 50 s. The presence of single amplimers was confirmed by melting-curve analysis and gel electrophoresis, and the data were analyzed as described by Nelson et al. (29) for ChIP (to obtain enrichments relative to input) and by Hagège et al. (18) for 3C (to obtain relative interaction frequencies relative to the “loading” and “intra-GAPDH” controls).
RNA FISH.
RNA FISH was performed as described previously (44), using four sets of five 50-mers (Gene Design, Japan). Each set targeted region b, c, d, or e of SAMD4A intron 1. In each 50-mer, roughly every tenth thymine residue was substituted by an amino modifier C6-dT coupled to the Alexa Fluor 488 or Alexa Fluor 555 reactive dyes (Invitrogen), and the five 50-mers in a set targeted <450 contiguous nucleotides. Then, after hybridization of the five 50-mers to one (fully stretched) target RNA, the distance between the outermost fluors is ∼150 nm. Since this distance is below the resolution limit of the microscope, the ∼25 fluors in each probe set appear as one diffraction-limited spot. (Of course, nascent transcripts are unlikely to be fully stretched.) Probes were purified using G-50 columns (GE Healthcare), ethanol precipitated twice, and concentrated using a Microcon-30 column (Millipore), and labeling efficiencies were calculated using the Base:Dye ratio calculator (Invitrogen; ∼8 fluors/100 nucleotides). For each experiment, HUVECs on coverslips were grown to ∼80% confluence, treated with TNF-α, fixed (17 min at room temperature) in 4% paraformaldehyde–0.05% acetic acid–0.15 M NaCl, washed three times in PBS, permeabilized (5 min at 37°C) in 0.01% pepsin (pH 2.0), rinsed in water treated with diethyl-pyrocarbonate, postfixed (5 min at 20°C) in 4% paraformaldehyde-PBS, and stored (overnight at −20°C) in 70% ethanol. Coverslips were dehydrated in 70, 80, 90, and 100% ethanol and hybridized (overnight at 37°C in a moist chamber) with 25 ng of labeled probes in medium containing 25% deionized formamide, 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 250 ng of sheared salmon sperm DNA/ml, 5× Denhardt's solution, 50 mM phosphate buffer (pH 7.0), and 1 mM EDTA. Next, the cells were washed once in 4× SSC (15 min at 37°C) and three times in 2× SSC (10 min at 37°C) and then mounted in Vectashield (Vector Laboratories) supplemented with 1 μg of DAPI (4′,6′-diamidino-2-phenylindole; Sigma)/ml.
The analysis depicted in Fig. 5 relies on the targets of the two probes uses lying on different RNA molecules (i.e., in the traveling and standing waves, respectively), even though they both lie within SAMD4A intron 1. (Note that exonic regions can be retained by “exon tethering” [12] as intronic RNA is degraded cotranscriptionally before the polymerase reaches the next exon by any number of mechanisms, including “recursive splicing” [6].) Various types of evidence indicate that the two targets are unlikely to lie on the same molecule. (i) The presence of a “trough” in the tiling microarray data summarized in Fig. 1A indicates that intronic RNA is degraded before the pioneering polymerase reaches exon 2 (if this intronic RNA persisted, there would be no trough). Similar troughs are seen in all five long genes analyzed in reference 44. (ii) The signal intensities in this microarray data indicate there are (at the very least) 3-fold more molecules of region b RNA compared to region c RNA after stimulation (with b:d and b:e ratios being even higher). This is because many polymerases generating the standing wave abort soon after transcribing region b (before they reach c, d, or e). Even if no degradation of intron 1 occurs before the polymerase reaches exon 2 (to generate one RNA molecule containing both targets), this still means that probe b would usually hybridize at the same allele to a different RNA molecule than probe c (or d or e). (iii) The half-lives of regions c, d, and e in intron 1 are between 3 and 6 min (measured using DRB and qRT-PCR [unpublished data]), which is comparable to those seen with intronic RNAs of other mammalian genes (19, 39). Since the pioneering polymerase takes ∼10 such half-lives to transcribe intron 1, much of this intron will inevitably be degraded well before the polymerase reaches exon 2.
Fig 5.
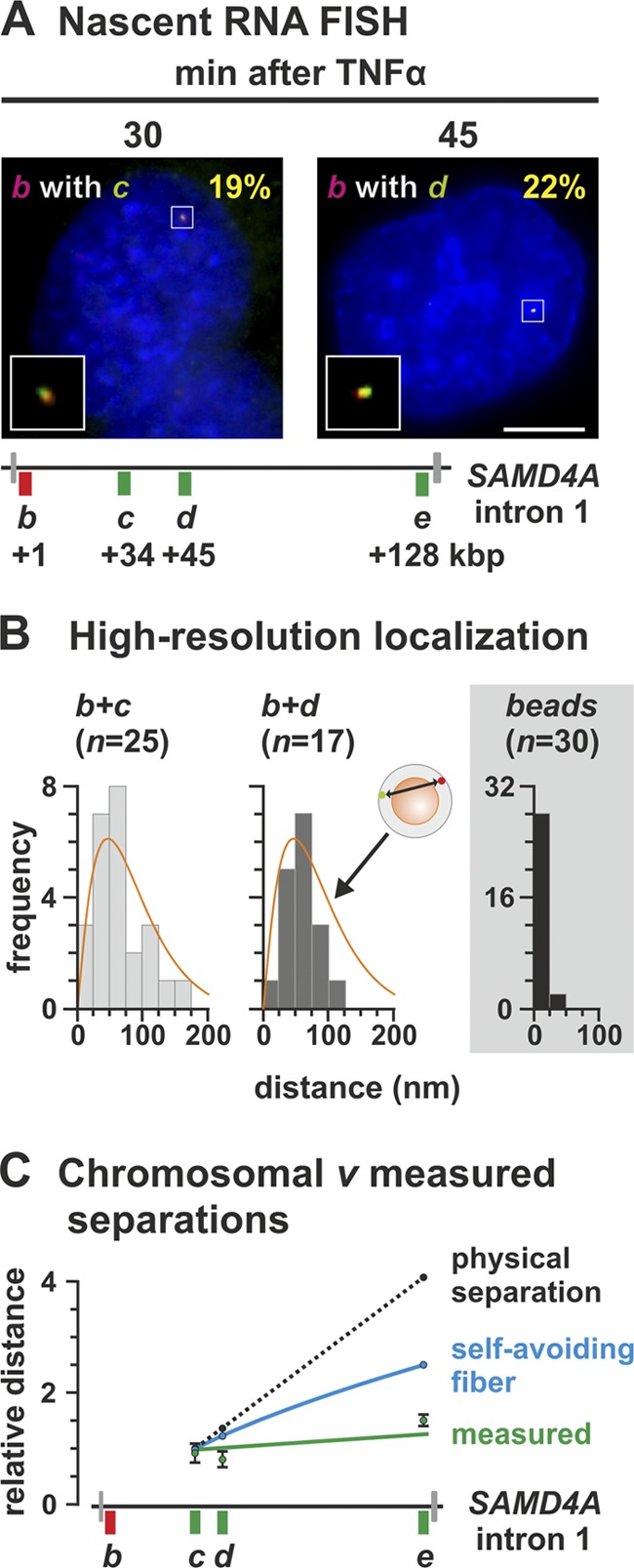
High-resolution microscopy of nascent transcripts in the standing and traveling waves. HUVECs were treated with TNF-α for different times, and nascent RNAs were detected by RNA FISH using intronic probes. (A) Typical RNA FISH images. Probe pairs b plus c and b plus d (positions shown in the diagram of SAMD4A intron 1) yield 19 and 22% cells with yellow foci (inset; ≥30% pixels with red signal also contain green signal or vice versa) marking colocalizing RNAs copied from one allele. Bar, 2 μm. (B) Single-molecule localizations. Gaussian curves were fitted to red and green foci, giving yellow foci such as those in panel B, and separations between peaks were determined with 27-nm accuracy. Few separations were <25 nm, and their distribution is consistent with foci being randomly distributed in a 35-nm shell around one 90-nm factory (diagram; orange curve from reference 33). A control for “perfect” colocalization is provided by 110-nm red/green fluorescent beads. n, the number of yellow foci analyzed. (C) Relative separations (± the SD; green line) between red and green peaks in yellow foci given by the probe pairs b plus c, b plus d, and b plus e (normalized to the mean value obtained with b plus c). The dotted black line shows what is expected if separations increased in proportion to the number of base pairs between b, c, d, and e. (The contour lengths of B-DNA between targets b plus c, b plus d, and b plus e are 11,120, 14,620, and 43,180 nm, respectively.) The blue line show what is expected if the template adopted a self-avoiding walk.
