

Abstract
Lipoteichoic acid (LTA), a glycerol phosphate polymer, is a component of the envelope of Gram-positive bacteria that has hitherto not been identified in Bacillus anthracis, the causative agent of anthrax. LTA synthesis in Staphylococcus aureus and other microbes is catalyzed by the product of the ltaS gene, a membrane protein that polymerizes polyglycerol phosphate from phosphatidyl glycerol. Here we identified four ltaS homologues, designated ltaS1 to -4, in the genome of Bacillus anthracis. Polyglycerol phosphate-specific monoclonal antibodies were used to detect LTA in the envelope of B. anthracis strain Sterne (pXO1+ pXO2−) vegetative forms. B. anthracis mutants lacking ltaS1, ltaS2, ltaS3, or ltaS4 did not display defects in growth or LTA synthesis. In contrast, B. anthracis strains lacking both ltaS1 and ltaS2 were unable to synthesize LTA and exhibited reduced viability, altered envelope morphology, aberrant separation of vegetative forms, and decreased sporulation efficiency. Expression of ltaS1 or ltaS2 alone in B. anthracis as well as in other microbes was sufficient for polyglycerol phosphate synthesis. Thus, similar to S. aureus, B. anthracis employs LtaS enzymes to synthesize LTA, an envelope component that promotes bacterial growth and cell division.
INTRODUCTION
Structural analysis of lipoteichoic acid (LTA) purified from Staphylococcus aureus, Bacillus subtilis, Enterococcus faecalis, and many other species revealed a polymer of glycerol phosphate linked to glycolipid, which provides for LTA anchoring in bacterial membranes (3, 5, 10, 38). Based on data from the in vivo and in vitro synthesis of LTA from radiolabeled precursors, a model was proposed whereby phosphatidyl glycerol is polymerized to generate the products polyglycerol phosphate and diacylglycerol (8, 11, 14). In agreement with this model, S. aureus LtaS, a polytopic membrane protein with an extracellular domain, is both necessary and sufficient for LTA synthesis from phosphatidyl glycerol (17). The expression of ltaS in S. aureus or ltaS homologues in B. subtilis and Listeria monocytogenes contributes to bacterial growth under physiological conditions (37°C) in the laboratory (17, 22, 34, 40, 42). These findings suggest that LTA synthesis may also be essential for the growth of Gram-positive bacteria during infection and that LTA inhibitors may be useful therapeutics for infectious diseases caused by Gram-positive microbes (17).
To investigate teichoic acids of Bacillus anthracis, the causative agent of anthrax disease (27), Molnár and Prágai used acid hydrolysis of bacterial envelope preparations but failed to detect either glycerol or ribitol, the constituents of LTA and wall teichoic acids (WTA), respectively (32). More recent work studied the contribution of the dltABCD operon to B. anthracis pathogenesis (12). Initially identified by Heaton and Neuhaus, the dltABCD operon of Lactobacillus casei was demonstrated to encode factors that catalyze the d-alanyl esterification of polyglycerol phosphate LTA as well as polyribitol phosphate WTA (18, 19, 35). An insertional lesion in the dltABCD operon of B. anthracis strain Sterne (pXO1+ pXO2−) caused the variant to display increased sensitivity to some antibacterial peptides as well as attenuation in a mouse model of respiratory anthrax (9). Unlike the B. anthracis Sterne parent, the dltABCD variant did not harbor esterified alanine in the cell wall envelope (12). Nevertheless, these studies did not identify the molecular nature of alanine esterification in the envelope of B. anthracis.
The secondary cell wall polysaccharide of B. anthracis is tethered to the cell wall envelope through murein linkage units (4, 23), a structure that in other Gram-positive bacteria is used for the immobilization of wall teichoic acid (2, 46). Although the genome of B. anthracis harbors tagO and other genes known to be involved in the synthesis of the cell wall linkage unit (23, 29), this microbe lacks the genes that are known to be required for wall teichoic acid synthesis (37, 45).
To explore the possibility that B. anthracis may synthesize LTA, we used in this study a bioinformatic approach and searched the genome of B. anthracis for homologues of S. aureus LtaS (LtaSSA). This search identified four polytopic membrane proteins with a conserved sulfatase (LtaS) domain, which were designated LtaS1, LtaS2, LtaS3, and LtaS4. Genetic analyses revealed that the ltaS1 and ltaS2 genes were both necessary and sufficient for LTA synthesis in B. anthracis and in other bacterial species. B. anthracis variants unable to synthesize LTA displayed multiple defects in envelope assembly that may account for the inability of mutant bacilli to sporulate and to separate dividing cells during their vegetative life cycle. These findings are in agreement with the general concept that LTA is an essential constituent of the envelope of Gram-positive organisms (33).
MATERIALS AND METHODS
Bacterial growth.
B. anthracis strain Sterne 34F2 and variants were grown in brain heart infusion (BHI), nutrient broth yeast extract (NBY), or Luria-Bertani (LB) medium at 37°C or 30°C when appropriate. S. aureus was grown in tryptic soy broth (TSB). Escherichia coli was grown in LB medium. Antibiotics were added to cultures as needed (spectinomycin, 200 μg/ml, and kanamycin, 20 to 50 μg/ml). Expression from the tetracycline-inducible promoter (Ptet) was induced by adding 150 ng/ml anhydrotetracycline to the medium, and expression via the spac promoter (Pspac) was induced by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG).
B. anthracis strains and plasmids.
The strains and plasmids used in this study are listed in Table 1. The alleles ltaS1::Spc, ltaS3::Kan, and ltaS4::Spc were obtained following transposon mutagenesis of B. anthracis Sterne with the bursa aurealis minitransposon (41). Transduction using bacteriophage CP-51 was performed for each marked allele using the B. anthracis Sterne wild-type or mutant background carrying unmarked mutations in ltaS genes (15). Newly transduced alleles were selected by growing bacteria at 30°C on appropriate selective medium and validated by sequencing the DNA of the transposon insertion site using inverse PCR as described previously (41). Unmarked B. anthracis variants (ΔltaS1, ΔltaS2, and ΔltaS3 mutants) were generated by allelic replacement using plasmid pLM4 as previously described (30). All strains were verified by DNA sequencing and immunoblotting with LtaS-specific antibodies. Mutants carrying insertions or deletions in the ltaS1 or ltaS3 gene displayed identical phenotypes, respectively. For simplicity, no distinction was made between these alleles and strains are referred to by the names listed in the “designation” column of Table 1. The E. coli-B. anthracis shuttle plasmid pJK4 was used to clone the ltaS genes with an IPTG-inducible promoter (24) and was used for both complementation studies and sufficiency experiments using E. coli as a host (see below). Primers used to amplify the ltaS open reading frames by PCR amplification from genomic DNA are listed in Table 2. Plasmid pPROEX (Invitrogen) was used to express the predicted soluble domains (as depicted in Fig. 1B) of LtaS1, LtaS2, LtaS3, and LtaS4 in E. coli. Primers used to amplify the corresponding genomic DNA are listed in Table 2.
Table 1.
