

Abstract
RANK ligand (RANKL), by mechanisms unknown, directly activates osteoclasts to resorb bone. Because c-Src is key to organizing the cell's cytoskeleton, we asked if the tyrosine kinase also mediates RANKL-stimulated osteoclast activity. RANKL induces c-Src to associate with RANK369–373 in an αvβ3-dependent manner. Furthermore, RANK369–373 is the only one of six putative TRAF binding motifs sufficient to generate actin rings and activate the same cytoskeleton-organizing proteins as the integrin. While c-Src organizes the cell's cytoskeleton in response to the cytokine, it does not participate in RANKL-stimulated osteoclast formation. Attesting to their collaboration, αvβ3 and activated RANK coprecipitate, but only in the presence of c-Src. c-Src binds activated RANK via its Src homology 2 (SH2) domain and αvβ3 via its SH3 domain, suggesting the kinase links the two receptors. Supporting this hypothesis, deletion or inactivating point mutation of either the c-Src SH2 or SH3 domain obviates the RANK/αvβ3 association. Thus, activated RANK prompts two distinct signaling pathways; one promotes osteoclast formation, and the other, in collaboration with c-Src-mediated linkage to αvβ3, organizes the cell's cytoskeleton.
INTRODUCTION
Osteoclasts are unique bone resorptive cells, necessary for maintaining skeletal homeostasis. A relative excess of their activity however, often eventuates in the common disorder osteoporosis. Thus, understanding the means by which osteoclasts resorb bone carries both physiological and clinical significance.
The magnitude of skeletal resorption reflects osteoclast number and the matrix-degrading capacity of the individual cell. Osteoclast abundance is governed by macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor κB ligand (RANKL) interacting with their respective receptors, c-fms and RANK, on monocyte/macrophage precursors. In fact, the two cytokines, in combination, are sufficient for osteoclast formation in vitro and in vivo.
The capacity of the mature osteoclast to resorb bone is largely dictated by cytoskeletal organization. Upon contact with mineralized substrate, the cell polarizes its resorptive machinery to its interface with bone and secretes skeleton-degrading molecules into a microenvironment isolated from the general extracellular space by “gasket-like” structures known as actin rings or sealing zones. The ability of osteoclasts to optimally resorb bone depends upon actin ring formation (reviewed in reference 25).
In 1991, Soriano et al. observed that the principal phenotype of c-Src-deleted mice resides in the skeleton (24). These animals develop severe osteopetrosis despite an abundance of osteoclasts. Thus, the disorder does not reflect failed osteoclast recruitment but defective function, specifically disorganization of the actin cytoskeleton (2). In fact, c-Src is the dominant Src family kinase expressed in the osteoclast and regulates the cell's cytoskeleton as an adaptor protein and through its enzymatic activity (7, 19, 23).
The integrin αvβ3 is a key regulator of the osteoclast cytoskeleton but does not promote osteoclastogenesis (18). Upon αvβ3 occupancy, c-Src is activated, leading to phosphorylation of Syk, whose recruitment to the integrin-stimulated signaling complex requires the costimulatory ITAM protein, Dap12 (29). Along with the adaptor, Slp-76, activated Syk phosphorylates the relatively osteoclast-specific guanine nucleotide exchange factor (GEF) Vav3, which transits the Rho GTPase, Rac, from its inactive GDP- to its active GTP-bound form (8, 9, 22). By impacting the Arp2/3 complex, Rac organizes the osteoclast cytoskeleton, including efficient actin ring configuration (6). Deletion of any of the signaling molecules in the αvβ3-activated sequence compromises the ability of osteoclasts to remodel their cytoskeleton and resorb bone.
During these studies, we found that M-CSF, which promotes proliferation of osteoclast precursors and inhibits apoptosis of the mature polykaryon, also organizes its cytoskeleton in a manner similar to that of the integrin (8, 30). In the present exercise, we turn to the key osteoclastogenic cytokine RANKL, whose absence in vivo obviates osteoclast formation. By mechanisms previously unknown, RANKL also organizes the cytoskeleton of mature resorptive cells, thereby stimulating their capacity to degrade bone (3). We find that RANKL's ability to optimally rearrange the osteoclast cytoskeleton depends upon induction of its receptor's association with c-Src, which links RANK to the αvβ3 integrin. The receptor/kinase complex activates the cytoskeleton-organizing molecules Syk, Slp-76, Vav3, and Rac, which are also induced by αvβ3. Despite the capacity of the tyrosine kinase to recognize TRAF6, c-Src-mediated induction of RANK to organize its cytoskeleton is independent of its osteoclastogenic signaling pathway. Thus, c-Src links RANK and αvβ3 and permits the complex to organize the osteoclast cytoskeleton.
MATERIALS AND METHODS
Mice.
Animals were housed in the animal care unit of the Washington University School of Medicine and were maintained according to guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International. All animal experimentation was approved by the Animal Studies Committee of Washington University School of Medicine.
Reagents.
Glutathione S-transferase (GST)–RANKL was expressed in our laboratory as described previously (14). Recombinant murine M-CSF was obtained from R&D Systems (Minneapolis, MN). Anti-Fas activating (CH11) antibody was from Upstate Biotechnology (Charlottesville, VA). The sources of antibodies are as follows: mouse anti-RANK monoclonal antibody (MAb) (IMG-128A) was from Imgenex (San Diego CA); MAb 327, directed against the c-Src protein, was a gift from A. Shaw (Department of Pathology, Washington University School of Medicine, St. Louis, MO); mouse anti-FLAG M2 MAb was from Sigma (St. Louis, MO); rabbit antihemagglutinin (anti-HA) antibody was from Covance (Princeton, NJ); rabbit anti-Src p-Y416, rabbit anti-β3-integrin, IκBα, phosphorylated IκBα, extracellular signal-regulated kinase (ERK), phosphorylated ERK, p38, phosphorylated p38, and rabbit anti-SLP-76 antibodies were from Cell Signaling (Beverly, MA); antiphosphotyrosine MAb 4G10 and rabbit anti-Vav3 antibody were from Upstate Biotechnology; mouse anti-NFATc1 and rabbit anti-Syk (N-19) antibody were from Santa Cruz Biotechnology (Santa Cruz, CA); cathepsin K antibody was from Millipore (Temecula, CA); mouse anti-CD95 antibody, was from BD Biosciences (Franklin Lakes, NJ); and rabbit anti-TRAF6 was from Enzo Life Sciences. The plasmid transfection reagent FuGENE 6 was purchased from Roche Applied Science. All other chemicals were obtained from Sigma.
