

Abstract
Linker for activation of T cells (LAT) is a transmembrane adaptor protein that is essential to bridge T cell receptor (TCR) engagement to downstream signaling events. The indispensable role of LAT in thymocyte development and T cell activation has been well characterized; however, the function of LAT in cytotoxic-T-lymphocyte (CTL) cytotoxicity remains unknown. We show here that LAT-deficient CTLs failed to upregulate FasL and produce gamma interferon after engagement with target cells and had impaired granule-mediated killing. We further dissected the effect of the LAT deletion on each step of granule exocytosis. LAT deficiency led to altered synapse formation, subsequently causing unstable T cell–antigen-presenting cell (APC) conjugates. Microtubule organizing center polarization and granule reorientation were also impaired by LAT deficiency, leading to reduced granule delivery. Despite these defects, granule release was still observed in LAT-deficient CTLs due to residual calcium flux and phospholipase C (PLC) activity. Our data demonstrated that LAT-mediated signaling intricately regulates CTL cytotoxicity at multiple steps.
INTRODUCTION
An essential component of the immune response is the ability to destroy virally infected or tumor cells. CD8 cytotoxic T lymphocytes (CTLs) have the capacity to eliminate these types of cells by two pathways: the Fas pathway and the granule pathway (17, 30). The Fas pathway is triggered by the interaction between Fas ligand (FasL) on CTLs and Fas on target cells, leading to caspase activation and apoptotic cell death. The granule pathway relies on directed granule exocytosis, a perforin-mediated directional transfer of granzymes from CTLs into target cells.
The process of granule exocytosis by CTLs has been well characterized (34). When CTLs encounter target cells, an immunological synapse (IS) is formed at the T-target cell contact zone. The IS consists of the T cell receptors (TCRs) and additional signaling proteins that accumulate in the center of the supramolecular activation cluster (cSMAC) and are surrounded by a peripheral ring enriched in adhesion molecules (pSMAC) (9, 23, 26, 35). Within minutes of TCR recognition, the microtubule organizing center (MTOC) of the T cell polarizes toward the target cell and remains positioned beneath the synapse (12). The lytic granules move in a minus-end direction along the microtubules toward the MTOC, eventually fusing with the plasma membrane of CTLs. The granules are then released from the secretory cleft, which is adjacent to the cSMAC, and delivered into target cells (34). Although this process has been well described, the signaling mechanisms that control granule movement and release are still under investigation. Published data show that calcium is critical for granule exocytosis and killing of target cells (6, 20). Studies using pharmacological inhibitors also demonstrate that signaling proteins in the TCR signaling pathway, such as phosphatidylinositol 3-kinase (PI3K), protein kinase C (PKC), and mitogen-activated protein kinase (MAPK), are important in CTL-mediated cytotoxicity (16, 25, 29). Recent data show that stronger TCR signals lead to increased death of target cells. In contrast, weak TCR stimuli fail to induce the recruitment of granules to the synapse, despite proper synapse formation and MTOC reorientation (1, 14), suggesting that MTOC polarization, granule transportation, and granule release are differentially regulated.
Linker for activation of T cells (LAT) is a transmembrane adaptor protein that plays an essential role in linking TCR engagement to downstream signaling events, such as Ras-Erk activation and calcium mobilization. Upon T cell activation, LAT is phosphorylated by the tyrosine kinase ZAP-70 and binds directly to Grb2, Gads, and phospholipase C-γ1 (PLC-γ1), as well as indirectly associating with SLP-76, Vav, and other signaling proteins (15, 19, 38). LAT binding to Grb2 leads to the membrane recruitment of Sos, a guanine nucleotide exchange factor (RasGEF), which activates Ras. Interestingly, TCR-mediated Ras activation also requires RasGRP1 (5). The interaction of LAT with PLC-γ1 and the Gads-SLP-76 complex is critical for the activation of PLC-γ1. Initiation of these signaling cascades eventually leads to the activation of transcription factors that regulate the genes required for T cell proliferation and effector functions. The importance of LAT in thymocyte development and T cell activation has been extensively studied. LAT-deficient Jurkat cells are defective in TCR-mediated MAPK activation and calcium flux (7, 37). Correspondingly, thymocyte development in LAT−/− mice is completely blocked at the DN3 stage (39). Furthermore, our recent study using LAT conditional knockout mice indicates that LAT deficiency in mature CD4 T cells dramatically diminishes TCR-mediated signaling, rendering T cells unresponsive to TCR stimulation (32).
Published studies have proven the essential role of LAT in thymocytes and CD4 T cells; however, the function of LAT in cytotoxic CD8 T cells is largely unknown. In the present study, we used LAT knock-in mice to investigate the role of LAT in the effector function of CTLs. Since T cell activation requires LAT, we deleted LAT after antigen-induced expansion and differentiation of naive CD8 T cells into CTLs. Our data demonstrated that LAT-deficiency severely impaired TCR-mediated signaling in CTLs. Consequently, these LAT-deficient CTLs were defective in gamma interferon (IFN-γ) production, FasL upregulation, and target cell killing through the granule-dependent pathway. We further dissected the effect of LAT deletion on each step of granule exocytosis and identified defects at multiple steps in this pathway. Our study demonstrated the important role of LAT in CTL cytotoxicity and showed that the CTL response is intricately controlled by multiple signaling events.
MATERIALS AND METHODS
Mice.
LAT−/− and LATf/f mice were generated as previously described (32, 39). LAT−/− mice were crossed with ERCre transgenic mice (kindly provided by Thomas Ludwig, Columbia University, New York, NY) to produce ERCre+ LAT−/− mice. LATf/f mice were crossed with OT-I transgenic mice (Jackson Laboratory, Bar Harbor, ME) to generate OT-I/LATf/f mice, which were then mated with ERCre+ LAT−/− mice to obtain ERCre+ LATf/− OT-I or LATf/− OT-I mice. These mice have been backcrossed onto the C57BL/6 background for more than 10 generations. All mice were used in accordance with National Institutes of Health guidelines. The procedures in the present study were approved by the Duke University IACUC. Mice were housed in a specific-pathogen-free facility.
Cytotoxicity assay.
A total of 5 × 105 ERCre+ LATf/− OT-I or LATf/− OT-I T cells (Thy1.2+) were adoptively transferred into Thy1.1+ Thy1.2+ mice at day 0. One day after transfer, these mice were infected with 1.5 × 104 Listeria-Ova intravenously. At days 7 and 8, they received 1.5 mg of tamoxifen (10 mg/ml in corn oil) intraperitoneally. At day 11, splenocytes from Thy1.1+ mice were loaded with 1 μM Ova peptide (SIINFEKL) and 6 μM Cell Tracker Orange (Molecular Probes) as target cells or unlabeled as negative controls. These cells were mixed at a ratio of 1:1, and 1.5 × 107 cells were transferred into mice that received OT-I cells. One hour later, the splenocytes were analyzed by fluorescence-activated cell sorting (FACS).
