

Abstract
Staphylococcus aureus is a significant human pathogen that is capable of infecting a wide range of host tissues. This bacterium is able to evade the host immune response by utilizing a repertoire of virulence factors. These factors are tightly regulated by various two-component systems (TCS) and transcription factors. Previous studies have suggested that transcriptional regulation of a subset of immunomodulators, known as the staphylococcal superantigen-like proteins (Ssls), is mediated by the master regulators accessory gene regulator (Agr) TCS, S. aureus exoprotein expression (Sae) TCS, and Rot. Here we demonstrate that Rot and SaeR, the response regulator of the Sae TCS, synergize to coordinate the activation of the ssl promoters. We have determined that both transcription factors are required, but that neither is sufficient, for promoter activation. This regulatory scheme is mediated by direct binding of both transcription factors to the ssl promoters. We also demonstrate that clinically relevant methicillin-resistant S. aureus (MRSA) strains respond to neutrophils via the Sae TCS to upregulate the expression of ssls. Until now, Rot and the Sae TCS have been proposed to work in opposition of one another on their target genes. This is the first example of these two regulators working in concert to activate promoters.
INTRODUCTION
Staphylococcus aureus is a major human pathogen responsible for a range of diseases from soft tissue infections to life-threatening infections such as toxic shock syndrome, pneumonia, sepsis, endocarditis, and osteomyelitis. The versatility of this bacterium is largely due to the regulated expression of an arsenal of virulence factors, including cell wall-associated factors, cytotoxins, and immunomodulators (16, 39). Among the immunomodulators, the staphylococcal superantigen-like proteins (Ssls) represent a family of 14 secreted proteins, each with a molecular mass of around 25 kDa, that have a variety of immunomodulatory properties, including inhibition of complement activation and neutrophil recruitment, as well as blocking of opsonization by IgG and IgA (17). Importantly, Ssl overproduction is associated with S. aureus pathogenesis in animal models of systemic infection (3, 7, 52).
The expression of virulence factors in S. aureus is tightly regulated by two-component systems (TCS) (i.e., accessory gene regulator [Agr] and S. aureus exoprotein expression [Sae] TCS), global regulators (i.e., Rot, SigB, SarA, and SarA homologues), and regulatory RNA molecules that interact to ensure the temporal expression of these factors (11, 15, 40). The best-characterized TCS in S. aureus is the Agr quorum-sensing system, which is composed of the agrBCDA structural genes and a 514-nucleotide regulatory RNA molecule, RNAIII, which is the main effector molecule of the Agr system (41). Upon reaching quorum, the Agr TCS is activated and the autoinducing peptide (AIP), encoded by agrD, is produced. AIP is processed and released by AgrB, upon which it is recognized by AgrC, the sensor kinase (41, 50). Subsequently, AgrC activates the response regulator AgrA, resulting in the expression of RNAIII (27, 38). RNAIII directly and indirectly controls the majority of Agr target genes by its antisense function (6, 37). The indirect regulation of virulence genes by RNAIII is mediated principally by posttranscriptional control of the transcription factor repressor of toxins (Rot), by which RNAIII prevents the translation of rot (8, 19). Rot is a member of the SarA family of DNA binding proteins involved in the repression of genes that encode various secreted proteins and activation of genes that encode cell-associated proteins (11, 30, 35, 47, 53, 54).
An additional critical TCS involved in the regulation of the staphylococcal virulon is the S. aureus exoprotein expression TCS, which is encoded by the sae locus (24). The sae locus consists of four open reading frames (saePQRS), of which saeR and saeS encode the response regulator and sensor histidine kinase, respectively (21). saeP encodes a poorly characterized putative lipoprotein, while saeQ encodes a membrane protein involved in the stabilization of SaeS (1, 26). The sae locus has two promoters, designated P1 and P3, of which P1 is positively autoregulated and negatively regulated by SigB and Rot (1, 18, 23, 25, 26, 30). Activation of P1 can occur through exposure to neutrophils and phagocyte products, such as low pH, H2O2, and the antimicrobial peptide α-defensin (18, 43, 55). Activation of the P3 promoter results in a transcript carrying saeR, saeS, and a truncated saeQ. This promoter demonstrates low, constitutive activity (18). The Sae TCS is primarily known for its requirement for the expression of cytotoxin-encoding genes (22, 24, 42, 46, 56). Consistent with this, the Sae TCS plays a critical role in S. aureus pathogenesis in several animal models of infection (25, 36, 42, 56, 57). In addition, the Sae TCS plays an essential role in S. aureus survival in human whole blood and in the avoidance of human neutrophil-mediated killing (56). Interestingly, the S. aureus strain Newman, a highly virulent and commonly used methicillin-sensitive strain, exhibits high sae expression levels, due to a single amino acid substitution in the first N-terminal transmembrane domain of SaeS that results in the change of a leucine at position 18 to proline (1, 4, 18, 48). This L18P amino acid substitution results in constitutive activation of the Sae TCS, which in turn is responsible for the altered phenotypes exhibited by strain Newman (31, 58), including the increased production of exoproteins observed in this strain (1, 18, 31, 32, 48).
We recently demonstrated that inactivation of agr in strain Newman resulted in the production of higher levels of Ssls than in the wild-type (WT) strain and that the ability of Agr to repress ssls was dependent on the RNAIII-mediated regulation of Rot (7). Surprisingly, we found that Rot regulates ssl expression by directly binding to and activating the ssl promoters. Recently, the Sae TCS has also been implicated in the regulation of ssls (44). In this study, we investigated the molecular mechanism by which Rot and the Sae TCS regulate ssl expression. Our genetic and biochemical data demonstrate that Rot and SaeR synergize to bind to the ssl promoters and activate the expression of these genes in S. aureus, a regulatory scheme that has not been previously reported.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The S. aureus strains used in this study are described in Table 1. S. aureus were grown in Roswell Park Memorial Institute medium (RPMI) supplemented with 1% Casamino Acids (CAS) (RPMI-CAS) with shaking at 180 rpm, unless otherwise indicated. When appropriate, tryptic soy broth (TSB) and RPMI-CAS were supplemented with chloramphenicol (CM) at a final concentration of 10 μg/ml.
Table 1.
Staphylococcus aureus strains
| Strain | Background | Description | Genotype | Reference or source |
|---|---|---|---|---|
| VJT 1.01 | Newman | Wild type | Wild type | 14 |
| VJT 2.84 | USA100 | USA100 | Wild type | NARSAa |
| VJT 2.59 | USA200 | UAMS-1 | Wild type | 20 |
| VJT 12.61 | USA300 | LAC | Wild type | 13 |
| VJT 4.79 | USA400 | MW2 | Wild type | 2 |
| VJT 2.86 | USA500 | USA500 | Wild type | 45 |
| VJT 15.49 | USA600 | USA600 | Wild type | NARSA |
| VJT 21.05 | USA700 | USA700 | Wild type | NARSA |
| VJT 21.06 | USA800 | USA800 | Wild type | NARSA |
| VJT 21.08 | USA1100 | USA1100 | Wild type | NARSA |
| VJT 7.17 | VJT 1.01 | Transduction of agr::tet from RN7206 into VJT 1.01 | agr::tet | 7 |
| VJT 9.98 | VJT 1.01 | Transduction of rot::Tn917 from RN10623 into VJT 1.01 | rot::Tn917 | 7 |
| VJT 16.99 | VJT 1.01 | Replacement of saeQRS with aad in VJT 1.01 | saeQRS::spec | This study |
| VJT 10.03 | VJT 1.01 | Transduction of rot::Tn917 from RN10623 into VJT 7.17 | agr::tet rot::Tn917 | 7 |
| VJT 17.27 | VJT 1.01 | Transduction of agr::tet from RN7206 into VJT 16.99 | agr::tet saeQRS::spec | This study |
| VJT 19.04 | VJT 1.01 | Transduction of rot::Tn917 from RN10623 into VJT 17.27 | agr::tet saeQRS::spec rot::Tn917 | This study |
| AH1263 | USA300 LAC | Erythromycin-sensitive clone | Wild type | 9 |
| AH1292 | USA300 LAC | Transduction of agr::tet from RN7206 into AH 1263 | agr::tet | 7 |
| VJT 28.25 | USA300 LAC | Transduction of saeS::bursa from HF6131 into AH1263 | saeS::bursa | This study |
| VJT 31.49 | USA300 LAC | Transduction of saeS::bursa from HF6131 into VJT 28.25 | agr::tet saeS::bursa | This study |
| HF6131 | RN6734 | Transduction of saeS::bursa | saeS::bursa | 1 |
| RN4220 | 8325-4 | Restriction-deficient cloning host | 28 |
NARSA, Network of Antimicrobial Resistance in Staphylococcus aureus.
