

Abstract
Mycobacterium tuberculosis serine/threonine protein kinases (STPKs) are responsible for orchestrating critical metabolic and physiological changes that dictate mycobacterial growth adaptation. Previously, we established that PknK participates in regulatory pathways that slow the growth of M. tuberculosis in a variety of in vitro stress environments and during persistent infection in mice. In the present study, we have elaborated on the mechanism of PknK-mediated regulation. Through transcription profiling of wild-type H37Rv and a ΔpknK mutant strain during logarithmic and stationary growth phases, we determined that PknK regulates the expression of a large subset of tRNA genes so that regulation is synchronized with growth phase and cellular energy status. Elevated levels of wild-type M. tuberculosis PknK (PknKMtb), but not phosphorylation-defective PknKMtb, in Mycobacterium smegmatis cause significant retardation of the growth rate and altered colony morphology. We investigated a role for PknK in translational control and established that PknK directs the inhibition of in vitro transcription and translation processes in a phosphorylation-dependent manner. Increasing concentrations of ATP or PknK exert cooperative effects and enhance the inhibitory function of PknK. Furthermore, truncation and mutational analyses of PknK revealed that PknK is autoregulated via intramolecular interactions with its C-terminal region. Significantly, the invariant lysine 55 residue was only essential for activity in the full-length PknK protein, and the truncated mutant proteins were active. A model for PknK autoregulation is proposed and discussed.
INTRODUCTION
Tuberculosis (TB) continues to be one of the world's leading infectious killers. With only a partially effective vaccine available and consistent annual increases in the percentage of drug-resistant strains causing disease, the global health impact is immense. One key aspect of Mycobacterium tuberculosis pathogenesis is its innate ability to alter its growth rate to enable survival in stress environments encountered during M. tuberculosis infection in macrophages (2). While the ability of bacterial cells to slow down growth under unfavorable conditions is not uncommon, it has enormous implications for M. tuberculosis. Through mechanisms not fully understood, M. tuberculosis is able to transition into a state of dormancy or persistence that forms the basis of the global risk of latent TB today (16). According to World Health Organization estimates, one-third of the world's population is infected with M. tuberculosis, 2 million of whom die annually (47) leaving a large reservoir of people who are asymptomatically infected with M. tuberculosis. Understanding the regulatory mechanisms that lead to mycobacterial persistence/latency is a current focus of TB research.
The central element in this adaptability is the ability to recognize and interpret external signals from specific environments and the conversion of these signals into transcriptional activation or repression. The M. tuberculosis signalome (signaling proteins) is enriched with “eukaryotic-like” Ser/Thr protein kinases (STPKs), a tyrosine kinase, paired two-component systems, and several orphan kinases, regulators, and phosphatases that orchestrate finely tuned and well-coordinated adaptive responses. The discovery of eukaryotic-like STPKs in M. tuberculosis (6) has reaffirmed the belief that bacterial pathogens are equipped with exquisite signal transduction mechanisms that not only impact host-pathogen interactions, but also enable metabolic and physiological adaptations within the bacteria.
The genome of M. tuberculosis encodes 11 STPK proteins named PknA through PknL (1, 6), 9 of which are transmembrane receptor kinases while 2 are cytoplasmic proteins (PknG and PknK) that are associated with the cell wall/membrane and are secreted during culture (only PknG) and infection (7, 27, 30, 46). M. tuberculosis STPKs are key regulators of mycobacterial growth. Two of the 11 STPKs, PknA and PknB, are essential for mycobacterial growth (13, 25), and at least 4 of them, PknH, PknI, PknJ, and PknK, have been implicated in the modulation of growth in culture and during infection (17, 24, 30, 37). Furthermore, convergence of PknH signaling pathways with the DevR-DevS (also known as DosR-DosS) two-component regulatory system (3) that defines the transcriptional adaptation of mycobacteria during hypoxia indicates that M. tuberculosis has sophisticated and highly coordinated networks that enable stress-induced growth and survival. Several studies have shown that PknA, PknB, PknD, PknH, PknK, and PknL phosphorylate proteins involved in cell division and transcription (reviewed in reference 38); however, there is very little information on the mechanisms of STPK-mediated regulation of mycobacterial growth.
The complex regulation of mycobacterial growth requires tight control of STPKs and regulatory proteins. Crystallization studies have indicated that ligand-induced N-lobe dimerization is a mechanism of mycobacterial STPK activation (15, 18, 29, 36, 48); however, at present, we do not understand how these structural changes regulate kinase activity or their downstream effects on mycobacterial growth and physiology. Typically, protein kinases are regulated by changes in their phosphorylation states, mostly within the activation loop, a 15- to 33-residue segment flanked by DFG and APE sequence motifs (22, 35), that result in modification of the active site to a conformation that is suitable for substrate binding and catalysis. An additional level of regulation is exerted by reversible phosphorylation of Ser/Thr amino acids outside the catalytic domain, and in most cases, these are regulatory events that, in the presence of coactivating secondary signals, modulate the activation process. Thus, understanding the mechanisms of kinase regulation becomes essential to fully comprehend the intricacies of mycobacterial growth regulation and its implications in persistence and latency.
In this context, we have focused on understanding the role of PknK in mycobacterial growth adaptation. Previously, we characterized an M. tuberculosis ΔpknK strain (LIX11) and established that in stressful environments, such as stationary phase, PknK functions to slow mycobacterial growth (30). In the present study, we compared the transcription profiles of LIX11 and H37Rv during logarithmic and stationary growth phases and determined that PknK regulates expression of tRNA genes as a function of growth, suggesting an involvement of PknK in translation control. We investigated the phosphorylation-dependent effects of M. tuberculosis PknK (PknKMtb) on the growth of fast-growing Mycobacterium smegmatis and on in vitro protein synthesis and established that PknK-mediated phosphorylation causes significant retardation of the M. smegmatis growth rate and colony development and, more importantly, inhibits in vitro protein synthesis via synergistic effects on transcription and translation processes. Finally, we provide evidence for an autoregulatory role of the PknK C terminus and propose a novel mode of kinase regulation.
MATERIALS AND METHODS
Strains, plasmids, and growth.
The strains and plasmids used in this study are listed in Table 1. Escherichia coli JM109 was used as the host strain for all molecular manipulations. E. coli cultures were grown in Luria Bertani (LB) broth or LB agar plates at 37°C. For pknKMtb overexpression, M. smegmatis was transformed with acetamide-inducible integrative plasmids pYA1662 and pYA1671 to yield the LIX79 and LIX80 strains overexpressing wild-type and phosphorylation-defective pknKMtb, respectively, and these strains were grown in LB broth supplemented with 0.05% Tween 80 with aeration or on LB agar plates at 37°C. Acetamide was added at a final concentration of 0.2% during exponential growth in liquid medium and in agar plates to induce pknKMtb expression. Broth-grown M. tuberculosis H37Rv and a ΔpknK deletion strain, LIX11 (30), were used for comparative expression profiling using microarrays. M. tuberculosis cultures were cultivated in Middlebrook 7H9 medium (Difco Laboratories) supplemented with 10% ADS (0.5% albumin, 0.2% dextrose, 0.085% saline) enrichment as described previously (30). When required, media were supplemented with 25 μg/ml kanamycin (Kan) and 50 μg/ml hygromycin (Hyg) for mycobacteria. All chemicals were obtained from Sigma-Aldrich, St. Louis, MO, unless otherwise indicated.
Table 1.
Bacterial strains and plasmids used in this study
| Strain/plasmid | Description | Source/reference |
|---|---|---|
| M. tuberculosis H37Rv | Virulent laboratory strain | ATCC 25618 |
| M. smegmatis mc2155 | ept-1 | W. R. Jacobs, Jr., Albert Einstein College of Medicine, NY |
| E. coli | ||
| JM109 | endA1 recA1 gyrA96 thi-1 hsdR17(rK− mK−) relA1 supE44 Δ(lac-proAB) [F′] traD36 proAB lacIqZΔM15 | Stratagene |
| LIX11 | H37Rv ΔpknK | 30 |
| LIX70 | mc2155::pJFR19 | This study |
| LIX79 | mc2155::pYA1662 | This study |
| LIX80 | mc2155::pYA1671 | This study |
| Plasmids | ||
| pETSUMO | Kanr; 6× His-SUMO expression vector | Invitrogen |
| pJFR19 | Hygr; amidase promoter; integrative vector | 5 |
| pYA1556 | pETSUMO::pknK(3333 bp) | 30 |
| pYA1635 | pETSUMO::phoP(744 bp) | This study |
| pYA1657 | pETSUMO::pknK(1-915 bp) | This study |
| pYA1658 | pETSUMO::pknK(1-2607 bp) | This study |
| pYA1659 | pETSUMO::pknK(3333 bp) K55M | This study |
| pYA1660 | pETSUMO::pknK(1-915 bp) K55M | This study |
| pYA1661 | pETSUMO::pknK(1-2607 bp) K55M | This study |
| pYA1662 | pknK gene in NdeI/XbaI site of pJFR19 | This study |
| pYA1671 | pknK K55M gene in NdeI/XbaI site of pJFR19 | This study |
RNA isolation and integrity.