Fig 1.

Model for the development of a subgene loop. (A) Diagram illustrating results obtained using microarrays (from reference 44). HUVECs were treated with TNF-α for different times, and total RNA was applied to a tiling microarray spanning 221-kbp SAMD4A (map on the left; exons shown as gray lines, TSS as a circle). Before stimulation (0 min), no signal is detected. After 10 min, signal (yellow) appears at the TSS, indicative of rapid and synchronous initiation. The pioneering polymerase (oval) then transcribes steadily to reach segment c after 30 min, and segment e after 60 min; this generates the yellow “traveling” wave that sweeps down the gene. After 10 min, additional polymerases continuously reinitiate but terminate prematurely (circular dotted arrow) to create a “standing” wave (green). Thus, the gene is transcribed by two polymerases that generate “traveling” and “standing” waves. (B) Working 3D model. Within 10 min of adding TNF-α, segment b has collided with a factory (sphere), and initiated; the polymerase in the factory is now elongating productively by reeling in b as it extrudes a transcript (red). By 30 min, this pioneering polymerase is transcribing c, and the promoter is now tethered close to the factory and thus likely to collide with another polymerase in the factory (arrow). By 31 min, the pioneer continues to reel in its template as it elongates (to generate the “traveling” wave), and the promoter has initiated again. However, this second polymerase will soon abort. Successive cycles of promoter attachment and detachment—and initiation and abortion (circular dotted arrow)—now follow (to generate the “standing” wave). While b and c are attached, a small subgene loop exists. By 60 min, the pioneer is transcribing e, as another polymerase transcribes b; the enlarged subgene loop now stretches from b to e.
Image analysis and high-resolution localization.
Images were collected using an Axioplan 2 inverted microscope (Zeiss) with a CoolSNAPHQ camera (Photometrics) via MetaMorph 7.1 (Molecular Devices). Yellow foci such as those in Fig. 5A were selected for analysis, and the distance between the red and green peaks constituting the focus determined (with 27-nm precision) after localizing each peak (with 16-nm precision). Thus, a 2D Gaussian intensity profile was statistically fit to a peak using regression analysis to minimize least-squares distances between intensity values (23, 43, 49). The pixel shift between fluorescence channels was assessed using 0.1-μm TetraSpeck beads (Molecular Probes) fluorescing at relevant wavelengths. Residual differences in alignment were accounted for, along with spot intensity, shape, and the signal-to-noise ratio to calculate distance uncertainty. All calculations were performed in MATLAB (MathWorks) using custom software routines.
siRNA assays.
Small interfering RNAs (siRNAs) targeting CTCF (siGENOME SMART pool; Dharmacon) were introduced into HUVECs as described previously (44) using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. The knockdown efficiency was assessed using Western blotting and ChIP; experiments were performed twice.
Statistical analysis.
P values (two-tailed) from unpaired Student t tests and Fisher exact tests were calculated using GraphPad; results were considered significant when P < 0.01.
RESULTS
Strategy.
When TNF-α is added to serum-starved HUVECs, SAMD4A is one of the first genes to respond. It is 221-kbp long and encodes a regulator of the inflammatory response. Transcription begins within 10 min, and then pioneering polymerases transcribe steadily at ∼3 kbp/min to reach the terminus after ∼85 min. This transcription cycle has been analyzed in detail (33, 44) using, among others, tiling microarrays (where the total RNA isolated every 7.5 min over a period of 3 h was hybridized to the array [44]); the diagram in Fig. 1A summarizes these results. Since exons represent such a small fraction of the gene (e.g., exon 1 is just 193 bp, compared to intron 1 of 134,000 bp), most signal detected by the array results from intronic (nascent) RNA. Within 10 min after stimulation, transcripts appear first at the promoter, to sweep down the gene; these transcripts (made by the “pioneering” polymerase) are depicted as a yellow “wave.” Once the pioneer leaves the promoter, additional polymerases initiate, but these soon abort within 10 kbp of the transcription start site (TSS). Successive cycles of initiation and abortion then continue to generate (intronic) transcripts within the first 10 kbp; these are depicted as the green “standing” wave. After ∼15 min, a “trough” develops between the two waves; this can only result if the intronic RNA is removed and degraded cotranscriptionally and rapidly and if the following polymerases soon abort (otherwise signal would fill the trough). The presence of polymerases and nascent (intronic) RNA at the points indicated in the diagram has been confirmed using ChIP both conventionally (see below) and coupled to high-throughput sequencing, “ChIP-on-chip,” RT-PCR, and RNA FISH (33, 44). Similar patterns are also seen on two other long, responsive genes: 312-kbp EXT1 and 458-kbp ZFPM2 (44).