Escherichia coli DH5α was used to propagate plasmids, and T7 lysY lacQ (New England BioLabs) was used as an expression strain for recombinant protein. E. coli was grown in Luria broth (LB), and when needed, the medium was supplemented with ampicillin at a final concentration of 100 μg/ml or kanamycin at a final concentration of 50 μg/ml.
Construction of mutants.
Strains used in this study are described in Table 1. S. aureus mutants lacking the agr locus, saeS, and rot were generated by transduction using phage 80α.
The S. aureus saeQRS::spec mutant strain was generated using the allelic replacement strategy previously described (5). Plasmids for allelic replacement of saeQRS were constructed using pGEM-T (Promega) and pKOR-1. Briefly, sequences flanking saeQRS were PCR amplified with primers VJT125 and VJT126 for the upstream fragment and primers VJT127 and VJT128 for a downstream fragment. The PCR amplicons were digested with XmaI and PstI and assembled into pGEMT-T (Promega). To generate the plasmid containing the saeQRS::spec construct, a spectinomycin resistance cassette (aad9) was amplified from plasmid pBT-S using primers VJT391 and VJT392 and subsequently digested with XmaI and subcloned into the pGEM-T saeQRS plasmid (an internal XmaI site was previously generated between both flanking sequences to facilitate the insertion of antibiotic resistance markers). A PCR amplicon of the resultant saeQRS::spec construct was then recombined into pKOR-1, resulting in the pKOR-1ΔsaeQRS::spec plasmid. Further allelic replacement was carried out in strain Newman according to previously described methods (5).
Plasmids.
Primers and plasmids used in this study are listed in Tables 2 and 3, respectively. Plasmids were isolated from E. coli and then transformed into the restriction-deficient S. aureus RN4220, followed by electroporation into the respective S. aureus strains.
Table 2.
Primers
| No. | Name | Sequence |
|---|---|---|
| 403 | rot-6XHis-3′-EcoRI | 5′-CCCCGAATTCTTAGTGATGGTGATGGTGATGCACAGCAATAATTGCGTTTAAAC |
| 420 | rot5′-XhoI | 5′-GGGGCTCGAGATGAAAAAAGTAAATAACGACACTG |
| 481 | saeR-XhoI-R | 5′-CCCCCTCGAGATGACCCACTTACTGATCG |
| 484 | saeS-EcoRI-F | 5′-CCCCGAATTCTTATGACGTAATGTCTAATTTGTG |
| 761 | saeS-F-XhoI | 5′-CCCCCTCGAGATGGTGTTATCAATTAGAAGTCA |
| 762 | saeS-R-BamHI | 5′-CCCCGGATCCTTATGACGTAATGTCTAATTTGTG |
| 745 | R-XhoI-3 × FLAG-rot | 5′-CCCCTCGAGCTTGTCATCGTCATCCTTGTAATCGATATCATGATCTTTATAATCACCGTCATGGTCTTTGTAGTC-CACAGCAATAATTGCGTTTAAACTA |
| 313 | rot-5′-F-NdeI | 5′-CCCCCATATGAAAAAAGTAAATAACGACACTG |
| 311 | rot-6 × His-3′-R-XhoI | 5′-CCCCTCGAGTTAGTGATGGTGATGGTGATGCACAGCAATAATTGCGTTTAAAC |
| 706 | saeR-R-BamHI | 5′-CCCGGATCCTTATCGGCTCCTTTCAAATTTATATC |
| 438 | saeR-5′-NdeI | 5′-CCCCCATATGACCCACTTACTGATCGTGGATG |
| 771 | saeS-dN92-R-XhoI | 5′-CCCCCTCGAGAAAGAAATTTATGAATTAAATCAATC |
| 762 | saeS-R-BamHI | 5′-CCCCGGATCCTTATGACGTAATGTCTAATTTGTG |
| 305 | p-ssl7-R-BIO | 5′-BIO-CCCCAGTACTATTCTCCCAATCTATTT |
| 374 | pSSL7-R-NdeI | 5′-CCCCATATGAGTACTATTCTCCCAATCTATTTA |
| 304 | p-ssl9-R-Bio | 5′-BIO-CCCCATTTTTTTGCTCCAATCTTAATG |
| 375 | pSSL9-R-NdeI | 5′-CCCCATATG-ATTTTTTTGCTCCAATCTTAATGTA |
| 82 | LukAB intra 3′-BIO | 5′-AGTATCACCATCAAGATTCTTC |
| 87 | LukAB intra 5′ | 5′-CCCCGAATTCAAAGAAGGATAATATTGAAAGG |
| 372 | LukAB intra 3′ | 5′-AGTATCACCATCAAGATTCTTC |
| 308 | p-ssl11-R-BIO | 5′-BIO-CCCC-AATTCTATGCTCCCAATTTTTAG |
| 345 | pssl11-F-PstI | 5′-CCCCCTGCAGTTAGGCACTGTGAAAGCGC |
| 323 | pssl11-R-no RBS-KpnI | 5′-CCCCGGTACCTTTTAGTCTATTTGATTTATTCTATTA |
| 341 | pssl7-F-PstI | 5′-CCCCCTGCAGGCAGACTAGTAATTGTAGGG |
| 319 | pssl7-R-no RBS-KpnI | 5′-CCCCGGTACCCTATTTATAAATTTTGTCTTAATATATT |
| 757 | pSSL7-F-saeR site | 5′-AAAAATAGTTAGAAAGAGGTTAATTCATA |
| 758 | pSSL7-R-saeR site | 5′-TATGAATTAACCTCTTTCTAACTATTTTT |
| 343 | pssl9-F-PstI | 5′-CCCCCTGCAGGAATGAAAGCTTAAGAAGCGG |
| 321 | pssl9-R-no RBS-KpnI | 5′-CCCCGGTACCCTTAATGTATTGGATTGTTATTATTA |
| 759 | pSSL9-F-saeR site | 5′-TTTATTTAGATGAGCGTTAATGTCAG |
| 760 | pSSL9-R-saeR site | 5′-CTGACATTAACGCTCATCTAAATAAA |
| 552 | P1-sae-R-PstI | 5′-CCCCTGCAGTTATTGTGGCAAAAGGTTTATAAA |
| 555 | P1-sae(full)-F-NdeI | 5′-CCCCATATGGCTAACTCCTCATTTCTTCAAT |
| 348 | sGFP-R-EcoRI | 5′-CCCGAATTCTTAGTGGTGGTGGTGG |
| 347 | psarA-F-PstI | 5′-CCCCTGCAGCTGATATTTTTGACTAAACCAAATG |
| 125 | NWMN_0674-0676 5′-1 AttB1 | 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTAATAAAAAGGATGGACATAGATG |
| 126 | NWMN_0674-0676 3′-1-Xma | 5′-TCCCCCCGGGAAAATGCAAAGACTAAAAAGAAG |
| 127 | NWMN_0674-0676 5′-2-Xma | 5′-TCCCCCCGGGACATTCTTTCTATTTATTGTGTG |
| 128 | NWMN_0674-0676 3′-2 AttB2 | 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTAGTTCAGATAAATATTTCCTTAC |
| 671 | prot-F-PstI | 5′-CCCCTGCAGACAGTAGATGCTCATCTTTTTTTAG |
| 517 | prot-R-no RBS-KpnI | 5′-CCCGGTACCAAATAAACTTGCTTTCTATTCAATTG |
| 391 | pSpec.B-Xma | 5′-TCCCCCCGGGAAAATTGAAAAAAGTGTTTCCACC |
| 392 | pSpec.A-Xma | 5′-TCCCCCCGGGCAGGTTATGACCATCTGTGCC |
Table 3.
