

Abstract
Acid sphingomyelinase (ASMase) converts the lipid sphingomyelin (SM) to phosphocholine and ceramide and has optimum activity at acidic pH. Normally, ASMase is located in lysosomes and endosomes, but membrane damage or the interaction with some bacterial and viral pathogens can trigger its recruitment to the plasma membrane. Rhinovirus and measles viruses each require ASMase activity during early stages of infection. Both sphingomyelin and ceramide are important components of lipid rafts and are potent signaling molecules. Each plays roles in mediating macropinocytosis, which has been shown to be important for ebolavirus (EBOV) infection. Here, we investigated the role of ASMase and its substrate, SM, in EBOV infection. The work was performed at biosafety level 4 with wild-type virus with specificity and mechanistic analysis performed using virus pseudotypes and virus-like particles. We found that virus particles strongly associate with the SM-rich regions of the cell membrane and depletion of SM reduces EBOV infection. ASM-specific drugs and multiple small interfering RNAs strongly inhibit the infection by EBOV and EBOV glycoprotein pseudotyped viruses but not by the pseudotypes bearing the glycoprotein of vesicular stomatitis virus. Interestingly, the binding of virus-like particles to cells is strongly associated with surface-localized ASMase as well as SM-enriched sites. Our work suggests that ASMase activity and SM presence are necessary for efficient infection of cells by EBOV. The inhibition of this pathway may provide new avenues for drug treatment.
INTRODUCTION
Ebolavirus (EBOV) is a negative-sense, single-stranded filamentous virus causing disease that is nearly 90% fatal in humans. Despite its severity, no approved vaccines or drug therapies exist to prevent or treat EBOV infection (13). An effective strategy for developing such treatments is to target key steps in virus entry into cells. The current view of EBOV entry is that the virus associates with cholesterol-rich lipid rafts (5) and coreceptors, such as integrins and DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin) (1, 50). Soon thereafter, other receptor proteins bind; these may be tissue- or cell-type specific and include tyro3, an Axl family member, and TIM-1 (27, 34, 49). The virus is then internalized by a macropinocytosis-like mechanism (45–47). Once inside the cell, the virus requires the pH-dependent lysosomal cathepsins B and L to cleave the surface glycoproteins prior to its pH-dependent fusion with cell membranes. Recently, a prefusion step requiring the late endosomal/lysosomal protein Niemann-Pick Type C1 (NPC1) was identified (7, 9). Although significant insights into the EBOV entry pathway and mechanism have been uncovered, gaps in understanding still exist, some of which could be exploited for drug development.
Much of the work that has been performed to determine the role of membrane cholesterol in the virus infection mechanism has used drugs such as cyclodextrin and nystatin to respectively deplete and sequester cellular cholesterol. These treatments reduce EBOV infection (5, 12); however, it has been demonstrated that sphingomyelin (SM), a major lipid raft component, is also depleted (19). Moreover, nystatin inhibits the recruitment of the sphingomyelin-processing enzyme acid sphingomyelinase (ASMase) (EC 3.1.4.12) from the lysosome to the outer leaflet of the plasma membrane (35). Therefore, the interpretation of these earlier EBOV entry experiments is more complex than was originally thought and requires further investigation.
SM is a mammalian membrane lipid that preferentially associates with cholesterol to form lipid rafts (43). During normal membrane recycling, SM is internalized and then routed through early endosomes, multivesicular bodies, and late endosomes. Then, SM is either recycled back to the plasma membrane via exocytosis or delivered to lysosomes, where it is hydrolyzed to ceramide and phosphocholine by ASMase (31). However, membrane damage and the binding of microbial pathogens can result in the translocation of lysosomal ASMase to the outer leaflet of the plasma membrane, where it cleaves surface-exposed SM (4, 51). The conversion of the SM in rafts to ceramide can result in raft enlargement, receptor clustering, membrane invagination, and macropinosome formation (22–24, 59), all of which promote the uptake of particles, including viruses, into cells.
Measles virus and rhinoviruses as well as the intracellular pathogens Neisseria gonorrhoeae and Trypanosoma cruzi all require ASMase function during entry (2, 14, 20, 21). This suggests that these pathogens may share a mechanism of ASMase-dependent cellular entry that could be exploited as a broad-spectrum intervention. Since EBOV, N. gonorrhoeae, and measles virus all use macropinocytosis for entry (15, 58), it is important to determine if EBOV also shares a dependence on ASMase activity, as this information will be useful in characterizing the poorly understood macropinocytic uptake mechanism.
Here, we investigated the roles of the SM-enriched rafts and ASMase in EBOV infection. We found that virus particles associate with the SM-rich regions of the cell membrane and that infection is strongly dependent on ASMase activity. Interestingly, virus particles induce the movement of ASMase to the cell surface at the site at which virus particles bind to the cell membrane. This outcome suggests that EBOV can recruit ASMase and SM to the site of cell binding and that this response is important in initiating infection.
MATERIALS AND METHODS
Cell culture.
HeLa (Ambion, Austin, TX), 293HEK (ATCC, Manassas, VA), 293FT (Invitrogen, Carlsbad, CA), and Vero E6 (CDC, Atlanta, GA) cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C at 5% CO2. 293FT cells (Invitrogen, Carlsbad, CA) were subcultured in 0.5 mg/ml G418. Healthy primary human fibroblasts (GM00038) and fibroblasts lacking detectable ASMase activity (GM0112) were purchased from the Coriell Cell Repository (Camden, NJ) and cultured in DMEM as per the provided protocol.
Reagents.
Vacuolin-1 was purchased from Calbiochem (Gibbstown, NJ). Desipramine, imipramine, and bafilomycin A were purchased from Sigma (St. Louis, MO). For the immunofluorescence assays staining for SM, lysenin and rabbit antilysenin were purchased from Peptides International (Louisville, KY). Goat anti-ASMase antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Donkey anti-rabbit Alexa Fluor 594, chicken anti-goat Alexa Fluor 647, 4′,6-diamidino-2-phenylindole (DAPI), Lipofectamine 2000, Stealth RNAi SMPD1 (HSS143988, referred to in the text as SMPD1 siRNA [small interfering RNA] sequence 6), and alamarBlue were purchased from Invitrogen (Carlsbad, CA). The oligonucleotide primers targeting the Zaire EBOV Mayinga nucleoprotein (NP) genes were purchased from IDT (Coralville, IA). The primers for PCR amplification of the SMPD1 gene were purchased from OriGene (Rockville, MD). The siRNA for SMPD1 (referred to in the text as SMPD1 siRNA sequences 1, 2, 3, and 5) were purchased from Qiagen (Valencia, CA). The polyethylenimine (PEI) for the large-scale cDNA transfections was from Polysciences, Inc. (Warrington, PA). pEBOV Zaire GP (a gift from Paul Bates, University of Pennsylvania, Philadelphia, PA), pVP40-green fluorescent protein (GFP) (a gift from Christopher Basler, Mount Sinai School of Medicine, New York, NY), and pVSV-G (BD Biosciences, San Jose, CA) were prepared with Qiagen kits or purified by CsCl gradient centrifugation (32). The recombinant vesicular stomatitis virus (VSV) in which the VSV-G gene was replaced by a gene encoding firefly luciferase (VSV-Luc) was provided by Sean Whelan, Harvard Medical School, Boston, MA. The recombinant, replication-competent Zaire EBOV with the GFP open reading frame (ORF) inserted into its genome (ZEBOV-GFP) was provided by Stuart Nichol and Jonathan Towner, CDC, Atlanta, GA. For measuring the mRNA levels in the cells, mRNA was purified using an Aurum Total RNA kit (Bio-Rad, Hercules, CA), and quantitative real-time PCR (qRT-PCR) was performed using the iScript One-Step RT-PCR kit with SYBR green (Bio-Rad, Hercules, CA).
Generation of ebolavirus-like particles (EBOVLPs).
To visualize EBOV particle entry into cells, virus-like particles were generated using GFP-tagged VP40 and EBOV GP (40). 293FT cells were plated at 90% confluence in 10-cm culture dishes 2 to 4 h prior to transfection. To generate EBOVLPs, each plate was transfected with 11.5 μg of pVP40-GFP and 0.5 μg of pEBOV GP using PEI. Forty-eight hours posttransfection, the supernatant containing EBOVLP was collected from 10 plates. The cell debris was pelleted at 1,000 rpm for 10 min in an Eppendorf 5810R centrifuge with standard swing-out rotor. The EBOVLPs were pelleted through 20% (wt/vol) sucrose at 82,700 × g for 3 h. The pellets were resuspended in 5 ml phosphate-buffered saline (PBS) or DMEM containing 10% FBS, aliquoted, and stored at −80°C until use.
Generation of EBOV GP pseudotyped VSV encoding luciferase (EBOV-VSV-Luc).
