

Abstract
Mutations in the gene encoding methyl-CpG-binding protein 2 (MeCP2) lead to disrupted neuronal function and can cause the neurodevelopmental disorder Rett syndrome. MeCP2 is a transcriptional regulator that binds to methylated DNA and is most abundant in neuronal nuclei. The mechanisms by which MeCP2 regulates gene expression remain ambiguous, as it has been reported to function as a transcriptional silencer or activator and to execute these activities through both gene-specific and genome-wide mechanisms. We hypothesized that posttranslational modifications of MeCP2 may be important for reconciling these apparently contradictory functions. Our results demonstrate that MeCP2 contains multiple posttranslational modifications, including phosphorylation, acetylation, and ubiquitylation. Phosphorylation of MeCP2 at S229 or S80 influenced selective in vivo interactions with the chromatin factors HP1 and SMC3 and the cofactors Sin3A and YB-1. pS229 MeCP2 was specifically enriched at the RET promoter, and phosphorylation of MeCP2 was necessary for differentiation-induced activation and repression of the MeCP2 target genes RET and EGR2. These results demonstrate that phosphorylation is one of several factors that are important for interpreting the complexities of MeCP2 transcriptional modulation.
INTRODUCTION
Methyl-CpG-binding protein 2 (MeCP2) is an epigenetic regulator of gene expression that is essential for normal brain development. MeCP2 binds to methylated cytosines in the mammalian genome and modulates transcription, thereby translating these epigenetic marks into changes in gene expression. Epigenetic regulation plays an instrumental role in the development and maturation of the mammalian brain. Accordingly, mutations in the X-linked gene MECP2 have severe consequences for neuronal development and can cause the neurodevelopmental disorder Rett syndrome (RTT) (2). MeCP2 is expressed in several cell types in the developing and adult brain, with the highest levels occurring in mature neuronal nuclei, where MeCP2 levels increase with postnatal age (5, 32). While it is clear that MeCP2 plays an important role in the function of mature neurons, defects in neuronal nuclear organization in MeCP2-null mice are apparent as early as embryonic day 15, suggesting that MeCP2 is also important at the early stages of neuronal development (33). As a result, immature or underdeveloped neurons are a hallmark of MeCP2 deficiency in the brain (4). Postnatal neuronal maturation requires developmental and activity-dependent changes in gene expression (38). The loss of MeCP2 function in the brain is predicted to interfere with the proper regulation of these gene expression changes and lead to the neurodevelopmental phenotypes associated with RTT.
However, the mechanisms by which disruption of MeCP2 function leads to improper neuronal development and RTT remain unclear. Initially, MeCP2 was characterized as a transcriptional repressor of methylated promoters, due to the association of highly methylated gene promoters with transcriptional silencing (21) as well as the presence of a domain in the MeCP2 protein capable of driving transcriptional repression in vitro (24). As a result, it was assumed that MECP2 mutation led to RTT because of a loss of developmentally important gene silencing. Contrary to this initial hypothesis, the expression changes that result from MeCP2 disruption or duplication suggest that MeCP2 plays an important role in activation of expression as well (7). Additionally, genomic analyses of MeCP2 binding have shown that MeCP2 binds throughout the neuronal genome, including active promoters and intergenic regions (34, 39), signifying that MeCP2 may be controlling gene expression by regulating chromatin structure and long-range chromatin interactions in addition to affecting expression through binding to proximal promoter regions. MeCP2 has thus emerged as a complex transcriptional modulator. What remains unclear is how these diverse functions of MeCP2 relate to the pathophysiology of RTT or how the seemingly opposing activities may be regulated.
Previous studies on the regulation of MeCP2 focused on its phosphorylation at two sites, serine 80 (pS80) and serine 421 (pS421). Phosphorylation of S80 in neurons is lost after stimulation of neuronal activity and has been reported to be required for proper chromatin association of MeCP2 (36). However, only limited expression changes were observed in cortical neurons expressing an S80A MeCP2 mutation, making the functional consequences of S80 phosphorylation unclear (36). Conversely, phosphorylation of S421 occurs in response to neuronal activity and is required for proper dendritic patterning and spine morphogenesis (41). There are conflicting reports on the role of pS421 MeCP2 in activity-dependent gene expression (10, 41), making the functional consequence of phosphorylation at this site unclear as well. Although the molecular functions of MeCP2 phosphorylation may be unclear, knock-in mice containing alanine mutations at S80, at S421, or at both S421 and S424 of MeCP2 all have distinct neurological defects (10, 20, 36), clearly demonstrating the importance of MeCP2 phosphorylation at these sites. None of these mouse models fully recapitulates the phenotypes seen with total MeCP2 disruption or overexpression (8, 11, 14, 31), highlighting the complexities in understanding the relevance of these modifications to RTT.
Posttranslational modifications, such as phosphorylation, are one potential mechanism to provide localized functional specificity to the widely distributed MeCP2. This may allow an individual MeCP2 molecule to act as either a transcriptional activator or a repressor, depending on the specific modifications it contains. However, similar to total MeCP2, pS421 MeCP2 is enriched throughout the genome, and activity-induced phosphorylation does not alter this genome-wide distribution (10). It may be that other posttranslational modifications are responsible for directing MeCP2 binding specificity. Additionally, phosphorylation of MeCP2 on serine 421 in response to psychostimulants occurs in a specific subset of neurons in the nucleus accumbens (12). Therefore, physiologically relevant phosphorylation of MeCP2 may be exquisitely cell type and activity dependent.
To gain further insight into their role in the regulation of MeCP2 functions, we sought to identify posttranslational modifications of MeCP2 in human SH-SY5Y neuroblastoma cells and to determine their functional relevance. We observed multiple phosphorylation, ubiquitylation, and acetylation sites on MeCP2 and further characterized the functional significance of phosphorylation of MeCP2 on S80 and S229 (pS229). Our results show that these phosphorylation events distinctly define the association of MeCP2 with specific protein cofactors. pS80 and pS229 MeCP2 were important for both activation and repression of the differentiation-induced MeCP2 target genes RET and EGR2, demonstrating the involvement of posttranslational modifications in the regulation of MeCP2 function.
MATERIALS AND METHODS
Cell culture.
SH-SY5Y neuroblastoma cells were maintained in minimal essential medium (MEM) supplemented with 15% fetal bovine serum (FBS), nonessential amino acids, and penicillin-streptomycin (Invitrogen) at 37°C with 5% CO2. Stable cell lines were generated as reported previously (33).
Sample preparation and MS/MS analysis.
FLAG-MeCP2 was purified from stably expressing SH-SY5Y cells according to the manufacturer's instructions (Sigma-Aldrich). Purified FLAG-MeCP2 was isolated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). In-gel digestion was performed with trypsin, chymotrypsin, or Glu-C (Staphylococcus aureus protease V8), and the resulting peptides were analyzed by tandem mass spectrometry (MS/MS) at the University of California, Davis, proteomics core facility (www.proteomics.ucdavis.edu).