Strains and plasmids used in this study
| Designation | Genotype | Description | Reference |
|---|---|---|---|
| Bacillus anthracis strains | |||
| WT | WT | B. anthracis Sterne 34F2 | |
| ltaS1 | ΔltaS1 | Sterne with an ltaS1 gene deletion | This work |
| ltaS1::Sp | Sterne with bursa aurealis insertion in ltaS1 | ||
| ltaS2 | ΔltaS2 | Sterne with an ltaS2 gene deletion | This work |
| ltaS3 | ΔltaS3 | Sterne with an ltaS3 gene deletion | This work |
| ltaS3::Km | Sterne with bursa aurealis insertion in ltaS3 | ||
| ltaS4 | ltaS4::Spc | Sterne with bursa aurealis insertion in ltaS4 | This work |
| ltaS1 ltaS2 | ltaS1::Spc-ΔltaS2 | ltaS1::Spc allele transduced in Sterne ΔltaS2 | This work |
| ltaS1 ltaS3 | ltaS1::Sp-ltaS3::Km | ltaS1::Spc allele transduced in Sterne ltaS3::Km | This work |
| ltaS1 ltaS4 | ΔltaS1-ltaS4::Spc | ltaS4::Spc allele transduced in Sterne ΔltaS1 | This work |
| ltaS2 ltaS3 | ΔltaS2-ltaS3::Km | ltaS3::Km allele transduced in Sterne ΔltaS2 | This work |
| ltaS2 ltaS4 | ΔltaS2-ltaS4::Sp | ltaS4::Sp allele transduced in Sterne ΔltaS2 | This work |
| ltaS3 ltaS4 | ltaS3::Km-ltaS4::Sp | ltaS4::Spc allele transduced in Sterne ltaS3::Km | This work |
| ltaS1 ltaS3-4 | ΔltaS1-ltaS3::Km-ltaS4::Sp | ltaS4::Spc and ltaS3::Km alleles transduced in Sterne ΔltaS1 | This work |
| Staphylococcus aureus strains | |||
| ANG499 pitet with no insert | spac-ltaSSA tet | S. aureus with IPTG (spac)-inducible ltaSSA | 17 |
| ANG499 pitet with ltaSSA | spac-ltaSSA tet-ltaSSA | S. aureus with both (spac) IPTG- and anhydrotetracycline (tet)-inducible ltaSSA | 17 |
| ANG499 pitet with ltaS1 | spac-ltaSSA tet-ltaS1 | S. aureus with IPTG (spac)-inducible ltaSSA and anhydrotetracycline (tet)-inducible ltaS1 | This work |
| Vectors and plasmids | |||
| pJK4 | No insert | Empty vector JK4 with no insert | 24 |
| pltaS1 | ltaS1 | Plasmid pJK4 carrying ltaS1 under the control of the IPTG-inducible (spac) promoter | This work |
| pltaS2 | ltaS2 | Plasmid pJK4 carrying ltaS2 under the control of the IPTG-inducible (spac) promoter | This work |
| pltaS3 | ltaS3 | Plasmid pJK4 carrying ltaS3 under the control of the IPTG-inducible (spac) promoter | This work |
| pltaS4 | ltaS4 | Plasmid pJK4 carrying ltaS4 under the control of the IPTG-inducible (spac) promoter | This work |
| pPROEX-sltaS1 | Soluble domain of LtaS1 (sLtaS1) | Plasmid used for production of recombinant sLtaS1 | This work |
| pPROEX-sltaS2 | Soluble domain of LtaS2 (sLtaS2) | Plasmid used for production of recombinant sLtaS2 | This work |
| pPROEX-sltaS3 | Soluble domain of LtaS3 (sLtaS3) | Plasmid used for production of recombinant sLtaS3 | This work |
| pPROEX-sltaS4 | Soluble domain of LtaS4 (sLtaS4) | Plasmid used for production of recombinant sLtaS4 | This work |
Table 2.
Sequences of primers used in this study
| Name | Sequence | Use |
|---|---|---|
| BAS2737 KO 1Kb UP XmaI F | TTTCCCGGGGTACGACGTTTTCGTGCTAAG | Allelic replacement of ltaS1 |
| BAS2737 KO 1Kb UP KpnI R | TTTGGTACCCGGAAGGGGAAGCTAAATAA | Allelic replacement of ltaS1 |
| BAS2737 KO 1Kb DN KpnI F | TTTGGTACCATTTCTCCACCTCTTTTGATCAT | Allelic replacement of ltaS1 |
| BAS2737 KO 1Kb DN EcoRI R | TTTGAATTCAACTTCTCAATCTTACTTCCCAAGAG | Allelic replacement of ltaS1 |
| BAS2737 XbaI F | TTTTCTAGAGGATGATCAAAAGAGGTGGAG | Expression of LtaS1 in E. coli |
| BAS2737 KpnI R | TTTGGTACCTTATTTAGCTTCCCCTTCCG | Expression of LtaS1 in E. coli |
| BAS2737 AvrII F | TTTCCTAGGGGATGATCAAAAGAGGTGGAG | Expression of LtaS1 in S. aureus |
| BAS2737 BamHI R | TTTGGATCCTTATTTAGCTTCCCCTTCCG | Expression of LtaS1 in S. aureus |
| BAS2737 BamHI F | TTTGGATCCGATGGAGATAATATGACGGAAGT | Cloning of sLtaS1 |
| BAS2737 HindIII R | TTTAAGCTTTTATTTAGCTTCCCCTTCCGG | Cloning of sLtaS1 |
| BAS5081 KO 1Kb UP XmaI F | TTTCCCGGGCAACCGGAAGGGATATTAAGTTTA | Allelic replacement of ltaS2 |
| BAS5081 KO 1Kb UP KpnI R | TTTGGTACCTTTCATTCATTTCCACCCTTTCTC | Allelic replacement of ltaS2 |
| BAS5081 KO 1Kb DN KpnI F | TTTGGTACCGAGTAATCTCAGCCTCTTTTTTTATA | Allelic replacement of ltaS2 |
| BAS5081 KO 1Kb DN EcoRI R | TTTGAATTCTGATATCACAGCGTAGCCAG | Allelic replacement of ltaS2 |
| BAS5081 XbaI F | TTTTCTAGACGACCAAGCCTGATAGAGAAA | Expression of LtaS2 in E. coli and B. anthracis |
| BAS5081 KpnI R | TTTGGTACCTTACTCAGCGCTCTTCTTTTCTT | Expression of LtaS2 in E. coli and B. anthracis |
| BAS5081 BamHI F | TCTTGGATCCGACGGTAGTAAGTTACAGGAAAC | Cloning of sLtaS2 |
| BAS5081 HindIII R | GTCCAAGCTTTTACTCAGCGCTCTTCTTTTCTTC | Cloning of sLtaS2 |
| BAS3608 KO 1Kb UP XmaI F | TTTCCCGGGGATTTCACAGCAACTACGTACTGT | Allelic replacement of ltaS3 |
| BAS3608 KO 1Kb UP KpnI R | TTTGGTACCTAAATAAAAAGTAAAAATGTGGAAGAAATATGTTAAAT | Allelic replacement of ltaS3 |
| BAS3608 KO 1Kb DN KpnI F | TTTGGTACCGAAAAATAATGTAAATATGAAAAGGGAGCAA | Allelic replacement of ltaS3 |
| BAS3608 KO 1Kb DN EcoRI R | TTTGAATTCGGATGCTATTAAGTGGAATGAAGATAT | Allelic replacement of ltaS3 |
| BAS3608 XbaI F | TTTTCTAGAAAGTTAAATAAATTGTATAAAGAAGGTGTAAAAATG | Expression of LtaS3 in E. coli |
| BAS3608 KpnI R | TTTGGTACCTTATTTTTCAGTTTCTTTAATTTTAGTTTGTACTTC | Expression of LtaS3 in E. coli |
| BAS3608 BamHI F | TCTTGGATCCGATAGTAGCAAATTACAAGAAACAGAAAA | Cloning of sLtaS3 |
| BAS3608 HindIII R | GTCCAAGCTTTTATTTTTCAGTTTCTTTAATTTTAGTTTGTACTTC | Cloning of sLtaS3 |
| BAS1327 XbaI F | TTTTCTAGAGAACTGTCATGCTTTTTAGGAGAG | Expression of LtaS4 in E. coli |
| BAS1327 KpnI R | TTTGGTACCCTATTCAAATTTCGTTGTCATCG | Expression of LtaS4 in E. coli |
| BAS1327 BamHI F | TTTGGATCCAGTGGGGACGGGTTCTCTG | Cloning of sLtaS4 |
| BAS1327 HindIII R | TTTAAGCTTCTATTCAAATTTCGTTGTCATCGTTC | Cloning of sLtaS4 |
Fig 1.