For in vitro cytotoxicity assays, splenocytes from ERCre+ LATf/− OT-I and LATf/− OT-I mice were primed with 1 μM Ova peptide. Two days after priming, T cells were cultured in the presence of 50 nM 4-hydroxytamoxifen (4-OHT) to induce LAT deletion and interleukin-2 (10 ng/ml) for another 3 to 5 days. EL4 and L1210/Kb were used as target cells. L1210/Kb cells (kindly provided by Hanne Ostergaard) are Fas deficient (10). Target cells loaded with 10 μM Ova peptide were labeled with 6 μM Cell Tracker Orange. Cells labeled with only 1 μM Cell Tracker Orange were used as negative controls. CTLs were mixed with 105 target cells at effector/target ratios of 4:1, 2:1, and 1:1 in a 96-well round-bottom plate at 37°C for 4 h. For FACS analysis, live target cells were gated as live/dead Pacific Blue-negative and phycoerythrin (PE; Cell Tracker Orange)-positive cells. Specific lysis was determined as 100 − [100 × (% of peptide-loaded targets/% of control targets in the presence of CTLs)/(% of peptide-loaded targets/% of control targets in the absence of CTLs)].
For the cytotoxicity assay in the presence of the PLC inhibitor, CTLs were pretreated with U73122 (Sigma) for 30 min at 37°C prior to incubation with L1210/Kb cells for 45 min. Inhibitors were kept at the same concentration during the killing assay.
FACS analysis.
To detect the expansion of OT-I T cells in vivo, single cell suspensions were prepared from spleens and stained with APC-Cy7–anti-CD8, APC–anti-TCRVα2, Pacific Blue–anti-Thy1.1, PE-Cy7–anti-Thy1.2, and 7-amino-actinomycin D (7AAD). For intracellular staining of IFN-γ, CTLs were restimulated with 1 μM Ova peptide for 5 h in the presence of Golgi-stop, fixed, permeabilized, and stained with APC–anti-IFN-γ. For the upregulation of FasL, 5 × 105 T cells were incubated with 5 × 105 EL4 cells preloaded with Ova peptide for 5 h and stained with APC–anti-CD8, PE–anti-FasL, and 7AAD. APC–anti-TCRβ, PE–anti-CD25, PE–anti-CD69, APC-Cy7–anti-CD62L, and APC–anti-CD44 were used to stain CTLs. Samples were analyzed on FACSCanto II (BD Biosciences). FACS plots shown were analyzed with FlowJo (Ashland, OR).
Conjugate assay.
OT-I CTLs were labeled with 5 μM Cell Tracker Violet (Molecular Probes) at 37°C for 20 min. L1210/Kb cells were labeled with 5 μM Cell Tracker Orange and were either pulsed with 10 μM Ova peptide or left untreated. A total of 105 CTLs were mixed with 105 L1210/Kb cells in a 96-well round-bottom plate and incubated at 37°C. At different time points, cells were pipetted to disrupt the nonspecific binding, fixed with 2% paraformaldehyde, and analyzed by FACS. Conjugates were defined as cells that were positive for both dyes.
gzmB substrate assay.
A granzyme B (gzmB) substrate assay was performed according to the GranToxiLux killing protocol (OncoImmunin) with some modifications. Briefly, L1210/Kb target cells were labeled with 5 μM Cell Tracker Orange and loaded with or without 10 μM Ova peptide. A total of 3 × 105 OT-I CTLs were mixed with 105 L1210/Kb cells, centrifuged in a 96-well round-bottom plate for 2 min, and then incubated at 37°C for 40 min in the presence of 40 μl of fluorogenic gzmB substrate solution. The gzmB activity was indicated by increased green fluorescence in target cells.
Western blotting.
For detecting LAT, perforin, and gzmB, CTLs treated with 4-OHT for 3 to 4 days were lysed, resolved by SDS-PAGE, and blotted with antiperforin (Kamiya Biomedical), anti-gzmB (R&D Systems), and two different anti-LAT antibodies. Anti-LAT (Ab#1) recognizes the cytoplasmic domain of LAT (38), and anti-LAT (Ab#2) recognizes a peptide in the central region of LAT, which is deleted in LATKO T cells (Cell Signaling, catalog no. 9166). For detection of phosphorylated proteins, CTLs were rested before being stimulated with anti-CD3 (5 μg/ml) and anti-CD8 (1 μg/ml). Cell lysates were analyzed by Western blotting with the following antibodies: pTyr (4G10), pLAT (Y191), pERK, ERK2, pAkt (Ser473, Thr308), pan-Akt, pPLCγ1, PLCγ1, pPKCθ, and PKCθ (Cell Signaling).
Calcium mobilization.
OT-I CTLs were loaded with Indo-1 for 30 min and then stained with PE anti-CD8. Calcium flux was initiated by the addition of anti-CD3-biotin (5 μg/ml) and anti-CD8-biotin antibodies (1 μg/ml), followed by cross-linking with streptavidin (25 μg/ml). Calcium flux was determined by monitoring the fluorescence emission ratio at 405/495 nm.
Immunofluorescence imaging and analysis.
For LAT staining, 2 × 105 CTLs were dropped onto poly-l-lysine-coated cover slides, fixed with 2% paraformaldehyde for 15 min at room temperature, permeabilized with ice-cold methanol for 10 min, and placed at −20°C for an additional 30 min. The cells were blocked with 2.4G2 with 5% donkey serum for 1 h at room temperature. LAT was detected by rabbit anti-mouse LAT antibody (Cell Signaling, catalog no. 9166), followed by Cy3-conjugated donkey anti-rabbit IgG (Jackson Laboratory). For conjugate immunofluorescence staining, 1.5 × 105 CTLs were mixed with 1.5 × 105 L1210/Kb cells preloaded with Ova peptide. The conjugates were formed by spinning down the cell mixture at 5,000 rpm for 30 s, followed by incubation at 37°C for various times. After incubation, the cells were dropped onto poly-l-lysine-coated cover slides, fixed, permeabilized, and blocked with 2.4G2 with 5% goat serum or donkey serum for 1 h at room temperature. For immunological synapse staining, the conjugates were fixed at 15 and 30 min. PKCθ was detected by rabbit anti-mouse PKCθ antibody (Cell Signaling), followed by Alexa Fluor 488-conjugated goat anti-rabbit IgG (Molecular Probes). LFA-1 was detected by rat anti-mouse LFA-1 antibody (eBioscience), followed by Cy5-conjugated goat anti-rat IgG (Jackson Laboratory). For MTOC and granule staining, conjugates were fixed at 15, 30, and 45 min. MTOCs were detected using mouse anti-α-tubulin antibody (Sigma), followed by Alexa Fluor 488-conjugated goat anti-mouse IgG (Molecular Probes). Granules were detected by APC-conjugated anti-gzmB antibody (eBioscience). Samples were examined with the 100× objective on a Zeiss Observer D1 station with a CoolSNAPHQ charge-coupled device (CCD) camera using Ex:485/20 and Em:520/35 filter sets for Alexa Fluor 488 and Ex:650/13 and Em:684/24 filter sets for APC (Semrock). Synapse images were recorded at 1-μm increments for the z-dimension and displayed as a projection view after three-dimensional reconstruction. MTOC and granule images are displayed as xy stack and z-projections.
Multichannel live cell video microscopy and data analysis.
The calcium imaging experiments were performed with the 40× objective lens on a Zeiss Observer D1 station equipped with a humidified environmental chamber and a CoolSNAPHQ CCD camera (Roper Scientific) and driven by MetaMorph Software. CTLs were loaded with Fura-2 (5 μM) for 30 min and washed with minimal imaging buffer before incubation with target cells. Calcium images were recorded at intervals of 30 s for 30 min with the filter sets Ex1:340/26, Ex2:387/11, and Em:510/84 (Semrock). For calcium elevation analysis in activated T cells, time zero was defined as the time point prior to contact with target cells. The baseline (value = 1.00) was then drawn by averaging the ratio of the fluorescence intensity (340/380 nm) before time zero. The integrated value of cytosolic calcium elevation was generated by summing the changes in relative calcium concentration at each time point within the first 10 min (30-s interval) of the calcium response in CTLs.
