

Abstract
Human immunodeficiency virus type 1 (HIV-1) infection can spread efficiently from infected to uninfected T cells through adhesive contacts called virological synapses (VSs). In this process, cell-surface envelope glycoprotein (Env) initiates adhesion and viral transfer into an uninfected recipient cell. Previous studies have found some HIV-1-neutralizing patient sera to be less effective at blocking VS-mediated infection than infection with cell-free virus. Here we employ sensitive flow cytometry-based infection assays to measure the inhibitory potency of HIV-1-neutralizing monoclonal antibodies (MAb) and HIV-1-neutralizing patient sera against cell-free and VS-mediated infection. To various degrees, anti-Env MAbs exhibited significantly higher 50% inhibitory concentration (IC50s) against VS-mediated infection than cell-free infection. Notably, the MAb 17b, which binds a CD4-induced (CD4i) epitope on gp120, displayed a 72-fold reduced efficacy against VS-mediated inocula compared to cell-free inocula. A mutant with truncation mutation in the gp41 cytoplasmic tail (CT) which is unable to modulate Env fusogenicity in response to virus particle maturation but which can still engage in cell-to-cell infection was tested for the ability to resist neutralizing antibodies. The ΔCT mutation increased cell surface staining by neutralizing antibodies, significantly enhanced neutralization of VS-mediated infection, and had reduced or no effect on cell-free infection, depending upon the antibody. Our results suggest that the gp41 CT regulates the exposure of key neutralizing epitopes during cell-to-cell infection and plays an important role in immune evasion. Vaccine strategies should consider immunogens that reflect Env conformations exposed on the infected cell surface to enhance protection against VS-mediated HIV-1 spread.
INTRODUCTION
The ability of HIV-1 to evade neutralizing antibody responses represents a major obstacle to the creation of an effective vaccine. The failure of HIV-1 vaccines is often attributed to the high sequence variability and conformational plasticity of the major neutralizing antigen, envelope glycoprotein (Env) (13, 39). The functional Env subunit is a trimeric spike made of gp120-gp41 heterodimers, which mediate viral entry during infection with both cell-free and cell-associated viral sources (5, 17). Cell-free infection of CD4+ T cells involves the release of viral particles from a productively infected cell, fluid-phase particle diffusion, viral attachment, and entry into an uninfected cell (28). Direct T cell-to-T cell infection occurs through contact between an infected donor T cell and an uninfected target T cell, resulting in the formation of an infectious cell-cell adhesion referred to as a virological synapse (VS) (17, 18). During VS-mediated infection, it has been proposed that virus particles may bud into the synapse and fuse directly with the plasma membrane at the synaptic space (34). However, a number of studies support a model for entry following VS formation involving two steps: (i) coreceptor-independent, coordinated transfer of viral particles into the target cell endocytic compartment (cell-to-cell transfer) followed by (ii) coreceptor-dependent fusion of the viral and cellular membranes within the endocytic compartment (VS-mediated infection) (3, 7, 29, 34). In support of this model, T cell VS was found to transfer into target cells immature HIV-1 particles which undergo viral membrane fusion only after proteolytic maturation of the viral core (7).
In cell-free virus particles, the gp41 cytoplasmic tail (CT) controls Env fusogenicity through inside-out allosteric mechanisms (16, 25, 45). These studies show that during virus particle production, the interaction of gp41 CT with Pr55Gag maintains Env in a prefusogenic conformation. After virus budding, cleavage of Pr55Gag and subsequent particle maturation relieve the inhibitory function of gp41 to activate Env fusogenicity. Thus, the gp41 CT plays an important role in regulating the fusogenic potential of Env during the virus life cycle. On cell-free HIV-1, the gp41 CT is important in regulating the exposure of both neutralizing and nonneutralizing epitopes on the Env ectodomain of mature virus particles (19). The fusogenicity of Env and the exposure of CD4-induced (CD4i) epitopes are enhanced in gp41 CT truncation mutants when tested with pseudovirion infection assays and cell-cell fusion assays (9, 46). During VS-mediated infection, the cell-surface Env functions first as a cell adhesion molecule and then as the viral membrane fusion apparatus (15). In this pathway, Env does not mediate membrane fusion until after the virus particle has undergone maturation (7). While the gp41 CT is not required for VS formation or subsequent infection (5, 10, 23), it does enhance the efficiency of cell-to-cell infection in nonpermissive cell types (10).
A number of broadly neutralizing monoclonal antibodies (MAbs) and peptide inhibitors have been tested for their ability to block cell-to-cell HIV-1 transfer or VS-mediated infection (5, 11, 21, 22, 33). To date, only antibodies that block Env-CD4 interaction have been shown to inhibit both cell-to-cell transfer and subsequent VS-mediated infection. Other neutralizing MAbs and entry inhibitors have been found to block infection from cell-associated HIV-1 after the transfer of virus across the VS. Using an indirect assay to measure increased HIV-1 DNA following coculture of donor and target cells, one study reported that VS-mediated infection could be inhibited by all neutralizing antibodies tested (21). Other studies have found that sera from HIV-1-positive patients are much less effective at blocking cell-to-cell transfer (5, 7) and VS-mediated infection (15) than cell-free HIV-1 infection. These studies on patient sera suggest that quantitative differences in neutralization sensitivity are likely to be found with some neutralizing MAbs.
Here we employ flow cytometry-based infection assays to screen a panel of broadly neutralizing MAbs and entry inhibitors for their ability to neutralize cell-free and VS-mediated infection. Flow cytometry-based assays allow us to directly measure HIV-1 infection specifically in target cells. In contrast to previous studies, we find that most anti-Env MAbs required significantly more antibody to block VS-mediated infection than cell-free virus. A pronounced effect was observed with the MAb 17b, which recognizes a CD4i epitope on gp120 (42). MAb 17b exhibited the greatest fold difference in 50% inhibitory concentration (IC50), with a 72-fold higher concentration required to block 50% of VS-mediated infection than cell-free infection. We further found that the relative resistance of VS-mediated infection to neutralization by both MAbs and neutralizing sera was partially overcome by truncation of the gp41 CT, while neutralization of cell-free virus was less affected by loss of the gp41 CT. These results support the hypothesis that the gp41 CT regulates conformations of HIV-1 Env at the cell surface to promote immune evasion during VS-mediated T cell infection.
MATERIALS AND METHODS
Viral constructs.
HIV-1 Gag-iGFP is a molecular clone based on pNL4-3 (2) with green fluorescent protein (GFP) inserted between the Gag MA and CA domains (14). Gag-iGFP ΔCT contains a premature stop codon in the Env reading frame causing a deletion of the C-terminal 144 amino acids (5). NL-GI contains GFP in place of Nef, and Nef is expressed from a downstream internal ribosome entry site (IRES) (6). NL-GI ΔCT was cloned by generating a PCR fragment of the C-terminal Env from the Gag-iGFP ΔCT plasmid. A PCR-based cloning strategy was used to precisely introduce the primary Env sequence into the NL-GI molecular clone. All PCR-amplified sequences were confirmed by sequence analysis. Primary CD4+ T cells were infected with NL-GI containing the NL4-3 envelope (X4-tropic) or the R5-tropic B-clade primary envelope (20) from pRHPA4259 clone 7 (SVPB14), obtained through the AIDS Research and Reference Reagent Program (ARRP), Division of AIDS, NIAID, NIH, from B. H. Hahn and J. F. Salazar-Gonzalez.