Aliquots for RNA isolation were taken from M. tuberculosis H37Rv and LIX11 cultures at day 3 (log phase) and day 30 (stationary phase) time points during growth, and total RNA was isolated using TRI reagent (Ambion Inc., TX), as described previously (31). Total RNA integrity was assessed using an RNA 6000 Nano Lab Chip on the 2100 Bioanalyzer (Agilent, Santa Clara, CA) according to the manufacturer's protocol. Total RNA purity was assessed with a NanoDrop ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies, Inc., DE). RNA was considered to be good quality when the 23S/16S rRNA ratios were greater than or equal to 1.0. RNA samples from two independent experiments were used for microarray analysis.
Labeling and microarray hybridization.
Poly(A) tails were added to the 3′ ends of RNA using the A-plus Poly(A) polymerase tailing kit (Epicentre Biotechnologies, Madison, WI). The samples were labeled using the Quick-Amp labeling Kit (Agilent). Five hundred nanograms of each sample was incubated with reverse transcription mixture at 42°C and converted to double-stranded cDNA (dscDNA) primed by oligo(dT) with a T7 polymerase promoter. The dscDNA was used as the template for cRNA generation by in vitro transcription and incorporation of the dyes Cy3-CTP and Cy5-CTP (Agilent). The cDNA synthesis and in vitro transcription steps were carried out at 40°C. The quality of labeled cRNA was assessed for yields and specific activity, followed by hybridization on a custom M. tuberculosis H37Rv 8x15k array designed by Genotypic Technology Pvt. Ltd. (Agilent microarray design identifiers [AMADID] 023057 and 026323; Agilent). Cy3- and Cy5-labeled samples (300 ng each) were hybridized using the Gene Expression Hybridization kit (Agilent) in Surehyb chambers (Agilent) at 65°C for 16 h. The hybridized slides were washed using Gene Expression wash buffers (Agilent) and scanned using the Microarray Scanner G2505C at 5-μm resolution. Data extraction from images was done using Feature Extraction software v 10.5.1.1 (Agilent), followed by analysis using GeneSpring GX version 10 software (Agilent). Normalization of the data was done in GeneSpring GX using (Lowess) normalization. In two-color microarray experiments, where two fluorescent dyes (red and green) have been used, intensity-dependent variation in dye bias may introduce spurious variations in the collected data. Lowess normalization is a technique that merges two-color data, applying a smoothing adjustment that removes such variation. Samples were grouped based on the replicates and genes showing up- or downregulation of >0.6-fold among the samples. The results are reported as the geometric means of log2 expression ratios ± standard deviations between the replicates. Quantitative reverse transcription-PCR (qRT-PCR) was performed to validate the microarray results as described previously (31), using sigA mRNA as an internal control for normalization.
Cloning and cell-free expression of M. tuberculosis PknK wild-type and mutant proteins.
The full-length pknK gene (3,333 bp) and the N-terminal regions encompassing 915 bp and 2,607 bp of pknK were amplified using M. tuberculosis H37Rv genomic DNA as the template and cloned into a pET SUMO expression vector (Invitrogen, Carlsbad, CA) to create plasmids pYA1556 (30), pYA1657, and pYA1658, respectively. The substitution mutation to replace lysine at position 55 with methionine (K55M) was generated in the plasmids pYA1556, pYA1657, and pYA1658 using PknK-K55M F and K55M R primers (27) and the QuikChange Lightning site-directed mutagenesis kit (Stratagene, La Jolla, CA) following the manufacturer's recommendations to produce plasmids pYA1659 to pYA1661. The plasmids pYA1659, pYA1660, and pYA1661 served as the template DNA for in vitro synthesis of PknK1-1110-, PknK1-305-, and PknK1-869-K55M mutant proteins, respectively. All clones and mutations were verified by DNA sequencing. For studies with purified PknK protein, we used a glutathione S-transferase (GST)-tagged PknK kinase domain (1 to 300 amino acids; referred to as PknK KD-GST) previously purified in our laboratory. Details of plasmid constructions are available upon request to V. Malhotra.
For cell-free synthesis of PknK proteins, highly pure plasmid DNA was extracted from overnight-grown cultures of the respective strains using the NucleoBond Xtra Maxi prep kit (Macharey-Nagel, Bethlehem, PA) and checked for integrity by electrophoresis on a 1% agarose gel. The S30 T7 high-yield protein expression system (Promega, Madison, WI) was used to synthesize PknK proteins, as described by the manufacturer. Briefly, all components of the cell-free expression system were premixed, aliquoted in 23-μl reaction mixtures, and stored at −80°C till further use. Two microliters of plasmid DNA (∼1 μg) was used as a template for in vitro transcription and translation (IVTT) in a 25-μl reaction volume and incubated at 37°C with shaking for 1 h. All steps were performed in an RNase-free environment. Proteins synthesized in this manner contained a 6×His SUMO tag at the N terminus of the protein. One-tenth volume of the PknK-IVTT reaction mixture was resolved on a 10% SDS-PAGE gel, transferred onto a nitrocellulose membrane, and subjected to immunoblotting with a monoclonal anti-His antibody (1:3,000; Sigma-Aldrich) and/or anti-PknK antibody as described previously (30). Detection was done with the SuperSignal West Dura chemiluminescent substrate (Thermo Scientific, Waltham, MA).
In vitro kinase assay.
PknK proteins were purified from the PknK-IVTT reactions by using the MagneHis protein purification system (Promega) according to the manufacturer's recommendations. Alternatively, the PknK-IVTT proteins were used directly after acetone precipitation. The proteins were assessed for kinase activity by incubation in 1× kinase buffer (Cell Signaling Technology, Danvers, MA) and 50 to 75 μCi [γ-32P]ATP (Perkin Elmer, Waltham, MA) in a 20-μl reaction volume for 1 h at 30°C. The reactions were stopped by the addition of 2× SDS sample buffer followed by boiling for 5 min. For transphosphorylation reactions, 10 to 12 μg of myelin basic protein (MBP) was used as a substrate. The samples were resolved on 4 to 15% SDS-PAGE gels, dried, and analyzed by phosphorimaging using a Typhoon TRIO+ scanner (Amersham Biosciences) or by autoradiography. In some cases, phosphorylation was detected by immunoblotting with anti-phosphoserine/phosphothreonine antibody (Millipore, Bedford, MA).
Assay for PknK-mediated inhibition of IVTT.
Ten microliters of in vitro-synthesized PknK wild-type or mutant proteins was acetone precipitated on ice for 15 min, centrifuged, and air dried. The precipitated PknK wild-type/mutant protein or 5 μg of the purified PknK KD-GST was exogenously added to a fresh IVTT reaction mixture (23 μl), along with 1 μg template DNA (pYA1635 or the Renilla reniformis luciferase gene [Rluc] control DNA). Reactions were supplemented with 1 mM ATP or ADP as needed and incubated at 37°C with shaking for 1 h. Control reactions without any exogenous addition of PknK or ATP were always performed in parallel. Synthesis of His-tagged PhoP was detected by immunoblotting with anti-His antibody, and the extent of inhibition was determined by densitometric analyses using AlphaEase imaging software (Alpha Innotech/Cell Biosciences, Santa Clara, CA).
Luciferase assay.
The production of Renilla luciferase from the Rluc control DNA in the S30 T7-coupled IVTT system was monitored by measuring the production of light after addition of the Renilla luciferase assay reagent (Promega). For experiments involving IVTT of firefly luciferase in rabbit reticulocyte lysates (RRL) (Promega) from either luciferase control DNA or presynthesized luciferase mRNA templates (Promega), the samples were diluted 1:20 in nuclease-free water, and luciferase synthesis was assessed by measuring production of light with the luciferase assay reagent (Promega), according to the manufacturer's recommendations. The reactions were analyzed in a Glomax 96 Microplate luminometer (Promega).
DLS.
Dynamic light scattering (DLS) measurements were performed using a 173° back-scattering Malvern Nano-ZS instrument, as described previously (45). Briefly, PknK KD-GST was subjected to in vitro kinase assay with 1 mM ATP or AMP-PNP (β,γ-imidoadenosine 5′-triphosphate, a nonhydrolyzable ATP analog) and kinase buffer as described above and diluted to 200 μl to give a final concentration of 1.25 μg/μl. The samples were filtered through a 0.45-μm Millipore Millex-HN filter to remove dust particles before DLS measurements. Repeated measurements were taken to ensure reproducibility.
Statistical analysis.
Statistical analyses were performed with a Student t test or one-way analysis of variance (ANOVA) using Graphpad Prism 5.0 software (GraphPad Software, San Diego, CA). A P value of <0.05 was considered statistically significant.
Microarray data accession number.
The microarray data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE37823 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37823).
RESULTS
PknK regulates M. tuberculosis tRNA synthesis in a growth-phase-dependent manner.