To assess the dependence of EBOV GP in infection, EBOV pseudotyped virus was generated using a recombinant VSV with the VSV-G gene replaced by the firefly luciferase gene, as previously reported (33). The pseudotypes were generated by expressing GP in cells and then inoculation with the recombinant VSV. The cells were transfected using PEI with 0.5 μg of pEBOV GP and 10 μg of pβ-gal (carrier plasmid). The cells were then inoculated with a VSV pseudotype, which had been previously made, bearing the GP of the Marburg virus (multiplicity of infection [MOI] = 1.0). The next day, the supernatant was removed. The cells were washed 3 times and resuspended in DMEM containing FBS. The cells were then incubated at 37°C for 48 h after virus inoculation, and the supernatant was collected for further use. As a control for the contamination of the inoculating virus, the cells were transfected with pβ-gal alone, and culture supernatant was collected and tested for activity by inoculation of fresh cells. The cells transfected with EBOV GP produced 1,000-fold-higher intensity signals than this control. All virus titers were determined by a limiting, serial 2-fold dilution onto HeLa cells with luciferase activity measurements taken 8 h postinoculation.
siRNA transfections.
HeLa cells were reverse transfected using Lipofectamine 2000 following the manufacturer's protocol in either 96- or 6-well plates. The cells were plated into wells containing the siRNA and transfection mixture. In preliminary work, the method was optimized to yield a >90% suppression of luciferase expression in a cell line stably made to express luciferase using a specific siRNA. At 24 h posttransfection, the cells were visually inspected for indications of siRNA toxicity. At 48 h posttransfection, the cells were inoculated with the EBOVLPs, EBOV GP pseudotyped virus, or ZEBOV-GFP (recombinant replication competent virus). The cells were also lysed for mRNA purification and subsequent RT-PCR. As a negative control, nontargeting siRNA (AllStars; Qiagen) was used. Additionally, Ctbp1 siRNA was used as a positive control, as previous work showed a 90% inhibition of ZEBOV-GFP infection when the expression of this protein was suppressed (47). The siRNA sequences used are shown in Table 1.
Table 1.
Sequences of siRNA used to suppress ASMase (SMPD1) mRNA expression
| siRNA name | Vendor | Catalog no. | Target sequence |
|---|---|---|---|
| Hs_SMPD1_1 | Qiagen | SI00011557 | TGGAATTATTACCGAATTGTA |
| Hs_SMPD1_2 | Qiagen | SI00011564 | CTGGTTTAGCTGGATATGGGA |
| Hs_SMPD1_3 | Qiagen | SI00011571 | CGGGCTGAAGAAGGAACCCAA |
| Hs_SMPD1_5 | Qiagen | SI03096121 | CTGCTGTGGGTAACCATGAAA |
| SMPD1_6 | Invitrogen | SMPD1HSS143988 | ACAAAGAGTAGCCAGACGGCAGGGC |
| Ctbp1_7 (+control) | Qiagen | SI04301325 | CACCGTCAAGCAGATGAGACA |
| AllStars (−control) | Qiagen | SI1027281 | Nontargeting |
Drug treatment and cytotoxicity assays.
Drug stocks (10 mM) were prepared in either molecular-grade water (imipramine and desipramine) or dimethyl sulfoxide (DMSO) (vacuolin-1), aliquoted, and stored at −80°C until use. A 10-μM stock of bafilomycin A, an inhibitor of the vacuolar ATPase, was prepared in DMSO. The working drug concentrations were determined by treating the cells with the drug in 2-fold serial dilutions starting at 100 μM. The cells were plated at 70% confluence in 96-well plates at least 4 h prior to drug treatment. The cells were then incubated with the drugs 1 h prior to addition of the virus. alamarBlue dye was used to determine the cytotoxicity of each drug treatment. The drug and dye were incubated with the cells for 24 h, and fluorescence was measured using a BMG Omega plate reader. To determine the impact of each drug on infection efficiency, total cell numbers were counted. A change of >20% in cell density or in alamarBlue fluorescence was used to indicate drug toxicity. No toxic effects were observed at the reported doses for any drug tested.
EBOV-GFP infection assay.
A recombinant, replication-competent Zaire EBOV that encodes GFP (ZEBOV-GFP) was used to measure the impact of the treatments on the infection. This virus has the GFP ORF inserted into the viral genome such that the protein is under the control of native virus promoter elements. The ZEBOV-GFP virus replicates similarly to the wild-type virus, and GFP expression can be detected 24 h after inoculation (11, 52). All the experiments with the replication-competent viruses were performed in a biosafety level 4 (BSL4) laboratory (Robert Shope Memorial BSL4 Laboratory, UTMB). All the infection assays were performed in 96-well plates with three or more replicates and were repeated in multiple independent experiments. The virus inoculation was performed 48 h after the siRNA transfection or 1 h after the drug treatment. The drugs remained on the cells at the indicated final concentrations for the duration of each assay.
Unless stated otherwise, an MOI of 0.3 was used for the virus inoculations. For many of the experiments, a lower MOI was tested, but this change yielded no impact on the overall outcome of the infection with each treatment. For both the siRNA and drug treatments, the virus was incubated with the cells for 24 h at 37°C, which corresponded to approximately one round of replication and allowed the accumulation of enough GFP to be detected. At the termination of the experiment, the cells were immersed in 10% formalin for at least 24 h. The cells were then rinsed 3 times with PBS, and the cell nuclei were stained with 0.4 μg/ml DAPI in PBS. The cells were then imaged using a Nikon TE2000 fluorescence microscope using a 10× objective lens and mounted with an EMCCD 16-bit camera (iXon, ANDOR). The images were analyzed using Cell Profiler image analysis software (Broad Institute, MIT, Boston, MA). The analysis pipeline is available upon request. Briefly, the cells identified by the nuclear DAPI staining were individually assessed for GFP fluorescence intensity. Control experiments were used to set the thresholds for signal over the background; the number of infected cells could thus be determined. The infection efficiency was typically expressed as the percentage of infected cells, either in the entire cell population or, when indicated, normalized to the infection of the untreated cells. All the data were analyzed by one-way analysis of variance (ANOVA) with Tukey's posttest using the GraphPad Prism statistical analysis software (version 5.00 for Windows, GraphPad Software, San Diego, CA).
Cell binding and internalization of VLPs.
For the mechanistic analyses, the impact of each treatment on the binding and internalization of the virus was the first step examined. HeLa cells were plated on 8-well chamber slides (Ibidi, Germany). A single drug concentration of 3 to 5 times the 50% effective concentration (EC50) was chosen; these concentrations were determined not to be cytotoxic using the alamarBlue cytotoxicity assay. The cells were preincubated for 1 h with 25 μM imipramine, 1 μM vacuolin-1, or vehicle prior to the inoculation with EBOVLPs, and the cells were then incubated for up to 90 min. Upon termination of the experiment, the medium was removed, and the cells were rinsed with PBS. The cells were fixed with 4% paraformaldehyde for 10 min at room temperature, rinsed 3 times with PBS, and stored at 4°C until antibody staining.
Luciferase infection assay of the EBOV GP-VSV core pseudotyped virus.
The pseudotyped viruses allowed the comparison of the relative impact of each treatment on the function of the EBOV GP compared with the GPs from the other viruses. HeLa cells were plated in 96-well plates (white walled; Corning) and incubated as above with each drug. After 1 h, the pseudotyped virus was added. Pseudotyped viruses bearing the glycoproteins of EBOV (EBOV-VSV-luciferase) or VSV (VSV-VSV-luciferase) were used. The cells were incubated with the drug and virus for 8 h at 37°C, after which time the medium was replaced with 100 μl luciferase assay buffer containing 0.2% Triton X-100 detergent (Promega). The cells were gently mixed to ensure lysis, and luciferase activity was measured (Veritas 96-well plate luminometer; Turner Biosystems). An additional reading of each plate was taken after 1 min to ensure that stable luciferase activity had been attained (32). The counts per second were analyzed by one-way ANOVA with Tukey's posttest using GraphPad Prism software.
qRT-PCR.
qRT-PCR was used to determine the relative fold change in the mRNA levels of specific genes after siRNA transfection and viral genomic RNA levels after infection. To determine the impact of the drug treatments on the viral RNA abundance, HeLa cells were plated in 6-well plates at 70% confluence 4 h prior to the addition of drug. The cells were preincubated for 1 h with 25 μM imipramine, 25 μM desipramine, 1 μM vacuolin-1 (each was 3 to 5 times the EC50 determined in the infection assays), or vehicle. ZEBOV-GFP was added at an MOI of 0.3 and incubated with the cells for 24 h at 37°C. At the termination of the experiment, 1 ml TRIzol (Invitrogen) was added to the cells. RNA purification was performed using a Bio-Rad Aurum Total RNA kit. qRT-PCR was performed using the iScript One-Step RT-PCR kit with SYBR green in a Bio-Rad CFX-96 RT-PCR machine. The following primers against EBOV NP amplified the region between nucleotides 2094 and 3609 of the viral genome (GenBank NC_002549.1): 5′ AGCGTGATGGAGTGAAGCGCC 3′ and 5′ TCCGGCATGGCAGCAAGTGT 3′. To determine the fold change in the ASMase mRNA expression, the transfected HeLa cells were treated as above, and the mRNA was purified. The primers against lysosomal ASMase (SMPD1; GenBank NM_000543) were purchased predesigned from Origene (catalog no. HP200515) with the sequences 5′ GCTGGCTCTATGAAGCGATGGC 3′ and 5′ AGAGCCAGAAGTTCTCACGGGA 3′. For both reactions, an annealing temperature of 59°C was used. A threshold cycle for signal detection was recorded, and the change in the threshold between the untreated and treated samples was calculated. GAPDH-specific primer sets (Qiagen) served as internal controls for the change in the total mRNA levels and to calculate the ΔΔCT values. The ΔΔCT and fold change in the expression values were determined using Bio-Rad CFX software (version 1.6). The expression values were further analyzed using the GraphPad Prism software to assess statistical significance (P < 0.05).