Cells and cell culture.
Human cell lines Jurkat E6-1 and MT4 (Arthur Weiss and Douglass Richman, respectively, ARRP) were maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 g/ml streptomycin, and 2 mM glutamine (complete RPMI medium). Primary CD4+ T cells were obtained from human peripheral blood through the New York Blood Center and isolated by negative selection with a Miltenyi CD4+ T cell isolation kit II (Miltenyi Biotec). Unactivated CD4+ T cells were maintained in complete RPMI medium containing 10 U/ml interleukin 2 (IL-2; ARRP). Cells were activated on day 0 by coculture with irradiated allogeneic peripheral blood mononuclear cells (PBMCs) in complete RPMI medium containing 50 U/ml IL-2 and 2 μg/ml phytohemagglutinin and used at 3 to 4 days postactivation.
Inhibitors and HIV-1-neutralizing patient sera.
The following inhibitors were tested for the ability to block cell-free and VS-mediated infection using 5-fold serial dilutions beginning at the given concentration, unless otherwise stated: 0.5 μg/ml Leu3a, an anti-CD4 antibody (BD Biosciences); 10 μg/ml IgG1 b12, an anti-gp120 MAb (b12; Dennis Burton and Carlos Barbas, ARRP); 30 μg/ml 17b and 120 μg/ml 17b, anti-gp120 MAbs against the CD4-induced binding site on gp120 (James E. Robinson, ARRP, and Tulane University stocks, respectively); 50 μg/ml 48d, E51, 21c, and 412d, anti-gp120 CD4-induced binding site MAbs (James E. Robinson, ARRP, and Tulane University); 50 μg/ml 4E10 and 2F5, anti-gp41 MAbs (Hermann Katinger, ARRP, and Polymun Scientific GmbH); 50 μg/ml 2G12, an anti-gp120 glycan-specific MAb (Hermann Katinger, ARRP); 50 μg/ml A32, an anti-gp120 MAb (James E. Robinson, ARRP); 50 μg/ml F425 A1g8, an anti-gp120 CD4-induced binding site MAb (Marshall Posner and Lisa Cavacini, ARRP); 50 μg/ml T20, a fusion inhibitor (Roche through ARRP); and 10 μg/ml AMD-3100 (bicyclam JM-2987), a CXCR-4 inhibitor (ARRP). HIV-1-neutralizing serum samples 1 and 2 and HIV-1 negative-control human serum (Luba Vujcic, ARRP) were used at a starting dilution of 1:50.
Cell-free infection assay.
Cell-free virus particles were produced in 293T cells (ATCC) by calcium phosphate transfection (27). Viral supernatants were quantified by p24 enzyme-linked immunosorbent assay. MT4 cells (0.125 × 106) were infected with 15 ng of wild-type (WT) NL-GI or 75 ng (batch 1) or 255 ng (batch 2) of NL-GI ΔCT per 1 × 106 cells to obtain up to 10% infection after 48 h in the absence of inhibitors. Virus supernatant and MT4 cells were preincubated separately with equal volumes of inhibitors for 30 min at 37°C before mixing. After 18 h, culture medium was replaced with complete RPMI medium containing 10 μM zidovudine (AZT; ARRP). At 48 h after mixing, cells were treated with trypsin-EDTA to dissociate cell-cell clusters, washed with phosphate-buffered saline (PBS), and fixed in 2% paraformaldehyde. Activated primary CD4+ T cells (0.15 × 106) were infected with 27 ng of CXCR4-tropic virus and 20 ng of CCR5-tropic virus per 1 × 106 cells. Inhibitors were added at the time of infection. Cells were trypsin treated and fixed after 36 to 48 h.
VS-mediated infection assay.
Jurkat (donor) cells were transfected by nucleofection (Amaxa Biosystems) with 5 μg NL-GI or NL-GI ΔCT DNA as previously described (5), cultured overnight in antibiotic-free medium, and purified by Ficoll-Hypaque density gradient centrifugation. Jurkat cells and MT4 (target) cells were dye labeled with 1 μM CellTrace Far Red DDAO-SE (Molecular Probes) for 10 min at 37°C or 20 μM CellTracker Blue CMF2HC (Molecular Probes) for 45 min at 37°C, respectively. Fluorescent labeling of both donor and target cells allowed unambiguous exclusion of donor-target doublets from the infection analyses. Donor or target cells (0.125 × 106) were preincubated separately with inhibitors for 30 min at 37°C before mixing at a ratio of approximately 1:1, cocultured at 37°C for 30 h, treated with trypsin, and fixed. In primary cell infection assays, primary activated donor CD4+ T cells were infected by spinoculation with 8.15 ng of X4-tropic or 6 ng of R5-tropic cell-free virus per 2.5 × 105 to 5 × 105 cells. After 24 h, cells were labeled with 2 μM eFluor 670 dye (eBioscience, Inc.) and mixed at a 1:1 ratio with activated uninfected target cells from autologous donors. Inhibitors were added at the time of mixing. Cells were trypsin treated and fixed after 36 to 48 h of coculture.
Cell-to-cell transfer assay.
Jurkat cells (donor cells) were transfected by nucleofection with 5 μg HIV-1 Gag-iGFP or Gag-iGFP ΔCT DNA (Amaxa Biosystems) as previously described (5) and cultured overnight in antibiotic-free medium. Unactivated primary CD4+ T cells (target cells) were resuspended in serum-free RPMI 1640 and stained with 1 μM CellTracker Orange CMTMR (Molecular Probes) for 45 min at 37°C, washed, and cultured overnight in complete RPMI medium containing 10 U/ml IL-2. Jurkat and CD4+ T cells were purified by Ficoll-Hypaque density gradient centrifugation. Donor and target cells were mixed at a ratio of approximately 1:1 and cocultured at 37°C for 3 h before they were treated with trypsin and fixed, as described above. Where inhibitors were used, donor and target cells were preincubated separately with equal volumes of inhibitors for 30 min at 37°C before mixing.
Calculations and statistics.
Unless otherwise stated, the percent inhibition was calculated for each well of a given experiment by finding the average of triplicate wells with no inhibitor (i.e., 0% inhibition) and normalizing all other values of the same experiment by the following equation: 100 − [(percent infected target cells with inhibitor/average percent infected target cells without inhibitor) × 100]. Statistical analysis and fitting were performed using Prism, version 5.0, software (GraphPad Software). Titration curves are of pooled data, where error bars represent the standard errors of the mean (SEMs) of at least two independent experiments. IC50 values were determined by nonlinear regression curve fits of pooled data from at least two independent titration experiments, and statistical significance between log10 IC50s was determined by unpaired two-tailed Student t test. Statistical significance between individual nonlinear regression curve fits for VS-mediated infection experiments with different frequencies of infected MT4 target cells (see Fig. S1 and S3C and D in the supplemental material) was determined by extra-sum-of-squares F test. P values of <0.05 represent statistically significant differences between individual nonlinear regression curve fits for each data set. P values of >0.05 indicate that one nonlinear regression curve adequately fits all data sets.
RESULTS
Relative resistance of VS-mediated infection to neutralization by patient sera and MAbs.
