

Abstract
Hantavirus glycoprotein precursor (GPC) is posttranslationally cleaved into two glycoproteins, Gn and Gc. Cells transfected with plasmids expressing either GPC or both Gn and Gc revealed that Gn is posttranslationally degraded. Treatment of cells with the autophagy inhibitors 3-methyladenine, LY-294002, or Wortmanin rescued Gn degradation, suggesting that Gn is degraded by the host autophagy machinery. Confocal microscopic imaging showed that Gn is targeted to autophagosomes for degradation by an unknown mechanism. Examination of autophagy markers LC3-I and LC3-II demonstrated that both Gn expression and Sin Nombre hantavirus (SNV) infection induce autophagy in cells. To delineate whether induction of autophagy and clearance of Gn play a role in the virus replication cycle, we downregulated autophagy genes BCLN-1 and ATG7 using small interfering RNA (siRNA) and monitored virus replication over time. These studies revealed that inhibition of host autophagy machinery inhibits Sin Nombre virus replication in cells, suggesting that autophagic clearance of Gn is required for efficient virus replication. Our studies provide mechanistic insights into viral pathogenesis and reveal that SNV exploits the host autophagy machinery to decrease the intrinsic steady-state levels of an important viral component for efficient replication in host cells.
INTRODUCTION
The ubiquitin-proteasome and autophagy-lysosome pathways are the two main routes for the clearance of proteins and organelles in most eukaryotic cells. The short-lived or misfolded proteins are mostly cleared by the ubiquitin-proteasome machinery, a cytoplasmic barrel-shaped multiprotein complex (4, 34). Ubiquitination of target proteins is the primary requirement for degradation by the proteosome machinery (33). However, organelles and aggregated proteins which are too large to pass through the narrow pore of the proteosome barrel are mostly cleared by the macroautophagy, a process generally referred to as autophagy (5, 19). Autophagy is an evolutionarily conserved mechanism by which unwanted intracellular material is sequestered within double-layered membrane-bound vesicles and targeted to lysosomes for degradation. Autophagy is a multistep process that is initiated by the formation of short-lived membrane crescents, known as isolation membranes, which upon expansion recognize cargo and form double membrane-bound structures known as autophagosomes (5, 19, 21). During the maturation phase of autophagy, autophagosomes fuse with lysosomes, and their contents are then degraded by the acidic lysosomal hydrolases. At least 31 autophagy genes (ATG genes) have been identified in yeast (Saccharomyces cerevisiae) that are required at different stages of autophagy. Many of these genes have homologues in mammalian cells (30, 35). Apart from the known major roles of autophagy in the degradation of unwanted cytoplasmic constituents, such as organelles and toxic aggregation-prone proteins, and in the recycling of nutrients and energy, this mechanism also defends host cells from the assaults of invading pathogens (5, 19, 21). For example, overexpression of BCLN-1, a component of class III phosphatidylinositol 3-kinase (PI3K) complex, in mouse brains during Sindbis virus infection reduced viral titers and virus-induced neuroapoptosis that resulted in the protection of mice against fatal Sindbis virus encephalitis (23). In Arabidopsis, the plant autophagy genes BCLN-1, PI3K/VPS34, ATG3, and ATG7 have been reported to decrease tobacco mosaic virus (TMV) replication (24). Similar to plants, Drosophila flies lack the adaptive immunity and rely on innate and intrinsic immune mechanisms to purge invading pathogens. Recently, it has been reported that autophagy plays a key role in protecting Drosophila from vesicular stomatitis virus (VSV) infection. The VSV glycoprotein serves as a pathogen-associated molecular pattern (PAMP) that triggers autophagosome development in Drosophila cells without the requirement of additional viral components (41). Using multifaceted experimental avenues, it has been shown that inactivation of autophagy gene ATG5 in virally infected neurons increases the susceptibility of mice to lethal Sindbis virus infection (31). Despite the role of autophagy in antiviral defense, viruses have evolved unique strategies to fight back the host autophagy response. For example, the alphaherpesvirus herpes simplex virus 1 (HSV-1)-encoded ICP34.5 protein antagonizes autophagy by directly binding to BCLN-1 (29) and inhibition of PKR signaling (9). The alphaherpesvirus HSV-1-encoded viral Bcl-2 (v-Bcl-2) gene product has been reported to bind to BCLN-1 and inhibit BCLN-1-mediated autophagy (16, 32).
Hantaviruses, members of the Bunyaviridae family, are enveloped negative-strand RNA viruses and category A pathogens that cause serious illness when transmitted to humans through aerosolized excreta of infected rodents (38). The two main diseases caused by hantavirus infection are hemorrhagic fever with renal syndrome (HFRS) and hantavirus cardiopulmonary syndrome (HCP), with mortalities of 15% and 50%, respectively (37, 38). Annually, 150,000 to 200,000 hantavirus infections are reported worldwide (13). The hantaviral genome is composed of three negative-sense genomic RNA segments, S, M, and L, that encode nucleocapsid protein (N), glycoprotein precursor (GPC), and viral RNA-dependent RNA polymerase (RdRp), respectively. The GPC is posttranslationally cleaved at a highly conserved WAASA site generating an N-terminal fragment (Gn) and a C-terminal fragment (Gc) (Fig. 1). Gn harbors a cytoplasmic tail domain of 142 amino acids, which carries out several functions, including the interaction with the nucleocapsid protein, suggesting a possible involvement in viral budding (11, 44). Pathogenic hantaviruses including Sin Nombre virus (SNV) use αvß3 integrins on endothelial cells for entry (7). Similar to many other viruses, pathogenic hantaviruses delay the type I interferon (IFN) response after infection and grow to higher titers. By delaying the IFN-mediated antiviral state, hantaviruses open a window of opportunity in which they replicate and spread within the endothelial cell layer (6, 39). The cytoplasmic tail domain of hantavirus glycoprotein Gn has been reported to inhibit RIG-I- and TBK-1-directed interferon responses by disrupting the formation of TBK1-TRAF3 complex (1, 2). Here, we show that Sin Nombre hantavirus Gn is a PAMP which triggers autophagy in cells independent of other viral components. Host autophagy machinery degrades Gn in cells. Interestingly, inhibition of autophagy dramatically suppresses SNV replication, suggesting that elevated levels of Gn inhibit virus replication by an unknown mechanism.
Fig 1.
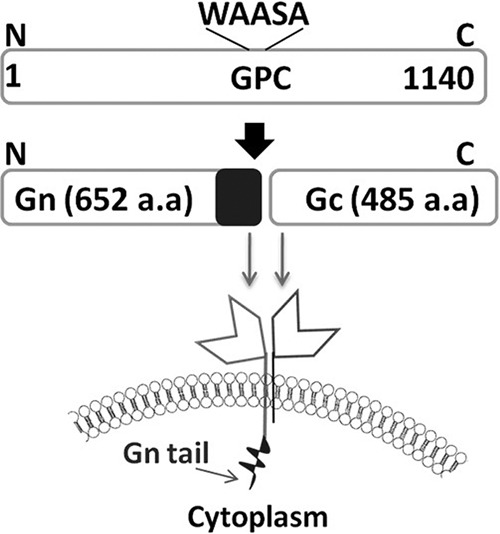
Schematic illustration of the hantavirus GPC and its maturation. GPC is cotranslationally cleaved in the endoplasmic reticulum by the host signal peptidase at a conserved WAASA motif, generating Gn and Gc. SNV GPC is 1,140 amino acids in length. The Gn of SNV consists of 652 residues (molecular mass, ≈75 kDa), with a predicted transmembrane domain and a long cytoplasmic tail domain of 142 amino acids (marked as a black rectangle at the C-terminal end of Gn). Gc is 488 residues in length (molecular mass, ≈55 kDa) and has a predicted shorter cytoplasmic tail of 8 amino acids. aa, amino acids.