Statistical analyses.
For all experiments, differences between groups were examined for statistical significance by using unpaired two-tailed Student t tests to assess the data.
RESULTS
Defective T cell cytotoxicity in vivo in the absence of LAT.
To study LAT function in CD8+ CTLs, we utilized LAT conditional knockout mice to delete the lat gene in mature effector T cells. In these mice (LATf/f), exons 7 to 11 of the lat gene, which encode the region containing critical tyrosine residues that bind Grb2, Gads, and PLC-γ1, are flanked by two LoxP sites. Cre-mediated deletion renders LAT nonfunctional. Meanwhile, deletion allows for the expression of a LAT-green fluorescent protein (GFP) fusion protein (32). This fusion protein is expressed from the LAT knock-in allele to allow for the accurate marking of T cells with LAT deleted. To achieve an inducible gene deletion and unified antigen specificity, we crossed ERCre and OT-I TCR transgenic mice onto a LATf/− background to generate ERCre+ LATf/− OT-I mice. LAT can be deleted in OT-I T cells in vivo by the injection of tamoxifen or in vitro by treatment with 4-OHT. LATf/− OT-I littermate mice were used as controls. For simplicity, we use “LATKO” to indicate cells from ERCre+ LATf/− OT-I mice and “WT” to describe cells from LATf/− OT-I controls after tamoxifen treatment.
We first tested whether LAT is required for CTL-mediated killing in vivo using the procedure as illustrated in Fig. 1A. OT-I cells from ERCre+ LATf/− OT-I and LATf/− OT-I mice (Thy1.2+) were adoptively transferred into Thy1.1+ Thy1.2+ mice. On the following day, these mice were infected with Listeria expressing ovalbumin to stimulate the proliferation and further differentiation of OT-I T cells. Mice without the transfer of OT-I T cells were used as controls to measure the cytotoxicity exhibited by endogenous Ova-specific T cells. Since LAT is required for T cell activation, we chose to delete LAT after the T cell expansion phase. Mice that received OT-I T cells were injected with tamoxifen on both days 7 and 8 after infection. As shown in Fig. 1B, the lat gene was efficiently deleted after tamoxifen injection as indicated by the expression of GFP in donor CD8 T cells (Thy1.2+). At day 11, 95% OT-I T cells in the mice with ERCre+ LATf/− OT-I cells were GFP+. The percentages of donor T cells were similar between LATKO and WT cells (Fig. 1C). We performed an in vivo cytotoxicity assay at day 11. Splenocytes from Thy1.1+ mice were loaded with 6 μM Cell Tracker Orange and 1 μM Ova peptide as target cells. Splenocytes without Cell Tracker labeling and the Ova peptide, which should not be killed by OT-I cells, were used as a negative control. These cells were mixed at a ratio of 1:1 and transferred into mice that had received OT-I cells. One hour after transfer, splenocytes from these mice were analyzed by FACS, and the percentage of specific killing was calculated accordingly. As shown in Fig. 1D and E, endogenous T cells could respond to Listeria infection, resulting in ∼34.8% lysis of target cells. Although 72.5% of target cells were killed in mice receiving WT OT-I cells, only 58.1% of them were lysed in mice receiving LATKO OT-I cells, indicating impaired killing by LATKO CTLs.
Fig 1.
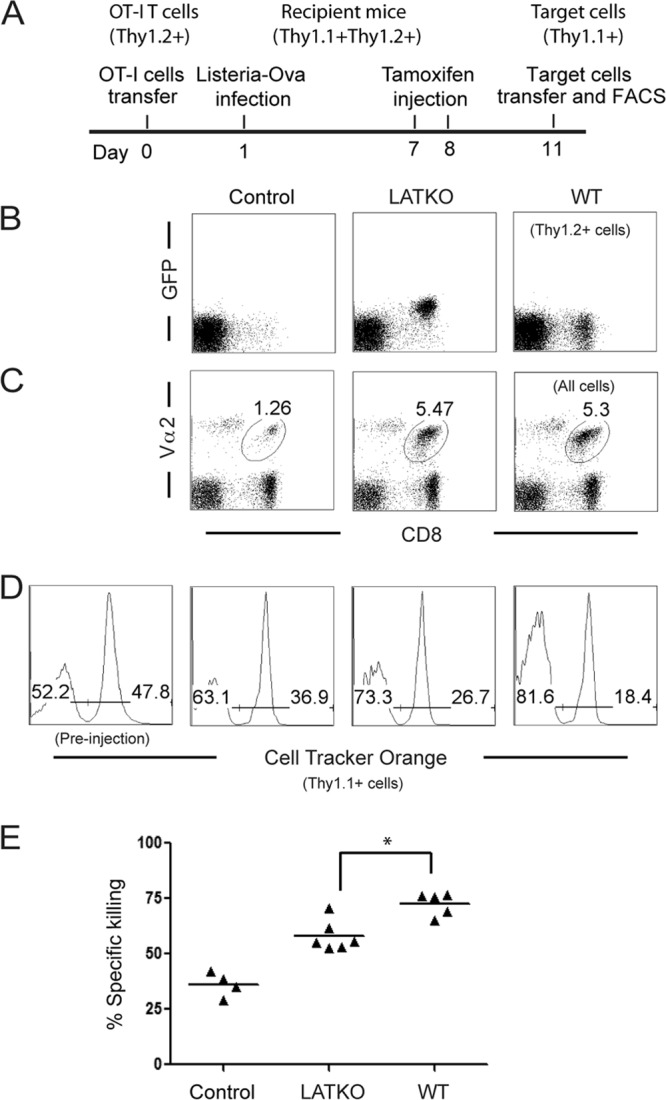
LAT in CTL cytotoxicity in vivo. (A) Timeline for in vivo killing assay. (B) Deletion efficiency of LAT. GFP expression in Thy1.2+ CD8+ T cells was analyzed by FACS. (C) Expansion of OT-I T cells. The percentage of donor CD8+ T cells (Thy1.2+) at day 11 after infection is indicated. (D) Cytotoxicity. As target cells, splenocytes from Thy1.1+ mice were loaded with 6 μM dye (Cell Tracker Orange) and 1 μM Ova peptide or left unloaded, mixed 1:1, and injected into mice that received OT-I CTLs. One hour later, splenocytes were analyzed by FACS. Plots were gated on live cells. (E) Percentage of specific killing as calculated in Materials and Methods. The data shown are one representative of two independent experiments (Number of mice used: Control, 4; LATKO, 6; WT, 5). Each dot represents one mouse. *, P < 0.05. Horizontal bars indicate the mean.