The model in Fig. 1B illustrates how the conformation of SAMD4A might change during a transcription cycle, assuming that elongating polymerases are immobile molecular machines housed in factories (33). By 31 min, two polymerases may be transcribing the long gene—the pioneer and a follower—which tether a subgene loop to the factory. After 60 min, this subgene loop has enlarged.
A subgene loop grows between transcribed regions.
We first validated that polymerases were bound to the appropriate segments of all three long genes at the relevant times using ChIP coupled to qPCR (ChIP-qPCR) and antibodies recognizing the N terminus of the major subunit of RNA polymerase II (RNAPII). As expected, binding reflects the presence of the standing and traveling waves, and treatment with a transcriptional inhibitor, DRB, abolishes the traveling wave (Fig. 2). When we used antibodies recognizing different phosphorylated isoforms of the polymerase (9, 44), similar data were obtained (not shown).
Fig 2.

RNA polymerase II positioning in SAMD4A, EXT1, and ZFPM2 during a transcription cycle. HUVECs were treated with TNF-α for 0 to 85 min, and ChIP followed by qPCR were applied. In some cases, the transcriptional inhibitor, DRB (50 μM), was added 25 min before harvesting. Diagrams on the left illustrate regions targeted by primer pairs used and their distance (in kbp) from the TSS. Bound RNA polymerase was detected using an antibody recognizing the largest subunit of the enzyme. The results (percent enrichments relative to the input ± the standard deviation [SD]; n = 6) are superimposed on data obtained previously using microarrays (44). Peak enrichments at different times coincide with the traveling (yellow) and standing waves (green). *, DRB significantly reduces enrichments seen (P < 0.01; unpaired two-tailed Student t test).
We next applied 3C to determine whether a subgene loop appears in SAMD4A and then enlarges using the TSS (segment b) as a reference point (Fig. 3A). At no time does the TSS contact points ∼10 kbp upstream or downstream of the gene (i.e., no band is seen when primer b is paired with primer a or i in the 3C-PCR). Before stimulation, the TSS is hardly transcribed and contacts no part of SAMD4A, except for region h at the terminus. This indicates that the TSS contacts the terminus to generate a “whole-gene” loop. (This loop will be analyzed in detail later.) After 10 min, the sole band is an emerging one seen with region c. Since region c is not maximally transcribed until 30 min after stimulation, we attribute this to a subset of polymerases initiating well before the majority in the population (since synchrony is not perfect). After 30 min, the contact with region c becomes prominent, and another with region d develops. After 60 min, contacts spread further 3′; after 85 min, they reach the end of the gene (so the subgene loop has grown to encompass the whole gene). A similar wave of evolving contacts is seen when HindIII replaces SacI during preparation of the 3C template, using 3C coupled to either conventional PCR (Fig. 3B) or qPCR (Fig. 3C). These interactions depend on transcription, since they are abolished by DRB (Fig. 3, gray panels).
Fig 3.

Stimulation induces an enlarging subgene loop to form in 221-kbp SAMD4A. HUVECs were treated with TNF-α for 0 to 85 min, and 3C was performed. In some cases, DRB (50 μM) was added 25 min before harvesting. Diagrams on the left illustrate regions targeted by 3C primers (green arrow for reference point, white for others). (A) Changing contacts detected using 3C and conventional PCR (3C-PCR) on templates generated using SacI. Frequent contacts between reference point b and the segment of the gene indicated are reflected by a band (from b plus a at the top through b plus c to b plus i). Contacts sweep down the gene in time with the traveling wave. DRB abolishes contacts. At 0 min, primers b plus h yield a band (indicative of a “whole-gene loop”) that disappears upon stimulation to reappear after 85 min. BAC, control 3C samples prepared using DNA from a bacterial artificial chromosome encoding SAMD4A; different primer pairs yield bands of comparable intensities. Intra-GAPDH and loading, unchanging (control) bands given by primers targeting two different restriction fragments in GAPDH or convergent primers targeting the same restriction fragment in SAMD4A. (B) Changing contacts detected using 3C-PCR on templates generated using HindIII. Primer i (green) was paired in turn with the primers indicated. Details and controls are as for panel A. Contacts sweep down the gene in time with the traveling wave, and a whole-gene loop is seen at 0 min. (C) 3C-PCR on templates generated using HindIII confirms the formation of enlarging subgene loops using primers from panel B. The positions of the traveling (green) and standing waves (yellow) are shown (data from reference 44). Interaction frequencies (arbitrary units [au] ± the SD; n = 4) are normalized relative to values given by “loading” and “intra-GAPDH” controls. Peak interactions coincide with peaks in the traveling wave, and a whole-gene loop is only seen at 0 and 85 min. *, difference significant (P < 0.01, two-tailed Student t test) compared to the 0-min sample or to the 30-min sample in the case of the 0-min sample.
The two other long, responsive genes yield similar 3C contacts that spread down the gene with time and which are indicative of an enlarging subgene loop (Fig. 4). However, in both cases no whole-gene loop equivalent to that in SAMD4A was seen at 0 min (Fig. 4). All of these results support the model in Fig. 1B.
Fig 4.