We performed antibody neutralization experiments for cell-free and VS-mediated infections by infecting a fluorescent dye-labeled T cell line with a GFP-expressing molecular clone of HIV-1, NL-GI (5). GFP detection serves as an indicator of de novo early HIV-1 gene expression in productively infected cells. We used flow cytometry to detect infection only in dye-labeled target cells to obviate the need to compensate for the large HIV-1 infection signal derived from input donor cells. Using this assay, we first examined two neutralizing patient serum samples that can completely neutralize cell-free NL-GI when used to infect the CD4+ T cell line MT4. When the same sera were titrated against homologous cell-associated HIV-1 challenge, inhibition was not complete at the concentrations capable of neutralizing cell-free infection (Fig. 1A). This titration of antisera is consistent with the levels of inhibition that we have previously reported (15).
Fig 1.

Relative neutralization resistance to neutralizing patient sera and MAbs during WT NL-GI VS-mediated infection. (A) Percent inhibition of the GFP-expressing HIV-1 molecular clone WT NL-GI during cell-free infection (solid lines, solid symbols) of the T cell line MT4 and VS-mediated infection (dashed lines, solid symbols) of MT4 cells (target cells) by Jurkat cells expressing WT NL-GI (donor cells) in the presence of 5-fold titrations of HIV-1-neutralizing patient sera and HIV-1-negative control human serum. (B) Representative plots of cell-free infection of MT4 cells by WT NL-GI with no inhibitor, the anti-CD4-blocking antibody Leu3a, or the CD4i antibody 17b. (C) VS-mediated infection of MT4 cells (target cells) by Jurkat cells expressing WT NL-GI (donor cells; not shown) with no inhibitor, Leu3a, or 17b. (D) Percent inhibition of WT NL-GI during cell-free infection (solid lines, solid symbols) and VS-mediated infection (dashed lines, solid symbols) by 5-fold titrations of MAb Leu3a, 17b, b12, or 4E10. Error bars represent the SEMs for at least two independent experiments using independently infected target cells or independently transfected donor cells on different days for cell-free or VS-mediated infections, respectively. P values represent statistically significant differences in log10 IC50s derived from nonlinear regression curve fits of pooled data. (E) Fold increase in IC50 of WT VS-mediated infection over WT cell-free infection for MAbs (light gray) and entry inhibitors (dark gray). An IC50 ratio of 1 (black line) indicates equal IC50s for a and b. P values represent statistically significant differences in log10 IC50s derived from nonlinear regression curve fits of pooled data: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To test whether certain epitopes may be more selective in their ability to distinguish cell-free versus cell-associated HIV-1, we titrated a panel of MAbs to examine their ability to block infection of the T cell line MT4 by cell-free or cell-associated WT NL-GI. We focused our initial analysis on four MAbs that target distinct regions of CD4 or Env: (i) Leu3a, an anti-CD4 HIV-1-blocking antibody, (ii) b12, an anti-gp120 antibody that binds to the CD4 binding site (CD4bs), blocking CD4-Env interaction and VS formation (31), (iii) 17b, an antibody which binds to the CD4i bridging sheet epitope in gp120 (42, 44), and (iv) 4E10, which binds the membrane proximal external region (MPER) of gp41 (40).
Cell-free WT infection (Fig. 1B) and VS-mediated WT infection (Fig. 1C) were both strongly inhibited in the presence of maximum concentrations of Leu3a (0.5 μg/ml). In contrast, at 30 μg/ml, 17b inhibited cell-free infection with a similar efficiency as Leu3a; however, it was a poor inhibitor of VS-mediated infection at the same concentration. Titrations of Leu3a, 17b, b12, and 4E10 were performed starting at maximum MAb concentrations of 0.5 μg/ml, 30 μg/ml, 10 μg/ml, and 50 μg/ml, respectively, which blocked >95% of cell-free HIV-1 infection in our assays. The variation in antibody concentration required for high levels of inhibition reflects the differential neutralization potency and binding affinity of these MAbs. We observed inhibition of both cell-free and VS-mediated infection of MT4 cells (Fig. 1D). The inhibition profile of the CD4 binding antibody Leu3a was the same for cell-free and cell-associated HIV-1. In contrast, VS-mediated infection required higher concentrations of b12, 17b, and 4E10 than cell-free infection to achieve comparable inhibition (Fig. 1D; see Table S1 in the supplemental material). Notably, at 30 μg/ml, 17b inhibited 97% of cell-free infection, whereas it inhibited 30% of VS-mediated infection. The differences in IC50s for cell-free and cell-associated viral inocula were statistically significant (P < 0.01 for b12 and 4E10; P < 0.001 for 17b). In independent repetitions of the VS-mediated neutralization experiments, variations in the level of WT NL-GI infection in the target cell population did not change the IC50 for VS-mediated infection (see Fig. S1 in the supplemental material).
Because of the relatively large defect of 17b in blocking VS-mediated infection, we tested a number of other CD4i antibodies for their ability to block cell-associated HIV-1. The CD4i MAb 48d showed 30% inhibition of cell-free infection at 50 μg/ml (see Fig. S2A in the supplemental material). Three additional CD4i MAbs, F425 A1g8 (4), E51 (49), and 21c (47), effectively inhibited cell-free infection (>85% inhibition at 50 μg/ml) and were therefore tested for the ability to also block VS-mediated infection. All three CD4i MAbs showed a statistically significant increase in IC50 during VS-mediated WT infection, as did the anti-gp41 MPER antibody 2F5 (26) (see Fig. S2A and Table S1 in the supplemental material). A small increase in the IC50 of the anti-gp120 carbohydrate antibody 2G12 (35) against VS-mediated infection was also observed, although this did not achieve statistical significance. This shift was less pronounced for the entry inhibitors AMD-3100 (36), a CXCR-4 antagonist, and T20 (43), a peptide fusion inhibitor, and significant only for T20 (P < 0.05). The nonneutralizing anti-gp120 MAb A32 (44) and the CD4i MAb 412d (48) did not block infection by cell-free or cell-associated HIV-1.
A side-by-side comparison of the MAbs shows the largest change in IC50 for 17b, with a 72.8-fold higher IC50 for VS-mediated infection than cell-free infection (Fig. 1E). Two MPER antibodies, 4E10 and 2F5, were intermediate in resistance, exhibiting 19.1- to 20.7-fold higher IC50s for VS-mediated infection than cell-free infection. The other CD4i antibodies, F425 A1g8, E51, and 21c, exhibited moderate resistance, indicated by the 10.4- to 13.3-fold higher IC50s for VS-mediated infection than cell-free infection. The MAb b12 showed a 7.9-fold change in IC50. Titrations with the carbohydrate-recognizing 2G12 antibody did not show a statistically significant resistance to neutralization. The small-molecule entry inhibitors showed only ∼2- to 3-fold increases. To various degrees, neutralization of VS-mediated WT NL-GI infection required a higher IC50 than neutralization of cell-free infection. The range of resistance suggests that variations in exposure of different epitopes may determine the degree of resistance to neutralization.