We previously demonstrated that PknK levels are elevated in stationary phase and that deletion of the pknK gene in H37Rv results in increased survival of the mutant under stationary-phase stress (30). To extend our investigations on the regulatory role of PknK, we used microarrays to compare the transcription profiles of the LIX11 mutant versus wild-type H37Rv under conditions of logarithmic-phase (3 days) and stationary-phase (30 days) growth in broth cultures. The rationale for using these time points was to maintain correlation with data reported earlier (30). Consistent with the idea that PknK plays a distinct role in stationary phase, twice as many genes (Fig. 1A, red dots) (n = 70, 32 up- and 38 downregulated, where n is the number of differentially expressed genes) were differentially expressed in the mutant during stationary phase than during log phase (Fig. 1A, blue dots) (n = 37, 7 up- and 30 downregulated). Several genes encoding conserved hypothetical proteins and those involved in lipid metabolism and cell wall processes were differentially regulated in the LIX11 mutant; however, the most significant change was observed in the category of tRNA genes (see Fig. S1 and Data sets S1 and S2 in the supplemental material). In the present study, we focused on the expression of tRNA genes in the LIX11 mutant. The tRNA genes, along with their changes in expression in the mutant relative to H37Rv in log and stationary phases, are listed in Table 2. Remarkably, of the 30 genes downregulated in LIX11 during log phase, 80% were tRNA genes (Fig. 1A, circled blue spots). To our surprise, we observed that tRNA expression in the LIX11 mutant was growth phase dependent. Among the 32 upregulated genes in stationary phase, 72% belonged to the tRNA gene category (Fig. 1A, circled red spots). We analyzed the expression of 5 randomly selected tRNA genes—glyU, lysT, gluT, glyV, and argW—in LIX11 and H37Rv by qRT-PCR. The differential expression of two tRNA genes, glyU and lysT, during the log and stationary phases and the upregulation of gluT, glyV, and argW during stationary phase in the LIX11 mutant validated the microarray results (Fig. 1B). Given that PknK levels are induced during stationary phase (30), these results suggest the involvement of PknK in downregulating tRNA expression in H37Rv during stationary phase and, more importantly, are reflective of the energy demand, rate of protein synthesis, and growth of the bacteria. Since all of these are low in stationary phase compared to log phase, these results further substantiate a PknK function in regulatory mechanisms that slow mycobacterial growth in stationary phase. Next, we determined the direct effects of PknK-mediated signaling and its implications in mycobacterial growth regulation.
Fig 1.

Graphical representation of the trend in tRNA expression in the ΔpknK mutant during logarithmic and stationary-phase growth. Differences between the transcriptional profiles of an M. tuberculosis ΔpknK strain (LIX11) and the wild-type H37Rv were determined as a function of growth using microarrays. (A) Scatter plot showing the distribution of genes (up- or downregulated) in the LIX11 mutant relative to the wild-type during logarithmic-phase (blue dots) and stationary-phase (red dots) growth based on their respective changes listed in Data sets S1 and S2 in the supplemental material. Note that each spot indicates a unique gene that is differentially expressed in the LIX11 mutant during log and stationary phases. The circled points highlight the biphasic expression of the tRNA genes in the mutant. A large subset of tRNA genes was downregulated in the mutant during logarithmic phase but upregulated during stationary phase. (B) qRT-PCR analyses of 5 randomly selected tRNA genes, glyU, lysT, gluT, glyV, and argW, in LIX11 and H37Rv during log and stationary phases. Expression was normalized using sigA mRNA as an internal control. The change in tRNA expression in the LIX11 mutant relative to H37Rv grown under specific growth conditions is presented as the mean and standard deviation (SD) of data from two independent experiments. The baseline expression of tRNA genes in H37Rv was set to 1.0 (dashed line). **, P < 0.01.
Table 2.
Differential expression of tRNA genes in M. tuberculosis ΔpknK mutant (LIX11) relative to H37Rv during logarithmic- and stationary-phase growth
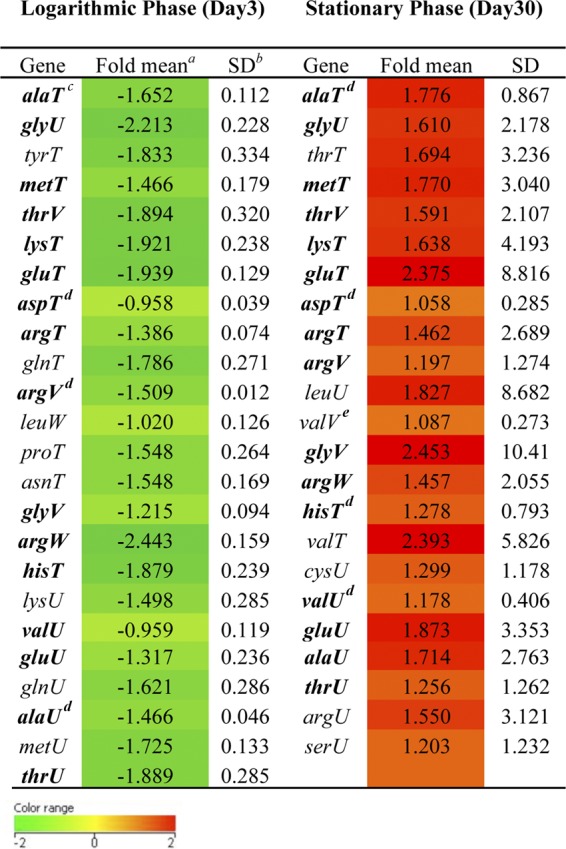
a Geometric mean of log2 ratio from two biological replicates.
b SD, standard deviation between the replicates. The red and green boxes indicate greater (more than 0.6) or lesser (less than −0.6) fold changes in LIX11 relative to H37Rv, respectively.
c The 16 tRNA genes common to the logarithmic (downregulated) and stationary (upregulated) phases are shown in boldface.
d Significant difference between LIX11 and H37Rv (P < 0.05) (Student t test).
e Significant difference between LIX11 and H37Rv (P < 0.01) (Student t test).
Overexpression of wild-type, but not phosphorylation-defective, pknKMtb in M. smegmatis inhibits growth rate and colony development.
To elucidate the underlying mechanisms of PknK-mediated regulation, we employed the approach of elevating intracellular PknK levels. We reasoned that increased cellular concentrations of PknK could substitute for activating signals and enable us to discern the direct downstream effects of PknK. Despite several attempts to transform M. tuberculosis H37Rv with plasmid pYA1662, which overexpresses the full-length pknKMtb gene from an acetamide-inducible promoter, we could not recover the transformants. As an alternative, M. smegmatis, which routinely serves as a surrogate host to express M. tuberculosis genes, was used in these experiments. We transformed M. smegmatis with empty vector and pYA1662 to construct the LIX70 and LIX79 (overexpressing pknKMtb) strains, respectively (Table 1). As shown in Fig. 2A, immunoblot analysis of induced (Ind) and uninduced (UI) cell lysates of LIX79 showed a significant increase in PknKMtb levels after 48 h of induction with acetamide (Fig. 2A, lane 5) compared to the uninduced control (Fig. 2A, lane 4). A small amount of leaky expression was observed in the uninduced LIX79 lysates (Fig. 2A, lane 4) but was considered insignificant in light of the level of overexpression after induction. Cell lysates of the LIX70 strain carrying the empty vector served as a negative control for pknKMtb expression (Fig. 2A, lanes 1 and 2).
Fig 2.

Increased intracellular levels of PknKMtb in M. smegmatis retard growth and colony formation in a phosphorylation-dependent manner. (A) Overexpression of pknKMtb in M. smegmatis. Shown is immunoblot analysis of UI and Ind cell lysates of LIX70 (vector control, lanes 1 and 2) and LIX79 (lanes 4 and 5) with anti-PknK antibody. The arrow indicates the induction of PknKMtb at 48 h postinduction in LIX79, which is absent in LIX70 lysates. PknKMtb synthesized by IVTT was used as a positive control for PknK (lane 6). A lower, nonspecific band was observed in all lanes (except lane 3, which is blank) and served as a loading control. (B) Effect of pknKMtb overexpression on M. smegmatis growth and survival. Viable counts for uninduced and induced LIX70 and LIX79 cultures were determined at 0-, 24-, and 48-h time points after induction with 0.2% acetamide. The results are presented as average CFU and SD from three independent experiments. The LIX79 induced cultures exhibited a 3.5-fold decrease in CFU compared to the uninduced control at 48 h postinduction. (C) PknKMtb-mediated effects on growth are phosphorylation dependent. Microscopic images depict the growth of single colonies from UI and Ind LIX70 (empty-vector control), LIX79 (overexpressing pknKMtb), and LIX80 (overexpressing phosphorylation-defective pknKMtb) cultures on solid medium (with or without acetamide). The induced cultures were always plated on acetamide-containing agar plates to maintain the effects of pknKMtb overexpression, and uninduced cells were plated on agar plates minus acetamide. Inhibition of growth was observed exclusively with PknKMtb overexpression (LIX79), but not with LIX80 cells overexpressing phosphorylation-defective PknKMtb. Representative images are presented (bars = 1 mm). (D) Elevated levels of PknKMtb retard growth and colony development significantly. Microscopic images (t = 24 h) of induced LIX79 and LIX80 cells plated on inducing medium followed by incubation for 4, 8, and 12 days reveal a lower growth rate of LIX79, but not LIX80, cells. Representative images are presented (bars = 1 mm).