Antibodies.
Chicken IgY antibodies to total, pS80 and pS229 MeCP2 were generated and affinity purified by Aves Labs using the peptides Total (RPNREEPVDSRTPVTERVS), pS80 (EASA[pS]PKQRRS), and pS229 (KMPFQA[pS]PGGKGE) conjugated to KLH. IgY fractions were collected from eggs and purified over an affinity column containing the corresponding peptide. For phospho-specific antibodies, an additional purification step using nonphosphorylated peptides was used to remove any non-phospho-reactive antibodies.
Phosphatase treatment.
Purified MeCP2e1-FLAG bound to anti-M2 anti-FLAG agarose (Sigma-Aldrich) or endogenous MeCP2 bound to protein A Sepharose (GE Healthcare) was resuspended in 1× alkaline phosphatase buffer (Roche) and treated with 50 U alkaline phosphatase (Roche) in the presence or absence of PhosSTOP phosphatase inhibitor cocktail (Roche) for 10 min at 37°C.
Lysate preparation and immunoprecipitation.
SH-SY5Y cells were lysed in HNB (0.5 M sucrose, 15 mM Tris-HCl [pH 7.5], 60 mM KCl, 0.25 mM EDTA [pH 8], 0.125 mM EGTA [pH 8], 0.5 mM spermidine, 0.15 mM spermine, 1 mM dithiothreitol [DTT]; Complete mini-EDTA-free protease inhibitor and PhosSTOP phosphatase inhibitor cocktails [Roche] were added prior to use) to isolate nuclei as previously described (29). Isolated nuclei were resuspended in high-salt buffer (20 mM HEPES [pH 7.9], 400 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% NP-40) and sonicated for five 2-s bursts at power level 3 using a Branson Sonifier with a microtip. The nuclear extract was diluted to a final NaCl concentration of 200 mM with low-salt buffer (20 mM HEPES [pH 7.9], 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% NP-40) and cleared by centrifugation at 16,000 × g for 10 min at 4°C. Total nuclear extract protein (500 μg) was immunoprecipitated with 4 μg of specific antibody or nonspecific IgY for 2 h at 4°C. Mouse cortex lysates were prepared from dissected cortices from E13.5 or E19.5 mouse embryos. Four cortices per time point were lysed in radioimmunoprecipitation assay (RIPA) buffer and sonicated for five 2-s bursts at power level 3 using a Branson Sonifier with a microtip and cleared by centrifugation at 16,000 × g for 10 min at 4°C. Total protein (500 μg for E13.5 or 1.5 mg for E19.5) was immunoprecipitated with 4 μg of specific antibody or nonspecific IgY for 2 h at 4°C. Immune complexes were purified with PrecipHen anti-IgY-conjugated agarose beads (Aves Labs) or protein A Sepharose (GE Healthcare) for 2 h at 4°C. Immune complexes were washed four times with 1× Tris-buffered saline (TBS) plus 0.1% NP-40, separated by SDS-PAGE, and analyzed by immunoblotting.
RNase and Benzonase treatment.
SH-SY5Y nuclear lysates were prepared as described above with the omission of EDTA and the addition of 5 mM CaCl2 to the high-salt buffer. Nuclear lysates were treated with either 250 U Benzonase or 100 μg RNase A and 500 U RNaseT1/mg total protein for 60 min at 25°C. Subsequent immunoprecipitations were performed as described above.
Quantitative PCR analysis of gene expression.
Total RNA was isolated using TRIzol reagent (Invitrogen) from SH-SY5Y cells and cell lines treated as indicated. cDNA was generated using a QuantiTect RT kit (Qiagen), and quantitative PCR was performed using SYBR green-based detection chemistry. Target gene expression was normalized to GAPDH. Primers used for quantitative PCR are listed in Table S1 in the supplemental material.
Tissue preparation and immunofluorescence.
Sagittal slices from 5-week-old wild-type (+/y) or MeCP2 null (tm1.1Bird/y) male mouse brains were fixed and costained with antibodies against phospho-MeCP2 and total MeCP2 (clone D4F3; Cell Signaling Technology) as previously described (28), substituting TBS for PBS and 1% bovine serum albumin (BSA) for calf serum.
Chromatin fractionation.
Micrococcal nuclease fractionation of SH-SY5Y nuclei was performed as previously reported (29). A total of 107 nuclei were isolated as described above and resuspended in 200 μl of nuclear buffer (20 mM Tris-HCl [pH 7.5], 70 mM NaCl, 20 mM KCl, 5 mM MgCl2, and 3 mM CaCl2 supplemented with protease inhibitors). The nucleus suspension was incubated with 225 U of micrococcal nuclease (Roche Applied Science) at 25°C for 4 min. The digestion was terminated by the addition of EDTA and EGTA to 5 mM each, the mixture was then centrifuged at 5,000 × g for 3 min, and the supernatant was designated the S1 fraction. The pellet was resuspended in 2 mM EDTA for 15 min at 4°C followed by centrifugation, and the supernatant and the pellet were designated the S2 and P fractions, respectively. For DNA analysis of S1, S2, and P fractions, aliquots were treated with lysis buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 5 mM EDTA, 0.5% SDS) for 1 h at 37°C and with phenol-CHCl3 extraction. Proteins were analyzed by SDS-PAGE.
Glutathione S-transferase (GST) pulldown assays.
GST-MeCP2e1 wild type and mutants were expressed and purified from Escherichia coli using glutathione agarose beads (Thermo Scientific) according to the manufacturer's instructions. Purified protein (5 μg) was then incubated with glutathione agarose and used in pulldown assays with 500 μg nuclear extract protein from SH-SY5Y cells prepared as described above. Protein complexes were washed four times with 1× TBS plus 0.1% NP-40, separated by SDS-PAGE, and analyzed by immunoblotting.
Chromatin immunoprecipitation.
SH-SY5Y cells were fixed in 1% formaldehyde for 10 min at room temperature, and nuclei were isolated as previously described (16). Chromatin from isolated nuclei was fragmented to an average size of ∼500 bp using a Bioruptor bath sonicator (Diagnode). Immunoprecipitation was performed from fragmented chromatin as previously described (16) using anti-chicken IgY PrecipHen beads (Aves Labs) to precipitate chicken IgY antibodies. Analysis of precipitated fragments was performed with quantitative PCR using SYBR green-based detection chemistry and is expressed as fold enrichment over ChIP with nonspecific IgY. Primers used for quantitative PCR are listed in Table S1 in the supplemental material.
RESULTS
Identification of MeCP2e1 posttranslational modifications by mass spectrometry and generation of phospho-specific MeCP2 antibodies.