Stimulation induces enlarging subgene loops to form in 458-kbp ZFPM2 and 312-kbp EXT1. HUVECs were treated with TNF-α for 0 to 85 min, and 3C was performed on templates prepared using HindIII. In some cases, 50 μM DRB was added 25 min before harvesting. Diagrams on the left illustrate regions targeted by 3C primers (green arrow for the reference point; white arrows for the others). (A) Changing contacts detected using 3C-PCR. Details and controls are as for Fig. 3B. Contacts sweep down the gene in time with the traveling wave, and DRB abolishes contacts. Primers b plus g yield no band at 0 min indicative of a whole-gene loop (or at any time, since the pioneering polymerase only reaches the termini well after 85 min). (B) Interaction frequencies determined by 3C-PCR. The positions of standing (green) and traveling (yellow) waves are indicated. Primer b (green) was used in turn with the primers indicated, and the results are given in the same row as the indicated primer. The interaction frequency (arbitrary units [au]] ± the SD; n = 4) is normalized relative to value given by “loading” and “intra-GAPDH” controls. Again, peak interactions (which are DRB sensitive) coincide with the traveling wave, and no whole-gene loop is seen in either gene. *, difference significant (P < 0.01, two-tailed unpaired Student t test) compared to the 0-min sample.
Localization of single nascent RNA transcripts in the standing and traveling waves.
3C shows that the two transcribed templates of one long gene are together, so we used an independent method—RNA FISH—to confirm that the nascent transcripts encoded by the contacting sequences also lie together. Here we use probe pairs (labeled with red or green fluors) targeting a short intronic RNA segment within each wave (Fig. 5A). As we have seen, the two targets lie on different RNA molecules generated by different polymerases. (See Materials and Methods for additional evidence that the two targets are unlikely to lie on one RNA molecule.) Previous work (33, 44) has shown that (i) the oligonucleotide probes used can detect a single intronic RNA efficiently to yield a diffraction-limited spot, (ii) HUVECs contain only two SAMD4A alleles (they are diploid and synchronized in the G0 phase of the cell cycle), (iii) ∼30% alleles in the population are being transcribed by a pioneering polymerase at any one time after stimulation, and (iv) a yellow spot indicative of colocalizing targets can only result from RNA copied from the same allele (since the spot area is so small compared to nuclear area that a green focus will only overlap by chance a red focus copied from a different allele in <1 nucleus in a thousand, assuming random distributions).
Targets for probe pairs b plus c and b plus d (at 30 and 45 min after stimulation, respectively) are copied from two DNA regions lying ∼33 and ∼44 kbp apart and yield yellow foci in ∼20% of cells (Fig. 5A). Since electron microscopy shows that nascent transcripts typically lie on the surface of a factory with an ∼90-nm protein-rich core (13, 14), we wanted to see whether these colocalizing transcripts lay this close together. Unfortunately, the conventional fluorescence microscope has a resolution of ∼250 nm, at best. Therefore, we used an approach (33) that allows resolution beyond the diffraction limit; we assume the red and green signals that yield a yellow focus mark two subdiffraction spots (one red, the other green), fit Gaussian curves to their intensities, and measure the 2D distance (with 27-nm accuracy) between peaks. There were few interpeak distances of ≤25 nm (the mean values were 64 and 60 nm for b plus c and b plus d, respectively), and the maximum was ∼160 nm (Fig. 5B). In other words, short interpeak distances are rare. Such a distribution is not that expected of red and green foci randomly distributed in a small (spherical) volume of analogous dimensions, where most interpeak distances lie close to zero (since so many red foci lie immediately above or below a green focus to give a short 2D distance). We then compared these experimental results to those obtained from a computer simulation of transcripts on the surface of a factory: red and green points were randomly distributed in a (variably sized) shell around a (variably sized) sphere, and then the 2D distance between points was measured. The best fit to the experimental data was given by points randomly distributed in a 35-nm shell around a 90-nm core (Fig. 5B, orange line), which is consistent with the known dimensions and location of transcripts determined by electron microscopy (13, 14). (Similar results are obtained with transcripts encoded by two co-associating genes on different chromosomes [33].) As a control, we imaged multicolor fluorescent beads that should colocalize “perfectly.” As expected, measured red-green distances were ≤25 nm (Fig. 5B, gray panel), the precision level of the method. As another control, overlapping red and green signals emitted by a mixture of red and green probes targeting just region c also had a significantly smaller mean separation than those given by b plus c or b plus d (data not shown).
We now argue that the separations seen above are inconsistent with a model involving the generation of two transcripts by two tracking polymerases. If such a model applied, and as the two nascent RNAs lie near their immediate templates, we would expect the separations seen with probe pairs b plus c, b plus d, and b plus e to increase in proportion to the number of base pairs between the DNA encoding the targets (Fig. 5C, black line). However, the observed separations (normalized relative to that seen with b plus c) remain essentially constant (Fig. 5C, green line). This result is consistent with the two targets being generated by polymerases lying a constant distance apart (i.e., fixed on the surface of one factory), with the intervening DNA forming an extending subgene loop. Note also that these results are also inconsistent with targets being encoded by a template that (i) follows a self-avoiding random walk (Fig. 5C, blue curve) or (ii) is tightly packed into a sphere between targets (if packed at the highest concentration ever seen in vivo [46], targets would still lie twice as far apart as observed [data not shown]).
Polymerases are poised on the promoters of three long genes before stimulation.
It is attractive to suppose that polymerases might be “poised” on the promoter of these long genes ready to facilitate a rapid response to stimulation (see also reference 15). ChIP with an antibody targeting the N terminus of the largest subunit of the enzyme shows it is indeed bound to the 5′ end of each gene before induction (Fig. 6) and that binding increases in synchrony with p65 binding to its cognate elements (data not shown). Since polymerases bound at “poised” promoters fire continually and unproductively to generate short sense and antisense transcripts (11, 36, 38), we used qRT-PCR to determine whether this was true here. Before stimulation, sense and antisense transcripts copied from the TSS were present at a 27:1 ratio; the ratios at positions bp −500 and bp 1750 were 5:1 and 73:1, respectively (Fig. 7A, white bars). This indicates that “noisy” initiations occur around the TSS and are biased in favor of sense transcription. At 30 min after stimulation, the amount of sense and antisense transcription in the region increases >8-fold, as the bias toward sense transcription remains much the same (at positions −500, the TSS, and +1750 the ratios become 17:1, 17:1, and 80:1, respectively) (Fig. 7A, gray bars). Therefore, the combination of engagement of poised polymerases and the bias toward sense transcription could facilitate a prompt response to TNF-α.