The effects of overexpressing pknKMtb on mycobacterial growth and physiology were assessed by determining the CFU of LIX70 and LIX79 at 0, 24, and 48 h postinduction. The uninduced (UI) controls were plated on noninducing medium; however, to maintain pknKMtb overexpression in induced (Ind) cells, the cultures postinduction were always plated on solid medium containing acetamide unless otherwise specified. The uninduced and induced LIX70 and LIX79 cultures grew similarly for 24 h postinduction (Fig. 2B). At 48 h postinduction, the induced LIX79 cultures exhibited an ∼3.5-fold decrease in viable counts relative to the uninduced LIX79 control (Fig. 2B). We noted that at 24 h postinduction, when there was no significant difference in the number of CFU of LIX70 (induced) and LIX79 (induced), the growth rates of the individual induced LIX79 colonies on solid medium containing the inducer were low. Figure 2C shows the growth of a single colony after 8 days of growth under inducing conditions (1 day in liquid medium followed by 7 days of growth on solid medium). While the uninduced LIX70 and LIX79 cells grew at a normal rate and showed similar colony morphologies, the LIX79 induced cells exhibited significant retardation in their growth rate and displayed aberrant “dwarfed” colony morphology (Fig. 2C, middle).
Studies by Kumar et al. have shown that replacement of lysine at position 55 in PknKMtb by methionine (K55M) abolishes PknK kinase activity (27); thus, an M. smegmatis strain overproducing PknKMtbK55M mutant protein was constructed (LIX80 [Table 1]). We confirmed the overexpression of the pknK K55M gene in induced LIX80 cell lysates by immunoblotting (see Fig. S2A in the supplemental material) and used it as a control in these experiments. The growth defect observed with LIX79 was absent in LIX80 (Fig. 2C, bottom), suggesting that PknKMtb-mediated effects on the growth and colony morphology of M. smegmatis are dependent on its ability to phosphorylate cellular proteins. Continued observation of the individual induced colonies over time (1 day in liquid medium followed by 4, 8, and 12 days of incubation on solid medium containing the inducer) revealed that the LIX79 induced cells were increasing in size (Fig. 2D); however, the rate of growth was significantly lower, and moreover, the colony exhibited vertical growth as opposed to the lateral growth observed with the LIX80 induced cells. Notably, this phenotype was reversed when induced LIX79 cells were grown on noninducing solid medium, confirming that the inhibitory effects of PknK were due to increased PknKMtb synthesis and phosphorylation activity in LIX79 (see Fig. S2B in the supplemental material). These observations corroborate our previous results with a ΔpknK mutant (30) and suggest that increased intracellular levels of PknK have a global effect on the macromolecular synthetic apparatus of the mycobacterium, resulting in an overall growth defect. However, the key question is whether PknK mediates these effects directly or indirectly through phosphorylation of other transcriptional regulators. Considering that microarray results implicated PknK in the regulation of tRNA synthesis in M. tuberculosis, we focused our investigation on a role for PknK in the regulation of protein synthesis.
Cell-free synthesis and functional activity of M. tuberculosis PknK wild-type and mutant proteins.
To determine whether PknK directly regulates protein synthesis, we used cell-free IVTT systems. The E. coli S30 T7 IVTT system that enables coupled transcription and translation of a gene under the control of a T7 promoter was used to synthesize PknK proteins. Plasmid pYA1556, carrying the pknK gene, whose expression is controlled by a T7 promoter, was used as a DNA template to produce His-tagged PknK1-1110 protein. Phosphorylation-defective PknK1-1110K55M was synthesized from plasmid pYA1659 (also under the control of a T7 promoter) to serve as a negative control in our investigations. Figure 3A shows the successful production of the PknK1-1110 (lane 1) and PknK1-1110K55M-IVTT (lane 2) proteins on a Western blot analyzed with anti-PknK antibody. To confirm that the proteins synthesized through the cell-free expression system were functionally active, we analyzed the kinase activities of IVTT-synthesized wild-type and mutant PknK proteins in an in vitro kinase assay using MBP as a substrate and [γ-32P]ATP. As shown in Fig. 3B, wild-type PknK1-1110-IVTT protein catalyzed the autophosphorylation and transphosphorylation of MBP (Fig. 3B, lane 3), and as expected, PknK1-1110K55M was completely devoid of any ATP binding or kinase activity (Fig. 3B, lane 5). In the absence of PknK, there was no phosphorylation of MBP, suggesting that the IVTT mixture had no endogenous kinase activity (Fig. 3B, lane 1). Thus, we concluded that the E. coli S30 T7 expression system could be successfully used for the production of catalytically active mycobacterial kinases that are stable and functional for subsequent use in biochemical enzymatic assays.
Fig 3.
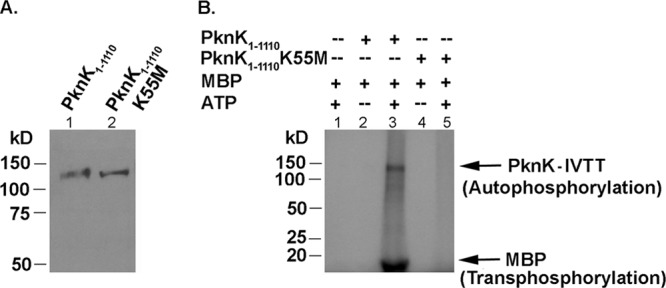
PknK proteins synthesized in a cell-free expression system are catalytically active. (A) IVTT-synthesized His-tagged PknK1-1110 (lane 1) and PknK1-1110K55M (lane 2) were resolved on a 10% SDS-PAGE gel and confirmed by Western blot analysis using the anti-His antibody. Recombinant PknK proteins of ∼130-kDa molecular mass were detected. (B) Kinase activity of IVTT-synthesized PknK proteins. In vitro kinase assays were carried out to test the auto- and transphosphorylation abilities of the wild-type or mutant PknK1-1110 kinase using MBP as a universal substrate protein in reactions with or without [γ-32P]ATP, followed by resolution on a 4 to 15% gradient SDS-PAGE gel. Radioactive signals were detected by phosphorimaging. The arrows indicate the bands corresponding to autophosphorylated PknK1-1110 and transphosphorylated MBP.
PknK inhibits in vitro transcription and translation in a phosphorylation-dependent manner.
The effect of exogenous addition of IVTT-synthesized PknK1-1110 on protein synthesis of bacterial cells was analyzed using the E. coli S30 T7 system. The choice of template plasmid DNA used in this assay, plasmid pYA1635 carrying the M. tuberculosis phoP gene (Rv0757) expressed from a T7 promoter, was serendipitous. As shown in Fig. 4A, immunoblot analysis with anti-His antibody revealed robust production of His-tagged PhoP upon addition of the plasmid pYA1635 to the IVTT reaction (Fig. 4A, lane 1). Addition of PknK1-1110 to the reaction did not affect the synthesis of PhoP (Fig. 4A, lane 2). In contrast, exogenous addition of PknK1-1110, together with 1 mM ATP, resulted in a significant decrease in PhoP detection (Fig. 4A, lane 3). No inhibition was observed in the presence of 1 mM ADP (Fig. 4A, lane 4), indicating a requirement for ATP for the inhibitory effect of PknK. It should be noted that all IVTT reactions have background levels of ATP as the energy source for the transcription and translation reaction; however, competition with endogenous ATP was ruled out, as inhibition occurred only in the presence of exogenously added PknK and 1 mM ATP. Addition of ATP alone to the active IVTT reaction did not have any inhibitory effect (see Fig. S3 in the supplemental material), reinforcing the idea that inhibition is specific to PknK and ATP. Densitometric analysis of the immunoblots from three independent experiments revealed significant inhibition (∼57%) of the phoP-IVTT reaction as a result of direct addition of PknK1-1110 and ATP (Fig. 4A, bottom). We also tested the phosphorylation-defective PknK1-1110K55M for its ability to inhibit PhoP protein synthesis. In contrast to the wild-type PknK, exogenous addition of PknK1-1110K55M and ATP did not have any inhibitory effect on PhoP synthesis (Fig. 4A, lane 5). These results suggest that PknK-mediated effects on IVTT are phosphorylation dependent and further explain the requirement for ATP for the inhibitory function of PknK.
Fig 4.

PknK inhibits in vitro transcription and translation via a phosphorylation-dependent mechanism. (A) PknK inhibits in vitro synthesis of target protein (PhoP) from plasmid pYA1635 in an E. coli S30 T7 IVTT system. IVTT assay mixtures were assessed for target protein production by immunoblotting with anti-His antibody in reactions with or without PknK and in the presence of 1 mM ATP or ADP. Bands corresponding to PhoP and PknK/PknK1-1110K55M are indicated by arrows. The experiment was repeated in triplicate, and one representative blot is presented. (Bottom) Densitometric analyses of three independent Western blots show significant inhibition of PhoP production due to exogenous addition of PknK and ATP (P < 0.01), but not with ADP or with a phoshorylation-defective PknK1-1110K55M mutant protein. (B) PknK-mediated inhibition is not gene specific. PknK inhibits in vitro synthesis of the Renilla luciferase protein. The amount of luciferase made in the assay after addition of IVTT-synthesized PknK1-1110 or purified PknK KD-GST was determined by measuring the production of light in a luminometer after addition of the luciferase assay reagent. Relative light units (RLU) are reported as the mean and SD from two independent experiments. **, P < 0.01; ***, P < 0.001. (C) PknK phosphorylates components of the IVTT machinery. An in vitro kinase assay of the E. coli T7 S30 mixture with wild-type or mutant PknK1-1110 protein was analyzed by immunoblotting with anti-phosphoserine/phosphothreonine antibody. Besides autophosphorylated wild-type PknK (indicated by the white arrow), signals indicating phosphorylation of the E. coli T7 S30 proteins (black arrows) were detected with the wild-type PknK, but not with the mutant K55M protein or a control reaction (without protein). A nonspecific band (∼60 kDa) detected in all three lanes served as a control for equal protein loading. (D) Effect of PknK on transcription versus translation. Five micrograms of PknK KD-GST and 1 mM ATP were added to an RRL translation system containing luciferase mRNA as the template or to an RRL-coupled IVTT system containing luciferase control DNA as the template. Synthesis of luciferase was monitored by the production of light. The results are presented as average RLU and SD from three independent experiments. PknK KD-GST significantly inhibited luciferase production in a coupled IVTT system (P < 0.001) compared to the “translation-only” system. ***, P < 0.001.