MeCP2 is expressed as two splice variants, MeCP2e1 and MeCP2e2, with human MeCP2e1 replacing the first nine amino acids of the MeCP2e2 amino terminus with a 21-amino-acid hydrophobic sequence (18). As MeCP2e1 is the predominant isoform expressed in the brain (18, 22), this isoform was used in a screen to identify sites of posttranslational modifications. FLAG epitope-tagged mouse MeCP2e1 (MeCP2e1-FLAG) was purified from stably expressing SH-SY5Y cells and analyzed by tandem mass spectrometry (MS/MS). Analysis of independent digestions with trypsin, chymotrypsin, and Glu-C allowed 90% coverage of MeCP2e1 (Fig. 1A). Six putative phosphorylation sites were identified and are summarized in Table 1. Some of these sites have been previously reported (41), while others are newly identified here. Multiple putative ubiquitylation sites were identified on lysine residues, with the most frequently observed peptides corresponding to ubiquitylation at lysine 271 (Table 1). Acetylated peptides were also identified by MS/MS, but it was not possible to definitively assign the modification to a specific amino acid due to their proximity (Table 1). Phosphorylation, ubiquitylation, and acetylation of MeCP2 in SH-SY5Y cells were confirmed by Western blot analyses of MeCP2 immunopurified from nuclear extracts (see Fig. S1 in the supplemental material). Some of the identified modification sites correspond to known RTT mutations, listed in Table 1. Although MeCP2e1 was used to generate the data in Table 1, the results are reported as the corresponding human MeCP2e2 amino acid positions, which are the standard used for reporting RTT mutations and modification sites. Despite relatively uniform coverage across the MeCP2e1 protein, most of the putative modification sites were clustered in the two functionally characterized domains of MeCP2, the methyl-CpG-binding domain (MBD) and the transcriptional repression domain (TRD) (Fig. 1B).
Fig 1.

Identification of posttranslational modifications in MeCP2e1. (A) Amino acid sequence of mouse MeCP2e1, showing residues identified by MS/MS in red. The green box indicates the methyl-DNA binding domain (MBD), and the blue box indicates the transcriptional repression domain (TRD). (B) Visual representation of the putative modification sites identified in this study in relationship to the defined domains of MeCP2 using human MeCP2e2 amino acid numbers. NTD, N-terminal domain; ID, interdomain; CTD, C-terminal domain.
Table 1.
Summary of modified MeCP2 residues identified by MS/MS analysis
| Modification | No. of modified peptides identified | RTT mutation | Frequency of mutation (%)a | Representative peptide |
|---|---|---|---|---|
| pS229 | 12 | 6 (0.14) | K.MPFQA[S]PGGK.G | |
| pS80 | 10 | K.AETSESSGSAPAVPEASA[S]PK.Q | ||
| pS401 | 6 | S401N | 5 (0.11) | K.APMPLLPSPPPPEPESSEDPI[S]PPEPQDLSSSICK.E |
| pS149 | 5 | K.VGDT[S]LDPNDFDFTVTGR.G | ||
| pS274 | 2 | R.KPG[S]VVAAAAAEAK.K | ||
| pS13 | 1 | R.LEEK[S]EDQDLQGLR.D | ||
| UbK271 | 18 | R.[K]PGSVVAAAAAEAK.K | ||
| UbK249 | 8 | K.GEGGGATTSAQVMVI[K].R | ||
| UbK321 | 5 | K.EVV[K]PLLVSTLGEK.S | ||
| UbK233 | 2 | K.MPFQASPGG[K]GEGGGATTSAQVMVIK.R | ||
| UbK 82 | 2 | K82R | 1 (0.03) | K.AETSESSGSAPAVPEASASP[K].Q |
| UbK119 | 2 | R.SAG[K]YDVYLINPQGK.A | ||
| UbK135 | 2 | K135E | 7 (0.17) | R.S[K]VELIAYFEK.V |
| UbK256 | 2 | R.[K]AEADPQAIPK.K | ||
| UbK12 | 2 | K12N | 2 (0.05) | R.LEE[K]SEDQDLQGLR.D |
| UbK130 | 1 | K.YDVYLINPQG[K].A | ||
| AcK305/307 | 3 | K305E/R | 5 (0.11) | [K]R[K]TRETVSIEVKEVVKPL |
| AcK321 | 2 | K.EVV[K]PLLVSTLGEK.S |
From RettBASE (mecp2.chw.edu.au).
Phospho-specific antibodies to MeCP2 modification sites identified by mass spectrometry were generated. Of five phosphorylation sites selected, we successfully generated MeCP2-specific antibodies to two, pS80 and pS229. These antibodies detected stably expressed wild-type MeCP2e1-FLAG from SH-SY5Y cells by immunoblotting but could not detect MeCP2e1-FLAG containing a serine-to-alanine mutation at the phosphorylation site (S80A or S229A) (Fig. 2A). Treatment of purified MeCP2e1-FLAG with alkaline phosphatase also abolished detection by the antibodies (Fig. 2B), together demonstrating that the pS80 and pS229 antibodies are specific to their intended phospho-epitopes. These data also suggest that the pS80 and pS229 are independent of each other, as disruption of one site does not affect phosphorylation of the other (Fig. 2A). Both the pS80 and pS229 phospho-specific antibodies were able to immunoprecipitate endogenous MeCP2 from SH-SY5Y nuclear lysates (Fig. 2C), suggesting that these modifications also occur in vivo. Quantitative dot blots of phospho- and non-phospho-peptides were used to measure the relative affinities of the antibodies to modified versus unmodified targets and demonstrated that the pS80 and pS229 MeCP2 antibodies have approximately 100- and 150-fold preferences for their phosphorylated targets, respectively (see Fig. S2 in the supplemental material). To explore the ability of MeCP2 to harbor multiple phosphorylations, pS80 or pS229 MeCP2 was immunoprecipitated from SH-SY5Y nuclear lysates, and the resulting precipitates were probed with the reciprocal phospho-specific antibody. The MeCP2 precipitated by anti-pS80 was detected with the pS229 MeCP2 antibody, while reciprocally, the pS229-precipitated fraction of MeCP2 was detected with the pS80 MeCP2 antibody (Fig. 2D). While this finding does not exclude the possibility of association of singly phosphorylated MeCP2 molecules, it suggests that MeCP2 can be simultaneously phosphorylated on both S80 and S229.
Fig 2.