To further investigate LAT function in CTLs in vitro, splenocytes from LATKO and WT mice were cultured in the presence of 1 μM Ova peptide for 2 days to activate OT-I CTLs; this was followed by treatment with 4-OHT for an additional 3 to 5 days to delete LAT. Two days after 4-OHT treatment, more than 99% of LATKO CTLs expressed GFP (Fig. 2A). Deletion of LAT exons 7 to 11 leads to the expression of LAT-GFP fusion protein (32). To confirm LAT deletion, we used two different antibodies: one (Ab#1) that recognizes both WT and LAT-GFP proteins and the other one (Ab#2) that detects only WT LAT. At day 3, WT LAT protein was undetectable by Western blotting (Fig. 2B), indicating that deletion of lat in vitro was efficient. To further determine the efficiency of LAT deletion at the single cell level, we performed immunofluorescence staining of WT and LATKO CTLs with an anti-LAT antibody that only recognizes WT LAT. As shown in Fig. 2, LAT staining was clearly seen on the plasma membrane of WT cells, while LAT staining of LATKO cells was similar to control staining with a secondary antibody alone (Fig. 2C). Among the >500 LATKO cells we examined, there was only one cell with membrane staining (data not shown). To further confirm complete deletion of LAT protein, we stained T-APC conjugates with anti-LAT and anti-α-tubulin, which stains MTOCs, to identify T cells that formed specific conjugates. As shown in Fig. 2D, LAT was clearly concentrated in the synapses of WT cells (105 LAT foci out of 133 polarized cells). We did not find any LAT foci in LATKO cells (71 polarized cells). Together, these data indicated that LAT protein was truly absent upon 4-OHT treatment in LATKO CTLs. In all of the subsequent experiments performed, >95% of LATKO CTLs expressed GFP. We also examined the expression of the TCR and activation markers on LATKO and WT cells. TCR expression remained unchanged in LATKO cells. CD25, CD44, and CD69 were expressed similarly in LATKO and WT cells. Downregulation of CD62L was slightly impaired in LATKO cells (Fig. 2E).
Fig 2.

Deletion of LAT in vitro. Splenocytes from ERCre+ LATf/− OT-I and LATf/− OT-I mice were primed with Ova peptide for 2 days before being treated with 4-OHT for an additional 3 to 5 days. (A) Deletion efficiency of LAT in vitro. GFP expression in CD8+ T cells was analyzed by FACS. (B) Absence of LAT protein in LATKO CTLs. WT and LAT KO lysates were analyzed by Western blotting using two polyclonal antibodies: Ab#1 recognizes the entire cytoplasmic domain, while Ab#2 only recognizes a peptide that is located in the deleted region. (C) The absence of LAT at the single cell level. CTLs (WT and LATKO) were fixed, permeabilized, and stained with anti-LAT antisera and Cy3-conjugated anti-rabbit secondary antibody. The negative control was LATKO staining with the secondary antibody only. (D) The absence of LAT at the synapse of LATKO CTLs. CTLs were incubated with L1210/Kb cells (green) preloaded with the Ova peptide for 30 min to allow for the formation of conjugates. Conjugates were stained with anti-α-tubulin to represent both MTOC (white) and anti-LAT antibody (red). (E) TCR expression and activation markers on CTLs. WT and LATKO CTLs were stained with anti-TCRβ, anti-CD25, anti-CD69, anti-CD62L, and anti-CD44 antibodies and analyzed by FACS. The data shown are one representative of three independent experiments.
To examine whether LAT deficiency affects CTL effector function, we first examined the cytotoxicity of LATKO CTLs in vitro using the EL4 cell line as target cells. EL4 cells were loaded with Cell Tracker Orange and Ova peptide. Consistent with the reduced killing observed in vivo, LATKO CTLs clearly showed a diminished cytotoxic capability toward the peptide-loaded EL4 cells (Fig. 3A). We then analyzed IFN-γ production upon stimulation with EL4 cells with or without the Ova peptide. Compared to WT cells, LATKO CTLs failed to produce significant amounts of IFN-γ (Fig. 3B). In addition to IFN-γ pathway, CTLs can also kill target cells through the Fas-mediated pathway in which triggering of the Fas pathway in target cells relies upon the upregulation of FasL on T cells. FasL was upregulated on WT CTLs upon re-encountering target cells. However, this upregulation was largely reduced in LATKO CTLs (Fig. 3B). To further determine whether granule-mediated killing is affected by LAT deficiency, we used a Fas-deficient cell line, L1210 expressing H2-Kb, as target cells. The cytotoxicity of LATKO CTLs was significantly decreased compared to WT CTLs, indicating that LAT is also important in granule-mediated killing (Fig. 3C).
Fig 3.

LAT in CTL cytotoxicity in vitro. (A) In vitro cytotoxicity against EL4 target cells. The data shown are from one representative of five independent experiments. (B) IFN-γ production and FasL upregulation. CTLs were restimulated by EL4 cells preloaded with (open) or without (filled) Ova peptide for 5 h. Intracellular staining of IFN-γ and surface expression of FasL on CD8+ cells were analyzed by FACS. The data are representative of three independent experiments. (C) In vitro cytotoxicity against L1210/Kb target cells. The data shown are from one representative of three independent experiments. (D) Granzyme B (GzmB) and perforin expression by Western blotting. The data are representative of three independent experiments. (E) Delivery of granules. L1210/Kb cells were labeled with Cell Tracker Orange with or without Ova peptide. CTLs were incubated with L1210/Kb cells in the presence of a cell-permeable fluorescein isothiocyanate-conjugated gzmB substrate for 40 min. The numbers in the figure indicate the percentage of L1210/Kb cells that received gzmB from CTLs. The data are representative of two independent experiments. n.s., not significant; *, P < 0.05; **, P < 0.005. Error bars represent means ± the standard errors of the mean (SEM).
Next, we examined the expression of perforin and gzmB, two effector molecules that are crucial for granule-mediated killing. The expression of gzmB was comparable between LATKO and WT CTLs, whereas perforin expression was slightly reduced in LATKO CTLs (Fig. 3D). To determine whether the granules in LATKO CTLs were delivered to target cells, we performed a FACS-based assay using a cell-permeable fluorogenic substrate of gzmB. This substrate can be cleaved in target cells in the presence of gzmB, which leads to the emission of fluorescent light for quantification of gzmB activity. Although gzmB activity was still detected in target cells incubated with LATKO CTLs, the intensity was much lower than the WT CTL-treated group (Fig. 3E). Altogether, these data demonstrated that LAT regulates IFN-γ production, FasL upregulation, and granule delivery.
LAT in the formation of the immunological synapse.
Our data showed that LATKO CTLs were capable of delivering granules to target cells albeit at a reduced level. Granule-dependent cytotoxicity of T cells involves several critical steps: conjugate formation, synapse formation, MTOC reorientation, granule polarization, and secretion. We sought to examine the roles of LAT in each step of CTL granule exocytosis. We first determined the efficiency of conjugate formation between LATKO CTLs and target cells. CTLs and target cells were labeled with two different dyes. Upon the formation of CTL-target cell conjugates, CTLs should stain for both dyes. As shown in Fig. 4A, LATKO and WT CTLs showed similar kinetics in the formation of conjugates at early time points and both peaked at 20 min, indicating that LATKO CTLs are capable of forming proper conjugates with target cells. At 20 min after mixing, the T cells gradually dissociated from target cells before or after killing, leading to a decline in the number of conjugates. For WT CTLs, 30.4, 24.6, and 13.7% of the conjugates were maintained at 30, 45, and 60 min, respectively. However, only 18.9, 9, and 3.3% of the conjugates were seen for LATKO CTLs at those time points, suggesting that LAT is required for the maintenance of stable conjugates. These data indicated that the initial conjugate formation between CTLs and APCs is LAT independent; however, LAT is necessary for the formation of stable conjugates that remain intact for an extended period of time.