To demonstrate that the action of PknK on protein synthesis is not gene specific, we determined the effect of PknK on the in vitro synthesis of the Renilla luciferase protein. The control DNA carrying the R. reniformis luciferase gene (Rluc) downstream from a T7 promoter was used as the template for the IVTT reaction with and without exogenous addition of PknK1-1110 and ATP. The Rluc protein levels were monitored by measuring production of light as a readout of luciferase synthesis in a luciferase assay system. We observed a 70% decrease in the production of light and therefore production of the luciferase protein when PknK1-1110 and ATP were added (Fig. 4B, lane 3). Thus, the effect of PknK1-1110 on IVTT is dependent not on the gene undergoing IVTT, but rather, on the mechanism of transcription and translation. Furthermore, inhibitory effects were also observed with exogenously added purified PknK KD-GST protein and ATP (Fig. 4B, lane 4). These results validate our observations with IVTT-synthesized PknK and also indicate that the kinase domain of PknK is sufficient for directing inhibition of in vitro protein synthesis.
In view of the above observations, we analyzed PknK1-1110-dependent phosphorylation of the IVTT components using an in vitro kinase assay followed by immunoblotting with anti-phosphoserine/phosphothreonine antibody. Although some background signal was observed in the control (IVTT minus PknK) and in PknK1-1110K55M reactions, there was a region of distinct kinase activity observed with the wild-type PknK1-1110 (Fig. 4C, lane 2, black arrows) that was absent with the K55M mutant (Fig. 4C, lane 3). We observed signal corresponding to autophosphorylated wild-type PknK1-1110 (Fig. 4C, lane 2, white arrow) that was absent in the K55M mutant protein reaction. Thus, we concluded that PknK inhibits in vitro protein synthesis through interactions and phosphorylation of specific components of the IVTT machinery.
PknK-mediated effects on in vitro transcription and translation are synergistic.
Bacterial cells have coupled transcription and translation mechanisms; thus, to fully understand the mechanism of PknK-mediated inhibition, it was imperative to analyze the effects of PknK on transcription and translation independently. For coupled transcription and translation, luciferase DNA was used as the template in the rabbit reticulocyte lysate (RRL) system that allows both transcription and translation, whereas presynthesized luciferase mRNA was used as a template for uncoupled RRL systems that only drive translation. Five micrograms of purified PknK KD-GST and 1 mM ATP were exogenously added to both assays and analyzed for any effects on luciferase production. PknK KD-GST drastically inhibited synthesis of luciferase from the DNA template (Fig. 4D, white bars). On the other hand, inhibition with luciferase mRNA as the template was moderate (∼35%) (Fig. 4D, black bars), indicating that the effects of PknK on translation alone contribute to, but do not solely account for, the significant inhibition of luciferase production, as observed in the coupled transcription and translation system. From these results, it is apparent that PknK inhibits both transcription and translation mechanisms so that the effects are synergistic and lead to an overall decrease in the efficiency of protein synthesis.
PknK and ATP concentrations are positive effectors of PknK-mediated inhibition of IVTT.
Kinase reactions are typically bimolecular reactions with ATP as one of the substrates. Since PknK undergoes autophosphorylation, PknK acts as a substrate, as well. We rationalized that the effective enzyme (here, PknK) and substrate (ATP or PknK) concentrations can positively or negatively affect the interactions with other substrate proteins in the IVTT mixture; thus, we investigated whether the inhibitory effect of PknK on IVTT was dose dependent. We studied the effects of PknK and ATP concentrations independently on the efficiency of IVTT using plasmid pYA1635 (phoP) as the DNA template. In agreement with the results shown in Fig. 4A, exogenous addition of PknK1-1110 and 1 mM ATP resulted in a decrease in PhoP synthesis (∼48%) compared to the minus-ATP control (Fig. 5A, left blot). Upon increasing the concentration of exogenous ATP to 2 mM, PknK1-1110 completely inhibited the synthesis of PhoP (Fig. 5A, left blot). On the other hand, there were no observable effects on PhoP synthesis by PknK1-1110K55M mutant protein even in the presence of 2 mM ATP (Fig. 5A, right blot) or by 2 mM ATP alone (see Fig. S3 in the supplemental material). These controls highlight the essential role of phosphorylation in PknK-mediated IVTT inhibition and remove any doubts about nonspecific inhibition. To eliminate any variations due to different preparations of PknK-IVTT proteins, we also tested a purified PknK KD-GST. Increasing concentrations of ATP in the presence of a constant amount of PknK KD-GST (5 μg) also led to a decrease in PhoP production (Fig. 5B, left blot). In fact, as with PknK1-1110-IVTT protein, there was no detectable protein synthesis in the presence of 2 mM ATP and PknK KD-GST (Fig. 5B, left blot).
Fig 5.

PknK and ATP concentrations are positive effectors of PknK-mediated inhibition of protein synthesis. (A) The effects of ATP concentrations on the ability of wild-type and mutant PknK1-1110 proteins to inhibit phoP-IVTT were determined by monitoring PhoP synthesis by immunoblotting with anti-His antibody. In the presence of 2 mM exogenous ATP, wild-type PknK1-1110 protein (left blot), but not the PknK1-1110K55M mutant protein (right blot), completely inhibited the synthesis of PhoP. (B) Effects of increasing concentrations of ATP in reaction mixtures containing 5 μg of PknK KD-GST (left blot) and of purified PknK KD-GST in reaction mixtures containing 1 mM ATP (right blot) on the efficiency of the phoP-IVTT reaction were determined as described for panel A.
To assess the effect of the PknK concentration, we varied the amounts of purified PknK KD-GST (1 to 10 μg) while keeping the amount of ATP constant at 1 mM. We chose 1 mM ATP for consistency with previous experiments. Increasing concentrations of PknK KD-GST led to a concomitant increase in the inhibition of PhoP synthesis (Fig. 5B, right blot). Together, these results indicate that both PknK and ATP levels exert cooperative effects that may govern intra- and/or intermolecular interactions between PknK, ATP, and the IVTT components that further dictate the inhibition of in vitro transcription and translation.
The PknK kinase domain forms oligomeric complexes: role of ATP.
In view of the dose-dependent effects of PknK on IVTT (Fig. 5), we wanted to determine if PknK existed as a monomer or a dimer in solution. Western blot analyses of the IVTT-synthesized full-length and kinase domains of wild-type PknK and K55M mutant proteins with an anti-PknK antibody revealed distinct changes in the molecular mass on simultaneously resolved native and denaturing gradient polyacrylamide gels. The linear masses of the His-tagged PknK1-1110 (∼130 kDa) and PknK1-305 (∼45 kDa) proteins on an immunoblot of the denaturing gel were as expected (Fig. 6A, left blot). On a Western blot of the native gel, no signal was detected with the full-length proteins, probably due to resolution constraints of the gel; however, a signal corresponding to more than twice the size of the linear kinase domain was observed for the wild-type and mutant PknK1-305 proteins, suggesting association between PknK KD proteins (Fig. 6A, right blot). To visualize these associations, we analyzed purified PknK KD-GST protein on a Coomassie-stained native gel and compared the sizes of PknK oligomers with respect to simultaneously resolved native protein standard markers. In contrast to a single band corresponding to the PknK KD-GST protein (∼60 kDa) obtained on a Coomassie-stained denaturing gel (Fig. 6B, left), we observed two bands of various larger sizes (between 150 kDa and 480 kDa) on the Coomassie-stained native gel, confirming that PknK KD forms oligomeric complexes (Fig. 6B, right). In accordance with our observations with the His-tagged PknK-KD1-305 (Fig. 6A, right blot), we observed a lower band between the 146-kDa and 242-kDa protein standards, suggesting the formation of a PknK-KD trimer (Fig. 6B, right). In addition, we noted the presence of a distinct higher band around the region corresponding to 400 to 480 kDa, indicating formation of higher-order oligomeric assemblies (Fig. 6B, right).
Fig 6.

PknK forms oligomeric complexes in solution. (A) Western blot (WB) analysis with anti-PknK antibody of IVTT-synthesized PknK1-1110 and PknK1-305 wild type and their K55M counterparts simultaneously resolved on native and denaturing 4 to 15% gradient polyacrylamide gels. Denaturing gel WB analysis showed the linear size of the proteins, whereas WB analysis of the native gel revealed the formation of an oligomer with the wild-type and mutant PknK1-305 proteins. (B) PknK KD-GST forms oligomers of various sizes in solution. A Coomassie-stained gel (C) shows the effect of ATP on the oligomerization pattern of PknK KD-GST (5 μg) after kinase assay with 1 mM ATP, followed by resolution on a native and denaturing 4 to 15% gradient polyacrylamide gel. The presence of ATP did not reveal any changes in the oligomerization pattern of PknK KD-GST. Lane M, native molecular mass protein marker. (C) Effects of ATP binding and hydrolysis on the PknK KD-GST particle size. Dynamic light scattering measurements were made on PknK KD-GST after in vitro kinase assay with 1 mM ATP or AMP-PNP (a nonhydrolyzable ATP analog) and compared with a PknK KD-GST (minus ATP) control. The data are presented as the change in diameter of the particle in solution. In comparison to the control, the particle size increased by ∼56% in the presence of ATP. The change in the diameter of the particle from 7.5 nm (control) to 11.7 nm (with ATP) is indicated by a shift on the x axis. In contrast, the change in particle size with AMP-PNP was minimal.