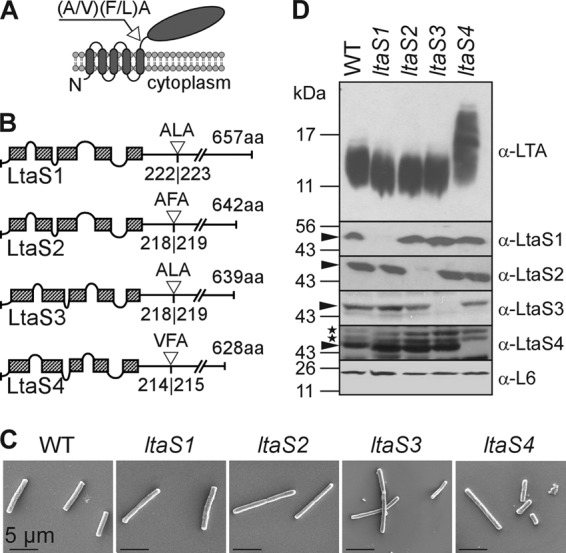
LtaS enzymes of B. anthracis. (A) Predicted topology of LtaS homologues in the plasma membrane of B. anthracis. LtaS proteins are embedded in the plasma membrane via five predicted transmembrane helices located at the N terminus of the proteins, allowing for extracellular localization of the larger catalytic domain at the C-terminal end of the protein. A single type I signal peptidase cleavage site is found following the last hydrophobic stretch, and signal peptidase cleavage effectively releases the catalytic domain (shown with arrow). (B) Linear models of LtaS enzymes of B. anthracis showing the amino acid sequences of predicted cleavage sites (shown with open triangle) and lengths of proteins. The hatched boxes represent hydrophobic regions that span the bilayer. aa, amino acids. (C) Scanning electron microscopy analysis of bacilli (wild type and ltaS1, ltaS2, ltaS3, and ltaS4 mutants) serially dehydrated and sputter coated with 80% platinum–20% palladium to 8 nm. Scale bar, 50 μm. (D) Synthesis of LTA in B. anthracis. Shown are immunoblot analyses of bacterial extracts isolated from the WT and ltaS1, ltaS2, ltaS4, and ltaS4 mutants by using antibodies specific for LTA, LtaS1, LtaS2, LtaS3, LtaS4, and ribosomal protein L6 (L6, loading control). The migration of protein size standards on SDS-PAGE gels is indicated in kilodaltons. Arrowheads point to processed LtaS proteins, and stars depict the positions of nonspecific cross-reactive immune species.
Inducible expression of ltaS1 in S. aureus.
Primers were used to amplify ltaS1 from B. anthracis and ligated with vector carrying the Ptet promoter as described earlier (17). The recombinant plasmid was inserted into the chromosome of S. aureus strain ANG499 (17), yielding the S. aureus strain with IPTG-inducible ltaSSA (Pspac-ltaSSA) and anhydrotetracycline-inducible ltaS1 (Ptet-ltaS1). Strains ANG513 (ANG499 Ptet with no insert) and ANG514 (ANG499 Ptet-ltaSSA) were used as controls (17). All primer sequences used for this study are listed in Table 2.
Immunoblot analyses.
B. anthracis cultures were grown in BHI and normalized to an optical density at 600 nm (OD600) of 5. Two milliliters of normalized culture was mixed with 1 ml of 0.1-mm glass beads to disrupt the bacterial envelope with a bead beater (BioSpec Products, Bartlesville, OK) by shearing five times for 2 min at 4°C. Glass beads and cell debris were sedimented by centrifugation at 200 × g for 1 min. Two samples of 0.5 ml of supernatant were transferred to two new tubes. One sample was used for analysis of LTA and centrifuged at 16,000 × g for 15 min. Sedimented material was suspended in 100 μl of sample buffer with 2% SDS and boiled for 20 min. The samples were subsequently centrifuged at 16,000 × g for 5 min, and SDS-solubilized LTA was subjected to SDS-PAGE and immunoblotting. Proteins in the second sample were precipitated with 10% trichloroacetic acid for 10 min on ice and sedimented at 16,000 × g for 10 min. Protein precipitates were washed with ice-cold acetone, sedimented (16,000 × g for 10 min), and air dried. Proteins were suspended in 100 μl of sample buffer with 2% SDS. Aliquots (10 μl) of samples were separated on 15% (vol/vol) polyacrylamide (PAA) gels and electrotransferred to polyvinylidene difluoride (PVDF) membranes. Polyglycerol phosphate was detected with LTA-specific monoclonal antibody (clone 55; HyCult Biotechnology, Uden, The Netherlands) and horseradish peroxidase (HRP)-linked anti-mouse antibodies (Cell Signaling Technology, Danvers, MA) at 1:500 and 1:5,000 dilutions, respectively. LtaS1, LtaS2, LtaS3, LtaS4, ribosomal L6, SrtA, and RpoA protein species were detected by incubation with polyclonal sera raised in rabbits using recombinant proteins purified in E. coli as described previously (1). Immunoreactive signals were detected by enhanced chemiluminescence detection.
Cultures of E. coli transformants harboring pJK4 without an insert or with either the ltaS1, ltaS2, ltaS3, or ltaS4 gene were diluted from overnight cultures and grown at 37°C for 16 h in the presence of 1 mM IPTG. Culture aliquots of E. coli harboring pJK4-ltaS1 were also collected after 1.5, 4.5, and 16 h following dilution without addition of IPTG. Cultures were all normalized to an OD600 of 5. Five milliliters of normalized culture was collected and bacteria sedimented by centrifugation at 16,000 × g for 10 min. Bacteria were lysed by boiling for 20 min in 500 μl of SDS sample buffer containing 2% SDS, and insoluble material was sedimented by centrifugation at 16,000 × g for 5 min. Sample aliquots (10 μl) were separated on 15% PAA gels, electrotransferred to PVDF membranes, and used for immunoblotting as described above.
Assessing bacterial growth and sporulation efficiency.
Growth of vegetative bacilli was compared between strains by monitoring the density of cultures and determining CFU counts of three independent cultures. Bacilli grown overnight were diluted 100-fold into 30 ml of LB medium and incubated at 37°C with shaking. Culture aliquots were withdrawn at defined time intervals, and OD600s were recorded. Aliquots were serially diluted in 10-fold increments and plated on LB agar for enumeration of CFU following overnight incubation of plates at 37°C. Sporulation was assessed by diluting 10-fold overnight cultures of bacilli grown in BHI into modified G medium (25) followed by incubation at 30°C with shaking. Aliquots of cultures were removed at 24 and 72 h and heated or not for 150 min at 65°C to kill vegetative bacilli. Both nonheated and heated aliquots were serially diluted and plated on solid medium for CFU enumeration. Sporulation efficiency was calculated as follows: (viable spores)/(total CFU) × 100.
Light microscopy.
Cells were fixed using 4% buffered formalin and observed. Images were obtained with a charge-coupled-device (CCD) camera on an Olympus IX81 microscope using 100× and 40× objectives. The lengths of bacilli was measured directly from acquired differential interference contrast microscopy (DIC) images using ImageJ and converted to lengths in micrometers using reference images with an objective micrometer. The data were displayed in a box and whisker plot.
Scanning electron microscopy.
Bacteria grown on solid medium were suspended in water, washed twice, and fixed for 30 min with 2% glutaraldehyde in phosphate-buffered saline (PBS) at room temperature, and they were then postfixed for 30 min in glutaraldehyde onto freshly prepared poly-l-lysine-coated glass coverslips. Samples were washed twice with PBS and subsequently serially dehydrated by consecutive incubations in 25% and 50% ethanol-PBS, 75% and 90% ethanol-H2O, and 100% ethanol (twice), followed by 50% ethanol-hexamethyldisilazane (HDMS) and 100% HDMS. After overnight evaporation of HDMS at room temperature, samples were mounted onto specimen mounts (Ted Pella, Inc., Redding, CA) and coated with 80% Pt–20% Pd to 8 nm using a Cressington 208HR sputter coater at 20 mA prior to examination with a Fei Nova NanoSEM 200 scanning electron microscope (FEI Co., Hillsboro, OR). The scanning electron microscope (SEM) was operated with an acceleration voltage of 5 kV, and samples were viewed at a distance of 5 mm.
Thin sectioning and transmission electron microscopy.
Bacteria grown on solid medium were harvested from plates, washed twice in water, and fixed with 2% glutaraldehyde–4% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) for 2 h. Fixed cells were washed three times with sodium cacodylate buffer and postfixed for 60 min in sodium cacodylate buffer containing 1% osmium tetroxide. Following these treatments, cells were washed twice with sodium cacodylate buffer and once with 0.04 M maleate buffer (pH 5.1) and subsequently stained for 60 min with 1% uranyl acetate in maleate buffer (pH 6.0). Cells were washed three times with maleate buffer prior to dehydration by successive incubations with 25%, 50%, 70%, 90%, and 100% ethanol and 100% propylene oxide. Cells were infiltrated by two consecutive incubations in 66% propylene oxide–33% Spurr resin and 50% propylene oxide–50% Spurr resin for 30 min, followed by 100% Spurr resin for 6 h, and finally incubated overnight at 60°C. Ninety-nanometer thin sections were cut using a Reichert-Jung (Scotia, NY) Ultracut E microtome and attached to copper grids (200 mesh). Thin sections were subsequently negatively stained using saturated uranyl acetate and lead citrate (Electron Microscopy Services, Hatfield, PA). Images were recorded using a Tecnai F30 (Philips/FEI) transmission electron microscope (field emission gun operating with a 300-kV accelerating voltage, using a magnification of ×15,000 to ×30,000) and a high-performance CCD camera with a resolution of 4,000 by 4,000 pixels. Images were captured using Gatan DigitalMicrograph software and processed using Adobe Photoshop (Adobe, San Jose, CA).