Fig 4.

Formation of CTL-target cell conjugates and the immunological synapse in LATKO CTLs. (A) Conjugate formation. CTLs were labeled with Cell Tracker Violet and mixed at a ratio of 1:1 with L1210/Kb cells labeled with Cell Tracker Orange with or without Ova peptide. At various time points, the cells were fixed and analyzed by FACS. CTL-target cell conjugates were defined as cells labeled with both fluorescent dyes. The graph shows one representative of three independent experiments. *, P < 0.05. Error bars represent means ± the SEM. (B) Quantitative analysis of cSMAC accumulation at the synapse of WT and LATKO CTLs. CTLs were incubated with L1210/Kb cells preloaded with the Ova peptide for 15 and 30 min to allow for the formation of conjugates. The conjugates were fixed, permeabilized, and stained with anti-PKCθ antibody to represent cSMAC and anti-LFA1 antibody to represent pSMAC. cSMAC accumulation was calculated as the ratio of the synaptic fluorescence intensity to the cytoplasmic fluorescence intensity. Each dot represents one CTL. Horizontal bars indicate the mean. n.s., not significant; **, P < 0.005. The data shown are from two independent experiments. (C and D) Immunological synapse patterns. The representative cells depicted here show the side and enface of cSMAC (C) and excluded pSMAC (D) of WT and LATKO CTLs.
Previous studies demonstrate that synapse maintenance requires continuous TCR-mediated signaling (11). Therefore, next we examined whether synapse formation is affected by LAT deletion. We chose PKCθ as a marker for the cSMAC and LFA-1 as a marker for the pSMAC (23). Conjugates were examined in the xy and xz planes to view the synapse from the side and enface, respectively. To evaluate the efficiency of cSMAC formation in the synapse, we calculated the accumulation of cSMAC as the ratio of the fluorescence intensity at the synapse to the fluorescence intensity in the cytoplasm at different time points. The intensity of cSMAC accumulation at the synapse of LATKO (average = 2.35, n = 46) and WT CTLs (average = 2.49, n = 44) was comparable 15 min after T cell-target cell engagement, indicating that cSMACs can form in the absence of LAT (Fig. 4B). Notably, by 30 min, the intensity of cSMAC accumulation significantly decreased in LATKO CTLs (average = 1.45, n = 34) compared to WT CTLs (average = 2.06, n = 40) (Fig. 4B). Further analysis of cSMAC fluorescence at the synapse of LATKO CTLs revealed a phenotype distinguished by multiple centers (Fig. 4C), indicating that synapse formation was altered by LAT-deficiency. We further examined pSMAC exclusion from the cSMAC. The percentage of pSMAC exclusion was similar between LATKO and WT CTLs (Fig. 4D, data not shown). These observations indicated that LATKO CTLs can form a mature immunological synapse; however, LAT is essential for the maintenance of a stable synapse.
LAT function in MTOC reorientation and granule movement.
The synaptic release of lytic granules in CTLs is guided by the reorientation of the MTOC. We next examined whether the MTOC in LATKO CTLs could be polarized toward the synapse upon TCR engagement. OT-I CTLs were mixed with Ova peptide-loaded L1210/Kb cells, fixed, and stained with anti-α tubulin and anti-gzmB to identify MTOCs and granules, respectively. To quantitate MTOC polarization, we evenly divided conjugated T cells into four zones as described previously (28) (Fig. 5A). In WT CTLs, MTOCs gradually moved to zone 1 after interacting with target cells, increasing from 26.7 to 72.5 to 81% at 15, 30, and 45 min, respectively (n ≥ 60 for each group, Fig. 5B). In contrast, the percentage of LATKO CTLs with MTOCs recruited to zone 1 was 8.9, 31.6, and 10.6% at the above time points (Fig. 5B). These results indicated that TCR-mediated synaptic MTOC reorientation in LATKO CTLs was inefficient and that this polarization was unstable.
Fig 5.

Impaired MTOC polarization and granule movement in LATKO CTLs. CTL-target cell conjugates were stained with anti-α tubulin and anti-gzmB to represent MTOCs (white) and granules (green), respectively. (A) The representative conjugates depicted here show MTOCs and granules localized at zones 1 to 4 in CTLs. (B) Quantitative analysis of MTOC localization in different zones of CTLs upon engagement of L1210/Kb cells for 15, 30, and 45 min. (C) Granule movement is controlled by MTOC polarization. The percentage of granules in zone 1 was quantitated in each CTL with MTOCs at different zones at 30 min. Each dot represents one CTL, and the value represents the percentage of granules localized to zone 1; horizontal bars indicate the mean (LATKO, n = 48; WT, n = 52). The data shown are from two independent experiments.
We also quantitated the percentage of granules located in zone 1 of CTLs to determine whether granule movement is controlled by the MTOC in LATKO CTLs. As shown in Fig. 5C, LATKO CTLs (n = 48) had a similar pattern of granule distribution related to MTOC localization as WT CTLs (n = 52) at 30 min. In CTLs with MTOCs immediately beneath the synapse (zone 1), the average percentage of granules in zone 1 was ∼59% (33 to 92%, n = 20) in LATKO CTLs and ∼63% (28 to 100%, n = 39) in WT CTLs. In CTLs with MTOCs partially polarized to the synapse (zone 2), the average percentage of granules in zone 1 was 41% (33.5 to 55.8%, n = 7) in LATKO CTLs and 31% (3.8 to 58.5%, n = 10) in WT CTLs. In CTLs with proximal and distal MTOC localization (zones 3 and 4), the average percentage of granules in zone 1 was 3.9% (0 to 23%, n = 21) in LATKO CTLs and 1.7% (0 to 5%, n = 3) in WT CTLs. Granules remained distal to the synapse if MTOCs failed to polarize in LATKO CTLs. These results suggested that granule movement was also controlled by MTOC reorientation in LATKO CTLs. We also performed live imaging to observe the movement of granules in T cells (see the videos in the supplemental material). CTLs were loaded with LysoTracker red, which stains granules, while EL4 cells loaded with Ova peptide were labeled with calcein, a dye that is retained in live cells and lost upon membrane permeabilization. Following CTL-target cell contact, the lytic granules in WT CTLs reoriented toward and were subsequently delivered into the target cells. In contrast, the granules in some LATKO CTLs remained distal to the synapse for more than 10 min, resulting in a failure of granule delivery to the target cell. Collectively, these data suggested that LAT functions to promote MTOC reorientation and, consequently, granule movement toward the synapse.
TCR-mediated signaling in LAT-deficient CTLs.
Our data demonstrated that, although LAT plays a significant role in each step of granule exocytosis, LATKO CTLs were still able to kill target cells. To analyze the TCR-mediated signaling events that regulate this residual cytotoxicity, we performed biochemical analyses of TCR-mediated signaling pathways in LATKO CTLs. LATKO and WT CTLs were stimulated with anti-CD3 plus anti-CD8 to assess the overall tyrosine phosphorylation of proteins. No significant difference was observed except that phosphorylated LAT was absent in LATKO CTLs (Fig. 6A). The lack of phosphorylated LAT was also supported by Western blotting with anti-pY191 of LAT (Fig. 6B), further confirming complete deletion of LAT. PLC-γ1 and MAPK activation, two important signaling events downstream of LAT, were dramatically diminished, as predicted, though they were not absent in LATKO CTLs. In addition, the activation of Akt, detected by both anti-pSer473 and anti-pThr308, was also reduced but not to the same extent as PLC-γ. Interestingly, PKCθ activation was largely intact.