Macrophage isolation and osteoclast culture.
Isolated bone marrow macrophages (BMMs) and splenocytes were differentiated into mature, multinucleated osteoclasts as described previously (10). Following 6 days of culture in M-CSF (30 ng/ml) and GST-RANKL (100 ng/ml), cells were stained for tartrate-resistant acid phosphatase (TRAP) activity (kit 387-A; Sigma). For prefusion osteoclast generation, BMMs or splenocytes were cultured in M-CSF and GST-RANKL for 3 days.
Staining of bone resorption pits.
BMMs were plated on bone slices and cultured with M-CSF and RANKL for 6 days to generate mature osteoclasts. The cells were then removed from bone slices with mechanical agitation. Bone slices were incubated with peroxidase-conjugated wheat germ agglutinin (Sigma) for 1 h and stained with 3,3′-diaminobenzidine (Sigma).
Actin ring reformation assay.
Mature osteoclasts, generated by exposure of bone-slice-residing BMMs to RANKL and M-CSF for 5 days, were washed twice with cold cytokine-free medium to disrupt actin rings followed by incubation at 37°C for 30 min. RANKL (100 ng/ml) was then added for the indicated time. Bone slices were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min. F-actin was stained with Alexa Fluor 488-phalloidin.
Plasmids and retroviral transduction.
c-Src-pMX-IRES-BSR was subcloned from c-Src-pMX-IRES-PURO, which contains a puromycin resistance sequence, into pMX-IRES-BSR by introducing the BamHI 5′ and XhoI 3′ restriction sites with a FLAG tag at the 3′ end. The ΔMYR (Δ76), ΔSH3 (Δ81-142), and ΔSH2 (Δ148-245) constructs were generated by standard molecular biology techniques. A total of 10 μg of the pMX retroviral vector was transfected transiently into Plat-E packaging cell line (20), using FuGENE 6 transfection reagent (Roche Applied Science). Virus was collected 48 h after posttransfection. BMMs or splenocytes were infected with virus for 24 h in the presence of 100 ng/ml M-CSF and 4 μg/ml Polybrene (Sigma). Cells were selected in the presence of M-CSF and 1 μg/ml blasticidin (Calbiochem) for 3 days prior to use as osteoclast precursors.
Western blotting and immunoprecipitation.
Forty micrograms of lysate was subjected to 8 to 12% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Filters were blocked in 0.1% casein in PBS for 1 h and incubated with primary antibodies followed by probing with fluorescence-labeled secondary antibodies (the Jackson Laboratory). Proteins were detected with the Odyssey infrared imaging system (LI-COR Biosciences). For immunoprecipitation, cells were cultured with M-CSF and RANKL for 2 to 3 days, starved in minimal essential medium alpha (α-MEM) without serum for 2 to 3 h, and treated with GST-RANKL for the indicated times. Eight hundred micrograms of lysate protein was incubated with 2 μg primary antibody overnight. Protein A or G-agarose (Sigma) beads were then added and incubated for 3 h. Beads were boiled in 2× sodium dodecyl sulfate sample buffer for 5 min. After centrifugation, proteins were separated by 8% or 12% sodium dodecyl sulfate-polyacrylamide gels and Western blotted as described above.
Gradient centrifugation.
Detergent-free density gradients were generated by the method of Macdonald and Pike (17). Fractions were immunoblotted as described above.
Active small GTPase assays.
Active Rac1 and RhoA were measured by pulldown assay (Thermo Scientific, Pierce, Rockford, IL).
Statistics.
All data are expressed as means ± standard deviations (SD), and statistical significance was calculated by Student's t test.
RESULTS
RANKL regulates the osteoclast cytoskeleton.
RANKL's induction of actin rings establishes the cytokine's capacity to organize the osteoclast cytoskeleton (3). To more precisely define this phenomenon, we determined RANKL's impact on the kinetics of these structures. Thus, mature bone-residing osteoclasts were washed in cold medium to disrupt their actin rings. The cells were then incubated, cytokine free, at 37°C for 30 min, after which they were exposed to RANKL or PBS for 15, 30, 60, or 120 min (13). Osteoclasts, maintained consistently in RANKL, progressively regenerate their actin rings, which are present in all cells after 2 h of cytokine exposure (see Fig. S1 in the supplemental material). In contrast, these cytoskeletal structures fail to appear in cells absent the cytokine. Hence, in addition to its osteoclastogenic properties, RANKL rapidly organizes the actin cytoskeleton of the mature resorptive polykaryon.
c-Src associates with RANK in a RANKL-dependent manner.
c-Src is a key regulator of actin dynamics in osteoclasts (5, 12, 19). It interacts with RANK at an undefined location within the receptor's carboxyl terminus, in dendritic cells, whose RANK content is an order of magnitude greater than that of osteoclasts (26). To explore the c-Src/RANK association in osteoclasts, spleen cells were exposed to RANKL and M-CSF for 3 days, which commits them to the resorptive cell's phenotype. These prefusion osteoclasts were cytokine starved and treated with RANKL over time. c-Src immunoprecipitates, immunoblotted for RANK and c-Src, establish that the two interact in a RANKL-dependent manner (Fig. 1A).
Fig 1.