Immunofluorescence assays.
For the immunofluorescence staining of the cells with antibodies, HeLa cells were fixed with freshly made 4% paraformaldehyde after the treatment and incubation with virus or VLPs. To restrict the staining to the plasma membrane, the cells were not permeabilized. For the SM raft staining, HeLa cells were blocked with 2% bovine serum albumin (BSA) for 15 min with the SM raft-binding protein, lysenin (1:50), prior to a 2-h incubation. The cells were rinsed and incubated with rabbit antilysenin primary antibody (1:1,000 in 2% BSA). The cells were rinsed again and incubated with donkey anti-rabbit Alexa Fluor 594 (1:200 in 2% BSA) for 1 h. The cell nuclei were stained with 0.4 μg/ml of DAPI for 30 min. For ASMase staining, the cells were blocked with 10% serum and incubated with goat anti-ASMase (1:50 in 2% chicken serum) overnight at 4°C. The cells were rinsed 3 times with PBS and then incubated with chicken anti-goat Alexa Fluor 647 for 1 h. The cells were visualized with a 100× oil objective lens using a Zeiss LSM 510 confocal immunofluorescence microscope.
Sphingomyelin depletion assay.
A sphingomyelin depletion assay was used to assess the requirement for sphingomyelin in EBOV entry. HeLa cells were plated on the day prior to treatment and incubated overnight at 37°C. Staphylococcus aureus SMase (Sigma) was diluted in Hanks balanced salt solution (HBSS) with calcium and magnesium and immediately applied to cells. Optimization of the SMase treatment showed that a 45-min incubation with 2 U SMase/ml at 37°C provided the peak reduction in the SM staining without the loss of cell viability; therefore, these conditions were used. After the enzyme treatment, the cells were washed twice with HBSS. The mock-treated cells were treated with HBSS only. The cells were then incubated with EBOV-GFP, the pseudotyped viruses (EBOV-VSV-luciferase or VSV-VSV-luciferase), or the GFP-tagged EBOVLPs for 1 h. Next, the cells were rinsed to remove the unbound virus. For the EBOV-GFP infection assays, the cells were incubated with concentrated virus (107 PFU/ml) that had been pelleted through sucrose and resuspended in PBS, and unbound virus was removed after 1 h, as above. After 24 h, the cells were fixed and imaged. For the VSV pseudotypes, the cells were incubated for a total of 9 h and analyzed for luciferase activity. For the binding assays using the GFP-tagged EBOVLPs, the cells were washed and fixed after 1 h of incubation with the VLPs.
RESULTS
EBOV associates with SM-enriched lipid rafts.
The lipid SM comprises 10% of the mammalian cell membrane. Unlike other lipids, SM preferentially associates with cholesterol to form SM and cholesterol-enriched lipid rafts in the plasma membrane (42). These lipid rafts form platforms that recruit cellular receptors to specific sites on the cell surface and aid in enhancing the signaling between these and other cellular proteins; lipid rafts have also been shown to be important for virus entry into cells (30). Methyl-β-cyclodextrin (MβCD), a compound known to extract cholesterol from cell membranes (5, 46, 47), inhibits EBOV infection; however, MβCD also extracts SM from cell membranes (19). Therefore, the impact of MβCD treatment on EBOV infection may be the result of the cholesterol extraction, SM extraction, or both. For this reason, we examined the role of SM and SM-associated enzymes in EBOV infection.
To determine if the depletion of cellular sphingomyelin reduces EBOV infection, HeLa cells were pretreated with 2 U/ml SMase in HBSS for 45 min, washed to remove the SMase, and inoculated with the GFP-expressing, replication-competent Zaire EBOV (ZEBOV-GFP) at an MOI of 0.3. After 1 h, the cells were washed to remove the unbound virus and incubated for 24 h to allow the infection and expression of the GFP infection marker. For the negative control, cells were incubated with HBSS only. Cells treated with SMase were more resistant to infection than those that received only the mock treatment (Fig. 1A). Next, we investigated if the effect of the SM depletion is specific to EBOV. HeLa cells were pretreated with SMase, as above, and were inoculated with luciferase-encoding VSV pseudotypes bearing either ZEBOV GP or VSV-G. The cells were then incubated for an additional 8 h before luciferase activity was measured. While VSV-G pseudotyped virus infectivity was unaltered, the EBOV GP pseudotype infectivity was reduced by >80% (Fig. 1B). To verify that SMase treatment reduced the amount of SM-rich rafts on the plasma membrane, HeLa cells were treated with SMase, as for infection assays, and then fixed in formalin. The cells were stained using the sphingomyelin-binding protein lysenin, followed by an antilysenin antibody (Fig. 1C), and the intensity of staining was measured. This analysis demonstrated that SMase treatment resulted in a 70% reduction in staining of SM-containing rafts and correlated with the reduction in ZEBOV and EBOV GP pseudotype infection reduction.
Fig 1.
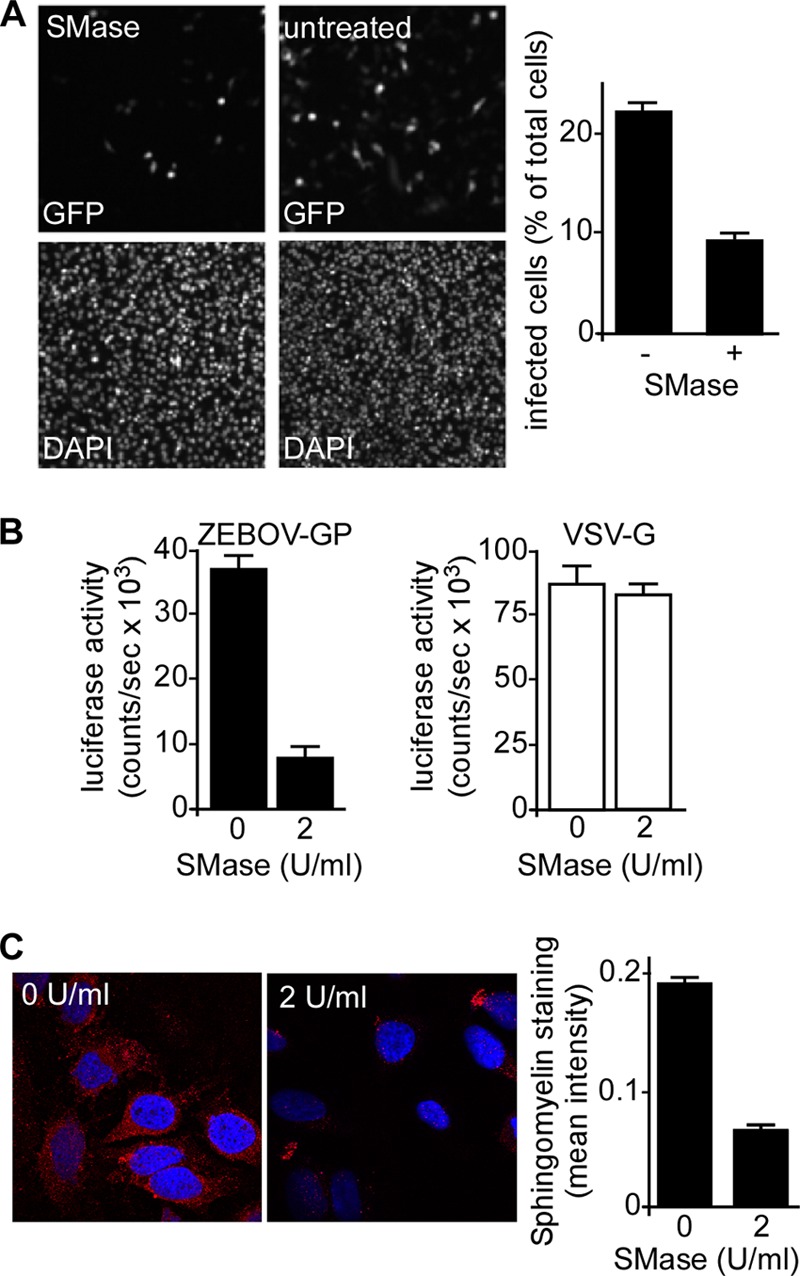
The depletion of cellular sphingomyelin reduces ZEBOV infection. HeLa cells were pretreated with 2 U/ml SMase in HBSS buffer for 45 min, washed to remove SMase, and inoculated with GFP-expressing replication-competent ZEBOV (ZEBOV-GFP) at an MOI of 5. After 1 h, the cells were washed to remove unbound virus and incubated overnight to allow the infection and expression of the GFP infection marker. The cells serving as the negative control were incubated with HBSS only. (A) Images of the cells treated with SMase and infected with virus, 24 h after virus inoculation. The infected cells express GFP (top). The cell nuclei were stained with DAPI (bottom). The number of infected cells per total cell count was calculated (right). (B) SMase treatment affects EBOV GP function. HeLa cells were pretreated with SMase as above and were inoculated with luciferase-encoding VSV pseudotypes bearing either ZEBOV GP or VSV-G (indicated). After 1 h, the unbound virus was removed, and the cells were incubated for an additional 8 h. Luciferase activity was then measured. (C) SMase treatment reduces sphingomyelin raft staining. HeLa cells were treated with SMase as in the infection assays and then fixed in formalin. The cells were stained using the sphingomyelin-binding protein, lysenin, followed by an Alexa Fluor 594 conjugated anti-lysenin antibody (red). The cell nuclei were stained with DAPI (blue). The untreated cells were incubated with HBSS only. The images were taken using a confocal microscope with a 100× oil objective lens. The intensity of the staining was determined by measuring the intensity of the staining per cell for 5 distinct images of treated and untreated cells with Cell Profiler software and is shown at right.