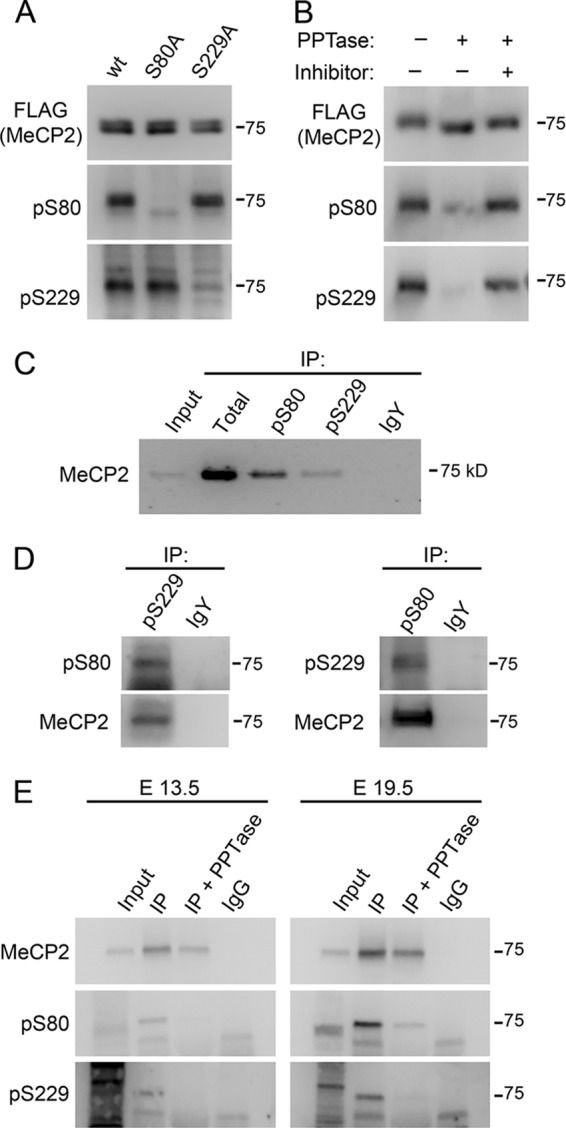
Characterization of MeCP2 phospho-specific antibodies. (A) Western blot of nuclear extracts from SH-SY5Y cell lines stably expressing the indicated MeCP2e1-FLAG construct with antibodies raised against pS80 and pS229 MeCP2 and the FLAG epitope. (B) Western blot of purified MeCP2e1 FLAG either mock treated or treated with alkaline phosphatase in the presence or absence of phosphatase inhibitors. (C) Western blot with anti-MeCP2 antibody of proteins immunoprecipitated from SH-SY5Y nuclear extracts with total, pS80, and pS229 MeCP2 as well as nonspecific IgY. Fifty percent of each immunoprecipitation was loaded. (D) Western blot of MeCP2 immunoprecipitated from SH-SY5Y nuclear extracts with anti-pS80 or -pS229 MeCP2 with the reciprocal antibody. (E) Western blot with the indicated antibodies of MeCP2 immunoprecipitated from mouse embryonic cortex lysates. Immunoprecipitates were mock treated (IP) or treated with alkaline phosphatase (+PPTase) prior to analysis.
While pS80- and pS229-specific antibodies detected phosphorylated MeCP2 from SH-SY5Y cells overexpressing exogenous MeCP2-FLAG or endogenous MeCP2 immunopurified from nuclear extracts, they were unable to detect endogenous MeCP2 from parental SH-SY5Y cell lysates by Western blot analyses (see Fig. S3A and B in the supplemental material). Similarly, both pS80 and pS229 MeCP2 could be detected from MeCP2 immunopurified from embryonic mouse cortex. Treatment of the immunopurified proteins with alkaline phosphatase ablated all signal from the pS80 and pS229 MeCP2 antibodies, demonstrating that these antibodies in fact detect phosphorylated MeCP2 and that these signals are not the result of low affinity interactions with unphosphorylated MeCP2 (Fig. 2E). pS80 but not pS229 MeCP2 was detected in adult brain extracts from wild-type mice and as a truncated form in extracts from Mecp2308/y mutant mice by Western blotting (see Fig. S3C in the supplemental material). These combined results indicate that endogenous pS80 and pS229 modifications are present in embryonic and mature mouse brain but exist on only a small subset of the total MeCP2 molecules.
Subcellular localization of phosphorylated MeCP2.
To examine the subcellular location of pS80 and pS229 MeCP2, sagittal slices from 5-week-old male mouse brains were costained with antibodies against total and phospho-MeCP2 and counterstained with DAPI (4′,6′-diamidino-2-phenylindole) to visualize heterochromatin. pS80 MeCP2 staining was observed in the same DAPI-positive heterochromatic chromocenters as total MeCP2 staining (Fig. 3A). However, pS80 MeCP2 staining showed a punctate distribution within the heterochromatic chromocenters that did not completely colocalize with total MeCP2 (Fig. 3B). The MeCP2 pS229 antibody was not effective in immunofluorescent staining in either tissue or cultured cells (data not shown).
Fig 3.

Subcellular distribution of phospho-MeCP2. (A) Immunofluorescent staining of the nucleus of a cortical neuron from 5-week-old mice with the indicated antibodies. DAPI was used to visualize heterochromatic foci (chromocenters). Bar, 5 μm. (B) Magnification of the region indicated by boxes in panel A containing a single heterochromatic chromocenter. Bar, 1 μm. (C) Gel electrophoresis of DNA from MNase fractions from SH-SY5Y nuclei. (D) Western blot analysis of MNase fractions from SH-SY5Y nuclei with antibodies against RNA polymerase II, MeCP2, and histone H1. (E) Western blot analysis of MNase nuclear fractions from SH-SY5Y cells stably expressing MeCP2e1-FLAG with the indicated antibodies.
Biochemical fractionation of nuclei was performed to examine the differences in subnuclear distributions of total and phosphorylated MeCP2. Micrococcal nuclease fractionation of purified nuclei generates three distinct nuclear fractions, S1, S2, and P (Fig. 2B). The S1 and P fractions primarily contain actively expressed genes and genes poised for expression, while the S2 fraction consists largely of constitutively silenced genes (30). Although the majority of MeCP2 appears to be associated with constitutively silenced heterochromatic chromocenters by immunofluorescence (Fig. 3A), the majority of endogenous MeCP2 from SH-SY5Y cells was found in the nuclease-sensitive S1 fraction enriched for actively expressed genes (Fig. 3C). This is in agreement with recently published results showing that the majority of MeCP2 from brain and liver is also found in the S1 fraction (37). There were no apparent differences in the distribution of total and phosphorylated MeCP2e1-FLAG between the fractions, as all MeCP2 forms were predominantly found in the active S1 fraction along with the known MeCP2 interacting protein Sin3A (25), while the MeCP2-interacting protein HP1α (1) was present in both the S1 and S2 fractions (Fig. 3D). To account for the differences in the amount of DNA present in each fraction, Western blot analysis was also performed using protein from each fraction to correspond to equal loading of DNA (see Fig. S4A and B in the supplemental material). No difference in MeCP2 distribution was found when the fractions were analyzed in this manner, showing that the enrichment of MeCP2 in the S1 fraction was not due to the unequal distribution of DNA across the fractions. Additionally, no difference in the predominantly S1 distribution was observed for the S80A or S229A MeCP2e1-FLAG phosphorylation site mutants (see Fig. S4C in the supplemental material). These results suggest that phosphorylated forms of MeCP2 are present in subnuclear locations similar to that of total MeCP2, together with RNA polymerase II and the known MeCP2-interacting proteins Sin3A and HP1α.
pS80 and pS229 MeCP2 interact with distinct MeCP2 protein cofactors.