c-Src associates with RANK in a RANKL-dependent manner. Splenocytes were exposed to RANKL for 3 days to generate prefusion osteoclasts (A, B, and C) or 5 days to produce fully differentiated, multinucleated osteoclasts (D, E, and F). Following 3 h or 1 h of incubation in cytokine-free medium, prefusion osteoclasts or mature osteoclasts, respectively, were exposed to RANKL with time. (A and D) c-Src immunoprecipitates (IP) were immunoblotted for RANK and c-Src. (B and E) Lysates of cytokine-starved cells, exposed to RANKL or carrier for 30 min, were subjected to density gradient centrifugation. Fractions were immunoblotted for RANK and c-Src. (C and F). Lysates were subjected to density gradient centrifugation, and c-Src immunoprecipitates of pooled fractions 12 to 15 were immunoblotted for RANK and c-Src. Results are representative of three independent experiments.
To explore the mechanisms by which RANKL promotes RANK/c-Src association, we performed density gradient centrifugation of lysates of cytokine-starved, prefusion osteoclasts exposed to RANKL or carrier. RANK and c-Src were identified by immunoblotting. In the absence of RANKL, its receptor and c-Src are in different locations within the gradient (Fig. 1B). With cytokine exposure, however, RANK moves to the c-Src-rich fractions, where the two proteins associate (Fig. 1C).
To determine if these phenomena obtain in fully differentiated osteoclasts, we maintained spleen cells in the presence of M-CSF and RANKL for 5 days, at which time they manifest the multinucleated phenotype. Like prefusion osteoclasts, RANKL induces the c-Src/RANK association and localizes the proteins in the same intracellular fractions from which they coprecipitate (Fig. 1D, E, and F).
c-Src SH2 and myristoylation domains dominate in RANK-mediated cytoskeletal organization.
c-Src impacts osteoclast function by phosphorylating effector molecules and via protein-protein recognition. To characterize the key c-Src adaptor domain(s) mediating RANKL-stimulated cytoskeletal organization, FLAG-tagged mutants of c-Src, lacking its amino-terminal myristoylation (ΔMYR), SH2 (ΔSH2), or SH3 (ΔSH3) regions, were retrovirally expressed in c-Src−/− splenocytes (Fig. 2A). The cells were differentiated into preosteoclasts, and equivalent levels of expression of the various constructs were ensured by anti-FLAG immunoblots (Fig. 2B). Similar results were obtained using the anti-c-Src327 MAb, which recognizes all constructs except that lacking the SH3 domain, which contains its epitope. Anti-FLAG immunoprecipitates of virgin cells and those treated with RANKL were immunoblotted for RANK and TRAF6 content. Although somewhat less than wild-type (WT) c-Src, the ΔSH3 mutant immunoprecipitates with RANK in a RANKL-dependent manner, but the ΔMYR and ΔSH2 mutants fail to effectively recognize the receptor (Fig. 2C). The inability of c-SrcΔSH2 to immunoprecipitate with RANK, following RANKL exposure, is not due to altered intracellular localization of the mutant kinase as density gradient analysis shows comigration with WT c-Src, as well as with mutant c-Src when its SH3 domain is absent (Fig. 2D). Deletion of the MYR motif, however, separates c-Src from RANK, but intracellular localization of the cytokine receptor is not affected by any c-Src mutation. Finally, we asked if the kinase activity of c-Src impacts its recognition of RANK. As seen in Fig. 2E, kinase-inactive, as well as constitutively active, c-Src associates with RANK as effectively as the WT in RANKL-treated cells.
Fig 2.

c-Src N-terminal myristoylated region and SH2 domain mediate RANK association. (A) WT and mutated c-Src constructs. (B) Lysates of c-Src−/− splenic macrophages, retrovirally transduced with empty vector or FLAG-tagged WT, ΔMYR, ΔSH3, or ΔSH2 c-Src, were immunoblotted with anti-FLAG or anti-c-Src MAb. (C) Cytokine-starved c-Src−/− preosteoclasts, retrovirally transduced with empty vector (Vect) or FLAG-tagged WT, ΔMYR, ΔSH3, or ΔSH2 c-Src, were maintained ± RANKL for 30 min. FLAG immunoprecipitates were immunoblotted for RANK, TRAF6, or FLAG. (D) Cytokine-starved c-Src−/− preosteoclasts, transduced with vector or FLAG-tagged WT, ΔMYR, ΔSH3, or ΔSH2 c-Src, were treated ± RANKL for 30 min. Lysates were subjected to gradient centrifugation, and the fractions were immunoblotted for FLAG or RANK. (E) c-Src−/− preosteoclasts, transduced with vector or WT, kinase-inactive (KD) or constitutively active (CA) c-Src, were maintained ± RANKL for 30 min. c-Src immunoprecipitates were immunoblotted for RANK or c-Src. Results are representative of three independent experiments.
c-Src regulates the osteoclast cytoskeleton but not osteoclast abundance.
Activated RANK is believed to exert its antiapoptotic effects, on osteoclasts, in conjunction with TRAF6 and c-Src. By this scenario, the SH3 domain of c-Src recognizes TRAF6 (26), which we confirm and establish is RANKL dependent (Fig. 2C). To determine if the component of c-Src proposed to mediate RANK-enhanced osteoclast survival is the same one organizing the cell's cytoskeleton, c-Src−/− spleen cells transduced with FLAG-tagged c-Src constructs were incubated with RANKL and M-CSF. Staining for tartrate-resistant acid phosphatase (TRAP) activity reveals no difference in numbers of c-Src−/− osteoclasts regardless of the construct with which they are transduced (Fig. 3A). As previously noted, however, c-Src−/− cells fail to spread, assuming a common phenotype of osteoclasts with cytoskeletal dysfunction (29). Expectedly, the mutant cell's abnormal facade is completely corrected by transduced WT c-Src. Absence of either the MYR or SH2 domains, however, does not rescue the deranged appearance of c-Src−/− osteoclasts, indicating both motifs are required for organization of the cell's cytoskeleton. While c-SrcΔSH3 reduces the percentage of spread osteoclasts, its disorganizing effect is substantially less than that from deletion of MYR or SH2. This conclusion is confirmed by the inability of ΔMYR- and ΔSH2-bearing c-Src−/− osteoclasts to resorb bone (Fig. 3B). To ensure that the cytoskeletal effects of the various c-Src mutants do not reflect altered differentiation, we exposed the various transductants to M-CSF and RANKL and temporally measured osteoclastogenic makers. In fact, levels of expression of NFAT2, the β3 integrin subunit, and cathepsin K are indistinguishable regardless of the c-Src construct employed (Fig. 3C).