Quantification of cell envelope thickness.
The thickness of the cell wall envelope was determined by examining at least 15 thin-sectioned cells observed on micrographs obtained by transmission electron microscopy. Sites along the cell envelope were randomly chosen and the number of pixels (thickness) measured using the measure tool in Adobe Photoshop. The conversion from pixels to thickness (nanometers) was achieved based on the number of pixels corresponding to the length of the scale bar (in nm) of the micrograph. Data were plotted in Graphpad Prism 5.0, and the Student t test (unpaired, 2-tailed) was used for statistical analyses.
RESULTS
ltaS genes of B. anthracis.
A genome search for homologues of the S. aureus ltaS gene in B. anthracis revealed four genes, BAS2737, BAS5081, BAS3608, and BAS1327, predicted to encode putative sulfatases. The corresponding proteins were found to share 51%, 42%, 41%, and 38% identity with LtaSSA, respectively (E values, 0 [BAS2737], 3e−159 [BAS5081], 2e−161 [BAS3608], and 7e−143 [BAS1327]). Sequence alignments and comparisons are provided in Table S1 and Fig. S1 in the supplemental material. The corresponding genes are designated herein ltaS1 (BAS2737), ltaS2 (BAS5081), ltaS3 (BAS3608), and ltaS4 (BAS1327) and code for predicted products of 657, 642, 639, and 628 amino acids, respectively (Fig. 1A). Several features of S. aureus LtaS are conserved in these proteins (Fig. 1A and B). All proteins contain hydrophobic domains predicted to span the plasma membrane five times, with short intervening loops (17). The fifth hydrophobic segment is followed by a predicted signal peptidase I recognition motif (Fig. 1A and B). This processing site has been confirmed for LtaSSA (39, 44), and a similar processing is observed for the LtaS enzymes of B. anthracis (see below).
B. anthracis mutants with mutations in individual ltaS genes.
To examine the function of ltaS genes, in-frame deletions in the ltaS1, ltaS2, or ltaS3 gene of B. anthracis strain Sterne (pXO1+ pXO2−) were generated by allelic replacement using a plasmid with a conditional replication defect (30) (ΔltaS1, ΔltaS2, and ΔltaS3 in Table 1). B. anthracis Sterne mutants with insertional lesions of the bursa aurealis element in ltaS1, ltaS3, or ltaS4 were identified in the Sphinx collection of transposon insertion mutants (41) (ltaS1::Sp, ltaS3::Km, and ltaS4::Sp in Table 1). These alleles were crossed back into the B. anthracis Sterne parent or ΔltaS mutant strain using bacteriophage CP-51 (15). B. anthracis variants with deletions or bursa aurealis insertions in single ltaS genes were not impaired for bacterial growth, as judged by measuring the optical density of replicating vegetative forms in liquid medium (data not shown). A comparison of bacterial shape and cellular morphology using scanning electron microscopy did not reveal any major difference between vegetative wild-type (WT) and ltaS mutant bacilli (Fig. 1C). Subtle differences in cell width or length were not accompanied by defects in cell separation or alteration in envelope thickness or sporulation (see Fig. 4 and 5D). Extracts derived from the vegetative forms of B. anthracis Sterne and its ltaS1::Sp, ΔltaS2, ltaS3::Km, and ltaS4::Sp mutants were separated on SDS-PAGE gels, transferred to nitrocellulose membranes, and examined for the presence of LTA using a polyglycerol phosphate-specific monoclonal antibody (16) (Fig. 1D). Extracts from ltaS1, ltaS2, and ltaS3 mutants harbored LTA molecules with abundances and mobilities on SDS-PAGE similar to those of the B. anthracis Sterne parent strain (Fig. 1D). Extracts of the ltaS4 mutant also harbored significant amounts of LTA; however, its polyglycerol phosphate molecules migrated with slower mobility and greater heterogeneity than the LTA of B. anthracis Sterne (Fig. 1D).
Fig 4.
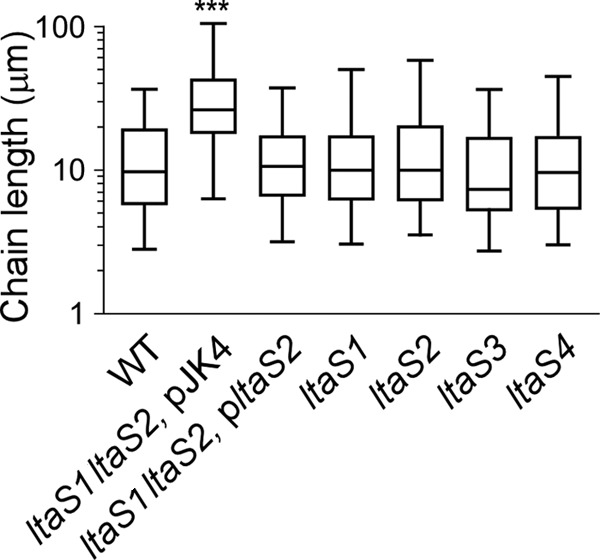
Box and whisker plot of the chain lengths of B. anthracis Sterne (WT) and variants with mutational lesions in ltaS1 and ltaS2 harboring or not pltaS2, as well as ltaS1, ltaS2, ltaS3, or ltaS4, grown on LB agar. Chain lengths were measured from DIC micrographs of vegetative bacilli (n = 100; micrographs not shown). ***, statistical significance was established with the unpaired, two-tailed Student t test (P < 0.001).
Fig 5.

Electron microscopy analysis of vegetative bacilli. (A) Scanning electron microscopy analysis of bacilli (WT, ltaS1 ltaS2 pJK4 [double mutant carrying the empty vector], and ltaS1 ltaS2 pltaS2 [complemented strain]) serially dehydrated and sputter coated with 80% platinum–20% palladium to 8 nm. Cells of wild-type and complemented mutant strains appear as rod-shaped bacilli, whereas the ltaS1 ltaS2 mutant bacilli remain tethered in long chains. Scale bar, 50 μm. (B) Thin-section transmission electron microscopy of the same cells as in panel A showing the irregular and deformed structure of the ltaS1 ltaS2 mutant compared to that of the WT and complemented strains. Scale bar, 200 nm. (C) Close-up of the cell envelope of thin-sectioned bacilli shown in panel B. The layers, from bottom to top, represent cytoplasm, membrane (thin darker line), envelope, and environmental milieu. Scale bar, 20 nm. (D) Measurement of the thickness of the cell envelope of bacilli. Measurements were performed using transmission electron micrographs as shown in panels B and C. Each dot represents one measurement.
To determine whether LtaS enzymes are expressed in vegetative forms of wild-type and mutant bacilli, we used PCR amplification to clone the coding sequences for the enzymatic domains of LtaS1, LtaS2, LtaS3, and LtaS4 into expression vectors. Thus, only the coding sequences following the predicted signal peptidase I recognition motif as shown on Fig. 1B were included in these clones. His-tagged proteins were purified by affinity chromatography for antibody production in rabbits. Antisera were used for immunoblotting experiments to detect LtaS enzymes in extracts of vegetative forms derived from B. anthracis wild-type and ltaS mutants (Fig. 1D). The extracts of B. anthracis Sterne vegetative forms harbored immunoreactive signals for each of the four enzymes LtaS1 to -4 (Fig. 1D). The electrophoretic mobilities of the four immunoreactive species on 10% SDS-PAGE were consistent with an observed molecular mass of approximately 50 kDa for each one of the four enzymes (Fig. 1D), in agreement with the calculated mass of the signal peptidase cleavage products corresponding to 49,452 Da (LtaS1), 48,749 Da (LtaS2), 48,674 Da (LtaS3), and 473,73 Da (LtaS4). Without processing, the proteins would otherwise display molecular masses of 74,634, 73,469, 73,406, and 72,227 Da, respectively (Fig. 1D). Each of the four ltaS1 to -4 mutant strains lacked an immunoreactive signal for the corresponding LtaS enzyme (Fig. 1D). Of note, deletion or insertion mutations in individual ltaS genes did not impact the abundance of the remaining three LtaS enzymes (Fig. 1D). As a control, similar intensities of immunoreactive signals for ribosomal protein L6 were detected in the extracts of wild-type and mutant B. anthracis strains (Fig. 1D).
LTA synthesis is abrogated in ltaS1 ltaS2 mutant B. anthracis.