MeCP2 interacts with a number of protein cofactors, including the transcriptional repressor Sin3A (25), heterochromatin protein 1 (HP1α, -β, and -γ) isoforms (1), members of the cohesin complex, including SMC3 (17), and the RNA- binding protein YB-1 (40). To determine the role of MeCP2 phosphorylation in these interactions, total, pS80 and pS229 MeCP2 was immunoprecipitated from SH-SY5Y nuclear extracts, and the coprecipitation of known interacting proteins was assessed.
Immunoprecipitation of total MeCP2 from SH-SY5Y nuclear extracts resulted in only weakly detectable association with Sin3A and HP1α, -β, and -γ and undetectable coimmunoprecipitation of SMC3 and YB-1, while immunoprecipitation with anti-pS80 or anti-pS229 MeCP2 resulted in enhanced coimmunoprecipitation of these cofactors despite the reduced amount of MeCP2 present in the precipitates (Fig. 4A, left). When the amounts of the precipitates analyzed were varied to achieve equal MeCP2 representation, Sin3A and YB-1 copurified with pS80 MeCP2, while SMC3, Sin3A, and HP1α, -β, and -γ copurified with pS229 MeCP2 (Fig. 4A, right). This indicates that pS80 and pS229 MeCP2 are each enriched for associations with distinct combinations of MeCP2 cofactors compared to the total MeCP2 fraction. Treatment of the nuclear extracts with RNase or Benzonase, a nuclease that digests both DNA and RNA, prior to immunoprecipitation did not disrupt the interactions with SMC3, Sin3A, and HP1α (Fig. 4B), indicating that these interactions represent protein complexes. The exception to this is YB-1, whose interaction with pS80 MeCP2 was disrupted by removal of RNA (Fig. 4B), as has been shown for its interaction with total MeCP2 (40). The digestion of DNA by Benzonase was confirmed by gel electrophoresis (see Fig. S5 in the supplemental material).
Fig 4.

pS80 and pS229 MeCP2 interact specifically with unique MeCP2 binding partners. (A) Western blot of proteins immunoprecipitated from SH-SY5Y cell nuclear extracts with antibodies against total, pS80, or pS229 MeCP2 or nonspecific IgY. Identical blots were performed using equal volumes of precipitate (left) or equal loading of MeCP2 (right). The percentage of each precipitate loaded is indicated below each lane. (B) Western blot of proteins immunoprecipitated from SH-SY5Y nuclear extracts which had been mock treated or treated with Benzonase (B) or RNase A and T1 (R) prior to immunoprecipitation with antibodies against pS80 or pS229 MeCP2 or nonspecific IgY. The percentage of each precipitate used is indicated below each lane. (C) Coomassie staining of SDS-PAGE-separated proteins from a pulldown assay using GST-tagged wild-type and mutant MeCP2e1. (D) Western blot of proteins from panel C with the indicated antibodies. (E) Western blot of proteins from a pulldown assay from SH-SY5Y nuclear extract using GST-tagged wild-type and mutant MeCP2e1 with the indicated antibodies.
To test whether phosphorylation of MeCP2 at specific sites is sufficient to promote increased interaction with distinct binding partners, phospho-mimetic mutations were generated at serine 80 (S80D) and serine 229 (S229D). Wild-type and phospho-mimetic mutant MeCP2e1 proteins were expressed and purified from E. coli as GST fusion proteins and used in GST pulldown assays with SH-SY5Y nuclear extracts. Wild-type GST-MeCP2e1 weakly interacted with HP1α from nuclear extracts (Fig. 4C and D). In contrast, S229D but not S80D GST-MeCP2e1 showed an increased affinity for HP1α compared to wild-type (Fig. 4D). Sin3a also showed an enhanced interaction with S229D GST-MeCP2 (Fig. 4D). However, S80D GST-MeCP2 did not show any difference from wt GST-MeCP2, despite the specific coimmunoprecipitation of pS80 MeCP2 and Sin3a (Fig. 4A). This suggested that pS80 is not sufficient to promote a specific interaction between the two proteins. Interestingly, all forms of GST-MeCP2e1 were also able to pull down endogenous MeCP2 in this assay (Fig. 4D), indicating the presence of multiple MeCP2 molecules in the same complex. While S229D GST-MeCP2 pulled down both HP1α and HP1β from nuclear extracts, it did not interact with HP1γ (Fig. 4E), suggesting that there may be more direct interactions of HP1α and HP1β than of HP1γ with MeCP2. Together, these data demonstrate that MeCP2 phosphorylation at specific sites can determine its interaction with unique cofactors.
pS229 MeCP2 regulates RA induced expression of the RET gene.
Differentiation of SH-SY5Y neuroblastoma cells as a model of neuronal differentiation was used to investigate the role of MeCP2 phosphorylation in the dynamics of differentiation-induced neuronal gene regulation. Induction of differentiation in neuroblastoma cell lines by treatment with retinoic acid (RA) or phorbol 12-myristate 13-acetate (PMA) results in changes in cell morphology and gene expression that are associated with neuronal differentiation (26, 27). These gene expression changes include induction of the receptor tyrosine kinase gene RET in an MeCP2-regulated manner (3).
As previously reported (3), treatment of SH-SY5Y cells with 10 μM RA resulted in increased RET expression from 4 to 24 h after treatment (Fig. 5A). Chromatin immunoprecipitation (ChIP) was used to examine enrichment of total and phosphorylated MeCP2 at the RET promoter and enhancer (Fig. 5B). Enrichment of total MeCP2 over a nonspecific antibody was observed at both regions of RET. Although a reduction of MeCP2 enrichment was observed specifically at the RET enhancer in response to 24 h RA treatment, consistent with previous reports (3), it did not reach statistical significance (Fig. 5C). While enrichment was relatively low for pS80 MeCP2 at both sites (Fig. 5D), pS229 MeCP2 was enriched specifically at the RET promoter but not at the RET enhancer (Fig. 5E). pS229 MeCP2 enrichment at the RET promoter was unaffected by RA treatment, suggesting that pS229 MeCP2 binding at the RET promoter was independent of RA-induced activation. Treatment with RA did not alter cellular levels of pS80 or pS229 MeCP2, as determined by Western blot analysis (see Fig. S6A in the supplemental material).
Fig 5.

MeCP2 regulates RA-induced expression of RET. (A) Changes in gene expression induced by treatment of SH-SY5Y cells with 10 μM RA for the indicated times. (B) Schematic of the RET gene showing the qPCR amplicons used for panels C to E. (C to E) ChIP with total MeCP2 (C), pS80 MeCP2 (D), and pS228 MeCP2 (E) antibodies analyzed by quantitative PCR at the indicated sites, expressed as enrichment over nonspecific IgY. (F) RA-induced changes in expression of RET in parental SH-SY5Y cells or SH-SY5Y cells stably expressing the indicated MeCP2e1-FLAG construct. Expression was measured by quantitative PCR and normalized to GAPDH expression. Data are means from three independent biological replicates. Error bars represent standard errors of the means (SEM). *, P < 0.05 compared to the corresponding result in the SH-SY5Y parental cell line.