MATERIALS AND METHODS
Cells and transfection.
HeLa and Vero E6 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum and penicillin-streptomycin (100 μg/ml) in a CO2 incubator. All plasmid DNA and small interfering RNA (siRNA) transfections were performed in six-well plates using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Human umbilical vein endothelial cells (HUVEC) were obtained from Lonza and were maintained in the EBM-2 medium containing all the growth factors required for endothelial cells (Lonza).
Plasmids.
All green fluorescent protein (GFP)-glycoprotein fusion proteins were cloned in pTriEX1.1 vector (Novagen) between the NcoI and XhoI sites. The plasmid pTGFP-GPC was generated by the fusion of GFP at the N terminus of the SNV glycoprotein precursor (GPC). GFP was PCR amplified from pTGFP plasmid (27) using a primer set shown in Table 1. The resulting PCR product, referred to as PCR 1 in Table 1, was gel purified and digested with NcoI and BamHI restriction enzymes. Vero E6 cells infected with Sin Nombre virus (strain 7773, a gift from Brian Hjelle, University of New Mexico) were lysed, and total RNA was purified and used to generate the cDNA. The resulting cDNA was used as the template along with a primer set shown in Table 1 to PCR amplify the open reading frame (ORF) encoding the GPC. This PCR product, referred to as PCR 2 in Table 1, was gel purified and digested with BamHI and XhoI restriction enzymes. The pTriEX1.1 vector was digested with NcoI and XhoI restriction enzymes and gel purified. The purified pTriEX1.1 backbone was mixed with two PCR products (PCR 1 and PCR 2) and ligated simultaneously. The ligation product was transformed into ECOS101 competent cells (Yeastern Biotech Co., Ltd.), and colonies harboring the plasmid of our interest were confirmed by restriction mapping and sequencing analysis. A similar strategy was used for the construction of a pTGPC-GFP plasmid, which expresses a GPC-GFP fusion protein having a GFP tag at the C terminus of the GPC precursor. The GPC precursor was PCR amplified from a pTGFP-GPC construct using a primer set shown in Table 1. The resulting PCR product (PCR 1) was digested with NcoI and BamHI restriction enzymes and gel purified. GFP was PCR amplified using the primer set shown in Table 1 (PCR 2) and digested with BamHI and XhoI restriction enzymes. The two digested PCR products were ligated to a pTriEx1.1 backbone digested with NcoI and XhoI, as mentioned above. The same strategy was used for the construction of all plasmids shown in Table 1. Briefly, the two PCR products (PCR 1 and PCR 2) were generated with appropriate primer sets shown in Table 1 and digested with appropriate enzymes, as mentioned in Table 1. The two PCR products were mixed with a pTriEX1.1 backbone that was digested with NcoI and XhoI and ligated.
Table 1.
Primer sequences for the plasmid constructs in the present study
| Plasmid no.a | Plasmid name | Primer sequenceb |
|
|---|---|---|---|
| PCR 1 | PCR 2 | ||
| 1 | pTGFP-GPC | Fp: 5′-CTCTATCCATGGTGAGCAAGGGCGAGGAGCTG-3′ | |
| Rp: 5′-AACTATGGATCCCTTGTACAGCTCGTCCATG-3′ | |||
| 2 | pTGPC-GFP | Fp: 5′-CTCTATCCATGGTAGGGTGGGTTTGCATCTTC-3′ | Fp: 5′-AGTCGAGGATCCGTGAGCAAGGGCGAGGAGCTG-3′ |
| Rp: 5′-AGTCAAGGATCCATTAGCTTTATTTTTTCTAC-3′ | Rp: 5′-AACTATCTCGAGTTACTTGTACAGCTCGTCCATG-3′ | ||
| 3 | pTGFP-Gn | Fp: 5′-CTCTATCCATGGTGAGCAAGGGCGAGGAGCTG-3′ | Fp: 5′-CTCTACGGATCCGTAGGGTGGGTTTGCATCTTC-3′ |
| Rp: 5′-AACTATGGATCCCTTGTACAGCTCGTCCATG-3′ | Rp: 5′-AGTCAACTCGAGCTATGCACTAGCTGCCCATATGATC-3′ | ||
| 4 | pTGn-GFP | Fp: 5′-CTCTATCCATGGTAGGGTGGGTTTGCATCTTC-3′ | Fp: 5′-AGTCGAGGATCCGTGAGCAAGGGCGAGGAGCTG-3′ |
| Rp: 5′-AGTCAAGGATCCTATGATCAGTTCAGTTGTTAAAAGG-3′ | Rp: 5′-AACTATCTCGAGTTACTTGTACAGCTCGTCCATG-3′ | ||
| 5 | pTGFP-Gc | Fp: 5′-CTCTATCCATGGTGAGCAAGGGCGAGGAGCTG-3′ | Fp: 5′-CTCTACGGATCCGATACCCCCTTAATGGAGTCC-3′ |
| Rp: 5′-AACTATGGATCCCTTGTACAGCTCGTCCATG-3′ | Rp: 5′-AGTCAACTCGAGCTAATTAGCTTTATTTTTTCTAC-3′ | ||
| 6 | pTGc-GFP | Fp: 5′-CTCTATCCATGGGATACCCCCTTAATGGAGTCC-3′ | Fp: 5′-AGTCGAGGATCCGTGAGCAAGGGCGAGGAGCTG-3′ |
| Rp: 5′-AGTCAAGGATCCATTAGCTTTATTTTTTCTAC-3′ | Rp: 5′-AACTATCTCGAGTTACTTGTACAGCTCGTCCATG-3′ | ||
| 7 | pTGFP-GnΔtail | Fp: 5′-CTCTATCCATGGTGAGCAAGGGCGAGGAGCTG-3′ | Fp: 5′-CTCTACGGATCCGTAGGGTGGGTTTGCATCTTC-3′ |
| Rp: 5′-AACTATGGATCCCTTGTACAGCTCGTCCATG-3′ | Rp: 5′-AGTCAACTCGAGCTATAATATAATTAAGGTGACTG-3′ | ||
| 8 | pTGFP-GnTail | Fp: 5′-CTCTATCCATGGTGAGCAAGGGCGAGGAGCTG-3′ | Fp: 5′-CTCTACGGATCCAAGATCCTAAGGTTGCTTACT-3′ |
| Rp: 5′-AACTATGGATCCCTTGTACAGCTCGTCCATG-3′ | Rp: 5′-AGTCAACTCGAGCTATGCACTAGCTGCCCATATGATC-3′ | ||
| 9 | pT-Gn | Fp: 5′-CTCTATCCATGGTAGGGTGGGTTTGCATCTTC-3′ | |
| Rp: 5′-AGTCAACTCGAGCTATGCACTAGCTGCCCATATGATC-3′ | |||
| 10 | pT-Gc | Fp: 5′-CTCTATCCATGGGATACCCCCTTAATGGAGTCC-3′ | |
| Rp: 5′-AGTCAACTCGAGCTAATTAGCTTTATTTTTTCTAC-3′ | |||
Plasmids 1 to 8 were generated by cloning two PCR products (PCR 1 and PCR 2) between the NcoI and XhoI sites of pTriEx1.1 plasmid. PCR 1 was digested with NcoI and BamHI restriction enzymes. Similarly, PCR 2 was digested with BamHI and XhoI. The two digested PCR products were then ligated to pTriEx1.1 digested with NcoI and XhoI. In plasmids 1 to 8, except for plasmids 4 and 6, PCR 1 was generated from pT-GFP plasmid and PCR 2 was generated from a pT-GPC construct (see Materials and Methods for details). For plasmids 4 and 6, PCR 1 was generated from pT-GPC, and PCR 2 was generated from pT-GFP. Construction of pT-GPC is described in detail in Materials and Methods. Plasmids 9 and 10 were constructed by generating a PCR product (PCR 1) from the pT-GPC plasmid, using the primers mentioned in the table. The PCR product was cloned in the pTriEx1.1 backbone between NcoI and XhoI restriction sites.