To test whether LtaS enzymes in B. anthracis fulfill redundant functions, we generated variants lacking any two of the four ltaS genes (Fig. 2A). Extracts of vegetative forms that had been derived from the isolated mutants were subjected to immunoblotting, which revealed that the ltaS1 ltaS2 mutant, but not the ltaS1 ltaS3, ltaS1 ltaS4, ltaS2 ltaS3, ltaS2 ltaS4, or ltaS3 ltaS4 variant, was defective for LTA synthesis (Fig. 2A). Of note, the electrophoretic mobility of the LTA of the ltaS1 ltaS3 and ltaS2 ltaS3 variants was similar to that of the B. anthracis Sterne parent (Fig. 2A). LTA molecules of ltaS1 ltaS4, ltaS2 ltaS4, and ltaS3 ltaS4 variants migrated more slowly than and at a speed similar to that of the LTA of the ltaS4 mutant (Fig. 2A). Further, a strain lacking three ltaS genes—ltaS1, ltaS3, and ltaS4–-produced LTA with the same electrophoretic mobility as that of the ltaS4 mutant (Table 1 and Fig. 2A). Repeated attempts to isolate a B. anthracis mutant lacking all four ltaS genes failed. The LTA synthesis defect of the ltaS1 ltaS2 mutant was restored following transformation with pltaS2, a plasmid expressing the ltaS2 gene, but not by the pJK4 vector plasmid (Fig. 2B). The growth of B. anthracis vegetative forms in laboratory media was not impaired in strains harboring ltaS1 ltaS3, ltaS1 ltaS4, ltaS2 ltaS3, ltaS2 ltaS4, ltaS3 ltaS4, or ltaS1 ltaS3 ltaS4 mutations (Fig. 2C). In contrast, mutants lacking the ltaS1 and ltaS2 genes displayed a significant defect in growth. The growth defect of ltaS1 ltaS2 mutants is due to the generation of daughter cells that are nonviable, as the plating of aliquots from mutant cultures produced 1,000-fold-fewer colonies on agar media than the plating of aliquots of B. anthracis Sterne cultures with equal OD600s (Fig. 2D). The defect in bacterial growth and the generation of viable daughter cells were restored to wild-type levels in the ltaS1 ltaS2 mutant harboring pltaS2 but not by the ltaS1 ltaS2 mutant with pJK4 vector control (Fig. 2D). Overproduction of LtaS1 from the pJK4 plasmid was poorly tolerated in all species (including E. coli and S. aureus), and thus, the complementation studies were performed with the pJK4-ltaS2 (pltaS2) plasmid exclusively.
Fig 2.
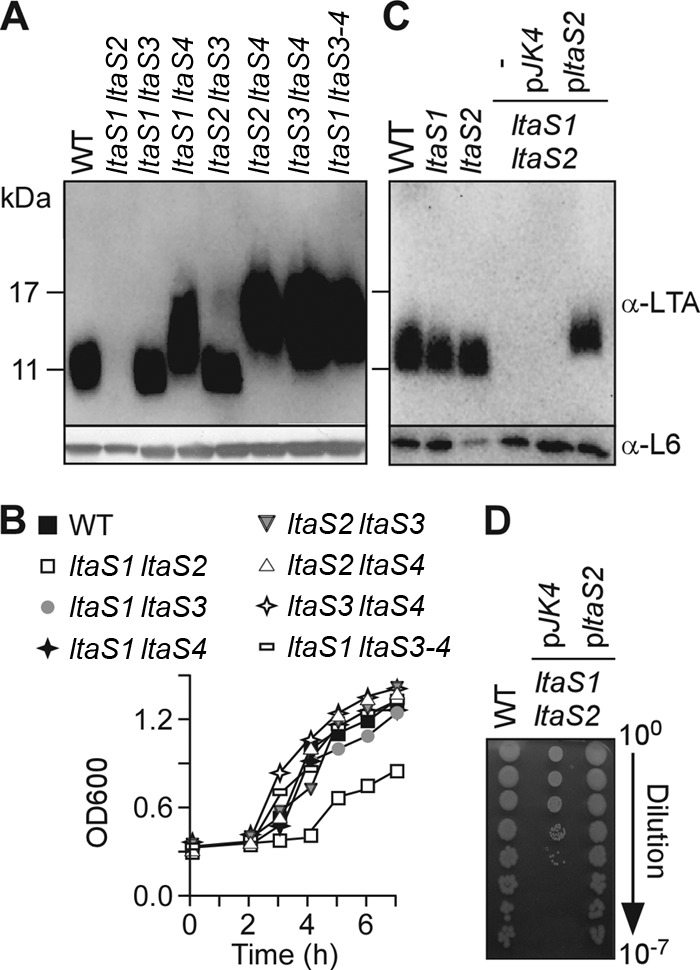
B. anthracis ltaS1 and ltaS2 are required for LTA synthesis and bacterial growth. (A) Immunoblot analyses of bacterial extracts isolated from the WT, the ltaS1 ltaS2, ltaS1 ltaS3, ltaS1 ltaS4, ltaS2 ltaS3, ltaS2 ltaS4, and ltaS3 ltaS4 double mutants, and the ltaS1 ltaS3 ltaS4 triple mutant by using antibodies specific for LTA, and ribosomal protein L6 (L6, loading control). The migration of protein size standards on SDS/PAGE gels is indicated in kilodaltons. (B) Growth curves of bacterial strains shown in panel A. Bacteria were diluted in BHI and grown at 37°C with shaking. Bacterial growth was monitored by optical density measurements of cultures at 600 nm (OD600). (C) Expression of ltaS2 on a plasmid restores LTA synthesis in the ltaS1 ltaS2 double mutant. Immunoblot analyses were performed as for panel A to compare LTA production among the WT, the ltaS1 or ltaS2 mutant, and the ltaS1 ltaS2 double mutant carrying no plasmid (minus), the plasmid with no insert (pJK4), or the plasmid carrying ltaS2 (pltaS2). (D) Expression of ltaS2 on a plasmid restores growth of the ltaS1 ltaS2 mutant. Bacterial growth was examined by plating serial dilutions (0- to 7-fold) for the WT, ltaS1 ltaS2 pJK4, and ltaS1 ltaS2 pltaS2.
LTA synthesis promoted by B. anthracis LtaS enzymes in heterologous hosts.
Expression of S. aureus ltaS from a plasmid with an inducible promoter in E. coli leads to the synthesis of polyglycerol phosphate LTA from phosphatidylglycerol (17). To test whether each of the four LtaS enzymes of B. anthracis is sufficient to promote synthesis of LTA, each gene was expressed from a plasmid in E. coli (Fig. 3A to C). Production of all the enzymes was observed upon addition of an inducer (IPTG) at mid-logarithmic growth; of note, LtaS4 was produced in moderate amounts compared to LtaS1, LtaS2, and LtaS3 (Fig. 3B). Immunoblotting experiments with bacterial extracts revealed the presence of polyglycerol phosphate in cells harboring pltaS2 and pltaS3 but not in vector control cells (Fig. 3A). LtaS4-mediated production of polyglycerol phosphate was never observed regardless of the medium or density of the cell culture. Expression of ltaS1 was poorly tolerated in E. coli, and polyglycerol phosphate production could be observed only in cultures grown in the absence of an inducer (IPTG) to mid-logarithmic growth (Fig. 3C). To analyze the function of ltaS1 further, we used an S. aureus mutant in which the ltaS gene was placed under the control of the IPTG-inducible Pspac promoter (17) and the B. anthracis ltaS1 gene was cloned under the control of the anhydrotetracycline-inducible Ptet promoter in the chromosome (Fig. 3D). When grown at 37°C in the presence of anhydrotetracycline (and in the absence of IPTG), S. aureus expressing B. anthracis ltaS1 formed colonies on TSB agar (Fig. 3E). As a control, the staphylococcal mutant without an insertion of ltaS at the Ptet promoter did not form colonies (Fig. 3E). This result is in agreement with earlier findings that LTA synthesis is essential for S. aureus growth at 37°C (17). A variant harboring the staphylococcal ltaSSA gene under the control of Ptet generated similar numbers of viable colonies as the mutant expressing B. anthracis ltaS1 (Fig. 3E). Staphylococcal extracts were subjected to immunoblotting with monoclonal antibody specific for polyglycerol phosphate. S. aureus Pspac-ltaS mutant cultures diluted into medium without the inducer IPTG produced only small amounts of immunoreactive LTA. This defect in LTA synthesis was alleviated when Pspac-ltaS mutants harbored a chromosomal insertion of Ptet-ltaSSA or of Ptet-ltaS1 (Fig. 3F). These two strains produced similar amounts of immunoreactive LTA; however, the polyglycerol phosphate of the Ptet-ltaS1 mutant strain migrated more slowly on SDS-PAGE than that of Ptet-ltaSSA mutant staphylococci, suggesting that the B. anthracis LtaS1 enzyme may generate LTA molecules with increased chain length compared to that of the LTA molecules produced by staphylococcal LtaS (Fig. 3F). Taken together, the results in Fig. 3 indicate that B. anthracis ltaS1 and ltaS2 encode polyglycerol phosphate synthases with redundant or partially overlapping functions, which may explain why each of the two genes can be deleted without affecting B. anthracis growth.