MeCP2 is also important for the transcriptional silencing of LINE-1 retrotransposons (23). Total, pS80, and pS229 MeCP2 enrichment at LINE-1 and ALU and the segmental duplications of chromosome 15q11-q13 was assessed by ChIP before and after treatment with RA or PMA (see Fig. S7 in the supplemental material). While total MeCP2 was enriched at various levels at all three elements before and after treatment, no significant enrichment of pS80 or pS229 MeCP2 was detected at any of these repetitive sites under any condition examined, demonstrating that while enrichment of total MeCP2 is found at many sites throughout the genome, enrichment of pS229 MeCP2 is specific to distinct sites, such as the RET promoter. To assess the requirement for pS229 and/or pS80 MeCP2 in the RA-induced changes in RET gene expression, SH-SY5Y cells stably overexpressing wild-type MeCP2 or the phospho-deficient pS229A or pS80A MeCP2 mutants as well as the parental SH-SY5Y cell line were utilized. Overexpression of wild-type MeCP2 resulted in the repression of RET expression in SH-SY5Y cells, consistent with previous reports (Fig. 5F) (3). The S80A and S229A mutants were both similar to the wild type in their ability to repress RET expression on SH-SY5Y cells (Fig. 5F). In contrast, overexpression of the S229A MeCP2 mutant but not S80A or wild-type MeCP2 caused a significant increase in RA-induced RET expression compared to that in the parental cell line after 24 h of treatment (Fig. 5F). These results suggest that while pS229 MeCP2 binding to the RET promoter does not change in response to RA treatment, phosphorylation of MeCP2 on serine 229 is required for optimal MeCP2-mediated control of RA induced RET expression.
Phosphorylated MeCP2 participates in the activation and repression of EGR2 in response to PMA-induced neuronal maturation.
PMA-induced differentiation of SH-SY5Y cells results in changes in cell morphology and gene expression, including expression the gene encoding the early growth response factor EGR2 (27, 35). Treatment with 16 nM PMA produced a rapid increase in EGR2 expression peaking at 1 h after treatment, followed by a decrease to an intermediate level by 8 h of treatment (Fig. 6A). MeCP2 enrichment was previously reported to occur at a specific regulatory element in the single intron of EGR2 (35). However, the role of PMA and the presence of phosphorylated forms of MeCP2 at this site have not been explored. To assess this, total MeCP2 and phospho-MeCP2 enrichment at the EGR2 promoter and intronic binding sites (Fig. 6B) before and after PMA treatment was examined by ChIP. Total MeCP2 enrichment was observed at a relatively uniform level (approximately 2-fold enrichment over IgY) at the EGR2 promoter and intron (Fig. 6C). Treatment with 16 nM PMA for 24 h caused an increase in total MeCP2 binding at the EGR2 intron but not at the promoter. In contrast to the PMA-induced enrichment of total MeCP2 at the EGR2 intron, enrichment of pS80 and pS229 MeCP2 at either the promoter or intron of EGR2 was not detected before or after PMA treatment (Fig. 6D and E). As with RA, PMA treatment for up to 24 h did not affect cellular levels of pS80 or pS229 MeCP2 detected by Western blotting (see Fig. S6B in the supplemental material).
Fig 6.

MeCP2 regulates PMA-induced expression of EGR2. (A) Changes in gene expression induced by treatment of SH-SY5Y cells with 16 nM PMA for the indicated times. (B) Schematic of the EGR2 gene showing the qPCR amplicons used for panels C to E. (C to E) ChIP with total MeCP2 (C), pS80 MeCP2 (D), and pS229 MeCP2 (E) antibodies analyzed by quantitative PCR at the indicated sites, expressed as enrichment over nonspecific IgY. (F) PMA-induced expression changes of EGR2 in parental SH-SY5Y or SH-SY5Y cell lines stably expressing the indicated MeCP2e1-FLAG construct. Expression was measured by quantitative PCR and normalized to GAPDH expression. Data are means from three independent biological replicates. Error bars represent SEM. *, P < 0.05 compared to the corresponding result in the SH-SY5Y parental cell line.
Despite the lack of phospho-MeCP2 binding to the promoter or intron of the EGR2 gene, the requirement of pS229 and/or pS80 MeCP2 in the transcriptional regulation of EGR2 was assessed using the SH-SY5Y cell lines stably expressing wild-type MeCP2 and the S80A and S229A mutants. Prior to PMA treatment, overexpression of wild-type MeCP2e1 caused a repression of EGR2 expression compared to the parental cell line (Fig. 6F), similar to the result observed with resting cells for RET (Fig. 5F). Expression of either the S80A or S229A MeCP2 mutant had the opposite effect, resulting in increased EGR2 expression in resting cells. After treatment with 16 nM PMA, cells overexpressing wild-type MeCP2 showed both increased induction of EGR2 at 1 h and increased repression at 24 h, compared to the parental cell line (Fig. 6F). In contrast, neither the S80A nor S229A MeCP2 mutant-expressing cell lines showed the increased EGR2 induction at 1 h that was observed with wild-type MeCP2. In addition, the MeCP2 phospho-deficient expressing mutant cells displayed a reduced ability to repress EGR2 expression after the induction peak, as EGR2 levels 24 h following PMA were significantly higher in both mutant cell lines (Fig. 6F). These results demonstrate that MeCP2 is capable of activating and repressing expression of EGR2 and that phosphorylation of S80 and S229 of MeCP2 is important for both effects on EGR2 expression in response to PMA.
DISCUSSION
One of the major dilemmas in understanding the role of MeCP2 in neuronal development and the etiology of Rett syndrome has been reconciling its demonstrated function as a gene-specific transcriptional silencer (25) with the observations that it binds throughout the genome (10, 34, 39) and is involved in the transcriptional modulation of active genes (7, 39). We hypothesized that posttranslational modifications of MeCP2 may play an important role in determining the transcriptional activities of MeCP2. In this study, we demonstrate a posttranslational modification map of MeCP2e1 in SH-SY5Y cells, including phosphorylation, acetylation, and ubiquitylation sites, most of which have not been reported previously. Phosphorylation of S80 and phosphorylation of S229 of MeCP2 are each important for regulating interactions between MeCP2 with distinct combinations of cofactors and are both present in active subfractions of the nucleus. pS229 MeCP2 specifically showed enriched binding to the RET promoter and was required for regulation of RET transcription in response to RA. We found that MeCP2 can act as an activator or repressor at the same gene and that phosphorylation on S80 and S229 influences the dynamic transcriptional regulation by MeCP2 in response to stimuli. These results reveal that the functions of MeCP2 are context dependent, influenced not only by posttranslational modifications but also by the neuronal activation state and individual genetic loci.