Plasmids
| Name | Description | Resistance | Reference or source |
|---|---|---|---|
| pOS1-PhrtAB | Empty vector control containing the hrtAB promoter | Cm | 51 |
| pOS1-PhrtAB-rot6 × His | hrtAB promoter controlling expression of rot with a C-terminal His tag | Cm | This study |
| pOS1-PhrtAB-saeRS | hrtAB promoter controlling expression of saeRS | Cm | This study |
| pOS1-Plgt | lgt promoter in an empty vector | Cm | 10 |
| pOS1-Plgt-saeS (Newman) | lgt promoter controlling expression of saeS amplified from strain Newman | Cm | This study |
| pOS1-Plgt-saeS (LAC) | lgt promoter controlling expression of saeS amplified from strain LAC | Cm | This study |
| pOS1-Plgt-rot3 × FLAG | lgt promoter controlling expression of rot containing a C-terminal FLAG tag | Cm | This study |
| pCM11 | sarA promoter and sod RBS controlling expression of sgfpa | Erm | 29 |
| pOS1sGFP promoterless | pOS1 containing the sgfp with no promoter | Cm | 7 |
| pOS1sGFP-Pssl7-sod RBS | ssl7 promoter controlling expression of sgfp | Cm | 7 |
| pOS1sGFP-Pssl9-sod RBS | ssl9 promoter controlling expression of sgfp | Cm | 7 |
| pOS1sGFP-Pssl11-sod RBS | ssl11 promoter controlling expression of sgfp | Cm | 7 |
| pOS1sGFP-PsarA-sod RBS | sarA promoter controlling expression of sgfp | Cm | This study |
| pOS1sGFP-Prot-sod RBS | rot promoter controlling expression of sgfp | Cm | This study |
| pOS1sGFP-PsaeP1 | sae P1 promoter controlling expression of sgfp | Cm | This study |
| pOS1sGFP-Pssl7-saeR-sod RBS | ssl7 promoter with mutated SaeR binding site controlling expression of sgfp | Cm | This study |
| pOS1sGFP-Pssl9-saeR-sod RBS | ssl9 promoter with mutated SaeR binding site controlling expression of sgfp | Cm | This study |
| pET41b | Empty expression vector with IPTG-inducible promoter | Amp | Novagen |
| pET41b saeSΔN92 | saeS deltaN92 expression vector | Amp | This study |
| pET41b saeR | saeR expression vector | Amp | This study |
| pET14b | Empty expression vector with IPTG-inducible promoter | Kan | Novagen |
| pET14b rot | rot expression vector | Kan | This study |
| pKOR1 | Empty vector used for allelic replacement | Cm | 5 |
| pKOR1 ΔsaeQRS::spec | pKOR1 vector containing saeQRS-homologous regions with aad insertion | Cm | This study |
sgfp, superfolder green fluorescent protein gene.
(i) Complementation plasmids.
For the construction of a plasmid expressing rot under the control of the hemin-inducible hrtAB promoter, a PCR amplicon containing the rot open reading frame (ORF) (NWMN_1655) into which codons for six histidine residues were inserted at the 3′ end just prior to the stop codon was made using primers 420 and 403. The PCR product was digested with EcoRI and XhoI and ligated into the E. coli/S. aureus shuttle vector pOS1-PhrtAB, which had been digested with the same restriction enzymes. The ligation product was then transformed into E. coli DH5α, and the resulting plasmid was designated pOS1-PhrtAB-rot6×His.
For the construction of a plasmid expressing saeRS under the control of the hemin-inducible hrtAB promoter, a PCR amplicon containing the saeRS ORF (NWMN_0675 and NWMN_0674, respectively) was made using primers 481 and 484. Further construction of the vector was carried out as done with the pOS1-PhrtAB-rot6×His vector. The resulting plasmid was designated pOS1-PhrtAB-saeRS.
For the construction of a plasmid constitutively expressing saeS, a PCR amplicon containing the saeS ORF (NWMN_0674) was made using primers 761 and 762 with either Newman genomic DNA (gDNA) or USA300 LAC gDNA as the template. The PCR products were digested with BamHI and XhoI and ligated into the E. coli/S. aureus shuttle vector pOS1-Plgt, which had been digested with the same restriction enzymes. The ligation products were then transformed into E. coli DH5α, and the resulting plasmids were designated pOS1-Plgt-saeS (Newman) and pOS1-Plgt-saeS (LAC).
For the construction of a plasmid constitutively expressing rot with a C-terminal FLAG tag, a PCR amplicon containing the rot ORF (NWMN_1655) was made using primers 745 and 313. The PCR product was digested with NdeI and XhoI and ligated into the E. coli/S. aureus shuttle vector pOS1-Plgt, which had been digested with the same restriction enzymes. The ligation product was then transformed into E. coli DH5α, and the resulting plasmid was designated pOS1-Plgt-rot-FLAG.
(ii) Generation of reporter plasmids.
The construction of ssl7, ssl9, and ssl11 reporter plasmids was described previously (7). For construction of a plasmid expressing the green fluorescent protein (GFP) gene under the control of the sarA promoter, a PCR amplicon was made using pCM11 as the template to amplify the sarA promoter and GFP gene using primers 347 and 348. The PCR product was digested with PstI and EcoRI and ligated into the E. coli/S. aureus shuttle vector pOS1sGFP, which had been digested with the same restriction enzymes. The ligation product was then transformed into E. coli DH5α, and the resulting plasmid was designated pOS1sGFP-PsarA-sod RBS.
For the construction of a plasmid expressing the GFP gene under the control of the sae P1 promoter, a PCR amplicon containing the P1 promoter region was made using primers 552 and 555. The PCR product was digested with PstI and NdeI and ligated into the E. coli/S. aureus shuttle vector pOS1sGFP, which had been digested with the same restriction enzymes. The ligation product was then transformed into E. coli DH5α, and the resulting plasmid was designated pOS1sGFP-Psae P1.
For construction of a plasmid expressing the GFP gene under the control of the rot promoter, a PCR amplicon containing the rot promoter region was made using primers 671 and 517. The PCR product was digested with PstI and KpnI and ligated into the E. coli/S. aureus shuttle vector pOS1sGFP, which had been digested with the same restriction enzymes. The ligation product was then transformed into E. coli DH5α, and the resulting plasmid was designated pOS1sGFP-Prot-sod RBS.
For construction of a plasmid expressing the GFP gene under the control of the ssl7 or ssl9 promoter in which the SaeR binding site has been mutated, a PCR splicing by overlap extension (SOE) approach was used, in which the 5′ ends were generated using primers 341 and 758 for the ssl7 promoter and primers 343 and 760 for the ssl9 promoter. The 3′ ends were generated with primers 319 and 757 for the ssl7 promoter and primers 321 and 759 for the ssl9 promoter. The 5′ and 3′ ends were mixed together as the template for a PCR using primers 341 and 319 for the ssl7 promoter and primers 343 and 321 for the ssl9 promoter. PCR products were digested with PstI and KpnI and then ligated into the E. coli/S. aureus shuttle vector pOS1sGFP, which had been digested with the same restriction enzymes. The ligation products were then transformed into E. coli DH5α, and the resulting plasmids were designated pOS1sGFP-Pssl7-SaeR-sod RBS and pOS1sGFP-Pssl9-SaeR-sod RBS.
GFP reporter assays.
GFP reporter assays were performed as described previously (7). Briefly, overnight cultures of S. aureus grown in RPMI-CAS plus CM were diluted 1:100 into 5 ml of RPMI-CAS plus CM. The optical density at 600 nm (OD600) and GFP fluorescence were measured at 0, 3, 5, 7, 9, and 24 h postsubculture using a Perkin-Elmer Envison 2103 multilabel reader.
Exoprotein profiles.
Exoproteins were produced and processed as described previously (3, 51). Briefly, S. aureus strains were grown in 5 ml RPMI-CAS overnight at 37°C with shaking at 180 rpm. The overnight cultures were then diluted 1:100 into 5 ml fresh RPMI-CAS in a 15-ml conical tube. Cultures were grown to stationary phase and then normalized to the same OD600. Bacterial cells were sedimented by centrifugation at 400 × g for 15 min, and proteins in the culture supernatant were precipitated using 10% (vol/vol) trichloroacetic acid (TCA) at 4°C overnight. The precipitated proteins were sedimented by centrifugation, washed, dried, resuspended in 1× SDS loading buffer, and boiled for 10 min. Proteins were separated using 15% SDS-PAGE and stained with Coomassie blue.
Immunoblots.
S. aureus cultures were grown in RPMI-CAS as indicated and normalized to the same OD600. Cells were then pelleted by centrifugation. Exoproteins were prepared from supernatants as described above. For cytoplasmic extracts, cell pellets were resuspended in PBS containing 0.1-mm glass beads. Cells were then disrupted using a FastPrep-24 tissue and cell homogenizer (MP Biomedicals, Solon, OH). The protein concentration was normalized for each sample using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). Proteins were resolved by 10, 12, or 15% SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated primary antibodies. An Alexa Fluor 680-conjugated anti-rabbit antibody was used as a secondary antibody. Membranes were then scanned using an Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE).