Fig 3.

c-Src N-terminal myristoylated region and SH2 domain principally organize the osteoclast cytoskeleton but do not regulate differentiation. (A) Spleen cells of Src−/− mice, transduced with c-Src domain deletion mutants, were cultured with M-CSF and RANKL for 7 days, after which the cells were stained for TRAP activity. The total number of osteoclasts (OC) and those spread were counted. Vect, vector. *, P < 0.05. Scale bar, 200 μm. The data are shown as means ± SD of triplicate samples. (B) Transduced osteoclasts, generated on bone, were removed after 5 days. Resorptive lacunae were stained by lectin (brown reaction product outlined), and pit area was determined. *, P < 0.05. Scale bar, 200 μm. The data are shown as means ± SD of triplicate samples. (C) c-Src−/− splenic macrophages, transduced with either empty vector or WT, ΔMYR, ΔSH3, or ΔSH2 c-Src, were cultured in RANKL and M-CSF over time. Osteoclast differentiation markers were determined by immunoblotting. Actin serves as a loading control. αFlag, anti-FLAG antibody. (D) c-Src−/− splenic macrophages, transduced as in panel C, were cultured in RANKL and M-CSF for 3 days. The cells were cytokine starved and exposed to RANKL with time. Signaling molecules were identified by immunoblotting. Actin serves as loading control. Results are representative of three independent experiments.
To further explore the means by which c-Src mediates cytoskeletal organization stimulated by the cytokine, we examined the mutants' impact on NF-κB, ERK, and p38, which are canonical signals activated via RANKL-RANK-TRAF6-dependent cascades. Again, deletion of any c-Src domain does not alter RANKL activation of these molecules (Fig. 3D). Collectively, these data provide evidence that the principal means by which c-Src mediates RANKL-stimulated cytoskeletal organization, compared to promoting survival and differentiation, differ.
RANKL activates cytoskeleton-organizing proteins.
c-Src is a proximal component of a canonical cytoskeleton-organizing complex activated by the αvβ3 integrin, in osteoclasts. To determine if the same obtains in the context of RANKL, we exposed c-Src−/− spleen-derived, transduced macrophages to M-CSF and RANKL for 5 days. These experiments were performed on the osteoclast's natural substrate, bone, which optimally activates cytoskeleton-organizing molecules in response to RANKL (see Fig. S2 in the supplemental material). The cells were cytokine starved and treated with RANKL for 20 min. c-Src activity, assessed by Tyr416 phosphorylation, is enhanced by the cytokine, in FLAG-tagged immunoprecipitates of c-Src−/− preosteoclasts transduced with the WT construct (Fig. 4A). The same occurs regarding phosphorylation of Syk, Slp76, and the relatively osteoclast-specific GEF Vav3, all of which contribute to αvβ3-stimulated cytoskeleton organization in osteoclasts (22, 29) (Fig. 4B, C, and D). Under each circumstance, except Syk, the magnitude of RANKL stimulation is somewhat less in c-SrcΔSH3-transduced than in WT-transduced mutant cells. On the other hand, none of these molecules are activated by RANKL in ΔMYR- or ΔSH2-expressing cells.
Fig 4.

RANKL activates cytoskeleton-organizing molecules principally via c-Src MYR and SH2 domains. (A) c-Src−/− spleen-derived macrophages, transduced with either FLAG-tagged WT Src, ΔMYR, ΔSH3, or ΔSH2 were cultured with RANKL and M-CSF for 5 days on bone powder. Serum- and cytokine-starved cells were exposed to RANKL (100 ng/ml) for 20 min. Phosphorylated c-SrcY416 and FLAG in FLAG immunoprecipitates (IP) (A), phosphotyrosine (p-Y) and SLP-76 in SLP-76 immunoprecipitates (B), phosphotyrosine and Syk in Syk immunoprecipitates (C), and phosphotyrosine and Vav3 in Vav3 immunoprecipitates (D) were determined by immunoblotting. Rac1-GTP (E) and RhoA-GTP (F) were detected by pulldown assay, and total GTPase was detected by immunoblotting. Densitometric data are expressed relative to vector (Vect)/RANKL−. The experiments were repeated twice.
RANKL's capacity to phosphorylate Vav3 in a c-Src-dependent manner suggests that GEF's target, Rac, which organizes the osteoclast cytoskeleton as an αvβ3-effector molecule, may also be activated by the cytokine (6, 8). RANKL increases GTP association with Rac but not RhoA, in c-Src−/− cells transduced with WT c-Src or c-SrcΔSH3 but not that of the tyrosine kinase lacking its MYR or SH2 domains (Fig. 4E and F). Similar to its upstream activators, Rac1-GTP is less abundant in c-SrcΔSH3- than WT-bearing cells. Thus, c-Src mediates RANKL-induced osteoclast cytoskeleton organization principally via its SH2 motif, with some contribution from its SH3 domain.
c-Src point mutants mirror domain deletion.
The capacity of c-SrcΔSH3 and c-SrcΔSH2 to continue to recognize target proteins suggests they are conformationally intact (Fig. 2C). To confirm such is the case, however, we expressed inactivating FLAG-tagged Myr (c-SrcG2A) (15, 21), SH3 (c-SrcW118K) (7), and SH2 (c-SrcR175L) (7, 27) point mutants in c-Src−/− splenocytes (see Fig. S3A in the supplemental material). Similar to their corresponding domain-deleted constructs, c-SrcW118K, but not c-SrcG2A or c-SrcR175L, continues to bind RANK (see Fig. S3B). The same parallelism holds regarding osteoclast number and spreading (see Fig. S4A in the supplemental material) as well as in vitro bone resorptive capacity (see Fig. S4B), expression of osteoclast differentiation markers (see Fig. S4C) and activation of osteoclastogenic signaling molecules (see Fig. S4D). In a like manner, Myr and SH2 domain point mutants fail to activate cytoskeleton-organizing proteins, while that of SH3 mirrors WT c-Src (see Fig. S5 in the supplemental material).
c-Src organizes the osteoclast cytoskeleton via RANK369–373.