We noticed that self-association of PknK occurred in the absence of ATP, and moreover, the K55M mutation that abolishes ATP binding and hydrolysis (Fig. 3B) did not hamper the associations of either the full-length or kinase domain K55M mutant protein (Fig. 6A). Similarly, with the purified PknK KD-GST protein, there was no change in the pattern with or without 1 mM ATP (Fig. 6B) or with increasing concentrations of ATP (data not shown). These results suggest that ATP binding may not be a prerequisite for PknK self-association; however, we cannot exclude the possibility that ATP binding and hydrolysis direct conformational changes required for activity. Due to the limited sensitivity of native PAGE to reveal subtle structural differences, we determined changes in particle size (here, PknK KD-GST) as a result of ATP or AMP-PNP (a nonhydrolyzable ATP analog) binding by dynamic light scattering. As shown in Fig. 6C, DLS measurements revealed a PknK KD-GST particle of 7.5-nm diameter in the absence of ATP. Binding and hydrolysis of ATP by PknK KD-GST resulted in a 56% increase in the size of the particle (∼11.7 nm) compared to PknK KD-GST (no ATP control) (∼7.5 nm). In contrast, changes in particle size as a result of binding a nonhydrolyzable ATP analog were minimal (∼16% increase). No changes were observed in the particle size of an unrelated, non-ATP binding protein with and without ATP (data not shown). Based on these data, we concluded that ATP binding does not affect intermolecular interactions between PknK KD molecules to form oligomeric complexes; however, ATP binding and hydrolysis may influence intramolecular interactions in PknK KD, resulting in conformation changes that increase its particle size.
The PknK C terminus exerts intrasteric control that autoregulates PknK activity.
The PknK C terminus is a multidomain regulatory region that contains an AAA motif encompassing an ATP/GTP binding site (P loop), a PDZ domain (a common structural domain of 80 to 90 amino acids found in the signaling proteins of bacteria, yeast, plants, and animals [27]), and a region consisting of superhelical (SUPR)- or tetratricopeptide (TPR)-like repeats (1, 28) (Fig. 7A); however, their roles in PknK function have not been elucidated. To investigate any regulatory role of the C terminus, we created and assayed wild-type and K55M mutant proteins of various lengths for their effects on the efficiency of the phoP-IVTT reaction in the presence of 1 mM ATP (Fig. 7A and B). In Fig. 4A, we demonstrated that the wild-type PknK1-1110 inhibits IVTT in an ATP-dependent manner. However, compared to its wild-type counterpart, the full-length K55M mutant protein was completely inefficient in inhibiting the IVTT reaction (Fig. 7B, top, and Fig. 4A). We attributed the lack of any inhibitory effect of PknK1-1110K55M to its inability to undergo ATP-induced activation. We concluded that, as a result of the substitution mutation, the K55M mutant is unable to bind and respond to ATP as an inducer of the conformational changes and autophosphorylation of PknK that are essential for its activity. In contrast, the PknK1-869K55M and PknK1-305K55M proteins inhibited the IVTT reaction, similar to their wild-type counterparts (Fig. 7B, middle and bottom). In light of these unanticipated results, we also investigated the in vitro kinase activities of the truncated K55M proteins. Both PknK1-869K55M and PknK1-305K55M showed consistent but faint autophosphorylation signals (see Fig. S4 in the supplemental material). These results highlight two novel aspects of PknK structure and activation that are illustrated in Fig. 7C: first, the lysine 55 residue is essential for PknK activity only in the full-length protein, and second, loss of the C-terminal region removes any steric hindrance that may have resulted from interactions between the C terminus and the catalytic site of the full-length protein, thereby leading to ATP binding and inhibitory activity despite the K55M mutation.
Fig 7.

Model for PknK activation and autoregulation: role of the C-terminal regulatory region. (A) Schematic linear representation of the various proposed domains in the full-length and truncated PknK proteins: a kinase domain (amino acids 26 to 283), an AAA motif (white box) encompassing the ATP/GTP-binding site (amino acids 368 to 375), a PDZ domain (amino acids 465 to 533), a helical region containing SUPR repeats (amino acids 777 to 1108), and a putative TPR-like motif (amino acids 904 to 929) (1, 27, 28; http://www.ebi.ac.uk/tools/pfa/iprscan and http://myhits.isb-sib.ch/cgi-bin/motif_scan). Truncated PknK proteins were constructed so that the PknK1-305 protein lacked the entire C-terminal regulatory region while the PknK1-869 lacked only the region containing the TPR-like helical motif (Δ241 amino acids). The K55M mutations in the proteins are marked with asterisks. (B) The PknK wild-type (WT) and mutant (K55M) proteins of various lengths were assayed for their inhibitory effects on IVTT of the target protein (PhoP) in the presence of ATP. CT, positive control for PhoP synthesis in the presence of pYA1635 (phoP) DNA alone. (C) Proposed model for PknK activation and autoregulation. For the sake of clarity, PknK is shown as a monomer; however, we have demonstrated that it can exist as oligomers of different sizes in solution. (1) Based on the results presented in this study, a hypothetical model for PknK activation (in terms of its ability to inhibit the IVTT reaction) is presented wherein, in the absence of any inducer, the interactions between the PknK C-terminal region and the N-terminal active site result in an inactive or autoinhibited kinase. Addition of a positive activator, such as ATP, results in conformational changes that relieve the autoinhibition and direct the autophosphorylation and activation of PknK. Similar structural changes resulting in PknK activation do not occur in the presence of ADP (arrow with an X). (2) Autophosphorylation of threonine residues in the PknK activation loop and some yet-unknown residues in the C-terminal region has been reported (27) and is depicted arbitrarily. The K55M mutation in the full-length protein excludes any possibility of ATP binding and hydrolysis and thus does not respond to structural activation by ATP, resulting in an inactive enzyme. Deletion of the C-terminal region in the truncated PknK1-869- and PknK1-305K55M proteins removes the autoregulatory control and allows ATP binding, resulting in an active protein. a.a, amino acids.
DISCUSSION
Our studies have focused on the functional characterization of PknK, a transcriptional regulatory STPK of M. tuberculosis. In this study, we elaborated on several structural and functional aspects of PknK-mediated growth regulation in vitro and in vivo. We determined that PknK mediates transcriptional reprogramming of M. tuberculosis by regulating tRNA expression during the logarithmic and stationary growth phases as a means to facilitate adaptation to changing growth environments. Furthermore, increases in PknKMtb levels and the resulting signaling events in M. smegmatis severely reduce the rate of growth and colony development. In accordance with its regulatory role in mycobacteria, we established that PknK directly inhibits in vitro transcription and translation through a phosphorylation-dependent mechanism and, in addition, provided valuable insights into the factors that enhance PknK-mediated inhibition of protein synthesis and contribute to PknK activation and regulation.
Although several STPKs, including PknK, have been implicated in the regulation of mycobacterial growth (13, 17, 24, 25, 30, 37), the molecular switch that governs the transition of M. tuberculosis from high to low growth rates is unknown. Given the induced levels of PknK and the increased survival of a ΔpknK mutant during stationary phase (30), we investigated its regulatory role by identifying genes that are regulated by PknK during the progression of M. tuberculosis from an “active” logarithmic phase to a “slow” stationary phase. Predictably, microarray results revealed a distinct regulatory role of PknK during stationary-phase growth. The differential expression of genes involved in cell wall processes and lipid metabolism in the LIX11 mutant validate our previous findings (30), which showed changes in the cell wall composition as a result of pknK deletion. A major highlight of these studies is the temporal expression of tRNA genes in the ΔpknK mutant. It is striking that of the 45 tRNA genes in M. tuberculosis (6), a significant number (>50%) were downregulated in the LIX11 mutant during log phase but upregulated in the stationary phase. Since M. tuberculosis is a G+C-rich organism, it is not surprising that the tRNA genes differentially regulated in LIX11 exhibited a codon bias for a G or C base in the third position. More importantly, a subset of 16 tRNA genes (up- or downregulated) was observed to be common between the logarithmic and stationary growth phases (Table 2). Although the precise mechanisms are presently unclear, this trend of tRNA expression underlines an important feature of PknK-mediated growth regulation. As with most bacteria, availability of nutrients and favorable growth conditions dictate the exponential growth of mycobacteria. Typically, such growth conditions are marked by high metabolic activity, leading to biosynthesis of macromolecules essential for growth and survival. It would then follow that during log phase, bacteria would exhibit an increased rate of protein synthesis and therefore require increased tRNA synthesis. Cells entering stationary phase are exposed to nutrient depletion, acidic pH, hypoxia, and oxidative stress, as well as other stress factors. Consequently, growth in stationary phase is slowed, primarily due to intracellular metabolic controls that would repress endogenous metabolism as a stress response mechanism. Repression of tRNA synthesis during stationary phase in the wild-type strain, but not in a pknK deletion strain, establishes PknK as a key component of such metabolic controls.