The neutralization of primary isolates of HIV-1 and infection of primary cells is often more difficult to achieve than neutralization of HIV-1 in cell lines with lab isolates of HIV-1 (37). Therefore, we tested the neutralization resistance of VS-mediated infection to 17b, which previously showed the highest fold increase in VS-mediated versus cell-free IC50, using primary activated CD4+ T cells as both donor and target cells. We compared NL-GI viruses containing the X4-tropic NL4-3 envelope (Fig. 2A and C) or the R5-tropic B-clade envelope (pRHPA4259 clone 7) derived from an acute-phase early clinical isolate (Fig. 2B and D) (20). The R5-tropic RHPA Env gene was cloned in cis into the GFP-expressing virus, rather than producing pseudovirions by cotransfection of the RHPA expression plasmid. MAb 17b effectively blocked the infection of primary cells with both cell-free X4-tropic or R5-tropic virus by 70 to 80%. In contrast, the same high antibody concentration inhibited ∼30% or less when directed against cell-associated HIV-1. In comparison, high concentrations (10 μg/ml and 25 μg/ml) of the CD4bs antibody b12 inhibited both modes of infection and both viral clones with similar efficiency (≥80% inhibition). Maximum inhibition of both modes of infection by b12 was achieved at 10 μg/ml. Because lower levels of b12 showed differences in neutralization efficiency of cell-free versus cell-associated HIV-1, a 5-fold titration series of b12 was performed in primary cells. Similar to the infection of the MT4 cells, cell-free infection in primary activated CD4+ T cells is more susceptible to b12 neutralization than VS-mediated infection (see Fig. S2B in the supplemental material). These results confirm our findings in the T cell line MT4 (Fig. 1D) that the relative resistance of VS-mediated infection to neutralization can also be observed when primary activated CD4+ T cells are infected with a primary R5-tropic virus isolate.
Fig 2.

Neutralization resistance of primary activated CD4+ T cells with CXCR4- and CCR5-tropic HIV-1 during VS-mediated infection. Representative flow cytometry plots of cell-free or VS-mediated infection of primary CD4+ T cells by HIV-1 containing the CXCR4-tropic NL4-3 envelope (A) or the envelope from a CCR5-tropic acute-phase early isolate (pRHPA4259 clone 7) (B) with no inhibitor, MAb b12, or MAb 17b. (C and D) Percent inhibition of cell-free or VS-mediated infection by HIV-1 containing the CXCR4-tropic NL4-3 envelope (C) or the envelope from a CCR5-tropic acute-phase early isolate (pRHPA4259 clone 7) (D). Error bars represent the SEMs for two independent experiments and at least four CD4+ T cell donors.
The gp41 CT regulates sensitivity to neutralizing MAbs during VS-mediated infection.
Because the cytoplasmic tail of gp41 is known to regulate the fusogenic capacity of Env (16, 25, 45, 46), we hypothesized that the exposure of certain epitopes may be regulated on the surface of cells through allosteric interactions with the gp41 CT, which are in turn influenced by the processing status of Pr55Gag (16, 19, 45). To directly test the role of the gp41 CT in modulating neutralization sensitivity, we performed antibody neutralization assays with an NL-GI viral mutant carrying a truncation of the gp41 CT that deletes the C-terminal 144 amino acids (ΔCT). When the viral clone carrying ΔCT Env was transfected into Jurkat cells, Western blotting showed the expected shift in molecular weight of ΔCT Env (Fig. 3A). Since the gp41 CT has been described as playing a role in endocytosis of Env as well as in regulating epitope exposure, we also performed cell-surface staining of the mutant using the neutralizing antibodies b12, 17b, and 4E10 and polyclonal neutralizing sera from two HIV-1-positive patients (Fig. 3B). The mutant showed higher levels of Env surface staining on HIV-1-positive cells expressing ΔCT NL-GI than cells expressing WT NL-GI. The WT Env-expressing cells stained variably and much weaker than ΔCT Env-expressing cells for the antibodies shown in Fig. 3B, as well as a number of other neutralizing antibodies examined in the study described in this paper (data not shown). The staining of WT Env by polyclonal patient antisera but not many monoclonal antibodies is suggestive of changes in Env epitope exposure on the surface of ΔCT-infected cells. From the antibody staining results, we conclude that the surface binding of most antibodies likely increases as a function of increased expression and/or enhanced epitope exposure on the ΔCT mutant.
Fig 3.
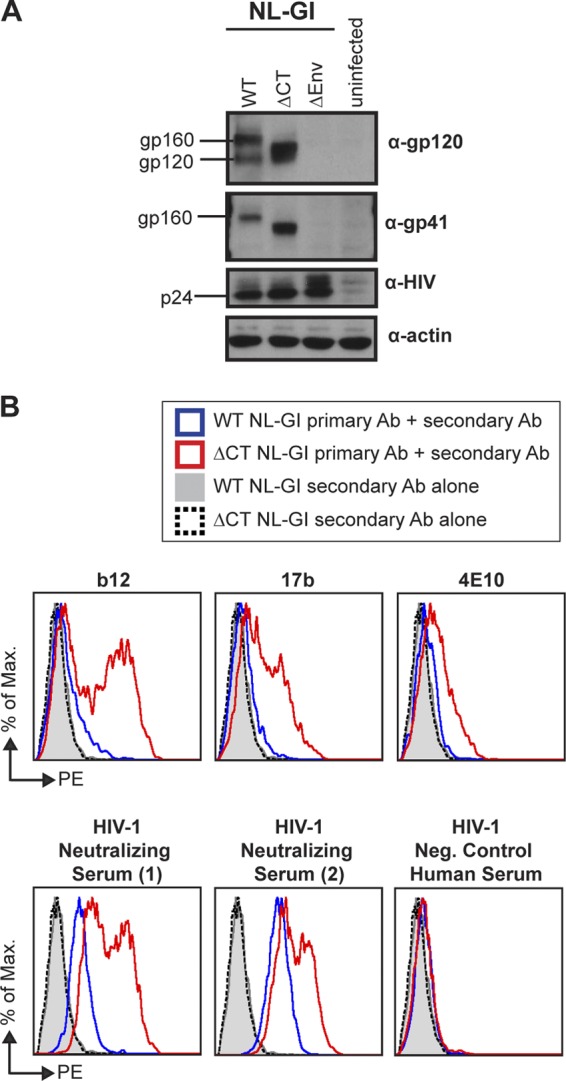
WT NL-GI and ΔCT NL-GI expression in donor Jurkat cells. HIV-1 protein expression in Jurkat cells nucleofected with WT NL-GI or ΔCT NL-GI and used as donor cells in VS-mediated infection experiments is shown. (A) Western blot of HIV-1-nucleofected Jurkat cell lysate using antibodies to detect gp120, gp41, p24, and actin. (B) Cell-surface immunostaining for HIV-1 envelope. Cells were stained with 15 μg/ml b12, 17b, or 4E10 or a 1:50 dilution of HIV-1-neutralizing or negative-control serum, followed by phycoerythrin (PE)-conjugated donkey anti-human IgG as the secondary antibody. Histogram plots show antibody binding to HIV-1-positive cells for WT NL-GI (blue line) compared to ΔCT NL-GI (red line), as well as WT NL-GI- and ΔCT NL-GI-expressing cells stained with secondary antibody alone (shaded gray histogram and black dotted line, respectively).