If SM is important for infection, EBOV particles should associate with the SM-containing rafts. Other work has shown that EBOV membrane fusion begins after a lag of 60 min and has a half time of 120 min (48), so HeLa cells were incubated with EBOVLPs for up to 90 min, as this time frame should encompass binding up to membrane fusion of virus entry into cells. Cells were then stained with lysenin but were not permeabilized to facilitate the observation of the events at the cell surface only. A strong association of EBOVLPs with SM-containing rafts was observed even at the earliest time point (30 min), with the number of these events increasing with time (Fig. 2). Interestingly, the intensity of SM staining at the cell surface corresponded to the number of the VLPs bound to the cells within the local area, suggesting that the VLPs were responsible for the accumulation of SM.
Fig 2.

EBOVLPs associate with sphingomyelin (SM) clusters and acid sphingomyelinase (ASMase) on the cell surface after VLP binding. GFP-tagged EBOVLPs (green) were incubated with HeLa cells for 30, 60, or 90 min, washed to remove unbound particles, and fixed in formalin. To identify only cell-surface interactions, the cells were not permeabilized before staining. SM was stained using the sphingomyelin-binding protein lysenin (red). ASMase was labeled using an anti-ASMase antibody (cyan), and the cell nuclei were stained with DAPI (blue). The cells were visualized with a confocal microscope with a 100× oil objective lens. The panels on the left show the merged images from each color channel with individual channels (except DAPI) shown in the panels to the right.
EBOV binding recruits lysosomal ASMase to the cell surface.
Acid sphingomyelinase (ASMase) is an enzyme that hydrolyzes SM both in lysosomes (where the enzyme is normally found) and on the outer leaflet of the plasma membrane. For ASMase to access the SM on the cell surface, lysosomal exocytosis must occur. ASMase acts by removing the SM phosphocholine head group to generate ceramide (57), which is hydrophobic and segregates to form rigid semicrystalline arrays. If these arrays form on the outer membrane leaflet, they promote large-scale membrane invagination, such as macropinosome formation, which can reach up to 400 nm in diameter (59). Recently, EBOV was shown to use macropinocytosis to enter cells. We therefore tested if, similar to SM, ASMase was associated with EBOVLPs.
The cells were incubated with VLPs for up to 90 min and then stained for ASMase. Strikingly, the ASMase staining was observed in clusters on the cell surface at the sites of VLP binding (Fig. 2). Because the cells were not permeabilized for this experiment, the ASMase must have been exposed on the cell surface at the site of the VLPs. Like SM, ASMase was found to be colocalized with EBOVLPs from the earliest time point (30 min) to the final time point (90 min), suggesting that, like SM, the ASMase can be rapidly recruited to the site of VLP binding on the cell surface. As with SM, there appeared to be a correlation between the number of EBOVLPs bound and the amount of ASMase association. These findings indicate that ASMase is either recruited to or is already present at the site of the VLP interaction with the cells. The substrate of ASMase, SM, is also present in the same clusters, close to the VLPs. Both conditions would be a prerequisite for ASMase to play a role in EBOV infection.
ASMase activity is required for EBOV infection.
Specific ASMase inhibitors, such as imipramine, desipramine, and amitriptyline, are water-soluble compounds that block the function of ASMase both at the cell surface and within lysosomes. Imipramine and desipramine were tested at concentrations up to 100 μM in HeLa, HEK293, and Vero cells with similar outcome. In HeLa cells, both drugs were potent inhibitors of infection and had EC50s of 3 to 5 μM (Fig. 3A to C). Furthermore, the EC50 for both drugs was independent of MOI (shown for imipramine in Fig. 3A and B). Only concentrations greater than 50 μM showed slight toxicity by the alamarBlue cytotoxicity assay. Amitriptyline was similarly potent (not shown). Each drug also worked equally well in HEK293 cells, with similar EC50s. In Vero cells, each drug was effective but less potent, with EC50s 2 to 3 times higher than those recorded for the HeLa cells (data not shown).
Fig 3.

EBOV infection is reduced by the ASMase inhibitors imipramine and desipramine. (A) HeLa cells were pretreated for 1 h with the ASMase inhibitor imipramine (25 μM) prior to the addition of the GFP-expressing, replication-competent EBOV at an MOI of 0.4 (ZEBOV-GFP, upper panels). The cells were fixed, and their nuclei were stained with DAPI (lower panels). Cells were then visualized using an epifluorescence microscope. The cells infected in the presence of the indicated concentrations of imipramine (open circles) were counted, and dose-response curves were fitted to the data using GraphPad Prism software. As each drug is water soluble, medium alone was used as a control (solid circles). (B) To test if drug impacted infection similarly at a low MOI, cells were again challenged with virus in the presence of imipramine (open circles) but at an MOI of 0.05. Medium-alone control is shown as solid circles, and data were processed as for panel A. (C) Cells were treated with desipramine (open circles) or medium alone (filled circles) at an MOI of 0.3. Data were processed as in panel A. (A to C) Representative experiments are shown with each point in triplicate. The EC50s for imipramine and desipramine were each calculated to be 3 to 5 μM and were the same for each MOI tested.
The impact of imipramine on virus propagation was also tested. For this, cells were incubated with virus for either 1 or 3 days at MOI of 0.15, 0.05, and 0.015. Since one round of replication is approximately 24 h, 3 days corresponds to 2 complete rounds of replication. During this time, the spread of virus was evident and appeared to be mostly by cell-free transmission, as most infected cells were separated from others and no difference in the ratio of cell clusters to single cells was seen (Fig. 4A). For permissive concentrations of drug (≤25 μM), a 10-fold increase in infected cell numbers occurred between day 1 and day 3. The increase was the same in the presence of the drug and in untreated cells. Importantly, the 50% inhibitory concentration (IC50) for the drug remained the same for each time point and virus dose and is consistent with the drug impacting the entry step of infection. Again, slight toxicity was seen with doses above 50 μM by alamarBlue, but only at 100 μM was cell density changed significantly, with a 15% reduction in cell numbers seen after the 3-day incubation.
Fig 4.