Fp, forward primer; Rp, reverse primer. Restriction sites are shown in boldface.
The pmRFP-LC3 plasmid (Addgene 21075) was a donation from Tamotsu Yoshimori (Research Institute for Microbial Diseases, Osaka University, Japan) (14). It encodes a monomeric red fluorescent protein (mRFP) fused to LC3.
siRNA knockdowns.
To substantiate the role of autophagy in Gn degradation, two essential autophagy genes, BCLN-1 (BCLN-1) and ATG7, were downregulated by siRNA transfection. SignalSilence BCLN-1 siRNA I, SignalSilence ATG7 siRNA I, and SignalSilence Control siRNA were purchased from Cell Signaling. BCLN-1 and ATG7 siRNA were cotransfected (100 nM final concentration) into monolayers of HeLa cells seeded in six-well plates using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Control siRNA was similarly transfected into control wells. Twenty-four hours after transfection, BCLN-1 and ATG7 siRNAs or the control siRNA was transfected once again together with plasmid pTGFP-Gn, plasmid pTGFP fused to Gn lacking the cytoplasmic tail (pTGFP-GnΔtail), or plasmid pTGFP fused to the cytoplasmic tail fragment of Gn (pTGFP-Gntail) (1 μg each). The effect of the siRNA knockdown on the expression levels of BCLN-1 and ATG7 proteins was verified by Western blotting using the respective antibodies 48 h after the first transfection. The effect of siRNA knockdowns on the stability of GFP-Gn, GFP-GnΔtail, and GFP-Gntail fusion proteins was quantified by the estimation of GFP signal using fluorescence-activated cell sorting (FACS) analysis. The GFP signal was compared with cells transfected with control siRNA.
Antibodies.
GFP (4B10) mouse monoclonal antibody (MAb), β-actin (8H10D10) mouse monoclonal antibody, BCLN-1 (D40C5) rabbit MAb, and rabbit ATG7 antibody were purchased from Cell Signaling Technologies. Goat anti-mouse IgG (whole molecule)-fluorescein isothiocyanate (FITC), goat anti-P62 (p0067), and rabbit anti-LC3 antibodies were purchased from Sigma.
Autophagy and proteosomal inhibitors.
To probe the roles of the two main degradation machineries, proteosome and autophagy, in Gn degradation, proteosomal inhibitor (MG132) and autophagy inhibitors (3-methyladenine [3-MA], LY-294002, and Wortmanin) were used. All inhibitors were added in fresh medium after the medium containing transfection mixtures was replaced at 4 to 5 h posttransfection of Gn constructs. MG132 and 3-methyladenine were obtained from Sigma and were used at final concentrations of 10 μM and 10 mM, respectively. LY-294002 and Wortmanin were purchased from EMD Calbiochem and were used at final concentrations of 50 μM and 100 μM, respectively. To determine whether lysosomal protease inhibitors pepstatin A (BP267110; Fisher Scientific) and E64d (13533; Cayman Chemicals) have a role in the degradation of the GFP-Gn fusion protein, we incubated HeLa cells with 10 μg/ml of both of these drugs for 4 h before transfection. Cells were transfected with a plasmid expressing GFP-Gn and incubated for another 4 h in medium lacking the drug. At 4 h posttransfection both of the drugs were added back at a concentration of 10 μg/ml. Cells were harvested at 12 and 24 h posttransfection and monitored by flow cytometry for GFP expression.
Flow cytometry.
FACS was used to analyze the expression levels of wild-type and mutant glycoproteins fused with GFP and to examine the effects of chemical inhibitors and siRNA gene knockdowns on their stability. Briefly, at 24 h posttransfection, cells were harvested with trypsin and fixed with 1% paraformaldehyde. Fixed cells were analyzed by a BD FACSCalibur Flow Cytometer, as previously reported (28). Total fluorescence signal of GFP-positive cells was calculated by multiplying the mean fluorescence value by the number of GFP-positive cells.
Confocal microscopy.
The colocalization of fluorescent signals of GFP-Gn with mRFP-LC3 in transfected HeLa cells was studied by confocal microscopy. HeLa cells were grown on coverslips in six-well plates and transfected with the combinations of plasmids mentioned above. Due to rapid degradation, the GFP-Gn signal was too low. We enhanced the GFP-Gn signal by staining with an anti-GFP primary antibody, followed by FITC-labeled secondary antibody. This staining procedure improved the signal significantly and allowed us to study the colocalization of Gn with LC3. At 24 h posttransfection, cells were washed twice with phosphate-buffered saline (PBS) and fixed for 15 min with 1% paraformaldehyde as previously reported (27). Autofluorescence was quenched by treating fixed cells with 50 mM ammonium chloride for 10 min. Cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min and then blocked for 30 min with 4% bovine serum albumin (BSA). Primary staining with anti-GFP was done overnight at 4°C, followed by three washes with PBS. Secondary staining was carried out for 1 h at room temperature, followed by three washes with PBS. Samples were mounted with UltraCruz mounting medium (Santa Cruz Biotechnology), and mounted slides were examined under a Ziess LSM 510 laser scanning microscope.
Western blotting.
Western blotting was performed to examine the expression of wild-type and mutant Gn proteins fused with GFP, to check the levels of LC3-I and LC3-II proteins upon Gn expression, and to confirm that siRNA knockdowns downregulated the levels of BCLN-1 and ATG7. At 24 h posttransfection, cells were washed with cold PBS and lysed with radioimmunoprecipitation assay (RIPA) lysis buffer containing protease inhibitor cocktail at 4°C. Protein samples separated on SDS-PAGE were transferred to polyvinylidene difluoride (PVDF) membrane, which was subsequently blocked for 1 h in blocking buffer (5% nonfat dry milk in PBS containing 0.1% Tween 20). The membrane was then incubated overnight at 4°C with the appropriate primary antibody. After three washes with PBS containing 0.1% Tween, the membrane was incubated for 1 h at room temperature with peroxidase-conjugated species-specific secondary antibody. The membrane was finally washed three times with PBS containing 0.1% Tween, and the bound proteins were detected with chemiluminescence detection reagents (Thermo Scientific). The luminescence was detected by a short exposure of the membrane to an X-ray film.
Virus infection and real-time PCR.