In principle, a “loss-of-function” phenotype resulting from a gene deletion can be complicated due to the activation of redundant, overlapping pathways, and in the case of well-coordinated regulatory effects of multiple transcriptional regulators and STPKs on mycobacterial growth, that is a distinct possibility. Moreover, the environmental signal for PknK activity is currently unknown. In view of these considerations and to elucidate the direct regulatory role of PknK, we evaluated the effects of pknK overexpression on mycobacterial growth and physiology. Increased intracellular PknKMtb levels and phosphorylation activity in a fast-growing, nonpathogenic M. smegmatis strain inhibited growth significantly and caused “dwarfed” colony morphology. Because PknK regulates the expression of genes involved in cell wall synthesis and lipid metabolism (see Fig. S1 in the supplemental material) (27), the defects in colony development are not surprising. It is noteworthy that pknKMtb overexpression did not affect cell viability significantly; rather, it severely reduced the rates of growth of the individual colonies. In addition, reversion of the inhibition phenotype when LIX79 induced cells were plated on noninducing medium suggests that intracellular PknK levels and activity are closely regulated. From the data presented, it is clearly evident that the cellular abundance of PknKMtb activity slows mycobacterial growth and, in addition, provides an explanation for our inability to obtain overexpression of pknK in the slow-growing M. tuberculosis. Together, the role of PknKMtb-dependent phosphorylation in the modulation of the growth rate and the regulation of tRNA synthesis during M. tuberculosis growth implicate PknK in mechanisms that control translation.
It is well known that phosphorylation of translation initiation and elongation factors is subjected to alteration during cell differentiation or bacteriophage infections (14, 21, 34, 41). In streptomycetes, ribosome-associated STPKs phosphorylate 11 ribosomal proteins and the elongation factor Tu (Ef-Tu), resulting in 30% loss of ribosomal activity (33). With respect to M. tuberculosis, evidence on the role of STPKs in translation control is limited. With the exception of Ef-Tu (42), phosphorylation of RNA polymerase subunits, transcription terminators, and ribosomal proteins has been speculated but has yet to be demonstrated (8, 40). The results presented in Fig. 4, 5, and 7 collectively provide evidence for a direct role of PknK in inhibition of in vitro protein synthesis. The cell-free expression system used in our studies provided an environment that eliminated interference from endogenous signals and transcriptional regulators and allowed direct assessment of PknK function. Additionally, IVTT enabled the synthesis of large proteins, such as PknK, that were stable, properly folded, and catalytically functional. This is the first instance of a successful demonstration of the synthesis of mycobacterial kinases using cell-free expression systems and their applicability to in vitro kinase assays.
Because transcription and translation occur simultaneously in bacteria, changes affecting one process automatically affect the other. Our data suggest that via phosphorylation of the IVTT components, PknK exerts inhibitory effects on transcription and translation processes, resulting in an overall decrease in the efficiency of protein synthesis. Although we have not identified the specific IVTT components that interact with PknK, our studies provide valuable insights into the factors that contribute to PknK function. PknK caused inhibition of in vitro transcription and translation only in the presence of exogenously supplied ATP. The role of ATP was 2-fold: first, it mediated PknK activation by directing conformational changes and autophosphorylation that were required for PknK's inhibitory function, and second, it acted as a positive effector of PknK action on the IVTT reaction. The increase in PknK activity or IVTT inhibition in the presence of increasing substrate (ATP or PknK) concentrations is consistent with the effects of excess substrate on enzyme kinetics. However, considering that the effective ATP and PknK concentrations or turnover are in a flux within the cells, it will be particularly relevant to investigate how these factors contribute to the regulation of PknK activity in changing growth environments. At least with PknK levels, it is sufficiently clear that an abundance of PknK in vitro and in mycobacteria, whether as a result of M. tuberculosis growth during stationary phase (30) or overexpression in M. smegmatis, inhibits protein synthesis, tRNA expression, and the growth rate. While the cell-free IVTT assay afforded a perturbation-free environment to illustrate PknK function, differential regulation of PknK activity due to environmental signals and transcriptional regulators must be considered to understand the mechanism of PknK-mediated growth regulation. The role of one such cellular signal, guanosine tetraphosphate (ppGpp), in the regulation of protein synthesis under stress conditions is well established (4, 44). Repression of tRNA and rRNA by ppGpp (23, 44) and its implications for the long-term persistence of M. tuberculosis (26, 39) necessitate further investigation of possible links between ppGpp signaling and PknK function.
Kinases are molecular switches that exist in either an “off” (inactive) state or an “on” (active) state. Ligand-induced N-lobe dimerization is emerging as the universal mechanism of STPK activation in M. tuberculosis (15, 18, 29, 36, 48). However, the soluble STPKs, PknG and PknK, exhibit structural uniqueness that may require other mechanisms of activation. As opposed to a monomeric PknB, PknG exists as a dimer in solution, and moreover, dimerization does not occur through the kinase domain but through the TPR domain at the C-terminal end (43). In addition, the ability of PknK-KD to form oligomeric complexes further highlights the structural differences between the M. tuberculosis membrane-bound receptor STPKs and soluble STPKs. PknK belongs to the MalT group of transcriptional activators that share motifs that classify them in a separate class of proteins called the signal transduction ATPases with numerous domains (STAND) proteins (28). STAND proteins are atypical AAA+ (12) proteins involved in signal transduction that integrate signals to assemble them into large scaffolds (11, 28, 32). Unlike E. coli MalT, where oligomerization is regulated by ATP and maltotriose (9, 10), intermolecular interactions of PknK did not appear to depend on ATP, indicating divergence from the MalT activation mechanism. However, the PknK C terminus contains regions that are homologous to MalT domains responsible for ATP binding and hydrolysis and maltoriose binding (9). Thus, it is possible that, in addition to the catalytic site, ATP binds to distinct sites in the C-terminal regulatory region and that a second ligand may be required in conjunction to regulate PknK activation.
The PknK C terminus has SUPR- or TPR-like domains that are typical protein interaction modules with unique regulatory functions. Currently, there is no structural information about PknK available that could aid in understanding the contributions of the PknK C-terminal regulatory region and its phosphorylation (27) towards PknK function. We demonstrated that the PknK C-terminal region autoregulates PknK activity, and we propose a model for PknK activation (Fig. 7C). We define PknK activation as the ability of PknK to inhibit the IVTT reaction, so an inactive PknK would have no effect on IVTT. We hypothesize that in the absence of an inducer, PknK exists in an inactive conformation with no inhibitory effects on IVTT. Addition of a positive activator, such as ATP, but not ADP, leads to autophosphorylation of PknK and induces conformational changes that facilitate PknK activation and, hence, inhibition of protein synthesis. Typically, the invariant lysine residue plays a role in positioning the ATP molecule and the substrate protein for catalysis (19). Previous results have shown a key role of lysine 55 in PknK activity (27). Our results confirm these observations and also show that lysine 55 is essential for the activity of the full-length PknK protein only. The loss of the C-terminal region, specifically the last 241 amino acids, resulted in truncated proteins that were active despite the mutation. It is possible that in the inactive mode the C terminus interacts with residues around the active site, including lysine 55, so its deletion renders the truncated K55M proteins active. We hypothesize that intramolecular interactions with the C terminus may regulate PknK activity by ensuring activation only in the presence of an activator ligand, like ATP. Such a mechanism of autoinhibition is favorable from a regulatory viewpoint, as it enables fine-tuning of an activation mechanism that would otherwise lead to wasteful dissipation of energy. Clearly, in light of PknK's role in slowing growth, it would not be beneficial to the cell for PknK to be constitutively active.
The autoinhibition mechanism for kinase regulation has not been described for any mycobacterial STPK but is highly conserved in eukaryotic kinases, such as the cyclic AMP (cAMP)-dependent protein kinase and the human twitchin kinase (20). Typically, part of the kinase contains an autoregulatory sequence that folds into the active site and blocks activity. The kinase is switched on by the binding of an allosteric regulator that removes the autoregulatory sequence from the active site. Protein kinase C was found to contain a pseudosubstrate near its N terminus, and truncation mutagenesis of the pseudosubstrate sequence rendered the kinase constitutively active (20). The autoregulatory sequence of PknK resides in the last 241 amino acids at the C terminus, thus implicating the TPR-like motif in PknK autoregulation. In summary, our findings open exciting avenues of research to address questions on PknK structure and function that will shed light on the mechanisms of PknK oligomerization and autoregulation and their implication in the regulation of M. tuberculosis growth and persistence.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Genotypic Technology Pvt. Ltd., Bangalore, India, for the microarray processing and data compilation reported in this article. We are grateful to Malini Rajagopalan for the kind gift of plasmid pJFR19. We thank Shaleen Korch, Praveen Alamuri, and Simran Banga for valuable suggestions and discussions throughout the project and Yiang Tian for help with dynamic light scattering experiments. We also acknowledge Gwendolyn Clay for the purified PknK KD-GST used in this study.