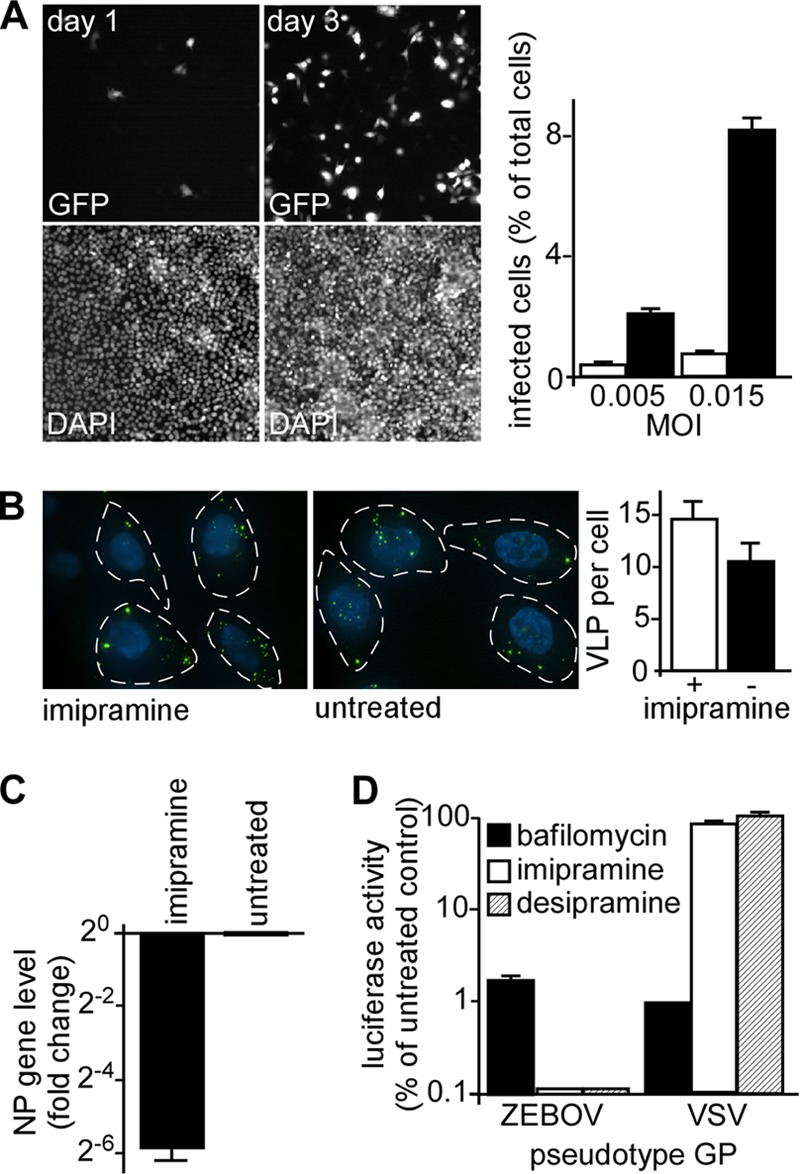
Imipramine impacts the entry step of EBOV infection. (A) The impact of prolonged incubation of virus with cells in the presence of drug was tested. Cells were incubated with EBOV-GFP at the indicated MOI, and then fluorescent cells (GFP, top panels) were counted on day 1 or day 3 postinoculation. Cell nuclei were stained with DAPI and counted (DAPI, lower panels). The number of infected cells was calculated as a percentage of the total number of cells (right) for day 1 (open bars) or day 3 (solid bars) for MOI of 0.005 and 0.015. (B) The effect of imipramine on VLP binding to cells was determined by incubating cells with VLP-GFP particles for 90 min. Cells were treated with imipramine at 25 μM (+) or left untreated (−). VLPs bound to cells (outlined by dashed lines and nuclei stained with DAPI, blue) were then counted for more than 20 cells per condition with the average ± standard deviation shown at right. Each was not significantly different (P > 0.12). (C) EBOV infection was measured by qRT-PCR. HeLa cells were pretreated with 25 μM imipramine dissolved in water for 1 h prior to the addition of virus and incubated with the drug overnight. The cells were then collected in TRIzol, and the relative change in NP vRNA was determined after normalization to GAPDH levels and to the mock-treated control. (D) Imipramine and desipramine affect a GP-specific infection step. HeLa cells were pretreated with 25 μM imipramine or desipramine for 1 h before the addition of the pseudotyped VSV bearing the GP of EBOV or VSV (as indicated) and encoding luciferase as the infection marker. After an 8-h incubation in the presence of each drug, luciferase activity was measured. Bafilomycin A, an inhibitor of pH-dependent virus infection, was used as a positive control. The data are shown relative to the untreated cells and are the averages of at least 3 replicates.
To test if imipramine altered binding of virus to cells, cells were incubated in the presence of drug and VLPs for 90 min. Cells were then imaged as optical z-sections, and the VLPs associated with each cell were counted. Cells treated with drug or left untreated had on average 14 and 11 particles per cell, respectively (Fig. 4B), which was not significantly different as determined by Student's t test (P > 0.12). We found that drug treatment did not alter the distribution of ASM or SM on the cell surface (not shown). These findings indicated that the effect of imipramine was not through altering cell binding and uptake.
As a measure of EBOV infection that was not dependent on GFP expression, we tested the impact of imipramine on viral RNA replication (Fig. 4C). HeLa cells were treated with 25 μM imipramine dissolved in water, and qRT-PCR was performed using primers targeting the EBOV nucleoprotein (NP) gene. Compared with untreated cells, the imipramine treatment reduced the EBOV NP RNA levels nearly 64-fold. The drug had little impact on GAPDH or actin mRNA levels.
ASMase requirement for ZEBOV infection is GP specific.
Next, we assessed if the requirement for ASMase in EBOV entry was GP specific. A recombinant vesicular stomatitis virus with the native VSV-G replaced by the ORF for firefly luciferase was used to generate pseudotyped viruses bearing the glycoproteins of EBOV or VSV. HeLa cells were preincubated for 1 h with 25 μM imipramine, 25 μM desipramine, or 25 nM bafilomycin A prior to the incubation with each pseudotyped virus. Each of the ASMase inhibitors was a potent inhibitor of the EBOV pseudotype, reducing luciferase activity 1,000-fold compared to background levels (Fig. 4D). For both imipramine and desipramine, the VSV-G pseudotype infectivity was unaffected. In comparison, bafilomycin A, an inhibitor of the vacuolar H+ V-ATPase pump, blocked all pseudotypes similarly. This is because both viruses must enter an acidified endosome, from which membrane fusion is triggered, to initiate infection. Importantly, the disproportionate impact on the EBOV GP pseudotype demonstrates that EBOV has a strict and specific dependence on ASMase activity for normal function of its GP to permit infection of cells.
ASMase siRNA-treated cells resist EBOV infection.
To further confirm the importance of ASMase for EBOV infection, siRNAs targeting the ASMase gene (SMPD1) were used to suppress ASMase expression. For 5 distinct siRNAs, ZEBOV-GFP infection was reduced by at least 67%, with two siRNAs being more potent and blocking infection by >83% in HeLa cells compared to the nontargeting negative control (Fig. 5A and B). Because ASMase antibodies did not perform well on Western blots, qRT-PCR was used to determine the impact of each siRNA on the SMPD1 mRNA levels. Compared to the nontargeting control (AllStars) siRNA, each siRNA reduced SMPD1 levels at least 4-fold. Again, as for drugs, treatment with the most active siRNA did not appear to alter VLP binding to cells (not shown). Attempts to restore ASMase activity in siRNA-treated cells were confounded by the toxicity of the overexpressed protein.
Fig 5.

EBOV infection is dependent on ASMase expression. (A) siRNA-mediated reduction of the ASMase mRNA results in reduced EBOV infection. HeLa cells were transfected with 5 siRNAs (labeled 1, 2, 3, 5, and 6) targeting distinct sites in the lysosomal and secreted forms of ASMase mRNA (SMPD1). At 48 h posttransfection, the cells were incubated with ZEBOV-GFP for 24 h (MOI, 0.3), and the infection was measured by counting the fluorescent cells and dividing by the number of total cells stained with DAPI. Ctbp1 siRNA was used as a positive control, as this siRNA was previously shown to inhibit ZEBOV-GFP infection (51). AllStars nontargeting siRNA (Qiagen) was used as a negative control, as were untreated cells (cells only). (B) At the time of infection, the ASMase mRNA expression in the HeLa cells was determined using qRT-PCR with SMPD1-specific primers. The fold change in the expression levels was determined relative to the GAPDH mRNA levels and to cells transfected with AllStars negative-control siRNA. (C) Primary human fibroblasts deficient in ASMase expression (−) or of normal phenotype (+) were incubated with ZEBOV-GFP for 24 h and fixed in formalin. The infection efficiency (%) was calculated by counting the GFP-fluorescent cells and dividing by the total number of cells stained with DAPI.
ASMase-deficient cells resist EBOV infection.
As another independent test of ASMase importance for EBOV infection, primary human fibroblasts defective in ASMase were used. These cells bear a double heterozygous mutation in the ASMase gene that results in loss of enzyme activity. The cells were challenged with ZEBOV-GFP, and infection was measured after 24 h. The infection was compared with human skin fibroblasts from a donor with no defect. Consistent with a role for ASMase in infection, the ZEBOV-GFP infection efficiency was reduced 3-fold in the ASMase-defective cells compared with the normal fibroblasts (Fig. 5C). Together with the drug and siRNA treatments, these findings indicate a strong dependence of EBOV infection on the presence and activity of ASMase in cells.
EBOV infection is inhibited by the lysosomal exocytosis inhibitor vacuolin-1.
The finding that ASMase is present on the cell surface at the site of the VLP interaction (Fig. 2) suggests that ASMase is recruited to this location or that VLPs preferentially associate with ASMase- (and SM)-rich regions on the plasma membrane. Because ASMase is normally located in lysosomes and would require exocytosis to become localized to the cell surface, the role of lysosomal exocytosis in EBOV infection was tested. Vacuolin-1 is a specific inhibitor of lysosomal exocytosis but does not affect endocytosis, including macropinocytosis (8). The treatment of HeLa cells with vacuolin-1 led to a potent inhibition of EBOV infection with an EC50 of 370 nM (Fig. 6A and B). This drug was extremely well tolerated, with doses as high as 100 μM showing no cytotoxicity by either the alamarBlue assay or the assessment of cell morphology and number (not shown). Similar to the results obtained with the ASMase inhibitors, this finding was independently confirmed by qRT-PCR to detect viral genome replication. Compared with the DMSO-treated control, vacuolin-1 reduced EBOV RNA more than 128-fold (Fig. 6C).