Fig 6.

Defective TCR signaling in LATKO CTLs. (A and B) TCR-mediated protein phosphorylation. CTLs were restimulated with anti-CD3 (5 μg/ml) and anti-CD8 (1 μg/ml) for the indicated time points before lysis. Cell lysates were analyzed by Western immunoblotting with anti-pTyr, phospho-specific, or pan antibodies against LAT, Erk, PLC-γ1, PKCθ, and Akt. The data shown are from one representative of three independent experiments. (C) Calcium mobilization. CTLs were loaded with the calcium indicator, indo-1. Calcium flux was induced by anti-CD3 and anti-CD8 cross-linking and analyzed by FACS. The data are representative of three independent experiments. (D) Time-lapse montages of both ratiometric Fura-2 images and the differential interference contrast of CTLs. CTLs were preloaded with Fura-2, mixed with Ova peptide-loaded L1210/Kb cells, and imaged at intervals of 30 s. The first time point (0′00″) marked the initial contact of a T cell with a target cell. Fura images are pseudo-colored, with warmer colors indicating higher intracellular calcium concentrations. Upper rows, WT CTLs; bottom rows, LATKO CTLs. (E) Quantitative analysis of calcium flux by live cell imaging. CTLs were pretreated with or without CTLA4-Ig and anti-ICAM-1 antibody for 30 min before incubation with target cells. The integrated value of calcium mobilized into the cytosol for each cell was generated by summing the changes in the relative calcium ratio (340/380 nm) at each time point within the first 10 min. Each dot represents one CTL. Horizontal bars indicate the mean. n.s., not significant; *, P < 0.05, **, P < 0.005. The data shown are from two independent experiments. (F) Calcium mobilization in the presence of U73122 (0.5 μM). (G) Effect of PLC inhibitor on CTL-mediated killing. CTLs were pretreated with U73122 (0.5 μM) or dimethyl sulfoxide (DMSO) for 30 min and then incubated with target cells in the presence of these inhibitors for 45 min before FACS analysis. The data shown are from one representative of three independent experiments.
Calcium flux is another downstream event, known to be critical for granule-mediated cytotoxicity as blockade of calcium completely inhibits granule release by CTLs (20). Previous studies show that in the absence of LAT, TCR-mediated calcium flux is abrogated in Jurkat T cells, thymocytes, and naive T cells (7, 32, 37). Therefore, we also examined TCR-mediated calcium flux in LATKO CTLs. CTLs were loaded with indo-1 and calcium flux was initiated by anti-CD3 and anti-CD8 cross-linking. As shown in Fig. 6C, calcium flux in GFP+ LATKO CTLs was dramatically diminished but not abrogated. To analyze calcium signaling at the single cell level, CTLs were loaded with Fura-2 and mixed with Ova peptide-loaded L1210/Kb cells for live cell imaging. Consistent with the FACS data (Fig. 6C), there was a detectable increase of Fura-2 fluorescence in LATKO CTLs, although it was much weaker than that seen in WT CTLs (Fig. 6D). We also quantitated Fura-2 fluorescent intensity in these CTLs. As shown in Fig. 6E, the integrated value of calcium mobilization into the cytosol in the first 10 min upon T-target cell interaction was much lower in LATKO CTLs (∼3, n = 24) than that in WT CTLs (∼8, n = 19). This residual calcium flux could be a result of costimulation or adhesion signaling (33). To examine this possibility, we pretreated target cells with CTLA4-Ig and anti-ICAM-1 antibody before imaging. With the blockade, calcium flux was significantly decreased in WT CTLs (∼5, n = 27) (Fig. 6E). However, it remained unchanged in LATKO CTLs (∼3, n = 24), suggesting that the residual calcium was not mediated by CD28 or LFA-1-mediated signaling.
These results suggested that upon engagement with target cells, LATKO CTLs were capable of mobilizing a small amount of intracellular calcium, which might be sufficient to allow for their residual cytotoxicity. To further determine whether this residual calcium mobilization is important for cytotoxicity by LATKO CTLs, we examined the killing capability of CTLs in the presence of U73122, a pharmacological inhibitor of PLC, a molecule upstream of calcium flux. CTLs were pretreated with the inhibitor for only 30 min to minimize toxicity and then used in the killing assay. At the concentration used (0.5 μM), U73122 abrogated calcium flux in LATKO CTLs (Fig. 6G); the percentage of killing by LATKO CTLs and WT CTLs was reduced by 77 and 67%, respectively. Inhibition by U73122 was dose-dependent (data not shown). Together, our data showed that TCR-mediated signaling was largely diminished by LAT deficiency; however, the residual activation of PLC-γ1 and calcium signaling enabled LATKO CTLs to kill target cells.
DISCUSSION
LAT plays an integral function in the complex network of TCR signaling. In this study, we investigated the function of LAT in CTLs by using an inducible deletion system. Our data revealed that the cytotoxicity triggered by LATKO CTLs was reduced compared to WT CTLs. LATKO CTLs exhibited multiple defects in effector functions, including IFN-γ production and FasL upregulation. In addition, every step of granule exocytosis was also affected by LAT-deficiency, such as immunological synapse maintenance and MTOC polarization. Importantly, these defects are not caused by reduced expression of the TCR at the cell surface, which is a phenomenon previously observed with long-term deletion of LAT (22, 32). In the present study, deletion of LAT by treatment with tamoxifen for less than a week did not cause any change in TCR surface expression. These defects are most likely due to the inactivation of downstream effector molecules. For examples, integrin, an adhesion molecule enriched in the pSMAC, is maintained in an inactive conformation with a low affinity for its ligand. “Inside-out” signals that originate upon engagement of the TCR or chemokine receptors result in conformational changes of integrins, thereby increasing their affinity (36). Our results showed that the conjugate maintenance and synapse stability between T cells and target cells was affected by LAT deficiency. It is possible that impaired inside-out signaling in LATKO CTLs failed to activate signaling molecules, such as Rap1, which associates with ADAP and SKAP55, to induce conformational changes of LFA-1 (21, 33). It is also possible that the activity of myosin IIA, an actin-based motor protein that is essential for the movement of microclusters and synapse persistence (13), is affected by LAT deficiency.
Deletion of LAT completely blocks thymocyte development and T cell activation (32, 39). Interestingly, although LATKO CTLs demonstrated multiple defects in cytotoxicity, their granule release was not totally blocked (∼30% granule release). One possibility is that there was a very small amount of undeleted LAT, which was sufficient to trigger the residual CTL killing. Our knock-in strategy allows us to accurately mark T cells with LAT exons deleted since LAT-GFP fusion protein is expressed from the LAT knock-in allele. As such, GFP+ CTLs should all have successfully deleted the LAT gene. We sorted out GFPhigh LATKO cells and assayed for cytotoxicity. The killing capability of the sorted cells was comparable to that of nonsorted ones (data not shown). Our Western blot analysis and immunofluorescence staining of LATKO CTLs showed undetectable levels of LAT or phospho-LAT, suggesting that in addition to gene deletion, there was no residual LAT protein in these cells. In addition, all our analyses were performed 3 to 5 days after tamoxifen treatment. At this time, it is unlikely that there was any LAT protein remaining without new protein synthesized. Nevertheless, despite that no LAT protein was detected in LATKO CTLs, it is difficult to prove experimentally that there is absolutely no LAT in these cells. Regardless, it is intriguing to observe that CTLs with extremely weak TCR signaling, as shown in our biochemical and calcium data, still retained cytotoxic ability (∼25% compared to WT cells). Given that T cell activation and interferon production were nearly abolished under the same inducible LAT deletion system, this finding suggested that the signaling strength governing CTL cytotoxicity differs from that required for other effector functions.