Having identified the cytoskeleton-organizing domain of c-Src recognizing RANK, we turned to the component of the receptor binding the kinase. This exercise is challenged, in primary osteoclastic cells, by the presence of endogenous RANK, which, upon cytokine stimulation, would compete with exogenous RANK for c-Src association. We therefore transduced WT bone marrow macrophages (BMMs) with retroviral vectors coding for various components of the RANK cytoplasmic region linked to the external domain of the human Fas (hFas) receptor (16, 28). The RANK cytoplasmic domains of these chimeric constructs were designed such that each contains only one of six intact putative TRAF binding motifs (PTMs) (28) (see Fig. S6 in the supplemental material). Mutation of all six PTMs served as a negative control. Lysates of BMMs transduced with each chimeric construct were immunoblotted with the hFas MAb, which does not recognize the murine protein, to ensure equivalent expression (Fig. 5A). The cells were cultured with M-CSF and RANKL for 6 days to activate endogenous receptors, resulting in formation of characteristic osteoclasts by each transductant (Fig. 5B). After actin ring disruption, on bone, the cells were incubated in cytokine-free medium at 37°C for 30 min, after which they were exposed to hFas MAb, which activates the chimeric receptors but not murine Fas. Actin ring reformation was determined over time. Only RANK PTMW3369–373 is sufficient to organize the osteoclast cytoskeleton as manifested by actin ring development (Fig. 5C).
Fig 5.

c-Src binds RANK369–373. (A) hFas expression in WT BMMs transfected with hFas/RANK WT, W1, W2, W3, W4, W5, W6, and P1-6, determined by immunoblotting. Results are representative of three independent experiments. Vect, vector; αhFas, anti-hFas antibody. (B) WT BMMs transfected with Fas/RANK constructs were exposed to M-CSF and RANKL for 3 days on plastic and stained for TRAP activity. Scale bar, 200 μm. (C) WT BMMs transfected with hFas/RANK constructs were cultured with RANKL and M-CSF on bone for 6 days. The cells were washed with cold medium to disrupt actin rings and stimulated with hFas MAb for 30 or 120 min. The percentage of osteoclasts with actin rings was determined. Scale bar, 100 μm. The data are shown as means ± SD of triplicate samples. (D) WT BMMs transduced with hFas/RANK constructs were cultured with M-CSF and RANKL for 3 days. They were cytokine starved and stimulated for 20 min by hFas MAb. c-Src immunoprecipitates (IP) and total cell lysate (TCL) were immunoblotted for hFas and c-Src. (E) WT BMMs transduced with hFas/RANK constructs were cultured with M-CSF and RANKL for 3 days. Cytokine-starved cells were stimulated for 5 or 20 min by hFas MAb. Lysates were immunoblotted for c-SrcY416 and c-Src. Vav3 immunoprecipitates were immunoblotted for phosphotyrosine and Vav3. Rac1-GTP and RhoA-GTP were detected by pulldown assay, and the total abundance of the GTPases was determined by immunoblotting. Results are representative of three independent experiments.
To determine if RANK's cytoskeleton-organizing motif, PTMW3, which binds TRAF6, also recognizes c-Src, hFas/RANK-transduced, serum- and cytokine-starved prefusion osteoclasts were maintained, with or without hFas activating antibody, for 20 min. c-Src immunoprecipitates were then immunoblotted for hFas. Similar to its cytoskeleton-organizing properties, RANK PTMW3 is the only mutant chimeric construct whose binding of c-Src is enhanced by the antibody (Fig. 5D). Furthermore, the quantities of c-Src present in hFas immunoblots are similar in vector- and WT chimera-transduced cells. Thus, unlike the αvβ3 integrin, the RANK cytoplasmic domain does not constitutively recognize the tyrosine kinase (29). Finally, confirming that RANK PTMW3 is the receptor's cytoskeleton-organizing motif, it is sufficient to activate c-Src, Vav3, and Rac (Fig. 5E).
c-Src/RANK association, in osteoclasts, is αvβ3 integrin dependent.
Osteoclasts lacking c-Src or the αvβ3 integrin exhibit features of cytoskeletal dysfunction, and we find the same obtains when RANK signaling is deficient in mature cells (see Fig. S1 in the supplemental material). These observations prompted us to ask whether the two molecules are functionally related in osteoclasts. Thus, we exposed WT and β3−/− BMMs to M-CSF and RANKL for 3 days. Following cytokine starvation, the cells were RANKL stimulated with time and c-Src immunoprecipitates were immunoblotted for RANK. Whereas, the cytokine rapidly induces c-Src/RANK recognition in WT cells, this association is markedly reduced absent αvβ3 (Fig. 6A). To confirm the integrin's role in mediating this interaction, we transduced β3−/− BMMs with empty vector or human β3 (hβ3). The human construct was chosen as its cytoplasmic motif is identical to that of its murine counterpart and an immunoprecipitating MAb recognizing its extracellular domain is available (29). Importantly, we have established that these transductants express physiological quantities of αvβ3 (11). hβ3- and vector-bearing cells were cultured with RANKL and M-CSF for 4 days, after which c-Src immunoprecipitates were again immunoblotted for RANK. WT hβ3 rescues the inability of β3−/− osteoclasts to spread while also restoring the RANKL-stimulated c-Src/RANK association (Fig. 6B). To determine if c-Src/RANK recognition requires activated αvβ3, we cytokine starved WT prefusion osteoclasts for 3 h. The cells were then maintained in suspension or plated on the integrin's ligand, vitronectin, for 30 min, which activates αvβ3-induced cytoskeleton-organizing molecules. As demonstrated by immunoblotting of c-Src immunoprecipitates for RANK, only liganded αvβ3 promotes association of the two proteins (Fig. 6C).
Fig 6.