Cloning, expression, and purification of recombinant proteins. (i) SaeR.
The saeR ORF (NWMN_0675) was PCR amplified from strain Newman genomic DNA with primers 706 and 438. After being digested by NdeI and XhoI, the PCR product was cloned into pET41b (EMD Biosciences, Darmstadt, Germany). The resulting plasmid was transformed first into DH5α and then into T7 lysY lacQ. The T7 lysY lacQ strain carrying the plasmid was grown in LB at 37°C with shaking at 180 rpm to an optical density at 600 nm of 0.6, followed by addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 0.1 mM. After overnight induction at 16°C, the cells were harvested and frozen at −80°C. SaeR was purified from the frozen cells using Ni-nitrilotriacetic acid (Ni-NTA) Superflow resin (Qiagen). The purified protein was concentrated using Centriprep spin columns (Millipore) and frozen at −80°C.
(ii) SaeSΔN92.
Except for the primers used for PCR amplification of the coding region, the cloning, expressing, and purification of the cytoplasmic domain of SaeS were carried out as described for SaeR above. The primers used were 771 and 762.
(iii) Rot.
The rot ORF (NWMN_1655) was PCR amplified from strain Newman genomic DNA with primers 311 and 313. After being digested with XhoI and NdeI, the PCR product was cloned into pET14b (EMD Biosciences, Darmstadt, Germany). The resulting plasmid was transformed first into DH5α and then into T7 lysY lacQ. The T7 lysY lacQ strain carrying the plasmid was grown in LB to an optical density at 600 nm of 0.6, and then 0.1 mM IPTG was added. After overnight induction at 16°C, the cells were harvested and frozen at −80°C. Rot was purified from the frozen cells using Ni-NTA Superflow resin (Qiagen).
Phosphorylation of SaeR for electrophoretic mobility shift assays (EMSA).
Purified SaeR was phosphorylated as described previously (49). Briefly, SaeR was mixed with purified SaeSΔN92 in phosphorylation buffer (10 mM Tris-HCl [pH 7.4], 50 mM KCl, 5 mM MgCl2, 10% glycerol). One millimolar ATP was then added, and the mixture was incubated at room temperature for 15 min before the protein was added to biotinylated DNA probes.
Generation of anti-Rot polyclonal antibody.
The rot gene was amplified, cloned, expressed, and purified as described above. The Rot polyclonal antibody was generated by Pacific Immunology by immunizing rabbits with the purified protein.
EMSA.
The primers used to generate EMSA probes are listed in Table 2. DNA probes of about 300 to 400 bp were PCR amplified using biotinylated primers, and 40 fmol of the biotinylated probes was mixed with various amounts of the desired purified recombinant protein in a final reaction volume of 20 μl in EMSA buffer (10 mM Tris-HCl [pH 7.4], 50 mM KCl, 5 mM MgCl2, 10% glycerol, 5 μg/ml salmon sperm DNA). After being incubated at room temperature for 15 min, samples were analyzed by 6% native polyacrylamide gel electrophoresis (10 mA/gel prerun for 30 min in Tris-borate-EDTA [TBE]). The gels were incubated in streptavidin DyLight (Thermo Scientific, Rockford, IL) diluted 1:1,000 in phosphate-buffered saline (PBS) plus 0.1% Tween and 5% bovine serum albumin (BSA) for 1 h and then visualized using the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE).
Immunoprecipitation.
The indicated S. aureus strain Newman mutants were grown to early stationary phase in 15 ml of RPMI-CAS and normalized to the same OD600. A 378-μl portion of 37% formaldehyde was added to each culture, and the tubes were rocked at room temperature for 20 min. Three milliliters of 2.5 M glycine was added to each culture, and the tubes were rocked at room temperature for an additional 5 min. Bacterial cells were sedimented by centrifugation at 400 × g for 15 min, and pellets were washed and frozen at −80°C until ready to use.
Pellets were thawed on ice and treated with lysostaphin to remove the cell wall. Protoplasts were then sonicated, and M2 anti-FLAG magnetic beads (Sigma-Aldrich, St. Louis, MO) were added to the lysates. Incubation of beads and lysates was carried out with end-over-end rotation at room temperature for 30 min. Beads were washed extensively, and bound proteins were eluted. An aliquot of this elution was mixed with SDS buffer for immunoblot analysis. The remaining elution product was treated with RNase A and proteinase K and then subjected to PCR analysis of the ssl7 and ssl9 promoters using primers 374/241 and 375/343, respectively.
Isolation of PMNs.
Blood samples were obtained from anonymous healthy donors as buffy coats from the New York City Blood Center. The New York City Blood Center obtained written informed consent from all participants involved in the study. Primary human neutrophils (PMNs) were isolated under endotoxin-free conditions as described previously (55).
Neutrophil reporter assay and CFU counts.
Purified PMNs were diluted to 1 × 106 cells/ml in RPMI-CAS. S. aureus reporter strains grown overnight in RPMI-CAS plus CM were subcultured 1:100 into 5 ml RPMI-CAS and grown to mid-log phase. Cultures were sedimented by centrifugation at 400 × g for 15 min and washed twice with sterile PBS. Cultures were subsequently normalized to an OD600 of 1.0. Bacterial cultures were incubated with neutrophils at a multiplicity of infection (MOI) of 20. GFP fluorescence was measured at 0, 3, 5, 7, and 18 h postsubculture using a Perkin-Elmer Envision 2013 multilabel reader. At 5 h postinoculation, cultures were serially diluted and plated on tryptic soy agar (TSA) for CFU enumeration.
RESULTS
Overproduction of Rot in clinical isolates is not sufficient for Ssl overproduction.
In S. aureus strain Newman, increased production of Rot resulted in the overproduction of Ssls (Fig. 1). However, when Rot was overproduced in several wild-type clinical isolates, we were unable to recapitulate the Ssl overproduction phenotype (Fig. 1). We observed that alpha-toxin levels were reduced in the majority of strains overproducing Rot, due to Rot-dependent repression of hla expression (34, 47), indicating that the overproduced Rot was functional in these strains. The inability of Rot to increase Ssl production in strains other than Newman indicated that while Rot is required for Ssl production (7), it alone is not sufficient to induce ssl expression in clinical isolates.
Fig 1.

Effect of overproduction of Rot in S. aureus clinical isolates. Immunoblot analysis of early-stationary-phase supernatants from various S. aureus clinical isolates containing a hemin-inducible Rot vector, grown in inducing (+ hemin) or noninducing (− hemin) conditions, is shown. Exoproteins were collected, precipitated, separated using SDS-PAGE, and transferred to nitrocellulose, and the indicated Ssls and Hla were detected by immunoblotting (IB). Corresponding whole-cell lysates were immunoblotted with an anti-6×His antibody to detect Rot-His.
A functional Sae TCS is necessary for the Ssl overproduction phenotype exhibited by agr-defective strain Newman.
Previous studies have revealed that a main difference between strain Newman and other S. aureus strains is the hyperactivity of the Sae TCS due to the L18P amino acid substitution in SaeS This substitution leads to the constitutive phosphorylation and activation of SaeR (1, 4, 18) and a series of Newman-specific phenotypes (31, 32, 48, 58). To determine if the Sae TCS is involved in the overproduction of Ssls exhibited by strain Newman, we generated an isogenic saeQRS mutant and examined the abundance of Ssls produced by this strain.
Exoprotein profile analysis revealed that deletion of saeQRS markedly affected exoprotein production compared to that by the wild-type Newman strain (Fig. 2A), a phenotype complemented by expressing saeRS in trans (Fig. 2A). In contrast, the exoprotein profile of an isogenic strain lacking rot was similar to that of wild-type Newman (Fig. 2A). Complementation studies revealed that overproduction of Rot into the rot mutant strain resulted in the enhanced appearance of a protein band at ∼25 kDa (Fig. 2A), which has been shown to correspond to the Ssls (3, 7, 52). Interestingly, Ssls were also overproduced, albeit to a lower level, when saeRS was overexpressed by the complementation plasmid (Fig. 2A). Immunoblot analysis demonstrated that as observed with Rot, the Sae TCS is also required for Ssl production, as the saeQRS mutant no longer produced Ssls (Fig. 2A), a phenotype complemented by expressing saeRS in trans.