We next compared the inhibition of cell-free ΔCT infection to the levels of inhibition obtained for cell-free WT infection in Fig. 1D and in Fig. S2A in the supplemental material. Because the infectivity of ΔCT viral particles was lower than that of WT, we tested multiple viral inputs for ΔCT and found that there was no change in the observed IC50 when the viral input of the ΔCT mutant was increased to infect a similar percentage of target cells as the WT virus (see Fig. S3A and B in the supplemental material). The higher ΔCT viral input that resulted in levels of infection similar to those for WT was therefore used in cell-free titration experiments. When comparing WT with ΔCT cell-free infections, we observed no significant differences in inhibition by Leu3a, b12, and 17b (Fig. 4A; see Table S1 in the supplemental material), as well as 2G12, E51, 21c, AMD-3100, and T20 (see Fig. S4A in the supplemental material). These MAb and inhibitors all displayed similar IC50s for WT and ΔCT cell-free infection, indicated by a fold change of ∼1 (Fig. 4C; see Table S1 in the supplemental material). For cell-free viral infections, 4E10 showed the largest fold change of 4.4, followed by F425 A1g8 and 2F5. These three MAbs required a higher IC50 (P < 0.05) to block infection by WT viral particles than that by ΔCT (Fig. 4A and Fig. S4A in the supplemental material). MAb 48d did not inhibit ΔCT cell-free infection at 10 μg/ml (see Fig. S4A in the supplemental material). Overall, we conclude that the gp41 CT truncation has only modest effects on the neutralization of cell-free HIV-1, which were dependent upon the epitope tested.
Fig 4.

Truncation of the gp41 CT enhances neutralization sensitivity to MAbs during VS-mediated infection. Percent inhibition of WT NL-GI (solid lines, solid symbols) and the gp41 cytoplasmic tail truncation mutant ΔCT (dashed lines, open symbols) by 5-fold titrations of MAbs Leu3a, b12, 4E10, and 17b during cell-free infection (A) and VS-mediated infection (B). Error bars represent the SEMs for at least two independent experiments using independently infected target cells or independently transfected donor cells on different days for cell-free or VS-mediated infections, respectively. Percent inhibition for WT infection is from Fig. 1D. P values represent statistically significant differences in log10 IC50s derived from nonlinear regression curve fits of pooled data. (C and D) Fold increase in IC50 of WT cell-free infection over ΔCT cell-free infection (C) or WT VS-mediated infection over ΔCT VS-mediated infection (D) for MAbs (light gray) and entry inhibitors (dark gray). An IC50 ratio of 1 (black line) indicates equal IC50s for a and b. P values represent statistically significant differences in log10 IC50s derived from nonlinear regression curve fits of pooled data: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
VS-mediated infection by WT-expressing donor cells (from Fig. 1D) and ΔCT-expressing donor cells was inhibited equally well by the anti-CD4 antibody Leu3a (Fig. 4B; see Table S1 in the supplemental material). In contrast, statistically significant differences in IC50s were observed during WT and ΔCT VS-mediated infection for b12 (P < 0.01), 4E10 (P < 0.05), and 17b (P < 0.01) (Fig. 4B). The other antibodies also showed significantly greater IC50s against WT VS-mediated neutralization (data from Fig. S2A in the supplemental material) than ΔCT VS-mediated neutralization, with the exception of F425 A1g8, which displayed a sizable shift that did not reach statistical significance in this analysis (see Fig. S4B and Table S1 in the supplemental material). In contrast, the entry inhibitors T20 and AMD-3100 did not show a statistically significant change in IC50. As with cell-free infection, we examined infection conditions that gave rise to different levels of infected target cells expressing ΔCT NL-GI and found that the infection level did not statistically change the IC50 for VS-mediated infection (see Fig. S3C and D in the supplemental material). Overall, during VS-mediated infection, the IC50s of ΔCT infection are reduced compared to those of WT infection (see Table S1 in the supplemental material), even though more Env epitopes are present at the cell surface (Fig. 3). MAb b12 exhibited the largest shift in IC50, consisting of a 29.7-fold increase for WT over ΔCT (Fig. 4D). MAb 17b showed an intermediate 7.7-fold increase in IC50, while the remaining MAbs and inhibitors showed smaller but statistically significant increases of up to ∼6-fold. When comparing the relative effect of ΔCT on cell-free versus cell-associated inocula, a greater overall effect was observed for most antibodies when testing cell-associated HIV-1 infection (Fig. 4C and D). Consistent with this, the fold change in IC50 of ΔCT VS-mediated infection over ΔCT cell-free infection (see Fig. S5 in the supplemental material) for anti-Env MAb was smaller than that in a similar comparison for WT NL-GI (Fig. 1E). Given the enhanced staining of ΔCT Env at the cell surface, the enhanced neutralization could be the result of either changes in epitope exposure or increased protein expression at the surface. Our result is supportive of changes in epitope exposure, as increased cell-surface Env would likely enhance MAb avidity at the synapse and make VS-mediated infection more difficult to inhibit. We interpret our data to suggest that truncation of gp41 CT generates greater exposure of VS-neutralizing epitopes at the cell surface, and this renders VS-mediated infection by ΔCT more sensitive to neutralizing antibodies.
Enhanced sensitivity of ΔCT to 17b occurs after cell-to-cell transfer.
Because VS-mediated infection of WT NL-GI strongly resisted neutralization by 17b compared to ΔCT NL-GI, we wished to further examine whether this effect was mediated by enhanced inhibition of the first step in the two-step model of VS-mediated HIV-1 entry, namely, cell-to-cell transfer. We therefore measured WT and ΔCT Gag-iGFP virus transfer into primary unactivated CD4+ T cells after 3 h of coculture. The Gag-iGFP construct allows fluorescent detection of viral transfer across virological synapses (14). Using this assay, Leu3a and b12, two MAbs that interfere with the CD4-Env interaction, both efficiently inhibited cell-to-cell transfer of both WT and ΔCT Gag-iGFP into target cells, while inhibition by 17b was minimal at 30 μg/ml (Fig. 5A and B). Titration curves show that Leu3a similarly inhibited both WT and ΔCT Gag-iGFP (Fig. 5C), confirming that the ΔCT mutation has no effect on the ability of Leu3a to block the gp120 binding site on CD4. The anti-gp120 MAb b12 also inhibited transfer of WT and ΔCT Gag iGFP at the highest concentration tested, 10 μg/ml (Fig. 5A and B); however, the ΔCT Gag-iGFP was more sensitive than WT Gag-iGFP to b12 neutralization (Fig. 5C). The enhanced sensitivity of ΔCT to b12 during VS-mediated infection (Fig. 4B) can be partially attributed to an enhanced ability to block the initial step of cell-to-cell transfer of HIV-1. In contrast, virus transfer levels were unaffected by 17b, despite increasing antibody concentrations (Fig. 5A and C), and the ΔCT Gag-iGFP mutation had no effect on neutralization by 17b (Fig. 5B and C). We conclude that the increased sensitivity of cell-associated ΔCT virus to 17b occurs after cell-to-cell transfer of HIV-1. Given our current model for endosomal uptake after VS formation, this inhibition is likely to occur within endosomal compartments in the target cell (7).
Fig 5.

Effect of the gp41 CT on cell-to-cell transfer of WT and ΔCT HIV-1. Representative flow cytomrtry plots of cell-to-cell transfer of the fluorescent HIV-1 WT Gag-iGFP (A) and ΔCT Gag-iGFP (B) from infected Jurkat cells (donor cells; not shown) to primary CD4+ T cells (target cells) with no inhibitor, Leu3a, b12, and 17b. (C) Percent inhibition of cell-to-cell transfer of WT Gag-iGFP (solid lines, solid symbols) and ΔCT Gag-iGFP (dashed lines, open symbols) by 5-fold titrations of MAbs Leu3a, b12, and 17b. Error bars represent the SEMs for at least two independent experiments using independently transfected donor cells on different days.