c-Src binding to RANK is αvβ3 dependent. (A) Cytokine-starved WT and β3−/− preosteoclasts were exposed to RANKL for the times shown. c-Src immunoprecipitates (IP) and total cell lysate (TCL) were immunoblotted for RANK, the β3 subunit, and c-Src. (B) β3−/− BMMs were retrovirally transduced with hβ3 or vector. After 4 days in RANKL and M-CSF, cytokine-starved cells were exposed to RANKL for the times shown. The cells were stained for TRAP activity (left panel). c-Src immunoprecipitates and TCL were immunoblotted for RANK, β3 subunit, and c-Src (right panel). Scale bar, 200 μm. (C) Cytokine-starved WT preosteoclasts were maintained in suspension (S) or plated on vitronectin (A) for 30 min. c-Src immunoprecipitates and TCL were immunoblotted for RANK and c-Src. Results are representative of three independent experiments.
c-Src links RANK and αvβ3.
It appears, therefore, that αvβ3 and RANK partner to activate a key cytoskeleton-organizing pathway in osteoclasts, suggesting the two molecules may associate. To determine if such is the case, we temporally exposed hFas/RANK-bearing WT osteoclasts to the hFas MAb. Immunoblotting of hFas immunoprecipitates for the β3 subunit demonstrates that activated RANK progressively associates with αvβ3 (Fig. 7A). Furthermore, this interaction is obviated in the absence of c-Src (Fig. 7B).
Fig 7.

c-Src links RANK and αvβ3. (A) WT preosteoclasts, transduced with WT hFas/RANK, were exposed to hFas MAb for the times shown. hFas immunoprecipitates (IP) were immunoblotted for β3 subunit and hFas. (B) WT and c-Src−/− preosteoclasts, transduced with WT hFas/RANK, were exposed to hFas MAb with time. hFas immunoprecipitates were immunoblotted for the β3 subunit and hFas. (C) Lysates of c-Src−/− preosteoclasts transduced with FLAG-tagged c-Src domain deletion constructs were immunoprecipitated with anti-FLAG MAb, and the product was immunoblotted for β3 subunit and FLAG. Vect, vector. (D) Lysates of c-Src−/− preosteoclasts transduced with FLAG-tagged c-Src point mutants were immunoprecipitated with anti-FLAG MAb, and the product was immunoblotted for the β3 subunit and FLAG. (E) HEK293T cells were transfected with FLAG-tagged c-Src constructs, HA-tagged RANK, and hβ3. HA immunoprecipitates were immunoblotted for hβ3 and HA. (F) Cytokine-starved c-Src−/− preosteoclasts transduced with FLAG-tagged c-Src point mutants and WT hFas/RANK were exposed to hFas MAb for 20 min. hFas immunoprecipitates were immunoblotted for β3 subunit and hFas. (G) Cytokine-starved, c-Src−/− prefusion osteoclasts, transduced with FLAG-tagged c-Src domain-deleted constructs, were maintained in suspension (S) or plated on vitronectin (A) for 30 min ± RANKL. FLAG immunoprecipitates were immunoblotted for RANK and FLAG. WB, Western blot. (H) Cytokine-starved, c-Src−/− prefusion osteoclasts, transduced with FLAG-tagged c-Src point mutants, were maintained in suspension (S) or plated on vitronectin (A) for 30 min ± RANKL. FLAG immunoprecipitates were immunoblotted for RANK and FLAG. The experiments were repeated 2 or 3 times with similar results.
The dependency of αvβ3/RANK association on c-Src raised the possibility that the kinase links the two receptors. In this circumstance, the component of c-Src binding the integrin should differ from that recognizing RANK. Thus, we transduced c-Src−/− splenic macrophages with FLAG-tagged c-Src deletion (Fig. 7C) or point mutants (Fig. 7D) and differentiated them into prefusion osteoclasts. Lysates were immunoprecipitated with FLAG MAb, and the product was immunoblotted for the β3 subunit. Similar to its association with RANK, c-Src requires an intact MYR domain to recognize β3. On the other hand, the β3/c-Src interaction, in osteoclasts, as in other circumstances (1), is unimpaired by absence or mutation of SH2 but aborted by ΔSH3 or c-SrcW118K.
These observations suggest a model whereby c-Src, by its SH3 region interacting with αvβ3 and its SH2 domain binding RANK, links the two receptors. Deletion or inactivation of either SH region should therefore obviate αvβ3/RANK association. Hence, we transfected HEK293T cells with WT hβ3, various combinations of FLAG-tagged c-Src deletion mutants, and HA-RANK. In fact, RANK and the integrin coprecipitate in the presence of WT c-Src but not in the absence of its MYR, SH2, or SH3 domain (Fig. 7E). To determine if the c-Src-mediated αvβ3/RANK association requires activation of the cytokine receptor, we transfected HEK393T cells with WT hFas/RANK (Fig. 7F). We once again ensured conformational integrity of the deletion constructs by expressing c-Src point mutants in the same cells. Whereas irrelevant IgG has no effect, activation with anti-Fas MAb yields hFas/RANK association only with WT c-Src. Alternatively, point mutation of any of the three domains obviates this interaction. Thus, αvβ3 and RANK are linked by c-Src only upon activation of the cytokine receptor.
Finally, we turned to the consequences of c-Src domain deletion (Fig. 7G) or mutation (Fig. 7H) on RANKL- or integrin-mediated RANK/c-Src binding. Once again establishing a central role for integrin occupancy, interaction between the cytokine receptor and tyrosine kinase is undetectable in all nonadherent transduced cells. While RANKL enhances RANK/c-Src binding in adherent WT cells, it fails to do so in the face of Myr or SH2 domain deletion or mutation. In keeping with partial recognition of c-Src by activated RANK, absent αvβ3 (Fig. 6A), RANKL modestly promotes this interaction in transduced cells lacking an intact SH3 motif.
DISCUSSION
The osteoclast cytoskeleton is a unique structure, which creates a resorptive microenvironment and delivers bone-degrading molecules into this extracellular space enclosed by actin rings. These circular sealing zones are the products of redistribution of podosomes, arranged in clusters in non-bone-residing cells. Upon attachment to mineralized substrate, these transient adhesion structures migrate to the bone-apposed plasma membrane, where in an actin-rich environment, they encompass the matrix surface to be degraded. Thus, actin ring formation, which requires RANKL, is key to organizing the osteoclast cytoskeleton to its resorptive conformation.