Fig 3.

B. anthracis LtaS1, LtaS2, and LtaS3 catalyze LTA synthesis in heterologous hosts. (A to C) Expression of B. anthracis LtaS1, LtaS2, and LtaS3 in E. coli promotes synthesis of polyglycerol phosphate. E. coli strains carrying pJK4 with no insert (minus) or with an insert (ltaS1, ltaS2, ltaS3, and ltaS4) were grown for 16 h in the presence of IPTG (A and B), and E. coli pJK4-ltaS1 was grown for 1.5, 4.5, and 16 h in the absence of IPTG (C). Extracts from normalized culture aliquots were separated by SDS-PAGE for immunoblot detection of LTA (A to C), LtaS enzymes, and RpoA (loading control) using specific antibodies (B). (D to F) Functional complementation of ltaS depletion in S. aureus with B. anthracis ltaS1 homologue. (D) Schematic representation of chromosomal organization in ltaS complementation strains. S. aureus cultures of strains carrying either no insert (minus), S. aureus ltaSSA, or B. anthracis ltaS1 at position X were grown overnight in the presence of IPTG. The following day, cultures were washed and diluted in fresh medium containing anhydrotetracycline only and analyzed for restoration of staphylococcal growth (E) as described for Fig. 2D and LTA production (F) by using antibodies specific for LTA and sortase A (SrtA; loading control). The values to the left of panels A, C, and F indicate the migration of protein size standards, in kilodaltons.
Microscopic visualization of wild-type and ltaS mutant B. anthracis Sterne.
Vegetative forms of B. anthracis Sterne and the four ltaS mutants (ltaS1 to -4) were subjected to scanning electron microscopy. Both wild-type and ltaS mutant strains appeared as rod-shaped cells or short chains, and they did not display major differences in their cell morphology (Fig. 1D). In contrast, cells of the ltaS1 ltaS2 double mutant appeared as filamentous chains. To measure the chain length of bacilli, strains were grown on plates and isolated colonies were suspended in liquid medium and analyzed by light microscopy (Fig. 4; 100 chains were measured for each strain). B. anthracis Sterne formed chains that were, on average, 9.86 (±0.66) μm in length. Chains of each single mutant displayed similar lengths, with perhaps the exception of the ltaS3 mutant, which seemed to display slightly shorter chains. Yet this difference did not reach statistical significance. The average chain length of the ltaS1 ltaS2 mutant with empty vector was increased to 24.69 (±1.51) μm (WT versus ltaS1 ltaS2 mutant, P < 0.0001). The presence of pJK4 was not responsible for the increased chain length (data not shown). The chain length phenotype of the ltaS1 ltaS2 mutant was reduced when ltaS1 ltaS2 bacilli were transformed with plasmid pltaS2 carrying wild-type ltaS2 (12.43 [±0.691] μm; WT versus ltaS1 ltaS2 pltaS2 mutant, P < 0.001). Of note, compared to B. anthracis Sterne, the dimensions of individual cells from the ltaS1 ltaS2 mutant were not altered. This increase in chain length was also observed by scanning electron microscopy (Fig. 5A).
Transmission electron microscopy of thin-sectioned vegetative forms was used to visualize the envelope of B. anthracis wild type and ltaS1 ltaS2 pJK4 and ltaS1 ltaS2 pltaS2 mutant cells (Fig. 5B and C). Images of the envelope of B. anthracis Sterne revealed a compact, layered appearance with an average diameter of 35.7 (±0.7) nm (n = 72) (Fig. 5D). The envelope of the ltaS1 mutant displayed comparable morphology, although its diameter was slightly increased (38.4 [±1.1] nm, n = 61; WT versus ltaS1 mutant, P = 0.035) (Fig. 5D). The envelope of the ltaS2 mutant appeared somewhat disordered, and its diameter was also increased (40.9 [±0.8] nm, n = 71; WT versus ltaS2 mutant, P < 0.001) (Fig. 5D). Finally, images of the envelope of the ltaS1 ltaS2 mutant revealed a general disorder in the otherwise organized structure of the B. anthracis envelope comprised of peptidoglycan layer, attached secondary cell wall polysaccharide, and S-layer (13, 31) (Fig. 5C). The diameter of the ltaS1 ltaS2 mutant envelope was determined to be 49.3 (±1.0) nm (n = 71), significantly increased in comparison with that of the wild-type or ltaS1 or ltaS2 mutant strain (P < 0.001; Fig. 5D). We investigated whether the ltaS1 and ltaS2 mutations affect the retention of S-layer proteins Sap and EA1 in the bacterial envelope, but we observed no difference with S-layer protein retention compared to that with the wild-type parent strain (data not shown).
LTA synthesis is required for B. anthracis sporulation.
When nutrients are limiting, B. anthracis vegetative cells differentiate into spores, a dormant structure carrying the genetic material of the bacterium (27). The transition from vegetative cells to spores is a series of morphological events that require major remodeling of this microbe's envelope (6). We wondered whether the ltaS genes might contribute to sporulation. B. anthracis Sterne, wild type and ltaS mutants, was grown in modified G medium at 30°C to trigger sporulation. The efficacy of spore formation was assessed by plating culture aliquots for colony formation with and without prior treatment at 65°C, a condition that kills vegetative bacilli but not spores. The efficiency of spore formation was calculated as the ratio of viable spores (heat-resistant CFU) to the total CFU count (no heat treatment) at 24 and 72 h following incubation in modified G medium. The data revealed that the ltaS1 ltaS2 mutant was unable to form heat-resistant spores, whereas mutants with defects in single genes (ltaS1, ltaS2, ltaS3, and ltaS4) did not display a defect (Fig. 6A).
Fig 6.

LtaS1 and LtaS2 are required for spore formation. (A) Sporulation efficiency of wild-type and mutant strains grown in modified G medium for 24 or 72 h and reported as percentage of heat-resistant organisms over total organisms (non-heat treated). Aliquots of cultures were removed at the indicated times, treated or not at 65°C, and plated on solid medium for enumeration of CFU. (B) Spore formation visualized by transmission electron microscopy following 16, 40, and 136 h of incubation of bacteria in modified G medium (WT, ltaS1 ltaS2 pJK4 [double mutant carrying the empty vector], and ltaS1 ltaS2 pltaS2 [complemented strain]).
To further characterize the defect in sporulation of the ltaS1 ltaS2 mutant, aliquots of B. anthracis cultures were subjected to transmission electron microscopy (Fig. 6B). Forty hours after the dilution of vegetative forms into G medium, B. anthracis Sterne cultures harbored only endospores. At the same time point and at 136 h, the ltaS1 ltaS2 mutant cultures harbored elongated vegetative bacilli but no spores. This defect was partially overcome when the ltaS1 ltaS2 mutant carried pltaS2, although the formation of spores was significantly delayed (Fig. 6B). In agreement with this result, the efficiency of spore formation in the ltaS1 ltaS2 (pltaS2) mutant strain was reduced to 63% and, in contrast to that of the wild type, did not increase over time (Fig. 6A). These data indicate that LTA synthesis of B. anthracis Sterne is absolutely required for spore formation.
DISCUSSION
LTA synthesis can be divided into the assembly of the glycolipid anchor, the polymerization of polyglycerol phosphate, and the decoration of LTA glycerol 2′OH with d-alanyl esters or N-acetylglucosamine (33, 38). In S. aureus, the membrane anchor moiety for LTA is composed of β-gentiobiosyldiacylglycerol [glucosyl-(1→6)-glucosyl-(1→3)-diacylglycerol (Glc2-DAG)] (7). Three enzymes—PgcA (α-phosphoglucomutase), GtaB (UTP:α-glucose-1-phosphate uridyl transferase), and YpfP (glycosyltransferase)—are required to produce Glc2-DAG (16, 20, 26). LtaA is required for moving Glc2-DAG across the membrane (16). Finally, LtaS polymerizes phosphatidylglycerol into polyglycerol phosphate while generating DAG as a secondary product (17).