HUVEC plated in six-well plates were cotransfected twice, with a 24-h interval, with the BCLN-1 and ATG7 siRNAs using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Twenty-four hours after the second siRNA transfection, HUVEC were infected with SNV at a multiplicity of infection (MOI) of 10. The virus was allowed to adsorb for 2 h, and then the virus inoculum was aspirated and replaced with fresh medium. At 12-h intervals, cells were collected by trypsinization, lysed, and stored at − 20°C until the end of the experiment. Total RNA was extracted from half of the cell lysate using an RNeasy kit (Qiagen). The concentration of total RNA was measured using a Nanodrop machine, and then equal amounts of RNA (100 ng) were used for cDNA synthesis using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen). Virus replication over time was monitored by the quantitative estimation of the viral S-segment RNA by real-time PCR analysis using Power SYBR green PCR Master Mix (Applied Biosystems). The primers used in real-time PCR analysis of the S segment were (forward primer) 5′-CTCAAAGATGCAGAAAGAGC-3′ and (reverse primer) 5′-ATGGTCATCAGGTTCAATCC-3′. As an internal control, β-actin was quantified using the forward primer 5′-AGCGAGCATCCCCCAAAGTT-3′ and the reverse primer: 5′-GGGCACGAAGGCTCATCATT-3′. The remaining half of the cell lysate was used to determine whether SNV infection induces autophagy after infection. Induction of autophagy was confirmed by the conversion of LC3-I to LC3-II by Western blot analysis using anti-LC3 antibody.
Immunoprecipitation and Western blotting.
HeLa cells seeded in 100-mm dishes were transfected with pTGFP or pTGFP-Gn plasmid, expressing wild-type Gn or GFP-Gn fusion proteins, respectively. Cells were grown in medium either containing or lacking the autophagy inhibitor 3-MA. Twenty-four hours posttransfection, cells were harvested and lysed with 500 μl of RIPA buffer containing 1× protease inhibitor cocktail (Thermo Scientific) and 1 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma). Fifty microliters of protein G agarose beads was washed and restored in PBS as a 50% slurry, followed by incubation with 0.5 ml of cell lysate for 10 min at 4°C on a rocker to reduce nonspecific binding of proteins to agarose beads. Beads were pelleted by brief centrifugation, and the supernatant was incubated overnight at 4°C with 5 μl of either mouse anti-SNV Gn antibody (Austral) or anti-GFP antibody (Cell Signaling Technologies). The next morning protein G agarose beads were added back into the solution, and the mixture was further incubated at 4°C for 6 h. Beads were pelleted down by a short centrifugation, followed by three washes with 1× PBS. One hundred microliters of sample buffer containing 2.5% β-mercaptoethanol (β-ME) was directly added to the washed beads. The mixture was heated at 95°C for 5 min, loaded on a 14% SDS-PAGE gel, and examined by Western blot analysis using either anti-Gn or anti-GFP antibody.
RESULTS
Hantavirus glycoprotein Gn is degraded in cells.
In SNV, the GPC precursor protein, encoded by the viral M segment RNA, is cleaved at a conserved WAASA site near the middle, generating an N-terminal fragment (Gn) of 652 amino acids and a C-terminal fragment (Gc) of 488 amino acids in length (Fig. 1) (25). Gn harbors a cytoplasmic tail domain of 142 amino acids at the C terminus. During virus assembly, both Gn and Gc are recruited to the virus envelope (Fig. 1). To monitor the expression of GPC in cells, we fused green fluorescent protein (GFP) at either the N or C terminus of GPC and expressed the fusion protein in HeLa cells. Since the GPC precursor is posttranslationally cleaved near the middle, the fusion of the GFP tag at the N terminus of the GPC precursor will report the expression of Gn in cells. Similarly, the GFP fusion at the C terminus of GPC will report the expression of Gc in cells. We transfected HeLa cells with these reporter constructs and monitored the GFP expression by FACS analysis at increasing time intervals. Since both Gn and Gc are expressed from a single mRNA, it is expected that both proteins are equally expressed in cells. Interestingly, we observed a poor GFP signal when the GFP tag was fused at the N terminus of the GPC precursor. On the other hand, a significantly stronger GFP signal was observed in cells when the GFP tag was fused at the C terminus of the GPC precursor (Fig. 2A). To rule out the possibility that GFP fusion at the N terminus of GPC precursor perturbed the GFP signal, we cloned Gn and Gc separately and expressed them in HeLa cells as GFP fusion proteins. Again, we observed a strong GFP signal in HeLa cells expressing a Gc-GFP fusion protein in comparison to cells expressing a Gn-GFP fusion protein (Fig. 2B and C). Fusion of GFP at either the N or C terminus of Gc had no effect on the GFP signal. However, the fusion of GFP at the N terminus of Gn slightly improved the GFP signal (Fig. 2C). These preliminary observations suggested that Gn is likely degraded in cells. Since Gn and Gc are recruited as heterodimers in the virus envelope (3), we asked whether coexpression of Gn and Gc rescues Gn expression in cells. We transfected HeLa cells with Gn-GFP reporter constructs along with another plasmid that expresses Gc lacking the GFP tag and monitored the GFP signal at increasing time intervals. We observed that coexpression of Gn and Gc slightly rescued Gn expression in the cells although the signal was still negligible in comparison to that of Gc (Fig. 2D). It is likely that formation of a Gn-Gc heterodimer in cytoplasmic membranes rescued the Gn glycoprotein from degradation, consistent with the idea that coexpression of Gn and Gc is important for the proper trafficking of Gn (42). However, similar studies with Gc revealed that coexpression of Gn and Gc had no effect upon the expression of Gc in cells, suggesting that Gc is significantly stable in comparison to Gn (Fig. 2E). To further confirm these results, all samples used in the experiments shown in Fig. 2A to E were examined by Western blot analysis using anti-GFP antibody. The expression of Gc at different time points was easily detected and was consistent with the GFP reporter assay (Fig. 2A, B, and E). However, due to the rapid degradation, Gn was not detectable by Western blot analysis, except that a weak signal was observed when both Gn and Gc were coexpressed (Fig. 2D). Since the results from FACS analysis of GFP fusion proteins were consistent with Western blot analysis, we preferred to monitor the expression of the desired proteins using FACS analysis in subsequent studies.
Fig 2.

The expression of hantavirus glycoproteins in HeLa cells. (A) HeLa cells in six-well plates were transfected with either pTGFP-GPC or pTGPC-GFP plasmid, expressing GPC and having a GFP tag at either the N terminus (i)or C terminus (ii), respectively. In each well a total of 4 μg plasmid DNA was transfected, containing 2 μg of expression plasmid and 2 μg of the empty vector. (B and C) HeLa cells in six-well plates were similarly transfected with the plasmids expressing either Gc or Gn and having the GFP tag at either the N or C terminus. Cells were harvested at different time intervals posttransfection (12, 24, 36, and 48 h), and protein expression was determined by monitoring the GFP signal, using FACS analysis. (D) HeLa cells in six-well plates were cotransfected with 2 μg each of pTGC and pTGFP-Gn plasmids, expressing untagged Gc and N-terminally tagged GFP-Gn fusion proteins, respectively. The Gn expression in cotransfected cells was compared with the cells expressing Gn alone. (E) Similar to the experiment shown in panel D, HeLa cells were cotransfected with plasmids expressing untagged Gn and N-terminally tagged GFP-Gc fusion proteins. Again, the expression of Gc in cotransfected cells was compared with the cells expressing Gc alone. Note that the data points in panels A through E were normalized to the 24-h time point in panel A. Note that in panels A to E, the samples at each time point were examined by Western blot analysis using anti-GFP antibody. The Western blots are shown inside each panel.
Deletion of the cytoplasmic tail domain does not rescue Gn degradation in cells.