Fig 6.

Changes induced by TNF-α on the binding of RNA polymerase II, CTCF, TFIIB, and nucleosomes to each end of SAMD4A, EXT1, and ZFPM2. HUVECs were treated with TNF-α for the times indicated, and ChIP was performed with antibodies against the largest subunit of RNA polymerase II, CTCF, TFIIB, and histone H3 (percent enrichments ± the SD; n = 6). The diagrams on the left illustrate targets of the primer pairs in promoters (S1, E1, and Z1), the SAMD4A gene body (S2), and 3′ untranslated regions (UTRs; S3, S4, E2, and Z2). The polymerase is found at all promoters, both before and after stimulation. It is also seen at the 3′UTR of SAMD4A after 85 min (as expected), but not at the 3′UTR of EXT1 or ZFPM2 (since pioneering polymerases only reach the end of these two long genes after the end of the experiment). CTCF binds to promoters throughout the time course with comparable enrichments. It also binds to S3 and S4 but not to S2, E2 or Z2. TFIIB binding to all promoters peaks after 10 to 30 min. Some TFIIB binds to the 3′UTR of SAMD4A but not to the middle of SAMD4A (as expected) or the 3′UTRs of EXT1 or ZFPM2. When 50 μM DRB is added to cells 25 min before harvesting (gray panel), less TFIIB binds to the SAMD4A 3′UTR. H3 levels are generally low, except for E1 and Z1 at 0 min, which changes on stimulation (perhaps as the nucleosomes reposition). *, difference significant (P < 0.01, two-tailed unpaired Student t test).
Fig 7.
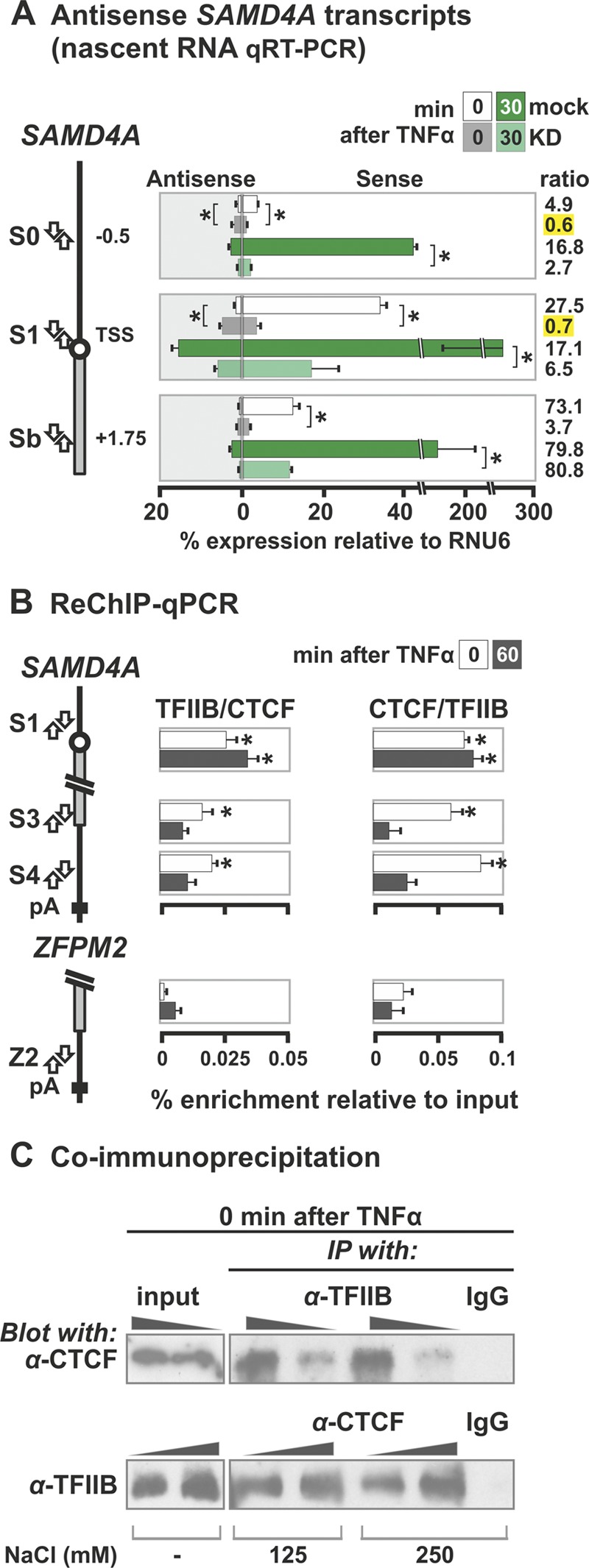
CTCF promotes sense transcription at the SAMD4A promoter and interacts with TFIIB. HUVECs were treated with TNF-α for 0 to 60 min. (A) Levels of sense and antisense transcripts at the SAMD4A promoter determined by qRT-PCR. HUVECs were treated with siRNAs that knock down CTCF (KD) or mock treated (mock) prior to stimulation with TNF-α for 0 or 30 min and isolation of total RNA. Next, the levels of sense and antisense transcripts from regions S0, S1, and Sb were quantified after first-strand cDNA synthesis (using sense or antisense primers indicated in the diagram). Knocking down CTCF reduces overall transcription levels at all three sites at both times; it also reduces the sense/antisense ratio in favor of antisense transcripts copied from S0 and S1 (yellow highlights). *, difference significant (P < 0.01, two-tailed unpaired Student t test; n = 4). (B) CTCF and TFIIB bind to the same promoter and 3′ end fragments of SAMD4A, assessed by sequential ChIP (ReChIP-qPCR), where an anti-TFIIB was used first and then an anti-CTCF (and vice versa). Then, the percent enrichments (relative to the input ± the SD; n = 6) of three SAMD4A regions were determined. The results indicate that bound CTCF and TFIIB on S1, S3, and S4 at 0 min but not at 60 min. The 3′UTR (Z2) of ZFPM2 does not bind CTCF or TFIIB and serves as a negative control. *, value significantly greater than the corresponding obtained with ZFPM2 (P < 0.01, two-tailed unpaired Student t test). (C) CTCF and TFIIB coimmunoprecipitate. Immunoprecipitations (IPs) were carried out (in 125 or 250 mM NaCl) using anti-TFIIB (top) or anti-CTCF (bottom), before CTCF (top) or TFIIB (bottom) were detected by immunoblotting (using loadings of 1× and 2×); a nonspecific immunoglobulin (IgG) and inputs (using loadings of 0.1× and 0.2×) are included as controls. CTCF is detected after pulling down TFIIB (top) and TFIIB after pulling down CTCF (bottom). This indicates the two interact in vivo.