Although the general morphological features of osteoclast polarization have been long appreciated, insights into the relevant molecular mechanisms are recent. Signals emanating from liganded αvβ3 activate a complex in which constitutively associated c-Src is the most proximal, established component (29). Inactivation of any of the complex-residing proteins yields poorly spread “crenated-appearing” osteoclasts due to cytoskeletal dysfunction. The promacrophage proliferative cytokine M-CSF, which also organizes the osteoclast cytoskeleton, does so by targeting the same signaling molecules as αvβ3 (8, 30). Furthermore, M-CSF-mediated activation of Rac, which is essential for osteoclast cytoskeletal remodeling and function, requires this integrin (8). Thus, αvβ3 and the macrophage-proliferating cytokine collaborate in organizing the osteoclast cytoskeleton in a c-Src-dependent manner.
The discovery that RANKL is the key osteoclastogenic cytokine provided the seminal insights into the mechanisms of bone resorption. This observation permitted generation of osteoclasts in vitro, enabling dissection of the means by which macrophages differentiate into mature resorptive cells. An unexpected outcome of these studies is the finding that RANKL also enhances osteoclast function by organizing the cell's cytoskeleton (3). However, the mechanisms mediating RANKL stimulation of the mature cell remained elusive.
We have established that like M-CSF, RANKL partners with αvβ3 to organize the osteoclast cytoskeleton. Perhaps the most compelling aspect of this scenario is the realization that this integrin-cytokine collaboration involves their interaction, wherein c-Src links the two receptors. These observations not only shed light on the heretofore elusive means by which RANKL stimulates the mature osteoclast to resorb bone but also provide insights into how c-Src mediates cytoskeleton organization in the bone resorptive process.
In osteoclasts, c-Src constitutively binds αvβ3, which activates the tyrosine kinase upon integrin occupancy. The same is not true regarding c-Src association with RANK (29). In fact, RANKL induces RANK/c-Src recognition, in authentic osteoclasts, only in the presence of αvβ3 activated by ligand, in an outside-in manner and is enhanced by the osteoclastogenic cytokine.
The biological implications of RANK/c-Src association are, however, controversial. RANK does not possess kinase activity, and Wong et al. conclude that its antiapoptotic properties reflect delivery of c-Src, via its SH3 domain, to the receptor by TRAF6, an adaptor protein essential for osteoclastogenesis (26). The logical consequence of this scenario should be decreased osteoclast number in the face of c-Src deficiency, which is, however, inconsistent with their enhanced abundance in c-Src−/− mice (2). Furthermore, absence of the SH2, SH3, or MYR domain does not dampen RANKL activation of its osteoclastogenic effector molecules. Despite their association, which we confirm, the c-Src/TRAF6 composite, therefore, does not appear to regulate RANK's osteoclastogenic properties. On the other hand, the requirement for the c-Src SH2 domain for RANK association supports the concept that the tyrosine kinase in osteoclasts functions exclusively to organize their cytoskeleton. While ΔSH2 profoundly affects signals consequent to RANK/c-Src association, the SH3 motif also participates, as evidenced by partial cytoskeletal disruption and some degree of impaired signaling in its absence.
Having established, with deletion and point mutants, that the SH2 domain of c-Src mediates RANK recognition, we asked which component of the cytokine receptor binds the kinase. Using chimeric constructs, in which RANK signaling is activated by hFas antibody, we find that a component of a single TRAF-interacting site is sufficient to organize the osteoclast cytoskeleton, promote RANK/c-Src recognition, and activate the same key cytoskeleton-organizing molecules as αvβ3 and c-Fms. Because SH2 domains classically bind phosphorylated tyrosines, we were surprised that no such residues are present in the region of RANK recognizing c-Src. While this observation suggests an indirect interaction, SH2 domains in other proteins may recognize binding partners by phosphotyrosine-free, surface-surface contact (4). The same noncanonical paradigm holds in the context of c-SrcSH3 recognizing the β3 cytoplasmic domain, which occurs in the absence of a proline-rich motif (1).
Our data suggest an interactive model between RANK, c-Src, and αvβ3 in organizing the osteoclast cytoskeleton (Fig. 8). In its nonliganded state, RANK resides in a plasma membrane region distinct from that containing c-Src. Upon RANKL stimulation, the receptor transits to a c-Src-rich domain, where in the presence of activated αvβ3, it associates with the SH2 motif of the tyrosine kinase. RANKL partially induces RANK/c-Src recognition in the absence of the c-Src SH3 region, which constitutively complexes the integrin and is necessary for optimal osteoclast cytoskeletal organization, likely because it links the two receptors. Thus, we propose that activated RANK prompts two distinct signaling pathways: one, which is c-Src and αvβ3 independent, promotes osteoclast formation, and the other, in which the tyrosine kinase and integrin are required, organizes the cell's cytoskeleton and stimulates its resorptive capacity.
Fig 8.