It has been previously reported that the cytoplasmic tail domain of Gn from New York 1 (NY-1) hantavirus, expressed from a plasmid, is ubiquitinated and degraded by the proteasomal machinery in cells (8). We asked whether the cytoplasmic tail domain of SNV Gn protein is mediating the degradation of the entire Gn molecule in cells. We expressed a Gn mutant, which is devoid of the cytoplasmic tail domain, as a GFP fusion protein in cells and compared its expression with the wild-type Gn at increasing time intervals using FACS analysis. In addition, the cytoplasmic tail was expressed separately as a GFP fusion protein in cells. As shown in Fig. 3A, it is clear that deletion of the cytoplasmic tail does not rescue the degradation of Gn. However, the tail alone was significantly expressed in comparison to the wild-type or mutant Gn. In addition, we noticed rapid degradation of all three proteins at 24 h posttransfection. To determine whether these proteins were degraded by the proteasome machinery, we added a proteasomal inhibitor (MG132) to cells 4 h posttransfection and monitored the GFP signal at increasing time intervals. As shown in Fig. 3B, we observed that MG132 slightly rescued the Gn protein from degradation. We did not notice any change in the stability of the Gn mutant in the presence or absence of MG132 (Fig. 3C). However, MG132 significantly protected the tail domain from degradation by 15-fold (Fig. 3D). After 24 h, the effect of the proteasome inhibitor wears off, as evident from the continued degradation of all three proteins. These observations suggest that although the proteosomal machinery degrades the Gn tail domain, neither the wild-type nor the mutant Gn is cleared by this degradation route in cells. In addition, the signal required for the degradation of Gn is not present in the tail region of SNV Gn. The amino acid residues that selectively mediate the degradation of other pathogenic hantavirus Gn tails have been reported (40).
Fig 3.
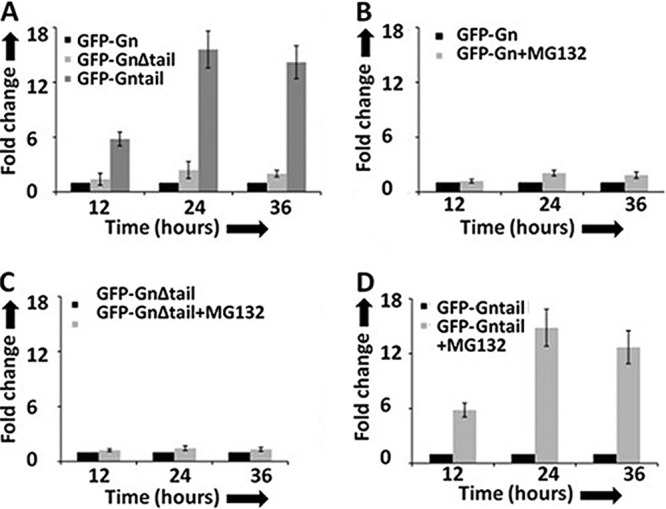
Gn tail domain does not regulate the expression of the entire Gn molecule. (A) HeLa cells in six-well plates were transfected with 4 μg of either pTGFP-Gn or pTGFP-GnΔtail or pTGFP-Gntail construct, expressing wild-type Gn, Gn devoid of the tail domain, and the Gn tail domain alone, respectively. Cells were harvested at increasing time intervals, and GFP signal was monitored by FACS analysis. (B, C and D) HeLa cells in six-well plates were transfected with 4 μg of either pTGFP-Gn (B) or pTGFP-GnΔtail (C) or pTGFP-Gntail (D), plasmid and the medium containing the transfection mixture was aspirated at 5 h posttransfection. The medium was replaced with either medium containing MG132 (10 μM final concentration) or normal medium lacking the MG132. Cells were harvested at increasing time intervals, and GFP expression was monitored as described in panel A. Fold change in panels A to D was calculated by normalizing the GFP signal to the level represented by the in the corresponding panel.
Gn is degraded by the autophagy machinery in cells.
Since Gn was not degraded by the proteasomal machinery, we next wanted to determine whether Gn is cleared by the autophagy-lysosome machinery. We again transfected HeLa cells with plasmids expressing either wild-type or mutant Gn or the tail domain alone, followed by the addition of either proteasome inhibitor (MG132) or autophagy inhibitors 3-methyladenine (3-MA), LY-294002, or Wortmanin, as described in Materials and Methods. Protein expression was quantitatively measured by FACS analysis at increasing time intervals. Compared to the proteasome inhibitor (MG132), the autophagy inhibitors significantly rescued from degradation both the wild-type and mutant Gn lacking the tail domain (Fig. 4A and B). However, the effect was much more pronounced for the wild-type Gn. Interestingly, none of the three autophagy inhibitors had any effect on the stability of the Gn tail domain (Fig. 4C). Since our previous results revealed that coexpression of Gn and Gc slightly prevented Gn from degradation (Fig. 2D), we monitored the effect of autophagy inhibitors on the stability of Gn in HeLa cells cotransfected with plasmids expressing both Gc and the Gn-GFP fusion protein. Interestingly, we noticed that cotransfection along with autophagy inhibitors offered double protection of Gn from degradation (Fig. 4D).
Fig 4.

Autophagy inhibitors protect the degradation of Gn in cells. HeLa cells in six-well plates were transfected with 2 μg of either pTGFP-Gn (A) or pTGFP-GnΔtail (B) or pTGFP-Gntail (C) construct, expressing GFP fused to either wild-type Gn (GFP-Gn), a mutant Gn devoid of the tail domain (GFP-GnΔtail), or the tail domain of Gn (GFP-Gntail), respectively. The medium containing transfection mixture was aspirated at 5 h posttransfection and replaced with fresh medium containing either 3-MA (10 mM final concentration), LY-294002 (LY; 50 μM final concentration), Wortmanin (Wort; 100 μM final concentration), MG132 (10 μM final concentration), or medium lacking the inhibitor (black bars). In each well a total of 4 μg of plasmid DNA was transfected, containing 2 μg of expression plasmid and 2 μg of the empty vector pT1.1. (D) Cells in six-well plates were cotransfected with 2 μg each of pTGFP-Gn and empty vector pT1.1 (black bars), pTGFP-Gn and pTGC plasmids lacking 3-MA (light gray bars), or pTGFP-Gn and pTGC plasmids containing 10 mM 3-MA (dark gray bars). The plasmid pTGC expresses untagged Gc. Fold change in panels A to D was calculated by normalizing the GFP signal to levels represented by the dark bar in the corresponding panel. (E) A Western blot showing Gn and GFP-Gn fusion protein using anti-Gn antibody. (F) Samples from panel E were examined by Western blot analysis using anti-GFP antibody. Western blot analysis (panels E and F) was carried out as described in Materials and Methods.
Due to the limited time effect of both autophagy and proteosome inhibitors, the degradation of all three proteins continued after 24 h of transfection. Since autophagy inhibitors selectively protected wild-type and mutant Gn from degradation, it is likely that Gn harbors a degradation signal upstream of the cytoplasmic tail region, which specifically targets Gn to autophagosomes for degradation.
To rule out the possibility that a GFP fusion at the N terminus of Gn does not lead to the degradation of Gn, we transfected HeLa cells with plasmids expressing either wild-type Gn or a GFP-Gn fusion protein, followed by incubation with autophagy inhibitor 3-MA. Cell lysates were immunoprecipitated using a monoclonal anti-Gn antibody, as described in Materials and Methods. The immunoprecipitated material was examined for the presence of Gn by Western blot analysis using either anti-Gn or anti-GFP antibody. This analysis revealed that both wild-type Gn and GFP-Gn fusion proteins were degraded in cells to an undetectable level (Fig. 4E). However, addition of 3-MA equally rescued both wild-type Gn and Gn-GFP fusion proteins (Fig. 4E), suggesting that the GFP tag at the N terminus of Gn does not play any role in the degradation of Gn. In addition, the fusion of a GFP tag upstream of the Gn start codon raises a question about the possible cleavage of the GFP tag by the N-terminal signal peptide of Gn protein after translocation through the endoplasmic reticulum. To clarify this issue, we analyzed the samples by Western blot analysis using anti-GFP antibody. As evident from Fig. 4F, the GFP tag is not cleaved from the GFP-Gn fusion protein. Fig. 2D shows a similar result. This is further supported by the slow electrophoretic mobility of the GFP-Gn fusion protein in comparison to wild-type Gn, consistent with their respective molecular masses. These results suggest that the N-terminal signal peptide is likely not cleaved after the translocation of Gn through the endoplasmic reticulum. However, further experimentation is required to delineate whether the N-terminal GFP tag affects the translocation of Gn through the endoplasmic reticulum.