Neutralization properties of HIV-1-neutralizing patient sera.
We next asked whether the ΔCT mutation could enhance the ability of neutralizing antibodies generated during natural HIV-1 infection to block cell-to-cell transfer and VS-mediated infection. Cell-free and VS-mediated infections using WT NL-GI in the presence of two well-characterized reference polyclonal serum samples found that cell-free infection with WT NL-GI could be inhibited up to 100% with increasing serum concentrations (data from Fig. 1A also shown in Fig. 6A). In contrast, both cell-to-cell transfer (Fig. 6B) and VS-mediated infection of WT (data from Fig. 1A also shown in Fig. 6C) were partially inhibited up to 50% at the maximum concentration tested, a 1:50 dilution. Truncation of gp41 CT had little to no effect on the neutralization of cell-free infection (Fig. 6A); however, cell-to-cell transfer of ΔCT was inhibited by an additional 25 to 30% compared to that for its WT counterpart (Fig. 6B). During VS-mediated infection, the patient sera blocked ΔCT by ∼30 to 40% more than WT at the maximum concentration (Fig. 6C). These results show that the relative resistance of VS-mediated infection to neutralizing patient sera is also partly controlled by the gp41 CT.
Fig 6.

Sensitivity of WT and ΔCT HIV-1 to neutralizing patient sera. Percent inhibition of WT HIV-1 (solid lines, solid symbols) and ΔCT HIV-1 (dashed lines, open symbols) during cell-free infection (A), cell-to-cell transfer (B), and VS-mediated infection (C) by 5-fold titrations of HIV-1-neutralizing patient sera and HIV-1-negative control human serum. Error bars represent the SEMs for at least two independent experiments using independently infected target cells for cell-free infection or independently transfected donor cells on different days for cell-to-cell transfer or VS-mediated infections. Percent inhibition for WT infection in panels A and C is from Fig. 1A.
DISCUSSION
Many studies have implicated cell-associated HIV-1 infection as an efficient route of viral spread (5, 8, 38). The results presented here further support the finding that this mode of infection plays a critical role in immune evasion. In vitro neutralization assays have generally found that VS-mediated infection is sensitive to neutralizing antibodies at high antibody concentrations (5, 21, 22, 33). However, studies with patient sera have indicated that VS-mediated HIV-1 infection can be considerably more difficult to neutralize (5, 15). Using quantitative, flow cytometry based assays, we measured the extent of VS-mediated neutralization by a panel of MAbs and found that many MAbs are less efficient at neutralizing cell-associated HIV-1, as indicated by statistically significant differences in IC50s. The CD4i MAb 17b and sera from HIV-1-positive patients were much more potent against cell-free HIV-1 than VS-mediated HIV-1 infection. The differences in the relative efficiencies of various MAbs at blocking cell-associated HIV-1 suggest to us that differential epitope exposure plays an important role in neutralization resistance. In addition, we examined a mutant with a truncation mutation in the gp41 CT and found that cell-associated HIV-1 was particularly sensitive to loss of the gp41 CT relative to cell-free HIV-1.
Our recent studies have revealed that particle maturation occurs after viral transfer across VS and that maturation is required to activate viral membrane fusion (7). Given the enhanced sensitivity of cells infected with a ΔCT virus to neutralization by some MAb, the deletion of the gp41 CT may abrogate the regulation of epitope exposure that occurs during VS-mediated infection. Our current model suggests that the VS entry pathway is distinct from cell-free infection, in that Env is bound to CD4 prior to particle assembly, well before it has been activated for fusion by the particle maturation process (15). In this model, the Env-CD4 interaction persists during viral budding, transfer into the endocytic compartment, and viral maturation within the endosome. Since viral maturation triggers viral membrane fusion (7), it follows that for antibodies to block this pathway, they must interact with neutralizing epitopes that are well exposed either at the cell surface or during the maturation process.
Conformationally sensitive neutralizing MAbs or components of polyclonal patient sera may fail to recognize their determinants during VS-mediated infection because (i) the epitopes are very transiently exposed within the endosomal compartment between Env maturation and membrane fusion, (ii) the preformed CD4-Env complex may sterically obstruct access to the epitope, or (iii) epitopes may be structurally hidden when maturation occurs after CD4-Env engagement. Truncation of gp41 CT may allow cell-surface Env to adopt an alternate conformation that may engage neutralizing antibodies more than the immature Env conformation found on the surface of WT infected cells.
Our studies on the inhibition WT HIV-1 infection by neutralizing antibodies, inhibitors (Fig. 1; see Fig. S2 in the supplemental material), and neutralizing patient sera (Fig. 6) are in agreement with previous studies that conclude that antibodies and inhibitors capable of blocking WT cell-free HIV-1 can also inhibit WT cell-associated HIV-1 infection at the highest concentration tested (5, 21, 22, 33). However, when specifically examining the IC50, we find that for most MAbs there were statistically significant increases in IC50s during VS-mediated infection compared to cell-free infection. In contrast, a previous study by Martin et al. (21) found no statistical difference between cell-free and VS-mediated IC50s for several MAbs and entry inhibitors, although increases in IC50 for several MAbs were noted by the authors for VS-mediated infection (21). This discrepancy between findings could be due to differences in assay specificity for the detection of new infection specifically in target cells. In contrast to our study design, Martin et al. used quantitative PCR to detect the relative increase in HIV-1 pol DNA when donor and target cells were mixed (21). This assay does not discriminate between integrated and unintegrated HIV-1 pol DNA in donor or target cells and may result in a larger variability because of a weak signal-to-noise ratio. Our use of a flow cytometry-based assay distinguishes donor and target cells by differential dye labeling, allowing all donor cells to be efficiently excluded from the infection analysis. We also detect de novo early HIV-1 gene expression from newly infected target cells using the NL-GI molecular clone, as opposed to a DNA intermediate that does not directly equate to de novo infection.
Prior studies also frequently used chronically infected cell lines (21, 22) that are also likely to carry mutations in HIV-1 accessory genes, such as vpu or nef. Given the known lack of vpu in the IIIB strain (30, 41), it is possible that tethered viruses that accumulate at the cell surface in the absence of vpu display Env in a form that is more similar to that found on cell-free virus. Here we use acutely transfected cell lines or acutely infected primary T cells to minimize these effects.
While this work was in review, a publication by Abela and coworkers (1) reported that antibodies and inhibitors directed toward gp120, but not CD4, coreceptor or gp41 were less effective at neutralizing cell-associated HIV-1 than cell-free virus. While employing different reporter cell-based assays and some flow cytometry-based assays, that study generally supports our findings that cell-to-cell transmission is often more difficult to inhibit than cell-free infection. However, CD4i MAbs and HIV-1-neutralizing patient sera, which showed some of the largest differences in our assay (Fig. 1), were not tested in that study. With different viral clones, they observed comparatively small effects for two antibodies against the MPER of gp41, 4E10, and 2F5. Based on this, they conclude that CD4 engagement is more difficult to block during synapse formation than postattachment entry steps that are targeted by anti-gp41 MAbs and inhibitors. Here we report significantly higher IC50s for the anti-gp41 MPER MAbs 4E10 and 2F5 for VS-mediated infection. The fold difference in IC50s is in fact larger than that for most of the other MAbs that we tested, including b12 (Fig. 1D and E; see Fig. S2 and Table S1 in the supplemental material). Additional studies are required to test whether such differences are due to differences in the Env genes examined and if these results are a general property of anti-gp41 MAbs or are unique to polyreactive MPER MAbs such as 4E10 and 2F5 (12, 24).