The SAMD4A whole-gene loop seen before stimulation is CTCF/TFIIB-dependent.
Loops formed by juxtaposition of the 5′ and 3′ ends of various genes have been observed (see, for example, references 30, 31, 40, and 42), and such a configuration is seen in SAMD4A before stimulation with TNF-α (but not in EXT1 or ZFPM2) (Fig. 3 and 4). This whole-gene loop is lost on stimulation (shown using 3C applied both conventionally and coupled to qPCR on templates generated with two different restriction enzymes in Fig. 3). One difference between SAMD4A and EXT1 or ZFPM2 lies in the nucleosome occupancy of their 5′ proximal regions; SAMD4A possesses such a nucleosome-free region both before and after stimulation, but one only appears in the two other long genes after TNF-α treatment (Fig. 6).
CCCTC-binding factor (CTCF) has been implicated in stabilizing chromatin loops (see, for example, references 26 and 28), so we examined whether it was involved. An in silico search (using the algorithm available from the School of Biological Sciences, University of Essex, Essex, United Kingdom [http://www.essex.ac.uk/bs/molonc/binfo/ctcfbind.htm]) uncovered potential CTCF-binding sites at both ends of SAMD4A, but only at the 5′ end of EXT1 and ZFPM2. Binding was confirmed by ChIP-qPCR (Fig. 6) (see also reference 44). Therefore, binding to both SAMD4A ends correlates with the formation of the whole-gene loop. (Note that CTCF monomers—in contrast to dimers—cannot mediate looping in vitro [25].) However, CTCF remains bound to these cognate sites throughout the 85 min we follow (Fig. 6), whereas the whole-gene loop disappears on stimulation (Fig. 3).
The general transcription factor, TFIIB, has also been implicated in stabilizing whole-gene loops in yeast (40). Like CTCF, it is bound before stimulation to both ends of SAMD4A, with significant enrichment at the 3′ end; again, this is not true of EXT1 or ZFPM2 (Fig. 6). Binding to the 3′ end of SAMD4A falls significantly upon stimulation (Fig. 6), correlating with the disappearance of the whole-gene loop. This suggests that CTCF and TFIIB might interact in vivo; both sequential ChIP (ReChIP) and coimmunoprecipitation experiments support this idea (Fig. 7B and C).
Knocking down CTCF using siRNAs (Fig. 8A) reduces binding to both ends of SAMD4A (as expected) and also reduces TFIIB binding (Fig. 8B). The CTCF knockdown also eliminates the 3C contact indicative of the whole-gene loop (Fig. 8C, compare m to KD). The presence of this loop depends on continued transcription (Fig. 8C, compare m to +DRB) and does not result from serum starvation (Fig. 8C, compare NS to m). Taken together, these results indicate that the polymerase, CTCF, and TFIIB, jointly stabilize the exceptional whole-gene loop in SAMD4A.
Fig 8.

TFIIB and CTCF stabilize the whole-gene loop in SAMD4A. HUVECs were treated with siRNAs to knock down CTCF (KD) or mock treated (m), grown (48 h), starved (18 h) in low-serum medium (nonstarved cells [NS] serve as a control), and harvested without TNF-α stimulation (except in panel D). (A) Western blot showing the siRNAs efficiently knock down CTCF without affecting β-tubulin. (B) ChIP-qPCR using antibodies targeting CTCF or TFIIB (percent enrichments ± the SD; n = 6) confirms that little CTCF in knocked-down cells is bound to three cognate sites in SAMD4A (S1, S3, and S4). CTCF knockdown leads to a concomitant loss of TFIIB binding. Serum starvation does not alter either binding profile. *, enrichment significantly different (P < 0.01; unpaired two-tailed Student t test). (C) 3C-PCR (using primers indicated in the diagram on the left) shows that the band indicative of the whole-gene loop disappears after knocking down CTCF or after DRB treatment (50 μM added 25 min before harvesting). BAC and loading controls are as described in Fig. 3B. –Ligase, control in which ligase was omitted from 3C reaction. The box (right) gives the 3C-PCR interaction frequencies (arbitrary units [au] ± the SD; n = 4) normalized relative to values given by loading and intra-GAPDH controls. *, significant difference (P < 0.01; two-tailed unpaired Student t test; n = 4). (D) CTCF knockdown reduces SAMD4A transcription induced by TNF-α (assessed using RNA FISH with intronic probes). Typical images collected 30 min after induction are shown (insets illustrate foci). Knocking down CTCF significantly reduces the number of SAMD4A (green) but not EXT1 foci (red) compared to the mock-treated control (P < 0.01; two-tailed the Fisher exact test). n, number of foci (percentages in brackets). Bar, 5 μm.
We also monitored the effects of knocking down CTCF on transcription 30 min after stimulation (using RNA FISH with probes targeting region c of SAMD4A and the equivalent region of EXT1). In mock-treated cells, 26 and 21% of the nuclei possessed at least one focus indicative of an active SAMD4A or EXT1 allele, respectively (Fig. 8D). After knockdown, substantially fewer nuclei (i.e., 15%) contained active SAMD4A alleles, while EXT1 activity remained unaffected (at 22%) (Fig. 8D). Finally, we checked (by qRT-PCR) whether knocking down CTCF reduced the sense and antisense transcripts at the SAMD4A promoter. Previously, we saw there was a strong bias toward sense transcription at or near the TSS. Knockdown reduced this strong bias (e.g., before stimulation, the 27:1 ratio between sense and antisense transcripts at the TSS shifted to 0.7:1 upon knockdown, and from 5:1 to 0.6:1 at bp −500) (Fig. 7A, green bars). This suggests that CTCF promotes selection of the appropriate strand and affects overall transcription levels of SAMD4A (but not EXT1), although we cannot rule out the possibility of this being an indirect effect of the knockdown.