These results represent the most comprehensive map of MeCP2e1 posttranslational modifications assembled to date and demonstrate that MeCP2 is a highly modified protein. It is interesting that the detected modification sites cluster in the MBD and TRD of MeCP2, which also harbor a majority of the RTT-causing point mutations. Additionally, most of the MeCP2e1 phosphorylation sites were found in close proximity to potential ubiquitylation sites, providing a potential for reciprocal regulation. Our studies were consistent with prior studies showing MeCP2 phosphorylation at pS80 and pS229 (6, 9, 41). A previously reported phosphorylation site, S421 (41), was not detected SH-SY5Y cells. This is likely due to the fact that MeCP2 posttranslational modifications are dependent on both cell type and activation state and therefore every cell will not necessarily contain all possible modifications. The diversity of the MeCP2e1 posttranslational modifications, as well as its genome-wide distribution (10, 34), is reminiscent of the complex regulation of the histone core proteins and their numerous modifications.
The presence of MeCP2 in the subnuclear fraction containing actively expressed genes is consistent with recent reports (37) and supports our prior genomic analyses of MeCP2 showing an overlap with Pol II binding and active gene promoters (39). The presence of MeCP2 in this fraction was unaffected by pS80 or pS229, despite their roles in both activation and repression of EGR2 and RET. This suggests that while MeCP2 is associated with a subnuclear fraction enriched for actively expressed genes, it may regulate expression of those genes in both a positive and a negative manner. The lack of MeCP2 in the nuclear fraction associated with silenced heterochromatin is unexpected, considering that MeCP2 colocalizes with heterochromatic foci by immunofluorescence and is required for the heterochromatic silencing of repetitive DNA elements (23). The heterochromatin fraction of MeCP2 may be only loosely associated and therefore easily dislodged by the fractionation process. Nonetheless, the appearance of MeCP2 in a nuclear fraction enriched for actively expressed genes adds further support to the observations that MeCP2 plays a role in regulating actively expressed genes.
Phosphorylation of MeCP2 on serine 80 and 229 each promotes the interaction of MeCP2 with unique combinations of binding partners, including Sin3A, YB-1, SMC3, and HP1. These phospho-specific interactions could underlie the formation of distinct MeCP2-containing complexes, each capable of regulating gene expression through different mechanisms. The interaction between Sin3A and MeCP2 was the basis for the characterization of MeCP2 as a transcriptional repressor (25). However, the preferential interaction of pS80 and pS229 MeCP2 with Sin3A compared to total MeCP2 suggests that not all MeCP2 molecules adopt this mechanism of regulation. SMC3 is a component of the cohesin complex, which is responsible for sister chromatid cohesion. The cohesin complex is also an important regulator of gene expression during development that participates in the formation of long-range chromatin loops (13). MeCP2 has also been reported to participate in long-range chromatin interactions (15), and this may be achieved in part through the specific interactions between SMC3 and pS229 MeCP2. HP1 proteins are responsible for the formation of transcriptionally silent heterochromatin, although more recent studies have also proposed roles for HP1 proteins in active gene expression (19). Repression and activation of expression are also mediated by phosphorylation of MeCP2 on serine 229, and this may be achieved through interactions with HP1 proteins. Although YB-1 appears to interact with MeCP2 in an RNA-dependent manner as previously described (40), the specific association of YB-1 and pS80 MeCP2 suggests that this interaction is not random and may be functionally relevant.
Our results demonstrate that MeCP2 plays three distinct roles in the regulation of EGR2 and RET expression. First, in untreated cells, MeCP2 acts as a repressor, reducing the basal expression of both genes, consistent with its described role as a transcriptional repressor (25). Second, MeCP2 can act as an activator by increasing the maximal expression of EGR2 in response to PMA. Third, MeCP2 can act to modulate the levels of expressed genes following the peak of activation. In this case, MeCP2 appears to be involved in maintaining the proper level of expression, in essence acting as the “brakes” to prevent overactivation of gene expression. The ability of MeCP2 to regulate transcription in these three ways reveals that it is a dynamic regulator of gene expression in response to stimuli, capable of both transcriptional repression and activation. Although the genome-wide binding of MeCP2 appears to be unchanged by neuronal activity, the broad distribution of MeCP2 hinders the high read coverage that may be required to see changes at discrete sites (10). The change in total MeCP2 enrichment observed at the EGR2 intron in response to PMA treatment may be indicative of many similar changes that occur throughout the genome in response to stimuli. However, these changes may occur at great distances from the genes they affect and may occur for only a subset of MeCP2 regulated genes.
Our study has also shown that the dynamic regulation of differentiation-induced transcription is mediated by MeCP2 phosphorylation. The effects of phosphorylation are not universal, however, and depend on a number of factors. While phosphorylation of MeCP2 on serine 80 and 229 is important for MeCP2 repression of RET in unstimulated cells, it appears to be dispensable for MeCP2-mediated repression of EGR2 under the same conditions. Alternatively, activation of EGR2 by MeCP2 in response to PMA requires phosphorylation at both sites. Finally, while phosphorylation of both serine 80 and 229 are required for MeCP2-mediated modulation of EGR2 transcription after expression levels have peaked, only serine 229 phosphorylation is required for modulation of RET expression levels. While phosphorylation of MeCP2 is involved in its dynamic regulation of transcription, the effects of phosphorylation on S80 and S229 are clearly complex and vary with both the type and duration of the stimuli as well as the specific gene being modulated.
How exactly pS80 and pS229 contribute to the regulation of specific gene expression remains unknown. The observation that MeCP2 binds throughout the genome has led to the hypothesis that phosphorylated forms of MeCP2 bind to distinct sites and provide regulatory specificity. Our data suggest that this may indeed be the case for MeCP2 phosphorylated on serine 229, which showed enrichment for binding to the RET promoter. It is possible that the chromatin environment directed by pS229 MeCP2 could impact the transcriptional response to stimulation similar to the manner in which modifications on histones direct chromatin states. While pS229 MeCP2 showed specificity in binding to the RET promoter and mutation of S229 affected RET transcript levels, neither global phosphorylation at pS229 nor binding of pS229 MeCP2 changed in a detectable manner in response to RA or PMA stimulation. It may be that phosphorylation of MeCP2 on S229 is relatively static and serves as a signal to recruit regulatory complexes to specific sites in the genome. The activity of these complexes may then be modulated by differentiation-induced signaling pathways to control gene expression. This hypothesis makes pS229 MeCP2 functions distinct from those of pS421, which dynamically increases with neuronal activity but is bound throughout the genome (9, 10).
In contrast to our results with the RET gene, we were unable to detect either pS80 or pS229 MeCP2 binding to proximal sites of EGR2 despite their requirement for PMA-induced expression of EGR2. It may be that the pS80 and pS229 MeCP2 binding sites responsible for EGR2 regulation are located elsewhere, perhaps even at great distances from the gene, as has been suggested in the regulation of many MeCP2 target genes (15, 39). It is important to note that we were unable to detect strong pS80 MeCP2 enrichment at any site we examined. This may mean that pS80 MeCP2, in contrast to previous reports (36), does not bind to chromatin. However, an alternate possibility is that the antibody used here is not suitable for chromatin immunoprecipitation under these conditions.