The anti-CD4 MAb Leu3a and the anti-CXCR4 inhibitor AMD-3100 did not show significant differences in IC50s between comparisons of WT cell-free and VS-mediated infection (Fig. 1D and E; see Fig. S2 in the supplemental material) or any other comparisons in our study. This is indicative of their functional targets, CD4 and CXCR4, which are accessible on the surface of the uninfected cell and are notably unaffected by conformational changes in Env. In contrast, T20 and all anti-Env MAbs tested except 2G12 displayed a significantly greater IC50 against WT VS-mediated infection than WT cell-free infection. Previous studies have not examined titrations of MAb 17b or neutralizing patient sera, which appear to show the strongest phenotypes. Our examination of a panel of CD4i MAbs suggests that not all CD4i MAbs are as extreme in differentially neutralizing ability as 17b. By definition, CD4i epitopes are sensitive to conformational changes in Env. The 17b epitope has been noted to be one of the most sensitive to conformational changes in gp120 (42, 44), perhaps more so than other CD4i epitopes, including those recognized by other CD4i MAbs that we tested. It is also notable that 2G12, which recognizes a glycan-based epitope and may be less sensitive to maturation-induced conformational changes in Env (19), is the only anti-Env MAb which did not display a statistically significant deficit against cell-associated HIV-1, although a 6.7-fold change was still observed (Fig. 1E). Overall, the data are consistent with our hypothesis that entry through the VS limits the exposure of conformation-sensitive epitopes of Env.
The data presented also indicate that the ΔCT mutation can enhance the sensitivity of VS-mediated infection at different stages of the viral entry process. Cell-to-cell transfer, the first step in the two-step model of VS-mediated entry, is sensitive to inhibition only by MAbs that block the Env-CD4 interaction and VS formation, such as Leu3a and b12 (Fig. 5). Consistent with the action of b12, changes in the gp120 ectodomain due to truncation of the gp41 CT enhance b12 binding and subsequent neutralization of cell-to-cell HIV-1 transfer. In contrast, the MAb 17b does not block cell-to-cell transfer (Fig. 5) but can inhibit VS-mediated infection (Fig. 4B) and therefore acts at a step after VS formation. Given our laboratory's observations that viral membrane fusion is triggered within an internal compartment after cell-to-cell transfer (7), 17b and other MAbs that do not block cell-to-cell transfer likely function within the endocytic compartment, before membrane fusion. Studies to visualize MAb localization within the target cell are ongoing.
Given the high plasma viremia sustained during natural infection and the relatively low levels of Env maintained on the surface of infected cells (32), it is likely that cell-free viral particles act as a dominant antigenic stimulus for B-cell responses. Consistent with this, we find that patient sera have a stronger ability to block cell-free but not VS-mediated infection (Fig. 1A). Many antibodies in sera are likely to be directed against cell-free virions and would therefore neutralize cell-free HIV-1 more effectively than cell-associated HIV-1. We suggest that resistance of cell-associated HIV-1 to certain neutralizing responses depends upon the ability of Env to regulate the exposure of key epitopes during VS-mediated infection.
All of the anti-Env MAbs that we tested displayed a significant shift in IC50 between WT and ΔCT VS-mediated infection (Fig. 4B; see Fig. S4B in the supplemental material), and the fold-change in IC50 was greater than that when cell-free virions were tested (compare Fig. 4C and D). Two antibodies, b12 and 17b, showed particularly large enhancement of sensitivity to neutralization during VS-mediated infection when the gp41 CT was deleted (Fig. 4D). The fold change in IC50 between WT and ΔCT VS-mediated infection ranged from 2 to 29.7, suggesting that truncation of the gp41 CT affects the IC50s of different MAbs to varying extents. In the context of our model, this is due to conformational changes in Env, which differentially affect the binding sites of all the anti-Env MAbs that we tested to varying extents. The fact that the IC50 of the peptide antagonist T20 is less affected than 2F5 and 4E10 by truncation of the gp41 CT during cell-free (Fig. 4C) and VS-mediated infection (Fig. 4D) may suggest that accessibility or steric issues related to the size of the inhibitor may also contribute to the relative differences in IC50s. When comparing WT and ΔCT VS-mediated infection, it is important to note that the levels of Env on the cell surface are considerably higher when the gp41 CT endocytosis YXXL signal is removed. Given a uniform increase in all epitopes on ΔCT Env, one might expect avidity effects to make the ΔCT VS more difficult to inhibit, increasing the IC50s of all MAbs equally. We did not see uniform differences across the different antibodies, suggesting that they are at least in part controlled by differences in epitope exposure between the WT and ΔCT states of Env.
The small changes in IC50 that we observed during cell-free infection with WT versus ΔCT viral particles suggest that functionally these particles display similar epitopes. However, the two MPER MAbs 2F5 and 4E10 displayed enhanced sensitivity of ΔCT cell-free virus neutralization over WT cell-free virus neutralization (Fig. 4C), indicating that the absence of gp41 CT can also have a significant impact on cell-free virus. A recent study by Joyner et al. (19) shows increased antibody binding of MPER MAbs 4E10 and 2F5 to immature, protease-deficient (PR−) virus particles compared to mature, wild-type virus. They conclude that the maturation process may cloak neutralizing epitopes on cell-free virus. In this study, differences in antibody binding could not be correlated with functional changes in neutralization sensitivity because immature particles are noninfectious (19). Furthermore, it remains an open question whether Env on the surface of infected cells during VS formation is conformationally similar to Env on protease-deficient, immature viral particles.
A study described in a previous report (9) employed gp41 CT truncations that retain an additional 20 amino acids on the intracytoplasmic side and observed 2- to 3-fold increases in neutralization sensitivity compared to that for WT virus to 0.5 μg/ml MAbs b12, 2G12, and 48d in a luciferase-based pseudovirion infection assay (9). The differences between these observations and our findings could be due to differences in the viral truncation mutant used or perhaps the expression of Env in trans when producing pseudovirus particles. The changes in IC50s that we observed when comparing WT and ΔCT VS-mediated infections were generally greater than those reported previously for cell-free virus. We do see some effects on the neutralization of cell-free HIV-1; however, the gp41 CT has a greater effect on epitope exposure and neutralization of VS-mediated infection. These studies and ours together suggest that the gp41 CT can modulate epitope exposure on the surface of both HIV-1-infected cells and virus particles.
VS-mediated infection has the potential to contribute significantly to in vivo HIV-1 spread and pathogenesis in lymphoid tissues that contain high numbers of susceptible CD4+ T cells. Current HIV-1 vaccine strategies are largely focused on targeting Env epitopes on cell-free HIV-1 or antigens representing native trimeric cell-free Env conformations. Our results provide evidence that unique conformations of cell-surface Env presented during VS-mediated infection should be a critical consideration in immunogen design, to prevent the selective neutralization escape of cell-associated virus at antibody concentrations capable of neutralizing cell-free infection. We also demonstrate a key role for the gp41 CT in controlling epitope exposure during infection through T cell virological synapses. If an antibody-based vaccine is to be effective at preventing HIV-1 infection, further characterization of the humoral response to cell-surface epitopes that participate in VS-mediated infection is essential.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the B. K. Chen lab and the Flow Cytometry Shared Resource Facility (Mount Sinai School of Medicine, New York, NY) for helpful comments and advice.