Fig 6.
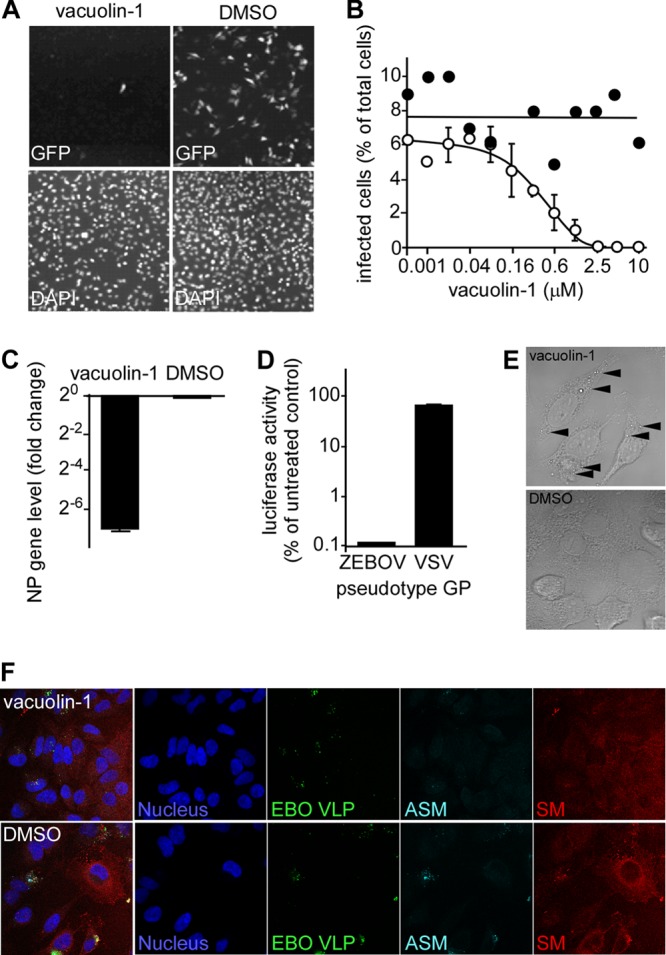
EBOV infection is reduced by the lysosomal exocytosis inhibitor vacuolin-1. (A) Vacuolin-1 inhibits EBOV infection. HeLa cells were pretreated with 1 μM vacuolin-1 in DMSO for 1 h and then incubated overnight with ZEBOV-GFP in the presence of the drug or DMSO (upper panels). The cells were fixed, stained with DAPI (lower panels), and then imaged. (B) The cells infected in the presence of the indicated concentration of vacuolin-1 (open circles) were counted and divided by the total number of DAPI-stained cells to give the percent infected. Sigmoidal dose-response curves were fitted to the data using GraphPad Prism software. DMSO was the drug vehicle and used as the negative control (filled circles). A representative experiment is shown. The EC50 was calculated to be 370 nM. (C) EBOV infection was measured by qRT-PCR. HeLa cells were pretreated with 1 μM vacuolin-1 for 1 h prior to a 24-h incubation with the drug and ZEBOV-GFP (MOI, 0.3). The expression of ZEBOV NP vRNA was measured and normalized to the GAPDH mRNA expression and to the drug vehicle (DMSO)-treated cells. (D) Vacuolin-1 inhibits a GP-specific step during infection. HeLa cells were pretreated with 1 μM vacuolin-1 for 1 h prior to an 8-h incubation with the drug and luciferase-expressing VSV pseudotypes bearing either the ZEBOV or VSV glycoproteins. (E) Vacuolin-1 causes vesicle accumulation within the cell cytoplasm. HeLa cells were treated with 1 μM vacuolin-1 or DMSO for 1 h, fixed, and then imaged using a microscope with differential interference contrast (DIC) optics and a 100× oil immersion lens. The arrowheads highlight examples of enlarged vesicles similar to lysosomes previously reported by others (10). (F) To confirm that vacuolin-1 treatment results in the recruitment of ASMase to the cell surface, HeLa cells were incubated with GFP-tagged EBOVLPs (green) for 90 min, washed, and fixed. The cells were not permeabilized to facilitate observation of the surface localization of each factor. SM was labeled using the sphingomyelin-binding protein lysenin (red). ASMase was stained with anti-ASMase antibody (cyan), and the cell nuclei were stained with DAPI (blue). The cells were imaged by confocal microscopy using a 63× objective lens.
The specificity of the exocytosis inhibitor was further tested by examining its activity against EBOV GP and VSV-G pseudotyped virus. As shown in Fig. 6D, 1 μM vacuolin-1 reduced the signal from the EBOV GP pseudotype to background levels, whereas the VSV-G pseudotype was unaffected. This finding reveals the strong and specific dependence of EBOV GP function on the exocytic activity in the cell. To confirm that the vacuolin-1 treatment was effective, HeLa cells were treated with 1 μM vacuolin-1 for 2 h prior to imaging by confocal microscopy. The treated cells produced numerous enlarged lysosomes, a characteristic of vacuolin-1 activity, in contrast to the mock-treated cells (Fig. 6E). Finally, vacuolin-1 has been shown to specifically inhibit ASMase translocation to the plasma membrane (4). HeLa cells were incubated with 1 μM vacuolin-1 for 1 h and then incubated with EBOVLPs for 60 min in the presence of drug. The cells were then fixed and stained with lysenin and anti-ASMase antibody. The cells were not permeabilized to facilitate the observation of the events occurring only at the plasma membrane. Compared with the mock-treated cells, vacuolin-1-treated cells had reduced ASMase staining. In addition, fewer EBOVLPs were bound to the drug-treated cells. These findings suggest that translocation of ASMase to the cell surface relies on exocytosis, as expected, but that the localization of ASMase or other exocytic cargo impacts the ability of virus to bind to cells.
DISCUSSION
SM is a major component of eukaryotic cell membranes that preferentially associates with cholesterol to form lipid rafts (42, 43). Previously, the role of rafts was shown to be important for EBOV infection but was thought to correlate to the cholesterol component of the raft (5). These studies involved treating the cells with MβCD and nystatin, which respectively extract and sequester cholesterol (5, 47). However, recently, it has been realized that the treatments used to extract cholesterol or disrupt rafts do not discriminate between the roles of cholesterol and SM. Using atomic force microscopy, it was shown that MβCD extracted SM along with cholesterol (19). Photoactivated localization microscopy with superhigh-resolution microscopy revealed that in HeLa cells treated with MβCD, the cholesterol-SM rafts disperse and raft-dependent signaling is lost. However, when cells are treated with SMase, cholesterol remains, while the SM is hydrolyzed to ceramide and can induce endocytosis (44). Furthermore, nystatin, a drug that is used to sequester cholesterol in cell membranes, blocked recruitment of ASMase to the plasma membrane and the subsequent hydrolysis of SM, which modulates raft size (35). Thus, MβCD extracts SM along with cholesterol (19), and nystatin, again thought specific for cholesterol, also impacts SM metabolism. Therefore, we sought to determine if EBOV infection was dependent on SM and ASMase activity.
First, we demonstrated that depleting plasma membrane SM by SMase reduced ZEBOV-GFP infection by more than 50%. This is a large change, given that cell membrane turnover and SM replenishment from intracellular stores and synthesis are rapid. Next, using pseudotyped viruses that differed only in their surface glycoprotein, it was shown that the requirement for SM is specific for EBOV GP function. Only EBOV infectivity was significantly reduced in SMase-treated cells, while the VSV-G pseudotyped virus was unaffected. This result indicates that EBOV GP has a specific requirement for SM at the plasma membrane whereas VSV-G does not.
Next, VLPs were used to examine the relationship of virus particle interaction with cells and the impact on SM and ASMase localization on the cell membrane. Using immunofluorescence microscopy to image nonpermeabilized cells, we showed that ASM, SM, and EBOVLPs colocalize in clusters at the cell surface. These clusters are seen as early as 30 min after inoculation of VLPs onto cells but increase over time, suggesting that ASMase is likely recruited to the plasma membrane by exocytosis.