When a CTL encounters a target cell, a mature immunological synapse is formed at the CTL-target contact zone, allowing the MTOC and granules to reorient and move toward the target cell. It has been shown that continuous TCR-mediated signaling is required for stabilizing cSMAC and pSMAC organization into the synapse, as well as polarizing the MTOC toward the synapse (11). Our conjugation assay showed that the initial conjugate formation between CTLs and target cells was not affected by LAT deficiency; however, the conjugates formed between target cells and LATKO CTLs were not as stable as those formed with WT CTLs. Our synapse data also showed that LAT deficiency affected synapse maintenance in CTLs. In addition, MTOC polarization in CTLs was unstable and inefficient in the absence of LAT, possibly due to the weak stability of conjugate and synapse formation. These observations suggested that mature synapses and polarized MTOCs could form without LAT; however, the maintenance of stable conjugates and synapses requires LAT-mediated signaling. An obvious question to be asked is that, with all of these defects, why are LAT-deficient CTLs still able to kill target cells? Naturally, CTLs executed an efficient killing process by forming relatively transient conjugates (in comparison to CD4 T cells) with target cells, delivering granules, and then quickly dissociating from target cells. This phenomenon was described at a quantitative level by Mark Davis's group. They show that only 3 Ova peptides are needed in the synapse to elicit OT-I T cell killing; however, ∼10 peptides are required to form a stable, mature immunological synapse (27). In contrast to CD4 helper T cells, which must have a stable synapse to maximize their proliferation and cytokine secretion, it might be an intrinsic advantage for CTLs to have brief contact with target cells. This transient interaction enables them to switch back to the hunting phase for the next target after each successful mission. This could explain why, although LATKO CTLs formed unstable conjugates and synapses with target cells, some lytic granules were delivered before CTLs dissociated from target cells.
Previous studies using inhibitors indicate that PI3K activity is critical for the cytotoxicity of CTLs and NK cells (8, 29). Our biochemical data showing that Akt phosphorylation was only slightly reduced in LATKO cells indicated that PI3K can be activated in the absence of LAT, although LAT is necessary for its optimal activation. A previous report also demonstrated the activation of PI3K/Akt in the absence of LAT in activated CD4 T cells (22). Even though LAT does not have a canonical p85-binding motif, phosphorylated LAT has been shown to bind the p85 subunit of PI3K (38). p85 also binds the cytoplasmic domain of CD28. However, the role of CD28-mediated PI3K activation in CTLs is limited. CTLs are able to eliminate target cells that do not express B7 molecules, indicating that T cell killing does not require the interaction of CD28 with B7 molecules. Blockade of CD28-mediated costimulation by CTLA4-Ig had no effect on the cytotoxic ability of LATKO CTLs (data not shown), further supporting a limited role of CD28 in CTL killing. Therefore, it is unlikely that CD28 signaling can be counted as the major source providing LAT-independent PI3K activation. It is unclear how PI3K is activated in the absence of LAT after TCR engagement. One possibility is that PI3K binds to adaptor proteins that contain YXXM motifs, such as TRIM (TCR-interacting molecule). It is also possible that p85 can be recruited to the membrane through the interaction between its proline-rich region and the SH3 domains of Lck or Fyn (24).
PLC-γ1-mediated signaling has been implicated in regulating the molecular mechanisms of granule exocytosis (4). Our data showed that PLC-γ1 phosphorylation and activity, albeit diminished, was still detected in LATKO CTLs and that this residual PLC activity appears to be important for cytotoxicity. Blockade of PKC or MAPK activity, the signaling pathways downstream of DAG, had only minor effects on the killing ability of LATKO CTLs (data not shown), suggesting that the residual cytotoxicity mediated by LATKO CTLs relies heavily on calcium signaling. Our data further demonstrated that inhibition of PLC by U73122 completely abrogated the residual calcium flux in LATKO CTLs and abolished their cytotoxicity. This provides a cohesive explanation that the residual PLC activity is sufficient to generate a minimal amount of cytosolic calcium elevation, which is sufficient to support the observed CTL killing.
Calcium signaling is directly involved in lytic granule release. One study determined that granule movement can occur in Ca2+-free medium; however, the release of granules and the killing of target cells can only happen after the addition of calcium (20). Whether intracellular Ca2+ regulates MTOC polarization has been a controversial issue. The minus end-directed motor protein dynein, which can bind calmodulin, has been shown to function in MTOC orientation in Jurkat cells (3). Kuhne's data demonstrated a correlation between calcium flux and MTOC polarization in Jurkat cells (18). However, recent data using single-cell photoactivation of the TCR reveal a different result where blocking calcium signaling has no effect on MTOC reorientation (28). Our data here show that, although TCR-mediated calcium mobilization was severely impaired in LATKO CTLs, their MTOC reorientation was only partially affected. The residual activity of PLC in LATKO CTLs might generate a small amount of calcium flux that is sufficient to activate downstream signaling molecules, including classical and novel PKC, protein kinase D, and the chimaerins. This is supported by our data showing that, in LATKO CTLs, the activity of PKCθ, a downstream signaling molecule of DAG, was relatively normal. Early studies using LAT-deficient Jurkat cells indicate that LAT is crucial for TCR-mediated PLCγ1 activation and calcium mobilization. It is not clear why TCR-mediated PLC-γ1 activation and calcium flux are not totally dependent on LAT in CTLs. It is likely that other molecules, such as LAB, exist in these OT-I CTLs to compensate for the loss of LAT. Our unpublished data showed that LAB is indeed expressed in CTLs. Whether it functions in these cells remains to be investigated using LAB- or LAT/LAB-deficient mice. It is also possible that residual PLC-γ1 activation and calcium flux are initiated through other signaling components, such as adhesion molecules and growth factors. LAT-independent Ras-MAPK activation has been documented previously (2). Similarly, ERK activation was observed in ZAP-70-deficient Jurkat cells (31). PLC-γ1 might be recruited to the plasma membrane through molecules other than LAT or other LAT-independent PLC isozymes may compensate for the residual activity of PLC-γ1.
In conclusion, our data demonstrate that in the absence of LAT, CTLs failed to produce IFN-γ and upregulate FasL, as well as exhibiting partial defects at each step of granule-mediated cytotoxicity. All of these results indicated the importance of LAT in CTL function. However, LATKO CTLs were still able to kill target cells at a reduced level, suggesting that CTLs may be programmed to adopt multiple pathways to perform their cytolytic function.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Duke University Cancer Center Flow Cytometry, DNA Sequencing, and Transgenic Mouse facilities for their excellent services.
This study was supported by National Institutes of Health grants AI048674 and AI056156.