Fig 2.

Contribution of Rot and the Sae TCS to Ssl production in strain Newman. The indicated Newman strains were grown to early stationary phase under either inducing (+ hemin) or noninducing (− hemin) conditions. Exoproteins were collected, precipitated, separated using SDS-PAGE, either stained with Coomassie blue (top panels) or transferred to nitrocellulose, and immunoblotted for the indicated Ssls (bottom panel). Corresponding whole-cell lysates were also immunoblotted for Rot. The arrow indicates Ssls.
To further examine the contribution of the Sae TCS in the regulation of the Ssls and whether this regulation is in conjunction with or independent of Rot, we generated mutants of strain Newman lacking both saeQRS and the agr loci. In this strain, an agr mutation causes overproduction of Ssls, a finding attributed to an increased abundance of Rot (7). Exoprotein profile analysis confirmed this, as the agr rot mutant no longer produced the Ssls and their production was restored by expressing rot in trans (Fig. 2B). As with the saeQRS mutant, virtually no exoproteins were produced by the agr saeQRS double mutant strain, a phenotype complemented by expressing saeRS in trans (Fig. 2B). This observation suggests that the overabundance of Rot in the agr mutant strain is not sufficient to support production of Ssls.
Immunoblot analysis of agr rot and agr saeQRS double mutant strains revealed that both Rot and the Sae TCS are required for the production of the Ssls (Fig. 2B). Consistent with this, the lack of Ssls in the agr rot saeQRS triple mutant was not rescued by producing either Rot or SaeRS (Fig. 2B). Taken together, these data suggest that Sae and Rot work synergistically to promote the production of Ssls.
The Sae TCS is required for the activation of the ssl promoters.
We have previously demonstrated that the regulation of ssl expression by Rot is due to the promoter activation by this transcription factor (7). To test whether Sae directly regulates the activation of the ssl promoters, the ssl7 and ssl9 promoters were fused to the GFP gene and the resulting transcriptional reporters were transformed into WT S. aureus strain Newman or the rot or saeQRS isogenic mutants. The strain lacking saeQRS was used to examine the effect of knocking out both the SaeS kinase and the SaeR response regulator. We observed that both Rot and the Sae TCS were required for the activation of the ssl promoters (Fig. 3A). Interestingly, the rot mutant was still able to activate the ssl9 promoter at low levels (Fig. 3A). This residual activation could be attributed to Sae TCS activity, as the rot saeQRS double mutant strain displayed no detectable activation (Fig. 3A).
Fig 3.
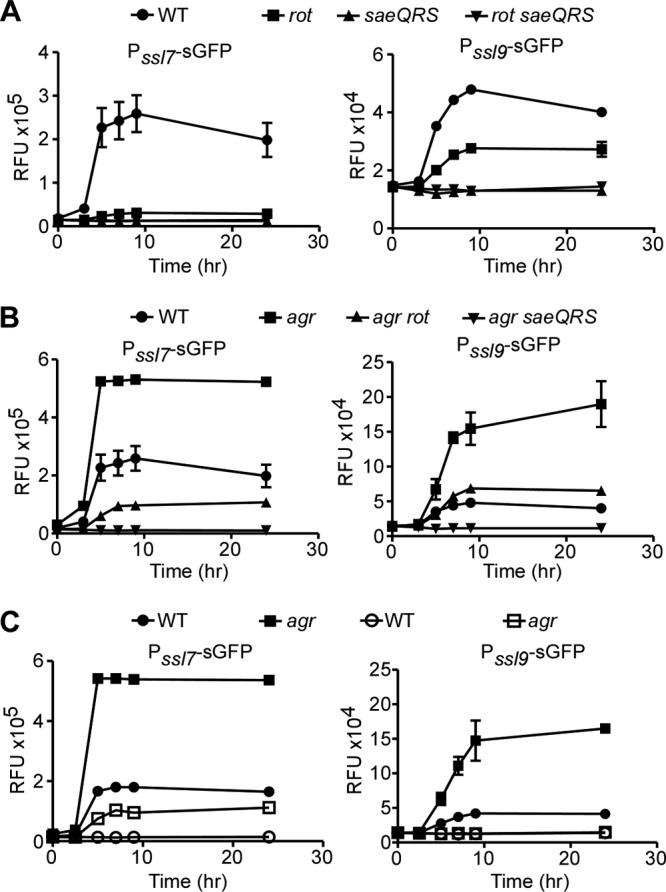
Role of the Sae TCS in the activation of the ssl promoters. (A and B) The indicated Newman strains were transformed with plasmids containing the ssl7 or ssl9 promoter controlling GFP gene expression, and GFP fluorescence was monitored at the indicated time points. Values represent averages from three independent experiments ± standard deviations (SD). (C) Reporter assay as done for panel A. Closed symbols indicate wild-type promoters, and open symbols indicate promoters in which the SaeR binding site has been mutated to impair SaeR binding.
In an effort to further elucidate the contribution of the Sae TCS to the activation of the ssl promoters, we transformed the reporter plasmids into an agr, agr rot, or agr saeQRS mutant strain. The agr mutant had higher levels of ssl promoter activation (Fig. 3B). This was attributed to increased levels of Rot present in an agr mutant, as an agr rot double mutant strain exhibited reduced promoter activation (Fig. 3B). In contrast, no promoter activation was observed in the agr saeQRS double mutant strain (Fig. 3B). Taken together, these data suggest that both Rot and the Sae TCS are necessary to attain full activation of the ssl promoters; however, in the absence of Rot, the Sae TCS can still elicit partial activation.
The requirement of the Sae TCS for the activation of the ssl promoters is consistent with the finding that these promoters harbor an SaeR binding site (7, 42, 49). To further examine the influence of SaeR on the activation of the ssl promoters, we generated transcriptional reporters in which the SaeR binding sites on the ssl7 and ssl9 promoters were disrupted. The ssl7 promoter contains a perfect SaeR binding site (GTTAA-N6-GTTAA), while the ssl9 promoter contains an imperfect SaeR binding site (TTTAA-N6-GTTAA). Mutation of the last adenine of the SaeR binding palindromic sequence (GTTAA) has been previously shown to disrupt binding of the regulator (49). Thus, we mutated the ssl7 and ssl9 promoters to generate a GTTAG-N6-GTTAA and a TTTAG-N6-GTTAA SaeR binding site, respectively. The transcriptional reporters harboring these mutated promoters were transformed into wild-type S. aureus or an agr mutant strain and promoter activation monitored as described above. We observed that mutation of the SaeR binding site resulted in a significant reduction in promoter activation (Fig. 3C, open symbols). These data strongly suggest that SaeR influences the activation of the ssl promoters, most likely due to direct binding of SaeR to these promoters.
SaeR and Rot directly and specifically interact with the ssl promoters.
To conclusively demonstrate that SaeR directly binds to the ssl promoters, electrophoretic motility shift assays (EMSA) were performed. Recombinant SaeR was purified from E. coli and phosphorylated with SaeS as described previously (49). We observed that SaeR interacted with ssl7, ssl9, and ssl11 promoter DNAs (Fig. 4A). In contrast, when EMSA where performed with SaeS alone, no shift was observed (data not shown). We also observed that Rot directly interacted with the ssl promoters (Fig. 4B). These interactions were specific, as SaeR and Rot were efficiently competed off when nonlabeled ssl promoter DNA was added but not when a nonpromoter control DNA was included (Fig. 4C).
Fig 4.