DISCUSSION
We analyzed the changing conformations of three long genes (221-kbp SAMD4A, 312-kbp EXT1, and 458-kbp ZFPM2) which respond to TNF-α (44). Within 10 min, pioneering polymerases initiate and then transcribe steadily to terminate >1 h later. Meanwhile, other polymerases initiate, but these soon abort. As a result, each of these long genes becomes transcribed by two polymerases, a pioneer and a follower, and these generate traveling and standing waves of nascent RNA (Fig. 1A). This system allows us to obtain new insights into the transcription cycle that are difficult, if not impossible, to obtain using continuously active, short genes.
We first examined the formation of subgene loops. If both elongating and initiating polymerases are transiently immobilized in one transcription factory (8, 32, 33), stimulation should induce a subgene loop to form and then enlarge (Fig. 1B). 3C analyses show this to be so (Fig. 3 and 4), and RNA FISH allied to subdiffraction localizations confirms that nascent RNAs copied from the contacting sequences lie sufficiently close together to be on the surface of one 90-nm factory (Fig. 5). Equivalent subgene loops are likely to be found in many human genes simply because ∼42% are longer than 30 kbp (4), the length of the smallest subgene loop we see. Since all parts of the transcription unit contact the 5′ end in the course of one transcription cycle, such subgene loops are likely to remain undetected by current approaches applied to unsynchronized genes. This model shares with others involving “transcriptional compartments” (48) or “active chromatin hubs” (3, 34) the idea that elongating polymerases interact (directly or indirectly) with the promoters where they initiated (see also reference 22).
We also investigated the whole-gene loop seen in SAMD4A before stimulation. CTCF, TFIIB, and ongoing transcription all seem to stabilize this loop. Knocking down CTCF reduces binding of both to both ends (Fig. 8B), eliminates the loop (Fig. 8C), and reduces productive elongation (Fig. 8D). It also reduces “noisy” transcription upstream of the promoter and relieves the bias toward sense promoter transcripts (Fig. 7A), a switch anticipated to reduce productive elongation (16). Since DRB treatment also eliminates the whole-gene loop (Fig. 8B), its existence depends on continuing transcription (either in the sense or antisense direction). However, here the polymerase seems to be bound only to the 5′ contacting partner and not to the 3′ one. It is then attractive to suppose that this whole-gene loop “poises” the gene so it can respond rapidly to TNF-α. An analogous role in increasing the efficiency of mRNA production has been suggested for such loops in yeast, the HIV-1 provirus, and other mammalian genes, either directly through close association with transcription/processing factors or indirectly through contacts with the nuclear pore and the mRNA export machinery (1, 20, 31, 35, 40–42, 50). Again, the use of a switchable and synchronized system allows us to monitor the appearance and (critically) the disappearance of this whole-gene loop.
In conclusion, all of these results are consistent with a model for transcription in which active polymerases are immobile molecular machines housed in factories, where components of the transcription machinery act as the critical molecular ties that loop the chromatin fiber (10, 20). In the case of the subgene loops in SAMD4A, EXT1, and ZFPM2, polymerases are probably the sole ties; in the whole-gene loop in SAMD4A, the polymerase (and perhaps CCTF and TFIIB) probably constitutes the tie at the promoter, while CTCF and TFIIB act at the terminus. Our data also highlight the cross talk between the poised, initiating (whether productively or unproductively), and elongating transcriptional machinery and the immediate and changing effects this has on the 3D structure of chromatin.
ACKNOWLEDGMENTS
We thank Tatsuhiko Kodama for RNA FISH probes, Davide Marenduzzo for discussions on the self-avoiding fiber calculations, and Shona Murphy and Clelia Laitem for siRNA reagents and advice.
This study was supported by the Biotechnology and Biological Sciences Research Council via the ERASysBio+/FP7 initiative (A.P.) and Wellcome Trust (A.P. and J.D.L.). A.P. is the Kemp Junior Research Fellow of Lincoln College; P.R.C. holds the EP Abraham Chair of Cell Biology and a Professorial Fellowship at Lincoln College.
Footnotes
Published ahead of print 14 May 2012
REFERENCES
- 1. Ansari A, Hampsey M. 2005. A role for the CPF 3′-end processing machinery in RNAP II-dependent gene looping. Genes Dev. 19:2969–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baù D, et al. 2011. The three-dimensional folding of the α-globin gene domain reveals formation of chromatin globules. Nat. Struct. Mol. Biol. 18:107–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blackledge NP, Ott CJ, Gillen AE, Harris A. 2009. An insulator element 3′ to the CFTR gene binds CTCF and reveals an active chromatin hub in primary cells. Nucleic Acids Res. 37:1086–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bradnam KR, Korf I. 2008. Longer first introns are a general property of eukaryotic gene structure. PLoS One 3:e3093 doi:10.1371/journal.pone.0003093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown JM, et al. 2006. Coregulated human globin genes are frequently in spatial proximity when active. J. Cell Biol. 172:177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burnette JM, Miyamoto-Sato E, Schaub MA, Conklin J, Lopez AJ. 2005. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics 170:661–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cai S, Lee CC, Kohwi-Shigematsu T. 2006. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat. Genet. 38:1278–1288 [DOI] [PubMed] [Google Scholar]
- 8. Chakalova L, Fraser P. 2010. Organization of transcription. Cold Spring Harbor Perspect. Biol. 2:a000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chapman RD, et al. 2007. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 318:1780–1782 [DOI] [PubMed] [Google Scholar]
- 10. Cook PR. 2010. A model for all genomes: the role of transcription factories. J. Mol. Biol. 395:1–10 [DOI] [PubMed] [Google Scholar]
- 11. Core LJ, Waterfall JJ, Lis JT. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322:1845–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dye MJ, Gromak N, Proudfoot NJ. 2006. Exon tethering in transcription by RNA polymerase II. Mol. Cell 21:849–859 [DOI] [PubMed] [Google Scholar]
- 13. Eskiw CH, Rapp A, Carter DR, Cook PR. 2007. RNA polymerase II activity is located on the surface of protein-rich transcription factories. J. Cell Sci. 121:1999–2007 [DOI] [PubMed] [Google Scholar]