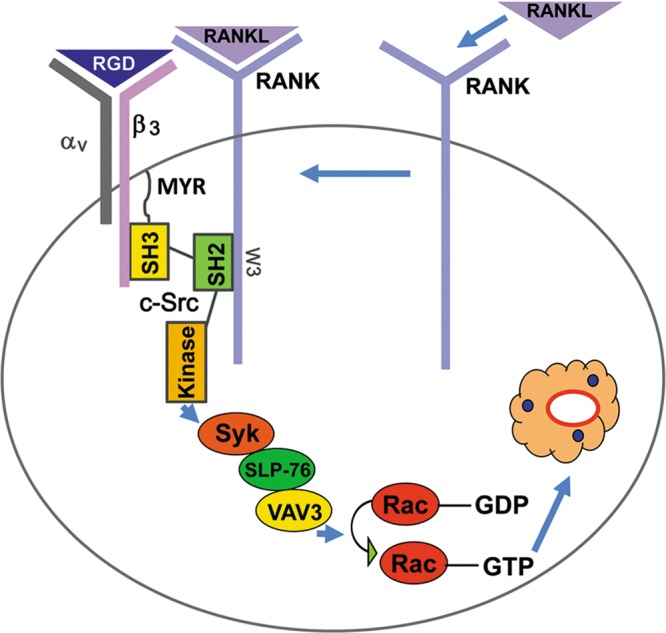
Model of RANK-mediated osteoclast cytoskeletal organization. Upon RANKL simulation, RANK migrates to c-Src-rich areas of the plasma membrane, where a 5-amino-acid, TRAF-6-interacting motif associates with the tyrosine kinase's SH2 domain in an activated αvβ3-dependent manner. The cytokine receptor and integrin form a complex mediated by c-Src, whose SH3 domain recognizes the β3 cytoplasmic tail. Liganded RANK and αvβ3 collaborate to induce a canonical signaling pathway involving c-Src, Syk Slp-76, Vav3, and Rac, which organize the osteoclast cytoskeleton.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by Shriners Hospitals for Children grant no. 8590 (S.L.T.) and National Institutes of Health grant no. AR032788, AR046523, AR054618, AR057037, and AR057235 to S.L.T. and AR047830 to X.F.
The contents of this project are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
We have no conflicting financial interests.
Footnotes
Published ahead of print 21 May 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Arias-Salgado EG, et al. 2003. Src kinase activation by direct interaction with the integrin b cytoplasmic domain. Proc. Natl. Acad. Sci. U. S. A. 100:13298–13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR. 1992. Requirement of pp60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J. Clin. Invest. 90:1622–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burgess TL, et al. 1999. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J. Cell Biol. 145:527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan B, et al. 2003. SAP couples Fyn to SLAM immune receptors. Nat. Cell Biol. 5:155–160 [DOI] [PubMed] [Google Scholar]
- 5. Chellaiah M, Fitzgerald C, Alvarez U, Hruska K. 1998. c-Src is required for stimulation of gelsolin-associated phosphatidylinositol 3-kinase. J. Biol. Chem. 273:11908–11916 [DOI] [PubMed] [Google Scholar]
- 6. Croke M, et al. 2011. Rac deletion in osteoclasts causes severe osteopetrosis. J. Cell Sci. 124:3811–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Destaing O, et al. 2008. The tyrosine kinase activity of c-Src regulates actin dynamics and organization of podosomes in osteoclasts. Mol. Biol. Cell 19:394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Faccio R, Takeshita S, Zallone A, Ross FP, Teitelbaum SL. 2003. c-Fms and the avb3 integrin collaborate during osteoclast differentiation. J. Clin. Invest. 111:749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Faccio R, et al. 2005. Vav3 regulates osteoclast function and bone mass. Nat. Med. 11:284–290 [DOI] [PubMed] [Google Scholar]
- 10. Faccio R, Zou W, Colaianni G, Teitelbaum SL, Ross FP. 2003. High dose M-CSF partially rescues the Dap12−/− osteoclast phenotype. J. Cell. Biochem. 90:871–883 [DOI] [PubMed] [Google Scholar]
- 11. Feng X, et al. 2001. A Glanzmann's mutation in b3 integrin specifically impairs osteoclast function. J. Clin. Invest. 107:1137–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grey A, Chen Y, Paliwal I, Carlberg K, Insogna K. 2000. Evidence for a functional association between phosphatidylinositol 3-kinase and c-src in the spreading response of osteoclasts to colony-stimulating factor-1. Endocrinology 141:2129–2138 [DOI] [PubMed] [Google Scholar]
- 13. Kim HJ, et al. 2006. Glucocorticoids suppress bone formation via the osteoclast. J. Clin. Invest. 116:2152–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lam J, et al. 2000. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Invest. 106:1481–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li L, et al. 2007. Variant estrogen receptor-c-Src molecular interdependence and c-Src structural requirements for endothelial NO synthase activation. Proc. Natl. Acad. Sci. U. S. A. 104:16468–16473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu W, et al. 2004. Functional identification of three receptor activator of NF-κB cytoplasmic motifs mediating osteoclast differentiation and function. J. Biol. Chem. 279:54759–54769 [DOI] [PubMed] [Google Scholar]
- 17. Macdonald JL, Pike LJ. 2005. A simplified method for the preparation of detergent-free lipid rafts. J. Lipid Res. 46:1061–1067 [DOI] [PubMed] [Google Scholar]
- 18. McHugh KP, et al. 2000. Mice lacking b3 integrins are osteosclerotic because of dysfunctional osteoclasts. J. Clin. Invest. 105:433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyazaki T, et al. 2004. Src kinase activity is essential for osteoclast function. J. Biol. Chem. 279:17660–17666 [DOI] [PubMed] [Google Scholar]
- 20. Morita S, Kojima T, Kitamura T. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7:1063–1066 [DOI] [PubMed] [Google Scholar]
- 21. Patwardhan P, Resh MD. 2010. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol. Cell. Biol. 30:4094–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reeve JL, et al. 2009. SLP-76 couples Syk to the osteoclast cytoskeleton. J. Immunol. 183:1804–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwartzberg PL, et al. 1997. Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src−/− mutant mice. Genes Dev. 11:2835–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soriano P, Montgomery C, Geske R, Bradley A. 1991. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 64:693–702 [DOI] [PubMed] [Google Scholar]
- 25. Teitelbaum SL. 2007. Osteoclasts: what do they do and how do they do it? Am. J. Pathol. 170:427–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wong BR, et al. 1999. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol. Cell 4:1041–1049 [DOI] [PubMed] [Google Scholar]
- 27. Xing L, et al. 2001. Genetic evidence for a role for Src family kinases in TNF family receptor signaling and cell survival. Genes Dev. 15:241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu D, et al. 2004. Development of a chimaeric receptor approach to study signalling by tumour necrosis factor receptor family members. Biochem. J. 383:219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zou W, et al. 2007. Syk, c-Src, the avb3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J. Cell Biol. 176:877–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP. 2008. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol. Cell 31:422–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.