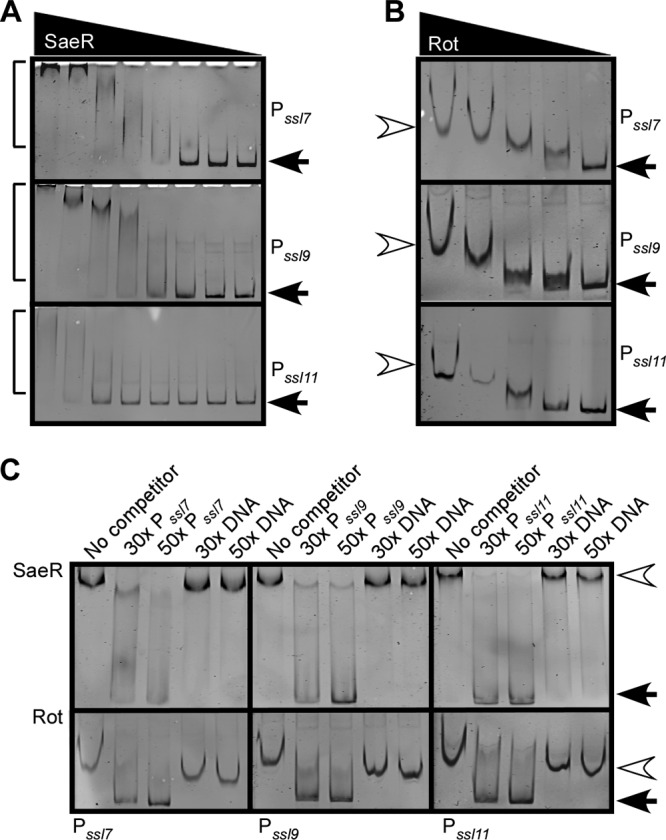
Binding of SaeR or Rot to the ssl7, ssl9, and ssl11 promoters. (A) EMSA of purified phosphorylated SaeR incubated with either the ssl7, ssl9, or ssl11 promoter containing a biotin tag. Two-fold serial dilutions of SaeR, starting with 150 pmol, were incubated with 40 fmol DNA. Protein-DNA complexes were separated by PAGE, and the DNA probe was visualized using streptavidin DyLight. Arrows indicate free probe, and brackets indicate shifted probe. (B) EMSA of purified Rot 2-fold serial dilutions, starting with 4 pmol, incubated with either the ssl7, ssl9, or ssl11 promoter as for panel A. Closed arrows indicate free probe, and open arrowheads indicate shifted probe. (C) EMSA in which 75 pmol of SaeR or 2 pmol of Rot was incubated with 40 fmol of the indicated biotinylated promoter DNA with a 30- or 50-fold molar excess of nonbiotinylated promoter DNA or nonbiotinylated control DNA. The EMSA reaction was performed and visualized as for panel A. Closed arrows indicate free probe, and open arrowheads indicate shifted probe.
Rot and SaeR form a complex with ssl promoters in vitro and in vivo.
Based on the genetic and biochemical data presented above, we hypothesized that Rot and SaeR are both able to occupy the ssl promoters concurrently. To test this hypothesis, ssl7, ssl9, or ssl11 promoter DNA was incubated with either Rot, phosphorylated SaeR, or both together and the samples analyzed by EMSA. While each protein alone was able to cause either a full shift (Rot) or a slight shift (SaeR) in the DNA migration, when present together, a supershift was observed (Fig. 5A). This supershift is indicative of both factors binding to the DNA concurrently, consistent with the finding that both Rot and SaeR are required for ssl promoter activation (Fig. 3).
Fig 5.

DNA binding properties of Rot, SaeR, and Rot/SaeR complexes. (A) EMSA of 2 pmol of purified Rot, 37.5 pmol of purified phospho-SaeR, or a mixture of Rot and phospho-SaeR incubated with 40 fmol of either the ssl7, ssl9, or ssl11 promoter containing a biotin tag. DNA was visualized using streptavidin DyLight. The arrow indicates free probe. (B) Immunoprecipitation of FLAG-tagged Rot from the indicated S. aureus Newman strains. The samples were separated using SDS-PAGE, transferred to nitrocellulose, and immunoblotted (IB) for FLAG to detect Rot or for SaeR (top) or used as temple for PCR amplification of the ssl7 and ssl9 promoters (bottom).
We next examined whether Rot and SaeR bind to ssl promoters in vivo. To this end, we performed immunoprecipitation studies using an agr rot double mutant strain containing a plasmid that produces a FLAG-tagged Rot. Whole-cell lysates were generated from cells previously cross-linked with formaldehyde to preserve protein-protein and protein-DNA complexes. Lysates were subsequently immunoprecipitated using an anti-FLAG antibody and the pulldown samples immunoblotted for SaeR. We found that immunoprecipitation of Rot resulted in the coisolation of SaeR (Fig. 5B). In addition, analysis of the immunoprecipitated material by PCR revealed that Rot could be found associated in vivo with the ssl7 and ssl9 promoters (Fig. 5B). Collectively, these data suggest that Rot and SaeR interact in vivo and form complexes with the ssl promoters.
Hyperactivation of SaeS results in increased production of Ssls.
We next investigated whether the saeSL18P allele in other S. aureus clinical strains would result in overproduction of the Ssls. To test this, we introduced a vector that constitutively expresses Newman saeSL18P into an isogenic mutant of USA300 strain LAC with agr and saeS mutated. As observed previously, inactivation of agr in strain LAC resulted in an increase in Ssl7 abundance (7), a phenotype dependent on a functional Sae TCS (Fig. 6, agr versus agr/saeS). Complementation studies demonstrated that expression of the saeSL18P allele induced the overproduction of Ssls, while expression of the saeSL18 allele rescued Ssl levels only to that of wild-type LAC (Fig. 6). These results demonstrate that constitutive activation of SaeS results in the overproduction of Ssls and that other S. aureus strains are capable of producing Ssls, provided that the Sae TCS is activated.
Fig 6.
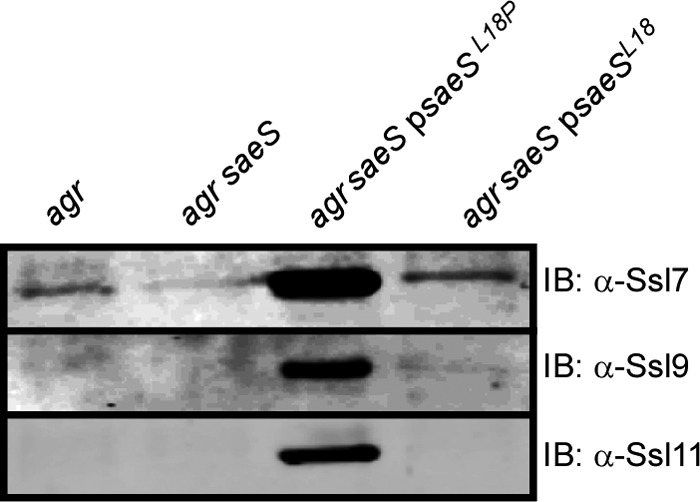
Effect of the SaeS-L18P mutant on the production of Ssls by USA300. The indicated USA300 LAC strains were grown to early stationary phase, and exoproteins were collected, precipitated, separated using SDS-PAGE, and transferred to nitrocellulose. The indicated Ssls were detected by immunoblotting.
S. aureus induces the activation of the ssl promoters in response to human neutrophils.
The data presented above demonstrate that the Sae TCS and Rot synergize to activate the expression of ssls. However, production of these proteins in vitro is minimal in S. aureus strains harboring the saeSL18 allele (Fig. 1 and 6). Previous studies have demonstrated that sae is upregulated in response to neutrophils and neutrophil products (18, 33, 43, 55). Specifically, the P1 promoter has been shown to be activated in the presence of α-defensins, key components in bacterial killing by neutrophils (18). Therefore, we sought to determine if this enhanced promoter activation, and thus expression of the Sae TCS, could induce ssl expression in clinical methicillin-resistant S. aureus (MRSA) strains following interaction with human neutrophils.
To test this hypothesis, we generated an sae P1 transcriptional reporter vector and transformed it into the MRSA pulsed-field gel electrophoresis type USA300. When this reporter strain was grown in culture in the presence or absence of primary human neutrophils, we observed an induction in the activation of the sae P1 promoter that occurred in a time- and neutrophil-dependent manner (Fig. 7A, sae P1 promoter). In addition to sae P1 activation, we also observed an increase in the activation of the ssl promoters in the presence of neutrophils in USA300 (Fig. 7A, Pssl7 and Pssl11). Importantly, neutrophil-mediated activation of sae P1 and ssl promoters was also observed in other MRSA clinical isolates (USA100, USA500, and USA800) (Fig. 7B). The USA300 saeQRS mutant strain was no longer able to activate the ssl11 promoter, regardless of whether neutrophils were present or not (Fig. 7C), indicating that the enhanced activation of the ssl promoters was attributable to the increased activation of sae. Rot was also found to be required for this neutrophil-mediated activation, as a rot mutant was unable to significantly enhance the activation of the ssl promoters, even in the presence of neutrophils (Fig. 7C). Importantly, the neutrophil-mediated activation of the sae P1 and ssl promoters was specific, since neutrophils did not influence the activation of the rot and sarA promoters (Fig. 7D). This neutrophil-mediated induction of the ssl promoters was not due to altered growth of the tested strains, since enumeration of CFU of USA300 and the isogenic saeQRS mutant strain grown for 5 h in the presence or absence of neutrophils demonstrated no difference in bacterial numbers (Fig. 7E). Taken together, these data demonstrate that clinically relevant MRSA strains sense neutrophils to activate the ssl promoters, a phenotype dependent on the Sae TCS and Rot.