B.P.O. was supported by the Arizona State University PREP program for Biomedical Research and Public Health Service grant GM071798 from the U.S. National Institutes of Health. This research was supported by Public Health Service grant AI46428 from the U.S. National Institutes of Health and Arizona State University Start-up Funding, awarded to J.E.C.-C.
Published ahead of print 1 June 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Av-Gay Y, Everett M. 2000. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 8: 238–244 [DOI] [PubMed] [Google Scholar]
- 2. Beste DJ, et al. 2007. Transcriptomic analysis identifies growth rate modulation as a component of the adaptation of mycobacteria to survival inside the macrophage. J. Bacteriol. 189: 3969–3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chao JD, et al. 2010. Convergence of Ser/Thr and two-component signaling to coordinate expression of the dormancy regulon in Mycobacterium tuberculosis. J. Biol. Chem. 285: 29239–29246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chatterji D, Ojha AK. 2001. Revisiting the stringent response, ppGpp and starvation signaling. Curr. Opin. Microbiol. 4: 160–165 [DOI] [PubMed] [Google Scholar]
- 5. Chauhan A, et al. 2006. Mycobacterium tuberculosis cells growing in macrophages are filamentous and deficient in FtsZ rings. J. Bacteriol. 188: 1856–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cole ST, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544 [DOI] [PubMed] [Google Scholar]
- 7. Cowley S, et al. 2004. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol. Microbiol. 52: 1691–1702 [DOI] [PubMed] [Google Scholar]
- 8. Cui T, Zhang L, Wang X, He ZG. 2009. Uncovering new signaling proteins and potential drug targets through the interactome analysis of Mycobacterium tuberculosis. BMC Genomics 10: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Danot O. 2001. A complex signaling module governs the activity of MalT, the prototype of an emerging transactivator family. Proc. Natl. Acad. Sci. U. S. A. 98: 435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Danot O. 2010. The inducer maltotriose binds in the central cavity of the tetratricopeptide-like sensor domain of MalT, a bacterial STAND transcription factor. Mol. Microbiol. 77: 628–641 [DOI] [PubMed] [Google Scholar]
- 11. Danot O, Marquenet E, Vidal-Ingigliardi D, Richet E. 2009. Wheel of life, wheel of death: a mechanistic insight into signaling by STAND proteins. Structure. 17: 172–182 [DOI] [PubMed] [Google Scholar]
- 12. Erzberger JP, Berger JM. 2006. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35: 93–114 [DOI] [PubMed] [Google Scholar]
- 13. Fernandez P, et al. 2006. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J. Bacteriol. 188: 7778–7784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frederickson RM, Mushynski WE, Sonenberg N. 1992. Phosphorylation of translation initiation factor eIF-4E is induced in a ras-dependent manner during nerve growth factor-mediated PC12 cell differentiation. Mol. Cell. Biol. 12: 1239–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gay LM, Ng HL, Alber T. 2006. A conserved dimer and global conformational changes in the structure of apo-PknE Ser/Thr protein kinase from Mycobacterium tuberculosis. J. Mol. Biol. 360: 409–420 [DOI] [PubMed] [Google Scholar]
- 16. Ginsberg AM, Spigelman M. 2007. Challenges in tuberculosis drug research and development. Nat. Med. 13: 290–294 [DOI] [PubMed] [Google Scholar]
- 17. Gopalaswamy R, Narayanan S, Chen B, Jacobs WR, Av-Gay Y. 2009. The serine/threonine protein kinase PknI controls the growth of Mycobacterium tuberculosis upon infection. FEMS Microbiol. Lett. 295: 23–29 [DOI] [PubMed] [Google Scholar]
- 18. Greenstein AE, Echols N, Lombana TN, King DS, Alber T. 2007. Allosteric activation by dimerization of the PknD receptor Ser/Thr protein kinase from Mycobacterium tuberculosis. J. Biol. Chem. 282: 11427–11435 [DOI] [PubMed] [Google Scholar]
- 19. Hanks SK, Hunter T. 1995. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9: 576–596 [PubMed] [Google Scholar]
- 20. Heierhorst J, et al. 2000. Protein Ser/Thr Kinases, p 297 In Conn MP, Means AR. (ed), Principles of molecular regulation. Humana, Totowa, NJ [Google Scholar]
- 21. Hershey JW. 1989. Protein phosphorylation controls translation rates. J. Biol. Chem. 264: 20823–20826 [PubMed] [Google Scholar]
- 22. Huse M, Kuriyan J. 2002. The conformational plasticity of protein kinases. Cell 109: 275–282 [DOI] [PubMed] [Google Scholar]
- 23. Jain V, Kumar M, Chatterji D. 2006. ppGpp: stringent response and survival. J. Microbiol. 44: 1–10 [PubMed] [Google Scholar]
- 24. Jang J, et al. 2010. Functional characterization of the Mycobacterium tuberculosis serine/threonine kinase PknJ. Microbiology 156: 1619–1631 [DOI] [PubMed] [Google Scholar]
- 25. Kang CM, et al. 2005. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 19: 1692–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klinkenberg LG, Lee JH, Bishai WR, Karakousis PC. 2010. The stringent response is required for full virulence of Mycobacterium tuberculosis in guinea pigs. J. Infect. Dis. 202: 1397–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar P, et al. 2009. The Mycobacterium tuberculosis protein kinase K modulates activation of transcription from the promoter of mycobacterial monooxygenase operon through phosphorylation of the transcriptional regulator VirS. J. Biol. Chem. 284: 11090–11099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leipe DD, Koonin EV, Aravind L. 2004. STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J. Mol. Biol. 343: 1–28 [DOI] [PubMed] [Google Scholar]
- 29. Lombana TN, et al. 2010. Allosteric activation mechanism of the Mycobacterium tuberculosis receptor Ser/Thr protein kinase, PknB. Structure 18: 1667–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malhotra V, Arteaga-Cortes LT, Clay G, Clark-Curtiss JE. 2010. Mycobacterium tuberculosis protein kinase K confers survival advantage during early infection in mice and regulates growth in culture and during persistent infection: implications for immune modulation. Microbiology 156: 2829–2869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Malhotra V, Tyagi JS, Clark-Curtiss JE. 2009. DevR-mediated adaptive response in Mycobacterium tuberculosis H37Ra: links to asparagine metabolism. Tuberculosis 89: 169–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marquenet E, Richet E. 2007. How integration of positive and negative regulatory signals by a STAND signaling protein depends on ATP hydrolysis. Mol. Cell 28: 187–199 [DOI] [PubMed] [Google Scholar]
- 33. Mikulik K, Zhoulanova E, Hoang QK, Janecek J, Bezouskova S. 1999. Protein kinase associated with ribosomes of streptomycetes. Folia Microbiol. 44: 123–130 [DOI] [PubMed] [Google Scholar]
- 34. Mikulik K, Zhulanova E. 1995. Sequencing of the tuf1 gene and the phosphorylation pattern of EF-Tu1 during development and differentiation in Streptomyces collinus producing kirromycin. Biochem. Biophys. Res. Commun. 213: 454–461 [DOI] [PubMed] [Google Scholar]
- 35. Nolen B, Taylor S, Ghosh G. 2004. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 15: 661–675 [DOI] [PubMed] [Google Scholar]
- 36. Ortiz-Lombardia M, Pompeo F, Boitel B, Alzari PM. 2003. Crystal structure of the catalytic domain of the PknB serine/threonine kinase from Mycobacterium tuberculosis. J. Biol. Chem. 278: 13094–13100 [DOI] [PubMed] [Google Scholar]
- 37. Papavinasasundaram KG, et al. 2005. Deletion of the Mycobacterium tuberculosis pknH gene confers a higher bacillary load during the chronic phase of infection in BALB/c mice. J. Bacteriol. 187: 5751–5760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pereira SF, Goss L, Dworkin J. 2011. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 75: 192–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Primm TP, et al. 2000. The stringent response of Mycobacterium tuberculosis is required for long-term survival. J. Bacteriol. 182: 4889–4898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prisic S, et al. 2010. Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U. S. A. 107: 7521–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robertson ES, Aggison LA, Nicholson AW. 1994. Phosphorylation of elongation factor G and ribosomal protein S6 in bacteriophage T7-infected Escherichia coli. Mol. Microbiol. 11: 1045–1057 [DOI] [PubMed] [Google Scholar]
- 42. Sajid A, et al. 2011. Interaction of Mycobacterium tuberculosis elongation factor Tu with GTP is regulated by phosphorylation. J. Bacteriol. 193: 5347–5358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scherr N, et al. 2007. Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 104: 12151–12156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srivatsan A, Wang JD. 2008. Control of bacterial transcription, translation and replication by (p)ppGpp. Curr. Opin. Microbiol. 11: 100–105 [DOI] [PubMed] [Google Scholar]
- 45. Tian Y, et al. 2009. Synthesis, nanostructure, functionality, and application of polyfluorene-block-poly(N-isopropylacrylamide)s. Macromolecules 43: 282–291 [Google Scholar]
- 46. Walburger A, et al. 2004. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304: 1800–1804 [DOI] [PubMed] [Google Scholar]
- 47. WHO 2009. Global tuberculosis control: epidemiology, strategy, financing. WHO, Geneva, Switzerland [Google Scholar]
- 48. Young TA, Delagoutte B, Endrizzi JA, Falick AM, Alber T. 2003. Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat. Struct. Biol. 10: 168–174 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.