Our data show that the characterizations of MeCP2 as a transcriptional repressor and as an activator are both correct. Not only can MeCP2 activate and repress transcription, but also, it can have both functions for the same gene at different times before and after response to stimuli. The precise function of MeCP2 at a given gene depends on the gene itself as well as the duration of the transcriptionally altering stimulus. When one considers the precise spatial and temporal signaling that underlies the development and maturation of the mammalian brain, it becomes easier to understand the confusion surrounding the role of MeCP2 as a transcriptional activator or repressor. We hypothesized that posttranslational modifications would determine the transcriptional activity of MeCP2. However, we were not able to correlate the phosphorylation events examined here with a single specific MeCP2 activity. Instead, phosphorylation appears to be only one factor that interacts with the signaling networks that determine the functional activity of MeCP2. Since our study focused on the functional relevance of only two of the many MeCP2 posttranslational modifications, it is likely that taking the full complement of MeCP2 modifications into account will allow greater success in assigning specific activities to modified forms of MeCP2.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a National Institutes of Health grant (2R01HD041462) and an ARRA supplement from the National Institute of Child Health and Human Development to J.M.L. and an International Rett Syndrome Foundation Fellowship to M.L.G.
Footnotes
Published ahead of print 21 May 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Agarwal N, et al. 2007. MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic Acids Res. 35:5402–5408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amir RE, et al. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23:185–188 [DOI] [PubMed] [Google Scholar]
- 3. Angrisano T, et al. 2011. Chromatin and DNA methylation dynamics during retinoic acid-induced RET gene transcriptional activation in neuroblastoma cells. Nucleic Acids Res. 39:1993–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Armstrong D, Dunn JK, Antalffy B, Trivedi R. 1995. Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 54:195–201 [DOI] [PubMed] [Google Scholar]
- 5. Balmer D, Goldstine J, Rao YM, LaSalle JM. 2003. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J. Mol. Med. (Berl.) 81:61–68 [DOI] [PubMed] [Google Scholar]
- 6. Bracaglia G, et al. 2009. Methyl-CpG-binding protein 2 is phosphorylated by homeodomain-interacting protein kinase 2 and contributes to apoptosis. EMBO Rep. 10:1327–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chahrour M, et al. 2008. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320:1224–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen RZ, Akbarian S, Tudor M, Jaenisch R. 2001. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27:327–331 [DOI] [PubMed] [Google Scholar]
- 9. Chen WG, et al. 2003. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302:885–889 [DOI] [PubMed] [Google Scholar]
- 10. Cohen S, et al. 2011. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 72:72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Collins AL, et al. 2004. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13:2679–2689 [DOI] [PubMed] [Google Scholar]
- 12. Deng JV, et al. 2010. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat. Neurosci. 13:1128–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dorsett D. 2011. Cohesin: genomic insights into controlling gene transcription and development. Curr. Opin. Genet. Dev. 21:199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guy J, Hendrich B, Holmes M, Martin JE, Bird A. 2001. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27:322–326 [DOI] [PubMed] [Google Scholar]
- 15. Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. 2005. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37:31–40 [DOI] [PubMed] [Google Scholar]
- 16. Im H, et al. 2004. Measurement of protein-DNA interactions in vivo by chromatin immunoprecipitation. Methods Mol. Biol. 284:129–146 [DOI] [PubMed] [Google Scholar]
- 17. Kernohan KD, et al. 2010. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev. Cell 18:191–202 [DOI] [PubMed] [Google Scholar]
- 18. Kriaucionis S, Bird A. 2004. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 32:1818–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwon SH, Workman JL. 2011. The changing faces of HP1: From heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription. Bioessays 33:280–289 [DOI] [PubMed] [Google Scholar]
- 20. Li H, Zhong X, Chau KF, Williams EC, Chang Q. 2011. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat. Neurosci. 14:1001–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meehan R, et al. 1992. Transcriptional repression by methylation of CpG. J. Cell Sci. Suppl. 16:9–14 [DOI] [PubMed] [Google Scholar]
- 22. Mnatzakanian GN, et al. 2004. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 36:339–341 [DOI] [PubMed] [Google Scholar]
- 23. Muotri AR, et al. 2010. L1 retrotransposition in neurons is modulated by MeCP2. Nature 468:443–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nan X, Campoy FJ, Bird A. 1997. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88:471–481 [DOI] [PubMed] [Google Scholar]
- 25. Nan X, et al. 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386–389 [DOI] [PubMed] [Google Scholar]
- 26. Oppenheimer O, Cheung NK, Gerald WL. 2007. The RET oncogene is a critical component of transcriptional programs associated with retinoic acid-induced differentiation in neuroblastoma. Mol. Cancer Ther. 6:1300–1309 [DOI] [PubMed] [Google Scholar]
- 27. Pahlman S, Ruusala AI, Abrahamsson L, Mattsson ME, Esscher T. 1984. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: a comparison with phorbolester-induced differentiation. Cell Differ. 14:135–144 [DOI] [PubMed] [Google Scholar]
- 28. Peddada S, Yasui DH, LaSalle JM. 2006. Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum. Mol. Genet. 15:2003–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reyes JC, Muchardt C, Yaniv M. 1997. Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J. Cell Biol. 137:263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rose SM, Garrard WT. 1984. Differentiation-dependent chromatin alterations precede and accompany transcription of immunoglobulin light chain genes. J. Biol. Chem. 259:8534–8544 [PubMed] [Google Scholar]
- 31. Shahbazian M, et al. 2002. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35:243–254 [DOI] [PubMed] [Google Scholar]
- 32. Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. 2002. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 11:115–124 [DOI] [PubMed] [Google Scholar]
- 33. Singleton MK, et al. 2011. MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol. Dis. 43:190–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Skene PJ, et al. 2010. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 37:457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Swanberg SE, Nagarajan RP, Peddada S, Yasui DH, LaSalle JM. 2009. Reciprocal co-regulation of EGR2 and MECP2 is disrupted in Rett syndrome and autism. Hum. Mol. Genet. 18:525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tao J, et al. 2009. Phosphorylation of MeCP2 at serine 80 regulates its chromatin association and neurological function. Proc. Natl. Acad. Sci. U. S. A. 106:4882–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thambirajah AA, et al. 2012. MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain. Nucleic Acids Res. 40:2884–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. West AE, Greenberg ME. 2011. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harbor Perspect Biol. 3:a005744 doi:10.1101/cshperspect.a005744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yasui DH, et al. 2007. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. U. S. A. 104:19416–19421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Young JI, et al. 2005. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. U. S. A. 102:17551–17558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou Z, et al. 2006. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52:255–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.