Gn triggers autophagy in cells.
The hallmark of autophagy execution is the lipidation of LC3 protein to generate a species known as LC3-II, which is commonly used as a marker for studying autophagy (15). We next wanted to determine whether Gn expression triggers autophagic signaling in cells. We monitored the LC3-II conversion in Gn-expressing HeLa cells at different time intervals using Western blot analysis. As shown in Fig. 5A, the appearance of LC3-II clearly demonstrates that Gn expression induces autophagosome formation in HeLa cells. In comparison, Gc expression did not induce such autophagosome formation in HeLa cells (data not shown). To further confirm that Gn is degraded in autophagosomes, we used confocal microscopy to visualize Gn in HeLa cells. It is evident from Fig. 5B that Gn colocalizes with the autophagy marker LC3, suggesting that Gn is likely targeted to autophagosomes for degradation. The strong colocalization of Gn with LC3 markers suggests that Gn is rapidly targeted to autophagosomes for degradation. To delineate whether autophagic degradation is occurring in cells expressing Gn, we monitored the expression of P62 protein in cells at different time intervals. Compared to untransfected cells, we observed a consistent decrease in P62 expression at the 24-h time point in cells transfected with a plasmid expressing Gn (Fig. 5C), suggesting that autophagic degradation is occurring in cells expressing Gn. Although these observations clearly illustrated that Gn is cleared by the autophagy machinery in cells, we wanted to determine whether the inhibition of autophagosome formation affects the stability of Gn in cells. We interrupted autophagy at both the nucleation and elongation phases by the simultaneous downregulation of two critical genes, BCLN-1 and ATG7, using siRNA. BCLN-1 and ATG7 are key components of the PI3K complex and ATG12 conjugation system, respectively. Downregulation of both of these genes was confirmed by Western blot analysis (Fig. 5D). HeLa cells cotransfected with plasmids expressing either wild-type or mutant Gn or the tail domain, along with either siRNA against BCLN-1 and ATG7 or a control siRNA, were harvested at 24 h posttransfection, and the GFP signal was monitored using FACS analysis. As shown in Fig. 5E, the inhibition of autophagosome formation significantly improved the stability of both the wild-type and mutant Gn. However, the effect was more pronounced with the wild-type Gn, consistent with our previous observations using the autophagy inhibitors (Fig. 4). Also, inhibition of autophagosome formation did not affect the stability of the Gn tail domain, consistent with our previous observations (Fig. 4). In addition, we used lysosomal protease inhibitors E64d and pepstatin A (36) to further confirm that Gn is degraded by the autophagy-lysosome route. HeLa cells transfected with a plasmid expressing Gn were incubated with E64d and pepstatin A, and Gn expression was monitored at different time intervals. As evident from Fig. 5F, these protease inhibitors protected Gn from degradation, confirming the degradation of Gn in autolysosomes. Taken together, these findings firmly establish that Gn is degraded by autophagy and that the degradation signal is located upstream of the tail region of Gn.
Fig 5.
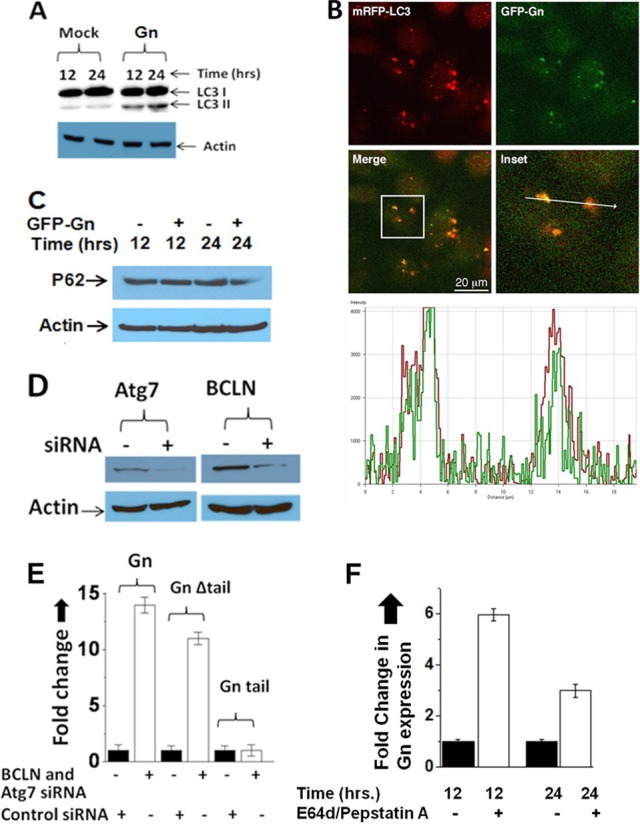
Gn degradation is mediated via the autophagy machinery. (A) Western blot showing the levels of LC3-II in mock- and Gn-transfected HeLa cells at 12 and 24 h posttransfection. (B) HeLa cells were cotransfected with plasmids expressing Gn-GFP and mRFP-LC3 fusion proteins. A confocal microscopy image shows that GFP-Gn colocalizes with the mRFP-LC3, which is an autophagy marker. The panel labeled Inset is a magnification of the boxed area in the merge panel. A line was drawn through the two puncta, and the fluorescence intensity under the line is represented in the bottom graph. The green line is the intensity of the GFP-Gn fluorescent signal, and the red line represents the intensity of the mRFP-LC3 fluorescent signal. The images were captured at 12-bit resolution; thus, the intensity ranges from 0 to 4,096. The signals for GFP-Gn and mRFP-LC3 show strong colocalization, as evidenced by the fact that both signals are significantly above the background, and both signals increase at the same location. The arrow in the inset is 20 μm in length. Similar results were obtained when Gn was expressed from a construct expressing the entire GPC precursor (data not shown). (C) Western blot showing the expression of P62 protein in HeLa cells, which were either mock transfected or transfected with a plasmid expressing GFP-Gn. Cells were lysed at 12 or 24 h posttransfection, and P62 expression was monitored using anti-P62 antibody. (D) Downregulation of Atg7 and BCLN by siRNA (as described in Materials and Methods) was confirmed by Western blot analysis using appropriate antibodies. (E) HeLa cells seeded in six-well plates were transfected with either a combination of BCLN-1 and Atg7 siRNAs or a control siRNA. At 24 h after transfection, BCLN and Atg7 siRNA or control siRNA was transfected once again together with either pTGFP-Gn, pTGFP-GnΔtail, or pTGFP-Gntail plasmid (2 μg each). At 24 h posttransfection GFP signal was examined by FACS analysis, and fold change related to the cells transfected with the control siRNA was calculated and plotted. (F) HeLa cells in six-well plates were transfected with a plasmid expressing GFP-Gn. Cells were maintained in medium either lacking or containing lysosome protease inhibitors E64d and pepstatin A. At 12 or 24 h posttransfection GFP signal was examined by FACS analysis, and fold change related to the cells lacking the drug was calculated and plotted.