Footnotes
Published ahead of print 7 May 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Beal AM, et al. 2009. Kinetics of early T cell receptor signaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity 31:632–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chau LA, Madrenas J. 1999. Phospho-LAT-independent activation of the Ras-mitogen-activated protein kinase pathway: a differential recruitment model of TCR partial agonist signaling. J. Immunol. 163:1853–1858 [PubMed] [Google Scholar]
- 3. Combs J, et al. 2006. Recruitment of dynein to the Jurkat immunological synapse. Proc. Natl. Acad. Sci. U. S. A. 103:14883–14888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Saint Basile G, Menasche G, Fischer A. 2010. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat. Rev. Immunol. 10:568–579 [DOI] [PubMed] [Google Scholar]
- 5. Ebinu JO, et al. 2000. RasGRP links T-cell receptor signaling to Ras. Blood 95:3199–3203 [PubMed] [Google Scholar]
- 6. Esser MT, Haverstick DM, Fuller CL, Gullo CA, Braciale VL. 1998. Ca2+ signaling modulates cytolytic T lymphocyte effector functions. J. Exp. Med. 187:1057–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Finco TS, Kadlecek T, Zhang W, Samelson LE, Weiss A. 1998. LAT is required for TCR-mediated activation of PLCγ1 and the Ras pathway. Immunity 9:617–626 [DOI] [PubMed] [Google Scholar]
- 8. Fuller CL, Ravichandran KS, Braciale VL. 1999. Phosphatidylinositol 3-kinase-dependent and -independent cytolytic effector functions. J. Immunol. 162:6337–6340 [PubMed] [Google Scholar]
- 9. Grakoui A, et al. 1999. The immunological synapse: a molecular machine controlling T cell activation. Science 285:221–227 [DOI] [PubMed] [Google Scholar]
- 10. He JS, Ostergaard HL. 2007. CTLs contain and use intracellular stores of FasL distinct from cytolytic granules. J. Immunol. 179:2339–2348 [DOI] [PubMed] [Google Scholar]
- 11. Huppa JB, Gleimer M, Sumen C, Davis MM. 2003. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat. Immunol. 4:749–755 [DOI] [PubMed] [Google Scholar]
- 12. Huse M, Quann EJ, Davis MM. 2008. Shouts, whispers and the kiss of death: directional secretion in T cells. Nat. Immunol. 9:1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ilani T, Vasiliver-Shamis G, Vardhana S, Bretscher A, Dustin ML. 2009. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat. Immunol. 10:531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jenkins MR, Tsun A, Stinchcombe JC, Griffiths GM. 2009. The strength of T cell receptor signal controls the polarization of cytotoxic machinery to the immunological synapse. Immunity 31:621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jordan MS, Singer AL, Koretzky GA. 2003. Adaptors as central mediators of signal transduction in immune cells. Nat. Immunol. 4:110–116 [DOI] [PubMed] [Google Scholar]
- 16. Juszczak RJ, Russell JH. 1989. Inhibition of cytotoxic T lymphocyte-mediated lysis and cellular proliferation by isoquinoline sulfonamide protein kinase inhibitors. Evidence for the involvement of protein kinase C in lymphocyte function. J. Biol. Chem. 264:810–815 [PubMed] [Google Scholar]
- 17. Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. 1996. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu. Rev. Immunol. 14:207–232 [DOI] [PubMed] [Google Scholar]
- 18. Kuhne MR, et al. 2003. Linker for activation of T cells, zeta-associated protein-70, and Src homology 2 domain-containing leukocyte protein-76 are required for TCR-induced microtubule-organizing center polarization. J. Immunol. 171:860–866 [DOI] [PubMed] [Google Scholar]
- 19. Liu SK, McGlade CJ. 1998. Gads is a novel SH2 and SH3 domain-containing adaptor protein that binds to tyrosine-phosphorylated Shc. Oncogene 17:3073–3082 [DOI] [PubMed] [Google Scholar]
- 20. Lyubchenko TA, Wurth GA, Zweifach A. 2001. Role of calcium influx in cytotoxic T lymphocyte lytic granule exocytosis during target cell killing. Immunity 15:847–859 [DOI] [PubMed] [Google Scholar]
- 21. Menasche G, Kliche S, Bezman N, Schraven B. 2007. Regulation of T-cell antigen receptor-mediated inside-out signaling by cytosolic adapter proteins and Rap1 effector molecules. Immunol. Rev. 218:82–91 [DOI] [PubMed] [Google Scholar]
- 22. Mingueneau M, et al. 2009. Loss of the LAT adaptor converts antigen-responsive T cells into pathogenic effectors that function independently of the T cell receptor. Immunity 31:197–208 [DOI] [PubMed] [Google Scholar]
- 23. Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. 1998. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 395:82–86 [DOI] [PubMed] [Google Scholar]
- 24. Okkenhaug K, Vanhaesebroeck B. 2003. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 3:317–330 [DOI] [PubMed] [Google Scholar]
- 25. Pardo J, et al. 2003. Differential implication of protein kinase C isoforms in cytotoxic T lymphocyte degranulation and TCR-induced Fas ligand expression. Int. Immunol. 15:1441–1450 [DOI] [PubMed] [Google Scholar]
- 26. Potter TA, Grebe K, Freiberg B, Kupfer A. 2001. Formation of supramolecular activation clusters on fresh ex vivo CD8+ T cells after engagement of the T cell antigen receptor and CD8 by antigen-presenting cells. Proc. Natl. Acad. Sci. U. S. A. 98:12624–12629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. 2004. T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 5:524–530 [DOI] [PubMed] [Google Scholar]
- 28. Quann EJ, Merino E, Furuta T, Huse M. 2009. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat. Immunol. 10:627–635 [DOI] [PubMed] [Google Scholar]
- 29. Robertson LK, Mireau LR, Ostergaard HL. 2005. A role for phosphatidylinositol 3-kinase in TCR-stimulated ERK activation leading to paxillin phosphorylation and CTL degranulation. J. Immunol. 175:8138–8145 [DOI] [PubMed] [Google Scholar]
- 30. Russell JH, Ley TJ. 2002. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 20:323–370 [DOI] [PubMed] [Google Scholar]
- 31. Shan X, et al. 2001. Zap-70-independent Ca2+ mobilization and Erk activation in Jurkat T cells in response to T-cell antigen receptor ligation. Mol. Cell. Biol. 21:7137–7149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shen S, Zhu M, Lau J, Chuck M, Zhang W. 2009. The essential role of LAT in thymocyte development during transition from the double-positive to single-positive stage. J. Immunol. 182:5596–5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith-Garvin JE, Koretzky GA, Jordan MS. 2009. T cell activation. Annu. Rev. Immunol. 27:591–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stinchcombe JC, Griffiths GM. 2007. Secretory mechanisms in cell-mediated cytotoxicity. Annu. Rev. Cell Dev. Biol. 23:495–517 [DOI] [PubMed] [Google Scholar]
- 35. Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. 2006. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 443:462–465 [DOI] [PubMed] [Google Scholar]
- 36. Takagi J, Petre BM, Walz T, Springer TA. 2002. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110:599–611 [DOI] [PubMed] [Google Scholar]
- 37. Zhang W, Irvin BJ, Trible RP, Abraham RT, Samelson LE. 1999. Functional analysis of LAT in TCR-mediated signaling pathways using a LAT-deficient Jurkat cell line. Int. Immunol. 11:943–950 [DOI] [PubMed] [Google Scholar]
- 38. Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE. 1998. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92:83–92 [DOI] [PubMed] [Google Scholar]
- 39. Zhang W, et al. 1999. Essential role of LAT in T cell development. Immunity 10:323–332 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.