Fig 7.

Human neutrophils and activation of the ssl promoters. (A) Reporter assay of S. aureus USA300 containing either the sae P1, ssl7, or ssl11 transcriptional reporter controlling GFP gene expression grown in the absence (closed circles) or presence (open circles) of primary human neutrophils (PMNs). (B) Reporter assay of MRSA USA100, USA500, or USA800 containing the sae P1 or ssl11 transcriptional reporter as for panel A. (C) Reporter assay of wild-type USA300 (WT) (circles), the saeQRS mutant (squares), or the rot mutant (triangles) containing the ssl11 transcriptional reporter grown as for panel A. (D) Reporter assay of WT USA300 containing a rot or sarA transcriptional reporter controlling GFP gene expression grown in the absence (no PMNs) or presence (+PMNs) of primary human neutrophils. (E) CFU counts of WT USA300 or saeQRS mutant growth with or without neutrophils for 5 h. Values represent averages from six (A to C) or three (D and E) independent donors ± SD.
DISCUSSION
S. aureus is a significant human pathogen capable of infecting most host tissues, a trait dependent on the evasion of the host immune response. Among the staphylococcal virulence factors, the Ssls are believed to be an important component of this host immune evasion strategy (3, 7, 17, 52). Our previous studies have demonstrated that the repression of ssl expression is due to RNAIII-mediated inhibition of Rot (7). However, while the overproduction of Rot in strain Newman leads to a robust increase in the production of the Ssls, the same phenotype is not observed when Rot is overproduced in clinically relevant S. aureus strains, indicating that Rot alone is not sufficient to enhance Ssl production in these strains (Fig. 1). We demonstrate in this study that both the Sae TCS and Rot are required to regulate ssl expression by directly binding and activating the ssl promoters.
The SaeR binding site has been recently identified (42, 49). Interestingly, each of the ssl promoters contains an SaeR binding site that overlaps with the −35 site (7). While some of the promoters contain an imperfect SaeR binding site, none of the mutations found are expected to abolish SaeR binding (49). The ssl7 promoter contains a perfect SaeR binding site (GTTAA-N6-GTTAA), while the ssl9 and ssl11 promoters contain imperfect sites (TTTAA-N6-GTTAA, and GTTAA-N6-TTTAA, respectively). We hypothesize that these differences in SaeR binding site could contribute to the observation that the ssl7 promoter exhibits more robust activation than the ssl9 promoter. This hypothesis is also supported by the finding that less SaeR is required to shift the ssl7 promoter than to shift the ssl9 or ssl11 promoter (Fig. 4A). Additionally, the introduction of a single nucleotide change in the SaeR binding site in the ssl9 promoter completely ablates activation (Fig. 3C), while in the ssl7 promoter, this single mutation greatly reduces but does not eliminate activation (Fig. 3C).
To our knowledge, the finding that SaeR and Rot work together to directly activate the expression of target genes has not been described previously. Interestingly, Rot has been found to repress the sae P1 promoter in vitro (30). We speculate that while Rot could inhibit P1 activation, the low, constitutive activity of the sae P3 promoter, which encodes a transcript containing both saeR and saeS, allows for enough phosphorylated SaeR to be available in the cell to activate the ssl promoters. Until now, these transcription factors have been shown to work in opposition to one another to regulate the expression of secreted and cell surface factors. However, with the ssls, these factors work together to provide a dual layer of positive regulation. This complex regulatory scheme may serve as insurance that the Ssls are produced both at the correct time during infection and in the appropriate context. For example, production of these factors could be critical early during infection, when Rot levels would be maximal and when S. aureus first encounters neutrophils. Ssls could protect S. aureus from the innate immune system until the bacterial density increases and the population reaches quorum, which is required for the activation of the Agr TCS and the subsequent expression of the S. aureus virulon.
While we demonstrate that both Rot and SaeR are required to activate the ssl promoters and that these factors both bind to the promoter DNA, the exact mechanism by which these two regulators work together is not known. We speculate that Rot may recruit SaeR to the ssl promoters, which in turn will recruit the RNA polymerase and activate gene expression (12). The notion that Rot facilitates the recruitment of SaeR to the ssl promoters is supported by the fact that less SaeR is required to create a supershift of the ssl7, ssl9, and ssl11 promoter DNAs in the presence of Rot than when Rot is absent (Fig. 4A and 5A). The Rot-mediated recruitment of SaeR could be due to direct interaction between Rot and SaeR. Alternatively, Rot could bend the DNA to better expose the SaeR binding site at these promoters. These possibilities are currently being explored.
Taken together, the data presented in this study reveal a new regulatory scheme by which S. aureus is able to activate the expression of a subset of genes through the coordinated efforts of two transcription factors previously demonstrated to work in opposition. The finding that neutrophils induce the expression of the ssls in clinically relevant strains of S. aureus further supports the role of these molecules in avoidance of the immune response and pathogenesis.
ACKNOWLEDGMENTS
We thank members of the Torres laboratory, Andrew Darwin, and Melanie Pearson for critical reading of the manuscript. We are also grateful to John Fraser for providing the anti-Ssl antibodies, to Taeok Bae for assistance with the SaeR phosphorylation and for providing the anti-SaeR sera, and to Anthony Richardson for providing the pBT-S plasmid.
Several MRSA isolates were obtained from the Network of Antimicrobial Resistance in Staphylococcus aureus (NARSA) program, which is supported under NIAID/NIH contract HHSN272200700055C. This work was supported by U.S. Public Health Service grant K22-AI079389 from the National Institute of Allergy and Infectious Diseases and New York University School of Medicine development funds to V.J.T. and by U.S. Public Health Service grant R01-A1090046 from the National Institute of Allergy and Infectious Diseases to J.M.V. M.A.B. was supported by an American Heart Association predoctoral fellowship (10PRE3420022), and S.L. was supported by NIH training grant 5T32-AI007180.