This work was supported by grants from the NIH, NIAID (AI074420), the Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease Award, and the Irma T. Hirschl/Monique Weill-Caulier Trust Career Scientist Award to B.K.C.
Footnotes
Published ahead of print 2 May 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Abela IA, et al. 2012. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog. 8:e1002634 doi:10.1371/journal.ppat.1002634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adachi A, et al. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blanco J, et al. 2004. High level of coreceptor-independent HIV transfer induced by contacts between primary CD4 T cells. J. Biol. Chem. 279:51305–51314 [DOI] [PubMed] [Google Scholar]
- 4. Cavacini L, et al. 2003. Conformational changes in env oligomer induced by an antibody dependent on the V3 loop base. AIDS 17:685–689 [DOI] [PubMed] [Google Scholar]
- 5. Chen P, Hubner W, Spinelli MA, Chen BK. 2007. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 81:12582–12595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen GB, et al. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671 [DOI] [PubMed] [Google Scholar]
- 7. Dale BM, et al. 2011. Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 10:551–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dimitrov DS, et al. 1993. Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol. 67:2182–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edwards TG, et al. 2002. Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J. Virol. 76:2683–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Emerson V, Haller C, Pfeiffer T, Fackler OT, Bosch V. 2010. Role of the C-terminal domain of the HIV-1 glycoprotein in cell-to-cell viral transmission between T lymphocytes. Retrovirology 7:43 doi:10.1186/1742-4690-7-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gupta P, Balachandran R, Ho M, Enrico A, Rinaldo C. 1989. Cell-to-cell transmission of human immunodeficiency virus type 1 in the presence of azidothymidine and neutralizing antibody. J. Virol. 63:2361–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haynes BF, et al. 2005. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308:1906–1908 [DOI] [PubMed] [Google Scholar]
- 13. Hoxie JA. 2010. Toward an antibody-based HIV-1 vaccine. Annu. Rev. Med. 61:135–152 [DOI] [PubMed] [Google Scholar]
- 14. Hubner W, et al. 2007. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol. 81:12596–12607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hubner W, et al. 2009. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323:1743–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang J, Aiken C. 2007. Maturation-dependent human immunodeficiency virus type 1 particle fusion requires a carboxyl-terminal region of the gp41 cytoplasmic tail. J. Virol. 81:9999–10008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199:283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jolly C, Sattentau QJ. 2004. Retroviral spread by induction of virological synapses. Traffic 5:643–650 [DOI] [PubMed] [Google Scholar]
- 19. Joyner AS, Willis, Crowe JE, Jr, Aiken C. 2011. Maturation-induced cloaking of neutralization epitopes on HIV-1 particles. PLoS Pathog. 7:e1002234 doi:10.1371/journal.ppat.1002234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li M, et al. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 79:10108–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martin N, et al. 2010. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J. Virol. 84:3516–3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Massanella M, et al. 2009. Antigp41 antibodies fail to block early events of virological synapses but inhibit HIV spread between T cells. AIDS 23:183–188 [DOI] [PubMed] [Google Scholar]
- 23. Monel B, et al. 2012. HIV cell-to-cell transmission requires the production of infectious virus particles and does not proceed through env-mediated fusion pores. J. Virol. 86:3924–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mouquet H, Warncke M, Scheid JF, Seaman MS, Nussenzweig MC. 2012. Enhanced HIV-1 neutralization by antibody heteroligation. Proc. Natl. Acad. Sci. U. S. A. 109:875–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murakami T, Ablan S, Freed EO, Tanaka Y. 2004. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 78:1026–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muster T, et al. 1993. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 67:6642–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pear WS, Nolan GP, Scott ML, Baltimore D. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U. S. A. 90:8392–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piguet V, Sattentau Q. 2004. Dangerous liaisons at the virological synapse. J. Clin. Invest. 114:605–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Puigdomenech I, Massanella M, Cabrera C, Clotet B, Blanco J. 2009. On the steps of cell-to-cell HIV transmission between CD4 T cells. Retrovirology 6:89 doi:10.1186/1742-4690-6-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ratner L, et al. 1985. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 313:277–284 [DOI] [PubMed] [Google Scholar]
- 31. Roben P, et al. 1994. Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. J. Virol. 68:4821–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rowell JF, Stanhope PE, Siliciano RF. 1995. Endocytosis of endogenously synthesized HIV-1 envelope protein. Mechanism and role in processing for association with class II MHC. J. Immunol. 155:473–488 [PubMed] [Google Scholar]
- 33. Sanchez-Palomino S, et al. 2011. A cell-to-cell HIV transfer assay identifies humoral responses with broad neutralization activity. Vaccine 29:5250–5259 [DOI] [PubMed] [Google Scholar]
- 34. Sattentau Q. 2010. Cell-to-cell spread of retroviruses. Viruses 2:1306–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scanlan CN, et al. 2002. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of alpha1→2 mannose residues on the outer face of gp120. J. Virol. 76:7306–7321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schols D, Este JA, Henson G, De Clercq E. 1997. Bicyclams, a class of potent anti-HIV agents, are targeted at the HIV coreceptor fusin/CXCR-4. Antiviral Res. 35:147–156 [DOI] [PubMed] [Google Scholar]
- 37. Seaman MS, et al. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J. Virol. 84:1439–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O. 2007. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 81:1000–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stamatatos L, Morris L, Burton DR, Mascola JR. 2009. Neutralizing antibodies generated during natural HIV-1 infection: good news for an HIV-1 vaccine? Nat. Med. 15:866–870 [DOI] [PubMed] [Google Scholar]
- 40. Stiegler G, et al. 2001. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on gp41 of human immunodeficiency virus type 1. AIDS Res. Hum. Retroviruses 17:1757–1765 [DOI] [PubMed] [Google Scholar]
- 41. Strebel K, Klimkait T, Martin MA. 1988. A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science 241:1221–1223 [DOI] [PubMed] [Google Scholar]
- 42. Thali M, et al. 1993. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 67:3978–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. U. S. A. 91:9770–9774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wyatt R, et al. 1995. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J. Virol. 69:5723–5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wyma DJ, et al. 2004. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J. Virol. 78:3429–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wyss S, et al. 2005. Regulation of human immunodeficiency virus type 1 envelope glycoprotein fusion by a membrane-interactive domain in the gp41 cytoplasmic tail. J. Virol. 79:12231–12241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xiang SH, Doka N, Choudhary RK, Sodroski J, Robinson JE. 2002. Characterization of CD4-induced epitopes on the HIV type 1 gp120 envelope glycoprotein recognized by neutralizing human monoclonal antibodies. AIDS Res. Hum. Retroviruses 18:1207–1217 [DOI] [PubMed] [Google Scholar]
- 48. Xiang SH, et al. 2005. Functional mimicry of a human immunodeficiency virus type 1 coreceptor by a neutralizing monoclonal antibody. J. Virol. 79:6068–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xiang SH, et al. 2003. Epitope mapping and characterization of a novel CD4-induced human monoclonal antibody capable of neutralizing primary HIV-1 strains. Virology 315:124–134 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.