Inactivation of the host autophagy machinery inhibits Sin Nombre virus replication in cells.
A possible degradation of Gn in cells infected with SNV has been previously reported (42). We wanted to determine whether degradation of Gn is due to SNV-induced autophagy in cells and illustrate if autophagic clearance of Gn has a role in SNV replication. We first interrupted autophagosome formation in HUVEC by the downregulation of the BCLN-1 and ATG7 genes using siRNA, as described in Materials and Methods. Both wild-type and autophagy-interrupted HUVEC were infected with SNV at an MOI of 10, and virus replication was monitored over time by quantitative estimation of viral S-segment RNA using real-time PCR analysis. As shown in Fig. 6B, SNV replication was significantly inhibited by the downregulation of the critical autophagy genes BCLN-1 and ATG7. A similar observation was made when autophagy was inhibited by chemical inhibitors 3-MA or LY-294002 (data not shown). In comparison, an active virus replication was observed in HUVEC transfected with control siRNA. Since the inhibition of autophagy machinery by the downregulation of the BCLN-1 and ATG7 genes is well established, it is highly likely that inhibition of SNV replication is due either to the inhibition of autophagosome formation in BCLN-1- and ATG7-downregulated cells or to the inhibition of autophagic degradation of Gn in autolysosomes. Some viruses are known to use autophagosome formation for productive replication in host cells (45). We next monitored the induction of autophagosome formation in the wild-type HUVEC after virus infection by detecting the conversion of LC3-I to LC3-II using Western blot analysis. As shown in Fig. 6A, SNV infection rapidly induced autophagosome formation in HUVEC. Interestingly, induction of autophagosome formation promoted virus replication (Fig. 6B). To define whether autophagosome formation or autophagic degradation of Gn is required for productive SNV replication, we examined virus replication in cells containing the lysosome protease inhibitors E64d and pepstatin A. These two protease inhibitors prevented the degradation of Gn in transfected cells (Fig. 5F). Interestingly, similar to BCLN-1 and ATG7 knockdown, these two inhibitors also inhibited virus replication in cells (Fig. 6B). The only commercially available antibody against SNV Gn failed to detect the low levels of Gn produced in infected cells. Therefore, we could not examine the steady-state levels of Gn in virus-infected cells. Taking into consideration the degradation of Gn by the host autophagy machinery in transfected cells and the reported lower intrinsic levels of Gn in SNV infected cells (42), it is likely that autophagic clearance of Gn is required for efficient virus replication. Inhibition of the host autophagy machinery, which likely prevents Gn degradation in virus-infected cells, dramatically hampers virus replication, suggesting that the elevated levels of Gn might inhibit virus replication by an unknown mechanism. Since we could not examine Gn expression in virus-infected cells, there is a remote possibility that induction of autophagosome formation in SNV-infected cells and not the autophagic degradation of Gn is required for productive virus replication.
Fig 6.
Effect of host autophagy response on virus replication. (A) SNV infection induces autophagy in infected HUVEC. HUVEC were infected with SNV at an MOI of 10, and then cells were collected at 12-h time intervals, lysed in RIPA buffer, and stored at −20°C. The total proteins in cell lysates were quantified using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). Equal amounts of proteins of each sample (10 μg) were loaded on an SDS-PAGE gel, and the LC3-II/LC3-I ratios were assessed by Western blotting. Autophagy was triggered as early as 12 h postinfection. (B) Induction of autophagy is essential for virus replication. HUVEC were transfected with either control siRNA or specific siRNA targeting the autophagy-essential genes BCLN-1 and Atg-7. At 48 h posttransfection, cells were infected with SNV at an MOI of 10. Virus production was monitored by quantifying S-segment RNA using real-time PCR. A similar experiment was performed with cells treated with lysosome protease inhibitors E64d and pepstatin A, as described in Materials and Methods. Virus replication in untreated cells resembled that of cells treated with a control siRNA and is not shown.
DISCUSSION
The hantavirus glycoprotein precursor (GPC) is expressed from a single transcript and is posttranslationally cleaved into two glycoproteins, Gn and Gc. It is expected that cells infected with hantaviruses contain equimolar concentrations of Gn and Gc. However, previous studies have revealed that the Gn concentration was significantly lower than that of Gn in SNV-infected cells (42). Our studies clearly demonstrate that although both Gn and Gc are expressed from a single transcript, Gn is selectively targeted for degradation by host autophagy machinery. Autophagic clearance of Gn likely maintains low intrinsic steady-state levels of Gn in virus-infected cells, which is required for efficient virus replication.
The growing evidence, including the enhanced replication of tobacco mosaic virus (TMV) by RNA interference (RNAi)-mediated silencing of several different autophagy genes and the reduction in alphavirus replication by enforced neuronal expression of BCLN-1, supports the role of autophagy in antiviral immunity (23, 24). Recent evidence suggests that autophagy transfers the viral nucleic acids from the cytoplasm to intercellular compartments containing Toll-like receptor 7 (TLR7) in plasmacytoid dendritic cells, which signals the induction of the type I interferon response (18). This observation suggests that autophagy may play a role in the delivery of endogenous viral nucleic acids to their cognate innate immune detectors (20). One of the well-characterized roles of hantavirus Gn is the inhibition of the type I interferon response (1, 2, 22, 26). The cytoplasmic tail domain of hantavirus glycoprotein Gn has been reported to inhibit RIG-I- and TBK-1-directed interferon responses by disrupting the formation of TBK1-TRF3 complex (1, 2). The delayed IFN-mediated antiviral state facilitates rapid hantaviral dissemination to neighboring cells, which is consistent with lower serum interferon levels in hantavirus-infected patients and hantavirus sensitivity to early interferon administration (43). Based on these reported observations, one would expect that rapid clearance of Gn by the host autophagy machinery is a host encounter to eliminate the viral suppressor of the type I interferon response. Consistent with this idea, it was expected that inactivation of autophagy and survival of Gn would promote viral replication in cells. However, we found that inhibition of the host autophagy response using two independent approaches adversely affected virus replication in cells.
In addition, the examination of the structural architecture of hantavirus envelope has revealed that hantavirus surface is formed of homotetrameric Gn complexes interconnected with Gc homodimers (10). Thus, the formation of infectious hantavirus particles requires more Gn than Gc. Moreover, the protruding part of the spike complex of hantavirus particles, required for the attachment to the cell surface receptor during entry, is composed of four Gn subunits. One would expect that rapid elimination of Gn during the virus replication cycle by the host autophagy mechanism will adversely affect virus assembly and will likely increase the chances for the production of less-infectious or noninfectious virus particles. On the basis of these known observations, it is expected that rescuing Gn degradation by the inactivation of the host autophagy machinery would facilitate virus replication in cells. Again inconsistent with these expectations, we found that inhibition of the host autophagy mechanism inhibited virus replication in cells, suggesting that elevated endogenous levels of Gn inhibit virus replication by an unknown mechanism. These observations suggest that induction of host autophagy by virus replication and clearance of Gn by this degradation route are viral strategies to decrease the endogenous steady-state levels of Gn. Taking these observations together, we propose that, similar to other viruses (12, 17, 45), hantaviruses require the host autophagy mechanism for efficient replication in the host cells.