As SM was required for EBOV infection and because SM, ASMase, and VLPs are associated together at the cell surface, the role of ASMase in EBOV infection was examined. Following the treatment of cells with ASMase inhibitors, such as desipramine and imipramine, infection was completely blocked in a dose-dependent manner. This block was specific, with only the EBOV GP pseudotyped virus being inhibited and VSV-G pseudotypes remaining unaffected. The drug data were further supported when primary cells deficient for ASMase expression or cells treated with ASMase-specific siRNA were tested. In both cases, these cells resisted infection by EBOV. For the siRNA, 5 independent siRNA reduced EBOV infection. The efficacy of the siRNA varied in terms of reduction of ASMase (SMPD1) mRNA levels (4- to 20-fold) but were similar in overall suppression of infection (70 to 80%). Aside from some ASMase mRNA still being present, the residual infection seen in these experiments may result from the long-lived secretory form of ASMase, which has a half-life of 48 h, is found nonspecifically associated with the plasma, and functions at neutral pH. In addition, the membrane-associated ASMase has similar properties and can function on the plasma membrane after exocytosis (38, 56). However, the ASMase inhibitor desipramine only inhibits lysosomal ASMase and not secretory SMase (29). So, desipramine completely blocking EBOV infection indicates that secreted ASMase may not be important. In addition to ASMase, several neutral sphingomyelinases (NSMases) are present within the cell. These differ from ASMase by cleaving SM at a neutral pH optimum. In particular, NSM2 is found at the inner leaflet of the plasma membrane (38) and is important in exosome biogenesis (39, 53). Preliminary work in our laboratory using NSM2 siRNA and chemical inhibitors of NSM suggests that NSM may also be important in EBOV entry, as it is for Sindbis (28) and measles (17) viruses. However, drugs specific for NSM showed toxicity, and siRNA gave sporadic efficacy, making it difficult to study.
Finally, we showed that EBOV infection is completely blocked by vacuolin-1, a specific inhibitor of lysosomal exocytosis and ASMase translocation to the plasma membrane (4, 8). This drug was specific for EBOV GP activity, as infection by VSV-G pseudotyped virus was unaffected after treatment. Vacuolin-1 induced swelling of lysosomes and accumulation of vesicles within the cell, which are both characteristic of its action. This treatment also resulted in fewer VLPs being bound to cells as well as a loss of ASMase accumulation on the cell surface. These findings indicate that ASMase migration to the cell surface is dependent on exocytosis and this movement correlates to the number of VLPs bound to the cell. We do not expect ASMase to be a receptor for EBOV, and additional work will be needed to determine if VLPs induce ASMase recruitment or if constitutive exocytic movement of ASMase from lysosomes to the cell surface is required for VLPs to bind cells. However, the findings suggest that some cellular component present in the exocytic pathway needs to be translocated to the cell surface with ASMase to aid in virus attachment to cells.
The finding that ASMase and SM are important for infection is compatible with previous findings regarding EBOV infection. Interaction of EBOV can occur through several receptor types. One of these, DC-SIGN (50), is known to interact with ASMase. In the case of the measles virus, binding DC-SIGN can increase ASMase activity, which in turn induces lysosomal exocytosis to bring additional ASMase to the surface, as well as CD150, an entry receptor for measles virus (2). Recently, it was shown that Niemann-Pick Type C1 (NPC1), a predominantly lysosomal transmembrane protein (37), acts as a virus receptor (7, 9). It is unknown if NPC1 could act at the cell surface to bind EBOV or if the vacuolin-1-sensitive exocytosis needed for EBOV infection could also bring NPC1 to the cell surface. Interestingly, NPC1 is responsible for trafficking SM from the late endosomes to lysosomes for degradation by ASMase and to the plasma membrane to replace lost SM (31). Additionally, in NPC1-deficient cells, ASMase activity is reduced by nearly 50%, and increasing ASMase activity by transfection of expression plasmids can ameliorate the NPC1 lipid trafficking defect (10). These data suggest a strong link between ASMase, SM, and NPC1 function.
Recruitment of ASMase to the plasma membrane by EBOV GP would have major consequences for virus infection, particularly if this occurs at the site of bound virus particles. ASMase hydrolyzes SM to ceramide and phosphocholine. Ceramide is an important messenger molecule, capable of initiating signaling cascades for either pro- or antiapoptotic events. Second, ceramide aids in raft expansion into large macrodomains in which cellular receptors cluster. Lastly, cleavage of the bulky phosphocholine head group from SM to release ceramide favors curvature of the membrane and invagination of the membrane toward the cleaved head group (54) and, subsequently, macropinosome formation (59). This outcome is consistent with the finding that EBOV enters cells via macropinocytosis (46, 47).
Other viruses have been shown to require ASMase activity for infection. Measles virus and rhinovirus (21) require ASMase for uptake into cells. Measles induction of ASMase activity is important for recruitment of the measles virus receptor CD150 from LAMP-1-positive vesicles to the cell surface. Similar to what was seen for EBOV, ASM also moved to the cell surface shortly after binding of measles virus to cells, colocalizing with CD150 (2). Rhinoviruses also induce recruitment of ASMase to the cell surface. This appears to be irrespective of the primary receptor used, with either LDL- or ICAM-1-binding rhinoviruses inducing ASMase migration from internal compartments. Again, ASMase activity was required for virus uptake into cells and infection was readily blocked by ASMase inhibitors (21). In contrast, Sindbis virus infection of neuroblastoma cells results in activation of SMase and precedes cell apoptosis (28). ASMase levels rapidly rise and peak 1.5 h after infection. Since UV-inactivated virus induces a similar outcome, the increase is likely due to a virus structural protein, but it is unclear if GP or another structural protein is involved. Obvious cell death through apoptosis was not seen in our work and could be due to cell type differences. However, EBOV infection of nonhuman primates has been reported to induce apoptosis in T cells, even though these cells are not productively infected and may lead to lymphopenia (18). It will be of interest to determine if ASMase activation is one mechanism of apoptosis induction for EBOV.
Invasion of mammalian cells by N. gonorrhoeae and the intracellular parasite T. cruzi (14, 16) also depends on ASMase activation and recruitment to the cell surface. For N. gonorrhoeae several proteins of the colony opacity-associated proteins appear to induce ASMase activity as early as 15 min after inoculation of bacteria onto cells (25). Inhibition of ASMase activity and genetic knockout prevent invasion of nonphagocytic (20) and phagocytic cells (25). The induction of ASMase activity is also seen together with induction of exocytosis, which is porin dependent (3), again showing similarities to what we observed with EBOV. T. cruzi invades cells by a calcium-dependent mechanism that is closely related to membrane wound repair (14). Cell membranes are repaired by exocytosis of ASMase and lipids from internal stores (51). The exocytic event is closely coupled with endocytosis as well as phagocytosis. The increase in membrane turnover aids in sealing membrane breaks. As EBOV appears to induce ASMase accumulation on the cell surface through exocytosis, a similar mechanism of induction may be involved but requires further work to be fully understood.
ASMase has been shown to be an important factor in the adaptive and innate immune responses. It regulates the secretion of cytotoxic granules in T cells (26), a cell type that is largely incapacitated during EBOV infection (55). Furthermore, SM degradation stimulates monocyte differentiation (36, 41). As monocytes and macrophages are the primary targets of EBOV infection (6), EBOV-GP-induced recruitment as ASMase may impact behavior and differentiation of each of these cell types to alter the immune response.
In summary, we have identified an important role for ASMase and its substrate, SM, in EBOV infection. This work was performed with replication-competent ebolavirus at BSL4 and was supported by mechanistic analysis using pseudotyped virus particles and VLPs. ASMase appears to be recruited to sites of VLP binding or may induce binding of VLPs to cells. ASMase can be brought to the cell surface by an exocytosis-related mechanism (4, 51). This may also bring other endosome-localized proteins to the cell surface. This mechanism is very similar to that seen for measles virus, whereby the measles virus receptor CD150 is brought to the cell surface. It will be important to determine if the mechanism of recruitment is the same, as this will have implications for development of broad-spectrum treatments for viruses and other pathogens that require ASMase exocytosis for infection. Indeed our present work shows that vacuolin-1, a potent inhibitor of exocytosis, blocks EBOV infection at submicromolar concentrations. It will be interesting to determine if this or related drugs can be used in preventing infection and spread of virus in vivo.
ACKNOWLEDGMENTS
This work was supported in part by funding to R.A.D. from the NIH grants, 5R01AI063513-02 and 2 U54 AI057156-06. M.E.M. was supported by a U.S. Army long-term health education and training program.
We thank Olena Shtanko and Krishna Narayanan for discussion of experimental design and reading the manuscript, respectively. Robert Maldonado worked on VLP binding to drug-treated cells and microscopy. We also acknowledge Jia Wang, Mohammad Saeed, and Zeming Chen at UTMB as well as Yasuf Hannun, Russell Jenkins, and Christopher Clarke of the Medical University of South Carolina for providing reagents and advice on experimental design.