The genome of B. anthracis contains homologues of pgcA (BAS4790), gtaB (BAS4789), and ypfP (BAS0483), but not ltaA. LtaA is a member of the major facilitator superfamily of proteins, of which B. anthracis encodes several different members (28). Interestingly, the glycolipid anchor of B. subtilis LTA assumes the same chemical structure as the membrane anchor of S. aureus LTA (5, 7). With the notable exception of ltaA, B. subtilis uses the same enzymes as described for the synthesis of the S. aureus glycolipid (21, 38). Similar to that of B. anthracis, the genome of B. subtilis encodes four LtaS homologues (17). Mutants lacking one of these genes, yflE (ltaSBS), grow more slowly than the wild type and display a defect in divalent cation homeostasis, an increase in chain length, and placement of aberrant septa, as well as enhanced cell bending and lysis (40). YflE (LtaSBS) has been referred to as the housekeeping synthase (40, 43). A mutant lacking all four paralogues of ltaS remains viable, albeit with severe morphological defects in cell structure and with elongated filaments of B. subtilis vegetative forms (40, 43). In vitro and in vivo biochemical analyses revealed that all four LtaS paralogues are active in cleaving phosphatidylglycerol to generate DAG. Three enzymes—YflE (LtaSBS), YfnI, and YqgS—generate polyglycerol phosphate in B. subtilis and in S. aureus (43). YfnI generates high-molecular-weight polyglycerol phosphate polymers, but their function is unknown (43). The fourth enzyme, YvgJ, adds a single glycerol phosphate moiety to Glc2-DAG, which earned the enzyme the designation LTA primase (42, 43). Of note, LTA primase expression is not required for the synthesis of glycolipid-linked LTA in either B. subtilis or L. monocytogenes (42, 43). Although the product of the LTA primase reaction is thought to be a precursor for LTA synthesis, it is also conceivable that the primase product is not a substrate for LTA synthesis and instead fulfills another function. Furthermore, there appear to be differences in the structures of the LTA products derived from YflE (LtaSBS), YfnI, and YqgS (43). YflE (LtaSBS) and YfnI generated significant amounts of LTA in S. aureus, yet only yflE (ltaSBS), not yfnI, supported the growth of ltaS-deficient S. aureus (17). B. subtilis mutants lacking ltaSBS and yfnI, but not mutants with mutations in single ltaS genes, are defective for LTA synthesis.
Here we report for the first time that B. anthracis synthesizes polyglycerol phosphate LTA molecules. Our findings are in disagreement with earlier work reporting that glycerol phosphate polymers are absent from the envelope of B. anthracis (32). The genome of B. anthracis harbors four homologues of S. aureus LtaS, and two of these, LtaS1 and LtaS2, are sufficient to synthesize polyglycerol phosphate LTA in B. anthracis and in heterologous hosts, S. aureus or E. coli. It is conceivable that one or both of the other two LtaS enzymes (LtaS3 and LtaS4) act as LTA primases (42). Alternatively, these enzymes may synthesize small amounts of LTA at unique stages or locations during the B. anthracis life cycle. We noted that ltaS1 ltaS2 mutants are defective in sporulation and vegetative growth. It seems plausible to us that LTA synthesis of B. anthracis is essential for the growth of this microbe under physiological conditions. If so, LtaS enzymes would represent a valuable target for the development of antibiotic compounds that can be used for the treatment and prevention of biodefense and emerging infectious disease threats (36). Considering the presence of the pgcA, gtaB, and ypfP genes in the chromosome of B. anthracis, we think it is likely that this organism also synthesizes Glc2-DAG membrane anchors for its LTA molecules. Our discovery of LTA in the envelope of B. anthracis suggests further that the physiological role of the microbe's dltABCD operon is likely the d-alanyl esterification of polyglycerol phosphates (12). Future work must address the chemical nature of the presumed glycolipid anchor and alanyl esterification of polyglycerol phosphates by purifying LTA molecules from the envelope of B. anthracis and determining their chemical structure.
It is intriguing that the phenotypic defects associated with specific ltaS mutants do not point to specific sequence features of the corresponding enzymes. LtaS homologues of B. anthracis, B. subtilis, and S. aureus share 38 to 78% identity at the amino acid level throughout their sequences (Table S1; Fig. S1). B. anthracis LtaS1 is most homologous to the housekeeping LTA synthase YflE (LtaSBS) and YfnI (the enzyme that produces large LTA polymers). LtaS2, LtaS3, and LtaS4 share the highest homology with YqgS (the enzyme involved in sporulation). Overall, LtaS2 and LtaS3 share the greatest amino acid sequence identity, 78%, yet the ltaS1 ltaS3 double mutant behaved mostly like the wild type, unlike the ltaS1 ltaS2 double mutant (Table S1; Fig. S1). In B. anthracis, LtaS1 and LtaS2 represent the housekeeping synthases. Defects in cell separation, growth, and sporulation are associated with failure to produce LTA in the ltaS1 ltaS2 double mutant. Similar phenotypic defects do not directly correlate with loss of LTA synthesis in B. subtilis; indeed, mutants displaying cell chaining, growth, and sporulation defects could still synthesize LTA (43). Perhaps this difference can be attributed to the fact that unlike B. anthracis, B. subtilis produces wall teichoic acid (WTA). An attempt to delete tagO, the gene that encodes the enzyme initiating synthesis of WTA in the yflE yfnI yqgS yvgJ quadruple mutant, was unsuccessful, suggesting that WTA and LTA may fulfill redundant functions in B. subtilis (40).
It has been suggested that round, spherical microbes (cocci) producing LTA—e.g., Staphylococcus spp., Streptococcus spp., and Lactococcus lactis—harbor a single ltaS gene. Organisms with more diverse cell shapes, e.g., ellipsoid microbes such as E. faecalis as well as rod-shaped Listeria spp., carry at least two ltaS genes. Finally, rod-shaped bacteria with complex developmental cycles (e.g., spore formation) such as B. subtilis encode four LtaS-like proteins. In all cases examined, abolishing LTA production by mutagenesis of cognate ltaS genes leads to morphological changes and cell division defects, suggesting that LtaS-mediated synthesis of LTA appears to fulfill a universal function for cell division.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Schneewind and Missiakas laboratories for discussions.
We acknowledge membership within and support from the Region V ‘Great Lakes' Regional Center of Excellence in Biodefense and Emerging Infectious Diseases Consortium (GLRCE, NIAID Award 1-U54-AI-057153). J.W.K. acknowledges support from the Molecular Cell Biology Training Grant (GM007183).