Although Gn is a strong repressor of the type I interferon response, its expression triggers the autophagy response in cells, suggesting that Gn is a PAMP which can trigger autophagy independent of other viral components by an unknown mechanism. Virus infection triggers a greater autophagy response than Gn expression induced by plasmid transfection in cells. This difference in the induction of the autophagy response could be due to poor transfection efficiency in comparison to virus infection in cells. It would be interesting to understand the mechanism for the induction of the autophagy response by Gn. Interestingly, the elevated autophagy response selectively targets Gn for degradation in autophagosomes. Previously, it has been reported that cells transfected with a plasmid expressing the 142-amino-acid-long cytoplasmic tail domain of the NY-1 hantavirus Gn revealed the ubiquitination and degradation of the tail domain. Although expressed separately, the degradation of the tail domain was linked to the degradation of the entire Gn molecule in cells. However, our studies clearly demonstrate that the cytoplasmic tail domain does not mediate the degradation of SNV Gn in cells. It is likely that Gn contains a degradation signal upstream of the tail domain, which is specifically recognized by the host autophagy machinery for the selective degradation of Gn in autophagosomes. Thus, it is probable that Gn has two unique domains in its structure, the cytoplasmic tail domain and a degradation domain. The cytoplasmic tail domain functions in the regulation of the type I innate immune response during infection. The degradation domain regulates Gn turnover in cells and maintains low steady-state levels of Gn required for efficient virus replication.
ACKNOWLEDGMENTS
This work was supported by research grants 5R21AI083672-02 and 5R01AI095236-02 from the NIH.
Footnotes
Published ahead of print 2 May 2012
REFERENCES
- 1. Alff PJ, et al. 2006. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 80:9676–9686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alff PJ, Sen N, Gorbunova E, Gavrilovskaya IN, Mackow ER. 2008. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. J. Virol. 82:9115–9122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Battisti AJ, et al. 2011. Structural studies of Hantaan virus. J. Virol. 85:835–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ciechanover A. 2006. The ubiquitin proteolytic system: from a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology 66:S7–19 [DOI] [PubMed] [Google Scholar]
- 5. Deretic V, Levine B. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Easterbrook JD, Klein SL. 2008. Immunological mechanisms mediating hantavirus persistence in rodent reservoirs. PLoS Pathog. 4:e1000172 doi:10.1371/journal.ppat.1000172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER. 1998. β3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. U. S. A. 95:7074–7079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Geimonen E, Fernandez I, Gavrilovskaya IN, Mackow ER. 2003. Tyrosine residues direct the ubiquitination and degradation of the NY-1 hantavirus G1 cytoplasmic tail. J. Virol. 77:10760–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He B, Gross M, Roizman B. 1997. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 94:843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hepojoki J, Strandin T, Vaheri A, Lankinen H. 2010. Interactions and oligomerization of hantavirus glycoproteins. J. Virol. 84:227–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hepojoki J, et al. 2010. Cytoplasmic tails of hantavirus glycoproteins interact with the nucleocapsid protein. J. Gen. Virol. 91:2341–2350 [DOI] [PubMed] [Google Scholar]
- 12. Jackson WT, et al. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156 doi:10.1371/journal.pbio.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jonsson CB, Figueiredo LT, Vapalahti O. 2010. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 23:412–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kimura S, Noda T, Yoshimori T. 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3:452–460 [DOI] [PubMed] [Google Scholar]
- 15. Klionsky DJ, et al. 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ku B, et al. 2008. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog. 4:e25 doi:10.1371/journal.ppat.0040025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kyei GB, et al. 2009. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 186:255–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. 2007. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315:1398–1401 [DOI] [PubMed] [Google Scholar]
- 19. Levine B. 2007. Cell biology: autophagy and cancer. Nature 446:745–747 [DOI] [PubMed] [Google Scholar]
- 20. Levine B, Deretic V. 2007. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7:767–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469:323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levine JR, et al. 2010. Antagonism of type I interferon responses by new world hantaviruses. J. Virol. 84:11790–11801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang XH, et al. 1998. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 72:8586–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y, et al. 2005. Autophagy regulates programmed cell death during the plant innate immune response. Cell 121:567–577 [DOI] [PubMed] [Google Scholar]
- 25. Lober C, Anheier B, Lindow S, Klenk HD, Feldmann H. 2001. The Hantaan virus glycoprotein precursor is cleaved at the conserved pentapeptide WAASA. Virology 289:224–229 [DOI] [PubMed] [Google Scholar]
- 26. Matthys V, Gorbunova EE, Gavrilovskaya IN, Pepini T, Mackow ER. 2011. The C-terminal 42 residues of the TULV Gn protein regulate interferon induction. J. Virol. 85:4752–4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mir MA, Duran WA, Hjelle BL, Ye C, Panganiban AT. 2008. Storage of cellular 5′ mRNA caps in P bodies for viral cap-snatching. Proc. Natl. Acad. Sci. U. S. A. 105:19294–19299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mir MA, Panganiban AT. 2008. A protein that replaces the entire cellular eIF4F complex. EMBO J. 27:3129–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Orvedahl A, et al. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35 [DOI] [PubMed] [Google Scholar]
- 30. Orvedahl A, Levine B. 2008. Autophagy and viral neurovirulence. Cell Microbiol. 10:1747–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Orvedahl A, et al. 2010. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7:115–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pattingre S, et al. 2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939 [DOI] [PubMed] [Google Scholar]
- 33. Richly H, et al. 2005. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120:73–84 [DOI] [PubMed] [Google Scholar]
- 34. Rubinsztein DC. 2006. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786 [DOI] [PubMed] [Google Scholar]
- 35. Sagar B, Marres HA, Hartman EH. 2010. Hypopharyngeal reconstruction with an anterolateral thigh flap after laryngopharyngeal resection: results of a retrospective study on 20 patients. J. Plast. Reconstr. Aesthet. Surg. 63:970–975 [DOI] [PubMed] [Google Scholar]
- 36. Sato K, et al. 2007. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 67:9677–9684 [DOI] [PubMed] [Google Scholar]
- 37. Schmaljohn CM. 1996. Molecular biology of hantaviruses. Plenum Press, New York, NY [Google Scholar]
- 38. Schmaljohn CS, Hooper JW. 2001. Bunyaviridae: the viruses and their replication, p 1581–1602 In Knipe DM, et al. (ed), Fields virology, 4th ed, vol 2 Lippincott, Williams, & Wilkins, Philadelphia, PA [Google Scholar]
- 39. Schonrich G, et al. 2008. Hantavirus-induced immunity in rodent reservoirs and humans. Immunol. Rev. 225:163–189 [DOI] [PubMed] [Google Scholar]
- 40. Sen N, Sen A, Mackow ER. 2007. Degrons at the C terminus of the pathogenic but not the nonpathogenic hantavirus G1 tail direct proteasomal degradation. J. Virol. 81:4323–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. 2009. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30:588–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spiropoulou CF, Goldsmith CS, Shoemaker TR, Peters CJ, Compans RW. 2003. Sin Nombre virus glycoprotein trafficking. Virology 308:48–63 [DOI] [PubMed] [Google Scholar]
- 43. Stoltz M, Ahlm C, Lundkvist A, Klingstrom J. 2007. Lambda interferon (IFN-λ) in serum is decreased in hantavirus-infected patients, and in vitro-established infection is insensitive to treatment with all IFNs and inhibits IFN-γ-induced nitric oxide production. J. Virol. 81:8685–8691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang H, Alminaite A, Vaheri A, Plyusnin A. 2010. Interaction between hantaviral nucleocapsid protein and the cytoplasmic tail of surface glycoprotein Gn. Virus Res. 151:205–212 [DOI] [PubMed] [Google Scholar]
- 45. Wong J, et al. 2008. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82:9143–9153 [DOI] [PMC free article] [PubMed] [Google Scholar]