Footnotes
Published ahead of print 9 May 2012
REFERENCES
- 1. Alvarez CP, et al. 2002. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 76:6841–6844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Avota E, Gulbins E, Schneider-Schaulies S. 2011. DC-SIGN mediated sphingomyelinase-activation and ceramide generation is essential for enhancement of viral uptake in dendritic cells. PLoS Pathog. 7:e1001290 doi:10.1371/journal.ppat.1001290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ayala P, Vasquez B, Wetzler L, So M. 2002. Neisseria gonorrhoeae porin P1.B induces endosome exocytosis and a redistribution of Lamp1 to the plasma membrane. Infect. Immun. 70:5965–5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bao JX, et al. 2010. Triggering role of acid sphingomyelinase in endothelial lysosome-membrane fusion and dysfunction in coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 298:H992–H1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bavari S, et al. 2002. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 195:593–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bray M, Geisbert TW. 2005. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int. J. Biochem. Cell Biol. 37:1560–1566 [DOI] [PubMed] [Google Scholar]
- 7. Carette JE, et al. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cerny J, et al. 2004. The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep. 5:883–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cote M, et al. 2011. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477:344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Devlin C, et al. 2010. Improvement in lipid and protein trafficking in Niemann-Pick C1 cells by correction of a secondary enzyme defect. Traffic 11:601–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ebihara H, et al. 2007. In vitro and in vivo characterization of recombinant Ebola viruses expressing enhanced green fluorescent protein. J. Infect. Dis. 196(Suppl 2):S313–S322 [DOI] [PubMed] [Google Scholar]
- 12. Empig CJ, Goldsmith MA. 2002. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 76:5266–5270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feldmann H, Geisbert TW. 2011. Ebola haemorrhagic fever. Lancet 377:849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fernandes MC, et al. 2011. Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J. Exp. Med. 208:909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frecha C, et al. 2011. Measles virus glycoprotein-pseudotyped lentiviral vector-mediated gene transfer into quiescent lymphocytes requires binding to both SLAM and CD46 entry receptors. J. Virol. 85:5975–5985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaspar EB, Mortara RA, Andrade LO, da Silva CV. 2009. Lysosomal exocytosis: an important event during invasion of lamp deficient cells by extracellular amastigotes of Trypanosoma cruzi. Biochem. Biophys. Res. Commun. 384:265–269 [DOI] [PubMed] [Google Scholar]
- 17. Gassert E, et al. 2009. Induction of membrane ceramides: a novel strategy to interfere with T lymphocyte cytoskeletal reorganisation in viral immunosuppression. PLoS Pathog. 5:e1000623 doi:10.1371/journal.ppat.1000623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Geisbert TW, et al. 2000. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest. 80:171–186 [DOI] [PubMed] [Google Scholar]
- 19. Giocondi MC, Milhiet PE, Dosset P, Le Grimellec C. 2004. Use of cyclodextrin for AFM monitoring of model raft formation. Biophys. J. 86:861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grassme H, et al. 1997. Acidic sphingomyelinase mediates entry of N. gonorrhoeae into nonphagocytic cells. Cell 91:605–615 [DOI] [PubMed] [Google Scholar]
- 21. Grassme H, Riehle A, Wilker B, Gulbins E. 2005. Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J. Biol. Chem. 280:26256–26262 [DOI] [PubMed] [Google Scholar]
- 22. Gulbins E, Dreschers S, Wilker B, Grassme H. 2004. Ceramide, membrane rafts and infections. J. Mol. Med. 82:357–363 [DOI] [PubMed] [Google Scholar]
- 23. Gulbins E, Kolesnick R. 2002. Acid sphingomyelinase-derived ceramide signaling in apoptosis. Subcell. Biochem. 36:229–244 [DOI] [PubMed] [Google Scholar]
- 24. Gulbins E, Li PL. 2006. Physiological and pathophysiological aspects of ceramide. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290:R11–R26 [DOI] [PubMed] [Google Scholar]
- 25. Hauck CR, et al. 2000. Acid sphingomyelinase is involved in CEACAM receptor-mediated phagocytosis of Neisseria gonorrhoeae. FEBS Lett. 478:260–266 [DOI] [PubMed] [Google Scholar]
- 26. Herz J, et al. 2009. Acid sphingomyelinase is a key regulator of cytotoxic granule secretion by primary T lymphocytes. Nat. Immunol. 10:761–768 [DOI] [PubMed] [Google Scholar]
- 27. Hunt CL, Kolokoltsov AA, Davey RA, Maury W. 2011. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 85:334–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jan JT, Chatterjee S, Griffin DE. 2000. Sindbis virus entry into cells triggers apoptosis by activating sphingomyelinase, leading to the release of ceramide. J. Virol. 74:6425–6432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jenkins RW, et al. 2011. A novel mechanism of lysosomal acid sphingomyelinase maturation: requirement for carboxyl-terminal proteolytic processing. J. Biol. Chem. 286:3777–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kiyokawa E, et al. 2005. Spatial and functional heterogeneity of sphingolipid-rich membrane domains. J. Biol. Chem. 280:24072–24084 [DOI] [PubMed] [Google Scholar]
- 31. Koivusalo M, Jansen M, Somerharju P, Ikonen E. 2007. Endocytic trafficking of sphingomyelin depends on its acyl chain length. Mol. Biol. Cell 18:5113–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kolokoltsov AA, Davey RA. 2004. Rapid and sensitive detection of retrovirus entry by using a novel luciferase-based content-mixing assay. J. Virol. 78:5124–5132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kolokoltsov AA, Fleming EH, Davey RA. 2006. Venezuelan equine encephalitis virus entry mechanism requires late endosome formation and resists cell membrane cholesterol depletion. Virology 347:333–342 [DOI] [PubMed] [Google Scholar]
- 34. Kondratowicz AS, et al. 2011. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. U. S. A. 108:8426–8431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lacour S, et al. 2004. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 64:3593–3598 [DOI] [PubMed] [Google Scholar]
- 36. Langmann T, et al. 1999. Transcription factors Sp1 and AP-2 mediate induction of acid sphingomyelinase during monocytic differentiation. J. Lipid Res. 40:870–880 [PubMed] [Google Scholar]
- 37. Lloyd-Evans E, et al. 2008. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14:1247–1255 [DOI] [PubMed] [Google Scholar]
- 38. Marchesini N, Hannun YA. 2004. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem. Cell Biol. 82:27–44 [DOI] [PubMed] [Google Scholar]
- 39. Marsh M, van Meer G. 2008. Cell biology. No ESCRTs for exosomes. Science 319:1191–1192 [DOI] [PubMed] [Google Scholar]
- 40. Martinez O, Valmas C, Basler CF. 2007. Ebola virus-like particle-induced activation of NF-kappaB and Erk signaling in human dendritic cells requires the glycoprotein mucin domain. Virology 364:342–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mathias S, Kolesnick R. 1993. Ceramide: a novel second messenger. Adv. Lipid Res. 25:65–90 [PubMed] [Google Scholar]
- 42. McIntosh TJ, Simon SA, Needham D, Huang CH. 1992. Interbilayer interactions between sphingomyelin and sphingomyelin/cholesterol bilayers. Biochemistry 31:2020–2024 [DOI] [PubMed] [Google Scholar]
- 43. McIntosh TJ, Simon SA, Needham D, Huang CH. 1992. Structure and cohesive properties of sphingomyelin/cholesterol bilayers. Biochemistry 31:2012–2020 [DOI] [PubMed] [Google Scholar]
- 44. Mizuno H, et al. 2011. Fluorescent probes for superresolution imaging of lipid domains on the plasma membrane. Chem. Sci. 2:1548–1553 [Google Scholar]
- 45. Mulherkar N, Raaben M, de la Torre JC, Whelan SP, Chandran K. 2011. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 419:72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nanbo A, et al. 2010. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 6(9):e1001121 doi:10.1371/journal.ppat.1001121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. 2010. Cellular entry of Ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 6(9):e1001110 doi:10.1371/journal.ppat.1001110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saeed MF, Kolokoltsov AA, Freiberg AN, Holbrook MR, Davey RA. 2008. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 4:e1000141 doi:10.1371/journal.ppat.1000141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shimojima M, et al. 2006. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 80:10109–10116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Simmons G, et al. 2003. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 305:115–123 [DOI] [PubMed] [Google Scholar]
- 51. Tam C, et al. 2010. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 189:1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Towner JS, et al. 2005. Generation of eGFP expressing recombinant Zaire ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology 332:20–27 [DOI] [PubMed] [Google Scholar]
- 53. Trajkovic K, et al. 2008. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319:1244–1247 [DOI] [PubMed] [Google Scholar]
- 54. van Blitterswijk WJ, van der Luit AH, Veldman RJ, Verheij M, Borst J. 2003. Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem. J. 369:199–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Warfield KL, et al. 2005. Induction of humoral and CD8+ T cell responses are required for protection against lethal Ebola virus infection. J. Immunol. 175:1184–1191 [DOI] [PubMed] [Google Scholar]
- 56. Zeidan YH, Hannun YA. 2010. The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr. Mol. Med. 10:454–466 [DOI] [PubMed] [Google Scholar]
- 57. Zeidan YH, Jenkins RW, Hannun YA. 2008. Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J. Cell Biol. 181:335–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zenni MK, et al. 2000. Macropinocytosis as a mechanism of entry into primary human urethral epithelial cells by Neisseria gonorrhoeae. Infect. Immun. 68:1696–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zha X, et al. 1998. Sphingomyelinase treatment induces ATP-independent endocytosis. J. Cell Biol. 140:39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]