Footnotes
Published ahead of print 8 June 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Anderson VJ, Kern JW, McCool JW, Schneewind O, Missiakas D. 2011. The SLH-domain protein BslO is a determinant of Bacillus anthracis chain length. Mol. Microbiol. 81: 192–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Araki Y, Ito E. 1989. Linkage units in cell walls of Gram-positive bacteria. Crit. Rev. Microbiol. 17: 121–135 [DOI] [PubMed] [Google Scholar]
- 3. Baddiley J. 1968. Teichoic acids in walls and the molecular structure of bacterial walls. Proc. R. Soc. Lond. B Biol. Sci. 170: 331–348 [DOI] [PubMed] [Google Scholar]
- 4. Choudhury B, et al. 2006. The structure of the major cell wall polysaccharide of Bacillus anthracis is species specific. J. Biol. Chem. 281: 27932–27941 [DOI] [PubMed] [Google Scholar]
- 5. Coley J, Duckworth M, Baddiley J. 1972. The occurrence of lipoteichoic acids in the membranes of Gram-positive bacteria. J. Gen. Microbiol. 73: 587–591 [DOI] [PubMed] [Google Scholar]
- 6. Driks A. 2002. Maximum shields: the assembly and function of the bacterial spore coat. Trends Microbiol. 10: 251–254 [DOI] [PubMed] [Google Scholar]
- 7. Duckworth M, Archibald AR, Baddiley J. 1975. Lipoteichoic acid and lipoteichoic acid carrier in Staphylococcus aureus H. FEBS Lett. 53: 176–179 [DOI] [PubMed] [Google Scholar]
- 8. Emdur L, Chiu T-H. 1975. The role of phosphatidylglycerol in the in vitro biosynthesis of teichoic acid and lipoteichoic acid. FEBS Lett. 55: 216–219 [DOI] [PubMed] [Google Scholar]
- 9. Fischer B, et al. 1996. Novel animal model for studying the molecular mechanisms of bacterial adhesion to bone-implanted metallic devices: role of fibronectin in Staphylococcus aureus adhesion. J. Orthop. Res. 14: 914–920 [DOI] [PubMed] [Google Scholar]
- 10. Fischer W. 1990. Bacterial phosphoglycolipids and lipoteichoic acids, p 123–234. In Hanahan DJ. (ed), Handbook of lipid research, vol 6 Plenum Press, New York, NY [Google Scholar]
- 11. Fischer W, Koch HU, Rösel P, Fiedler F, Schmuck L. 1980. Structural requirements of lipoteichoic acid carrier for recognition by the poly(ribitol phosphate) polymerase from Staphylococcus aureus H: a study of various lipoteichoic acids, derivatives, and related compounds. J. Biol. Chem. 255: 4550–4556 [PubMed] [Google Scholar]
- 12. Fisher N, et al. 2006. The dltABCD operon of Bacillus anthracis Sterne is required for virulence and resistance to peptide, enzymatic, and cellular mediators of innate immunity. J. Bacteriol. 188: 1301–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fouet A. 2009. The surface of Bacillus anthracis. Mol. Aspects Med. 30: 374–385 [DOI] [PubMed] [Google Scholar]
- 14. Glaser L, Lindsay B. 1974. The synthesis of lipoteichoic acid carrier. Biochem. Biophys. Res. Commun. 59: 1131–1136 [DOI] [PubMed] [Google Scholar]
- 15. Green BD, Battisti L, Koehler TM, Thorne CB, Ivins BE. 1985. Demonstration of a capsule plasmid in Bacillus anthracis. Infect. Immun. 49: 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gründling A, Schneewind O. 2007. Genes required for glycolipid synthesis and lipoteichoic acid anchoring in Staphylococcus aureus. J. Bacteriol. 189: 2521–2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gründling A, Schneewind O. 2007. Synthesis of glycerol phosphate lipoteichoic acid in Staphylococcus aureus. Proc. Nat. Acad. Sci. U. S. A. 104: 8478–8483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heaton MP, Neuhaus FC. 1992. Biosynthesis of D-alanyl-lipoteichoic acid: cloning, nucleotide sequence, and expression of the Lactobacillus casei gene for the D-alanine-activating enzyme. J. Bacteriol. 174: 4707–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heaton MP, Neuhaus FC. 1994. Role of the D-alanyl carrier protein in the biosynthesis of D-alanyl-lipoteichoic acid. J. Bacteriol. 176: 681–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jorasch P, Warnecke DC, Lindner B, Zahringer U, Heinz E. 2000. Novel processive and nonprocessive glycosyltransferases from Staphylococcus aureus and Arabidopsis thaliana synthesize glycolipids, glycophospholipids, glycosphingolipids, and glycosylsterols. Eur. J. Biochem. 267: 3770–3783 [DOI] [PubMed] [Google Scholar]
- 21. Jorasch P, Wolter FP, Zahringer U, Heinz E. 1998. A UDP glucosyltransferase from Bacillus subtilis successively transfers up to four glucose residues to 1,2-diacylglycerol: expression of ypfP in Escherichia coli and structural analysis of its reaction products. Mol. Microbiol. 29: 419–430 [DOI] [PubMed] [Google Scholar]
- 22. Karatsa-Dodgson M, Wörmann ME, Gründling A. 2010. In vitro analysis of the Staphylococcus aureus lipoteichoic acid synthase enzyme using fluorescently labeled lipids. J. Bacteriol. 192: 5341–5349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kern J, Ryan C, Faull K, Schneewind O. 2010. Bacillus anthracis surface-layer proteins assemble by binding to the secondary cell wall polysaccharide in a manner that requires csaB and tagO. J. Mol. Biol. 401: 757–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kern JW, Schneewind O. 2008. BslA, a pXO1-encoded adhesin of Bacillus anthracis. Mol. Microbiol. 68: 504–515 [DOI] [PubMed] [Google Scholar]
- 25. Kim HU, Goepfert JM. 1974. A sporulation medium for Bacillus anthracis. J. Appl. Bacteriol. 37: 265–267 [DOI] [PubMed] [Google Scholar]
- 26. Kiriukhin MY, Debabov DV, Shinabarger DL, Neuhaus FC. 2001. Biosynthesis of the glycolipid anchor in lipoteichoic acid of Staphylococcus aureus RN4220: role of YpfP, the diglucosyldiacylglycerol synthase. J. Bacteriol. 183: 3506–3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koch R. 1876. Die Ätiologie der Milzbrand-Krankheit, begründet auf die Entwicklungsgeschichte des Bacillus anthracis. Beitr. Biol. Pflanzen 2: 277–310 [Google Scholar]
- 28. Law CJ, Maloney PC, Wang DN. 2008. Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 62: 289–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lazarevic V, Karamata D. 1995. The tagGH operon of Bacillus subtilis 168 encodes a two-component ABC transporter involved in the metabolism of two wall teichoic acids. Mol. Microbiol. 16: 345–355 [DOI] [PubMed] [Google Scholar]
- 30. Marraffini LA, Schneewind O. 2006. Targeting proteins to the cell wall of sporulating Bacillus anthracis. Mol. Microbiol. 62: 1402–1417 [DOI] [PubMed] [Google Scholar]
- 31. Mesnage S, Tosi-Couture E, Gounon P, Mock M, Fouet A. 1998. The capsule and S-layer: two independent and yet compatible macromolecular structures in Bacillus anthracis. J. Bacteriol. 180: 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Molnár J, Prágai B. 1971. Attempts to detect the presence of teichoic acid in Bacillus anthracis. Acta Microbiol. Acad. Sci. Hung. 18: 105–108 [PubMed] [Google Scholar]
- 33. Neuhaus FC, Baddiley J. 2003. A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67: 686–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oku Y, et al. 2009. Pleiotropic roles of polyglycerolphosphate synthase of lipoteichoic acid in growth of Staphylococcus aureus cells. J. Bacteriol. 191: 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peschel A, et al. 1999. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 274: 8405–8410 [DOI] [PubMed] [Google Scholar]
- 36. Peters NK, Dixon DM, Holland SM, Fauci AS. 2008. The research agenda of the National Institute of Allergy and Infectious Diseases for antimicrobial resistance. J. Infect. Dis. 197: 1087–1093 [DOI] [PubMed] [Google Scholar]
- 37. Read TD, et al. 2003. The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature 423: 81–86 [DOI] [PubMed] [Google Scholar]
- 38. Reichmann NT, Gründling A. 2011. Location, synthesis and function of glycolipids and polyglycerolphosphate lipoteichoic acid in Gram-positive bacteria of the phylum Firmicutes. FEMS Microbiol. Lett. 319: 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schallenberger MA, Niessen S, Shao C, Fowler BJ, Romesberg FE. 2012. Type I signal peptidase and protein secretion in Staphylococcus aureus. J. Bacteriol. 194: 2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schirner K, Marles-Wright J, Lewis RJ, Errington J. 2009. Distinct and essential morphogenetic functions for wall- and lipo-teichoic acids in Bacillus subtilis. EMBO J. 28: 830–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tam C, Glass EM, Anderson DM, Missiakas D. 2006. Transposon mutagenesis of Bacillus anthracis strain Sterne using bursa aurealis. Plasmid 56: 74–77 [DOI] [PubMed] [Google Scholar]
- 42. Webb AJ, Karatsa-Dodgson M, Gründling A. 2009. Two-enzyme systems for glycolipid and polyglycerolphosphate lipoteichoic acid synthesis in Listeria monocytogenes. Mol. Microbiol. 74: 299–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wörmann ME, Corrigan RM, Simpson PJ, Matthews SJ, Gründling A. 2011. Enzymatic activities and functional interdependencies of Bacillus subtilis lipoteichoic acid synthesis enzymes. Mol. Microbiol. 79: 566–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wörmann ME, Reichmann NT, Malone CL, Horswill AR, Gründling A. 2011. Proteolytic cleavage inactivates the Staphylococcus aureus lipoteichoic acid synthase. J. Bacteriol. 193: 5279–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xia G, Peschel A. 2008. Toward the pathway of S. aureus WTA biosynthesis. Chem. Biol. 15: 95–96 [DOI] [PubMed] [Google Scholar]
- 46. Yokoyama K, Miyashita T, Arakai Y, Ito E. 1986. Structure and functions of linkage unit intermediates in the biosynthesis of ribitol teichoic acids in Staphylococcus aureus H and Bacillus subtilis W23. Eur. J. Biochem. 161: 479–489 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.