Footnotes
Published ahead of print 8 June 2012
REFERENCES
- 1. Adhikari RP, Novick RP. 2008. Regulatory organization of the staphylococcal sae locus. Microbiology 154: 949–959 [DOI] [PubMed] [Google Scholar]
- 2. Anonymous. 1999. Four pediatric deaths from community-acquired methicillin-resistant Staphylococcus aureus—Minnesota and North Dakota, 1997–1999. MMWR Morb. Mortal. Wkly. Rep. 48: 707–710 [PubMed] [Google Scholar]
- 3. Attia AS, Benson MA, Stauff DL, Torres VJ, Skaar EP. 2010. Membrane damage elicits an immunomodulatory program in Staphylococcus aureus. PLoS Pathog. 6: e1000802 doi:10.1371/journal.ppat.1000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. 2008. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 190: 300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55: 58–63 [DOI] [PubMed] [Google Scholar]
- 6. Benito Y, et al. 2000. Probing the structure of RNAIII, the Staphylococcus aureus agr regulatory RNA, and identification of the RNA domain involved in repression of protein A expression. RNA 6: 668–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benson MA, et al. 2011. Staphylococcus aureus regulates the expression and production of the staphylococcal superantigen-like secreted proteins in a Rot-dependent manner. Mol. Microbiol. 81: 659–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boisset S, et al. 2007. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev. 21: 1353–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5: e10146 doi:10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bubeck Wardenburg J, Williams WA, Missiakas D. 2006. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 103: 13831–13836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheung AL, Bayer AS, Zhang G, Gresham H, Xiong YQ. 2004. Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus. FEMS Immunol. Med. Microbiol. 40: 1–9 [DOI] [PubMed] [Google Scholar]
- 12. Cho H, Jeong DW, Li C, Bae T. 2012. Organizational requirements of the SaeR binding sites for functional P1 promoter of the sae operon in Staphylococcus aureus. J. Bacteriol. 194: 2865–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diep BA, et al. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367: 731–739 [DOI] [PubMed] [Google Scholar]
- 14. Duthie ES, Lorenz LL. 1952. Staphylococcal coagulase; mode of action and antigenicity. J. Gen. Microbiol. 6: 95–107 [DOI] [PubMed] [Google Scholar]
- 15. Felden B, Vandenesch F, Bouloc P, Romby P. 2011. The Staphylococcus aureus RNome and its commitment to virulence. PLoS Pathog. 7: e1002006 doi:10.1371/journal.ppat.1002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foster TJ. 2005. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3: 948–958 [DOI] [PubMed] [Google Scholar]
- 17. Fraser JD, Proft T. 2008. The bacterial superantigen and superantigen-like proteins. Immunol. Rev. 225: 226–243 [DOI] [PubMed] [Google Scholar]
- 18. Geiger T, Goerke C, Mainiero M, Kraus D, Wolz C. 2008. The virulence regulator Sae of Staphylococcus aureus: promoter activities and response to phagocytosis-related signals. J. Bacteriol. 190: 3419–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geisinger E, Adhikari RP, Jin R, Ross HF, Novick RP. 2006. Inhibition of rot translation by RNAIII, a key feature of agr function. Mol. Microbiol. 61: 1038–1048 [DOI] [PubMed] [Google Scholar]
- 20. Gillaspy AF, et al. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect. Immun. 63: 3373–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giraudo AT, Calzolari A, Cataldi AA, Bogni C, Nagel R. 1999. The sae locus of Staphylococcus aureus encodes a two-component regulatory system. FEMS Microbiol. Lett. 177: 15–22 [DOI] [PubMed] [Google Scholar]
- 22. Giraudo AT, Cheung AL, Nagel R. 1997. The sae locus of Staphylococcus aureus controls exoprotein synthesis at the transcriptional level. Arch. Microbiol. 168: 53–58 [DOI] [PubMed] [Google Scholar]
- 23. Giraudo AT, Mansilla C, Chan A, Raspanti C, Nagel R. 2003. Studies on the expression of regulatory locus sae in Staphylococcus aureus. Curr. Microbiol. 46: 246–250 [DOI] [PubMed] [Google Scholar]
- 24. Giraudo AT, Martinez GL, Calzolari A, Nagel R. 1994. Characterization of a Tn925-induced mutant of Staphylococcus aureus altered in exoprotein production. J. Basic Microbiol. 34: 317–322 [DOI] [PubMed] [Google Scholar]
- 25. Goerke C, et al. 2005. Role of Staphylococcus aureus global regulators sae and sigmaB in virulence gene expression during device-related infection. Infect. Immun. 73: 3415–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jeong DW, et al. 2011. Identification of the P3 promoter and distinct roles of the two promoters of the SaeRS two-component system in Staphylococcus aureus. J. Bacteriol. 193: 4672–4684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koenig RL, Ray JL, Maleki SJ, Smeltzer MS, Hurlburt BK. 2004. Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region. J. Bacteriol. 186: 7549–7555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kreiswirth BN, et al. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305: 709–712 [DOI] [PubMed] [Google Scholar]
- 29. Lauderdale KJ, Malone CL, Boles BR, Morcuende J, Horswill AR. 2010. Biofilm dispersal of community-associated methicillin-resistant Staphylococcus aureus on orthopedic implant material. J. Orthop. Res. 28: 55–61 [DOI] [PubMed] [Google Scholar]
- 30. Li D, Cheung A. 2008. Repression of hla by rot is dependent on sae in Staphylococcus aureus. Infect. Immun. 76: 1068–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luong TT, et al. 2011. Staphylococcus aureus ClpC divergently regulates capsule via sae and codY in strain Newman but activates capsule via codY in strain UAMS-1 and in strain Newman with repaired saeS. J. Bacteriol. 193: 686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mainiero M, et al. 2010. Differential target gene activation by the Staphylococcus aureus two-component system saeRS. J. Bacteriol. 192: 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Malachowa N, et al. 2011. Global changes in Staphylococcus aureus gene expression in human blood. PLoS One 6: e18617 doi:10.1371/journal.pone.0018617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McNamara PJ, Bayer AS. 2005. A rot mutation restores parental virulence to an agr-null Staphylococcus aureus strain in a rabbit model of endocarditis. Infect. Immun. 73: 3806–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McNamara PJ, Milligan-Monroe KC, Khalili S, Proctor RA. 2000. Identification, cloning, and initial characterization of rot, a locus encoding a regulator of virulence factor expression in Staphylococcus aureus. J. Bacteriol. 182: 3197–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montgomery CP, Boyle-Vavra S, Daum RS. 2010. Importance of the global regulators Agr and SaeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS One 5: e15177 doi:10.1371/journal.pone.0015177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morfeldt E, Taylor D, von Gabain A, Arvidson S. 1995. Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J. 14: 4569–4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morfeldt E, Tegmark K, Arvidson S. 1996. Transcriptional control of the agr-dependent virulence gene regulator, RNAIII, in Staphylococcus aureus. Mol. Microbiol. 21: 1227–1237 [DOI] [PubMed] [Google Scholar]
- 39. Nizet V. 2007. Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J. Allergy Clin. Immunol. 120: 13–22 [DOI] [PubMed] [Google Scholar]
- 40. Novick RP. 2003. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 48: 1429–1449 [DOI] [PubMed] [Google Scholar]
- 41. Novick RP, Geisinger E. 2008. Quorum sensing in staphylococci. Annu. Rev. Genet. 42: 541–564 [DOI] [PubMed] [Google Scholar]
- 42. Nygaard TK, et al. 2010. SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J. Infect. Dis. 201: 241–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Palazzolo-Ballance AM, et al. 2008. Neutrophil microbicides induce a pathogen survival response in community-associated methicillin-resistant Staphylococcus aureus. J. Immunol. 180: 500–509 [DOI] [PubMed] [Google Scholar]
- 44. Pantrangi M, Singh VK, Wolz C, Shukla SK. 2010. Staphylococcal superantigen-like genes, ssl5 and ssl8, are positively regulated by Sae and negatively by Agr in the Newman strain. FEMS Microbiol. Lett. 308: 175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roberts RB, et al. 1998. Molecular epidemiology of methicillin-resistant Staphylococcus aureus in 12 New York hospitals. MRSA Collaborative Study Group. J. Infect. Dis. 178: 164–171 [DOI] [PubMed] [Google Scholar]
- 46. Rogasch K, et al. 2006. Influence of the two-component system SaeRS on global gene expression in two different Staphylococcus aureus strains. J. Bacteriol. 188: 7742–7758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Said-Salim B, et al. 2003. Global regulation of Staphylococcus aureus genes by Rot. J. Bacteriol. 185: 610–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schafer D, et al. 2009. A point mutation in the sensor histidine kinase SaeS of Staphylococcus aureus strain Newman alters the response to biocide exposure. J. Bacteriol. 191: 7306–7314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sun F, et al. 2010. In the Staphylococcus aureus two-component system sae, the response regulator SaeR binds to a direct repeat sequence and DNA binding requires phosphorylation by the sensor kinase SaeS. J. Bacteriol. 192: 2111–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. 2011. Peptide signaling in the staphylococci. Chem. Rev. 111: 117–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Torres VJ, et al. 2010. Staphylococcus aureus fur regulates the expression of virulence factors that contribute to the pathogenesis of pneumonia. Infect. Immun. 78: 1618–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Torres VJ, et al. 2007. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe 1: 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tseng CW, Stewart GC. 2005. Rot repression of enterotoxin B expression in Staphylococcus aureus. J. Bacteriol. 187: 5301–5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tseng CW, Zhang S, Stewart GC. 2004. Accessory gene regulator control of staphyloccoccal enterotoxin d gene expression. J. Bacteriol. 186: 1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Voyich JM, et al. 2005. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol. 175: 3907–3919 [DOI] [PubMed] [Google Scholar]
- 56. Voyich JM, et al. 2009. The SaeR/S gene regulatory system is essential for innate immune evasion by Staphylococcus aureus. J. Infect. Dis. 199: 1698–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Watkins RL, Pallister KB, Voyich JM. 2011. The SaeR/S gene regulatory system induces a pro-inflammatory cytokine response during Staphylococcus aureus infection. PLoS One 6: e19939 doi:10.1371/journal.pone.0019939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zielinska AK, et al. 2011. Defining the strain-dependent impact of the staphylococcal accessory regulator (sarA) on the alpha-toxin phenotype of Staphylococcus aureus. J. Bacteriol. 193: 2948–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]