- 14. Eskiw CH, Fraser P. 2011. Ultrastructural study of transcription factories in mouse erythroblasts. J. Cell Sci. 124:3676–3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrai C, et al. 2010. Poised transcription factories prime silent uPA gene prior to activation. PLoS Biol. 8:e1000270 doi:10.1371/journal.pbio.1000270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Flynn RA, Almada AE, Zamudio JR, Sharp PA. 2011. Antisense RNA polymerase II divergent transcripts are P-TEFb dependent and substrates for the RNA exosome. Proc. Natl. Acad. Sci. U. S. A. 108:10460–10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Glover-Cutter K, Kim S, Espinosa J, Bentley DL. 2008. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat. Struct. Mol. Biol. 15:71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hagège H, et al. 2007. Quantitative analysis of chromosome conformation capture assays 3C-qPCR. Nat. Protoc. 2:1722–1733 [DOI] [PubMed] [Google Scholar]
- 19. Hicks MJ, Yang CR, Kotlajich MV, Hertel KJ. 2006. Linking splicing to Pol II transcription stabilizes pre-mRNAs and influences splicing patterns. PLoS Biol. 4:e147 doi:10.1371/journal.pbio.0040147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iborra FJ, Escargueil AE, Kwek KY, Akoulitchev A, Cook PR. 2004. Molecular cross-talk between the transcription, translation, and nonsense-mediated decay machineries. J. Cell Sci. 117:899–906 [DOI] [PubMed] [Google Scholar]
- 21. Kagey MH, et al. 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467:430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kolovos P, Knoch TA, Grosveld FG, Cook PR, Papantonis A. 2012. Enhancers and silencers: an integrated and simple model for their function. Epigenet. Chromatin 5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larkin JD, Publicover NG, Sutko JL. 2011. Photon event distribution sampling: an image formation technique for scanning microscopes permits tracking of subdiffraction particles with high spatial and temporal resolution. J. Microsc. 241:54–68 [DOI] [PubMed] [Google Scholar]
- 24. Li G, et al. 2012. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148:84–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacPherson MJ, Sadowski PD. 2010. The CTCF insulator protein forms an unusual DNA structure. BMC Mol. Biol. 11:101 doi:10.1186/1471-2199-11-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Majumder P, Boss JM. 2010. CTCF controls expression and chromatin architecture of the human major histocompatibility complex class II locus. Mol. Cell. Biol. 30:4211–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mapendano CK, Lykke-Andersen S, Kjems J, Bertrand E, Jensen TH. 2010. Crosstalk between mRNA 3′ end processing and transcription initiation. Mol. Cell 40:410–422 [DOI] [PubMed] [Google Scholar]
- 28. Mishiro T, et al. 2009. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 28:1234–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nelson JD, Denisenko O, Bomsztyk K. 2006. Protocol for the fast chromatin immunoprecipitation ChIP. method. Nat. Protoc. 1:179–185 [DOI] [PubMed] [Google Scholar]
- 30. O'Reilly D, Greaves DR. 2007. Cell-type-specific expression of the human CD68 gene is associated with changes in Pol II phosphorylation and short-range intra-chromosomal gene looping. Genomics 90:407–415 [DOI] [PubMed] [Google Scholar]
- 31. O'Sullivan JM, et al. 2004. Gene loops juxtapose promoters and terminators in yeast. Nat. Genet. 36:1014–1018 [DOI] [PubMed] [Google Scholar]
- 32. Papantonis A, Cook PR. 2011. Fixing the model for transcription: the DNA moves, not the polymerase. Transcription 2:41–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Papantonis A, et al. 2010. Active RNA polymerases: mobile or immobile molecular machines? PLoS Biol. 8:e1000419 doi:10.1371/journal.pbio.1000419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patrinos GP, et al. 2004. Multiple interactions between regulatory regions are required to stabilize an active chromatin hub. Genes Dev. 18:1495–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perkins KJ, Lusic M, Mitar I, Giacca M, Proudfoot NJ. 2008. Transcription-dependent gene looping of the HIV-1 provirus is dictated by recognition of pre-mRNA processing signals. Mol. Cell 29:56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Preker P, et al. 2008. RNA exosome depletion reveals transcription upstream of active human promoters. Science 322:1851–1854 [DOI] [PubMed] [Google Scholar]
- 37. Schoenfelder S, et al. 2010. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 42:53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seila AC, et al. 2008. Divergent transcription from active promoters. Science 322:1849–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Singh J, Padgett RA. 2009. Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 16:1128–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Singh BN, Hampsey M. 2007. A transcription-independent role for TFIIB in gene looping. Mol. Cell 27:806–816 [DOI] [PubMed] [Google Scholar]
- 41. Tan-Wong SM, Wijayatilake HD, Proudfoot NJ. 2009. Gene loops function to maintain transcriptional memory through interaction with the nuclear pore complex. Genes Dev. 23:2610–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tan-Wong SM, French JD, Proudfoot NJ, Brown MA. 2008. Dynamic interactions between the promoter and terminator regions of the mammalian BRCA1 gene. Proc. Natl. Acad. Sci. U. S. A. 105:5160–5165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thompson RE, Larson DR, Webb WW. 2002. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82:2775–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wada Y, et al. 2009. A wave of nascent transcription on activated human genes. Proc. Natl. Acad. Sci. U. S. A. 106:18357–18361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Y, Fairley JA, Roberts SG. 2010. Phosphorylation of TFIIB links transcription initiation and termination. Curr. Biol. 20:548–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weidemann T, et al. 2003. Counting nucleosomes in living cells with a combination of fluorescence correlation spectroscopy and confocal imaging. J. Mol. Biol. 334:229–240 [DOI] [PubMed] [Google Scholar]
- 47. Yaffe E, Tanay A. 2011. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet. 43:1059–1065 [DOI] [PubMed] [Google Scholar]
- 48. Yao J, Ardehali MB, Fecko CJ, Webb WW, Lis JT. 2007. Intranuclear distribution and local dynamics of RNA polymerase II during transcription activation. Mol. Cell 28:978–990 [DOI] [PubMed] [Google Scholar]
- 49. Yildiz A, et al. 2003. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science 300:2061–2065 [DOI] [PubMed] [Google Scholar]
- 50. Yun K, So JS, Jash A, Im SH. 2009. Lymphoid enhancer binding factor 1 regulates transcription through gene looping. J. Immunol. 183:5129–5137 [DOI] [PubMed] [Google Scholar]