

Abstract
Viral mutational escape from CD8+ cytotoxic T lymphocytes (CTLs) is typically considered to be a dichotomous process and uncommon during chronic HIV-1 infection. Ex vivo passaging of HIV-1 from persons with chronic infection, however, revealed the evolution of many fixed substitutions within and around CTL-targeted regions, with an associated increase in replicative capacity. This indicates an evolution of mutations during chronic HIV-1 infection that trade replicative fitness for incomplete evasion of CTLs, or “partial escape.”
TEXT
HIV-1-specific CD8+ cytotoxic T lymphocytes (CTLs) exert a major selective pressure that shapes viral sequences (22) through evolution to avoid CTL antiviral activity (3, 18). Mutational escape from CTLs generally is considered an all-or-nothing phenomenon. Because persistence of a CTL response is driven by antigenic stimulation (allowing a waning of responses against cleared pathogens), mutational escape of an epitope typically has been associated with decay of the CTL response against that epitope (6, 13). In line with this scenario, it has been noted that rapid viral escape and CTL retargeting occur during early infection (6, 14, 18), while chronic infection is marked by stability of both epitope sequences and CTL targeting (16) or much-delayed epitope escape mutation (12, 13).
However, the generation of escape mutations can be limited by structural and functional constraints on viral replicative capacity (RC), and increasing data indicate that the options for evasion of CTLs targeting some epitopes are associated with substantial RC costs (7, 15, 19, 21, 26, 27). The reported examples of this phenomenon involve immunodominant epitopes restricted by human leukocyte antigen (HLA) alleles that are associated with superior immune containment of HIV-1 (B*13, B*27, B*57), suggesting that both CTL antiviral pressure and RC loss associated with escape contribute to the benefits of these HLA types. In these cases, escape tends not to be associated with decay of the CTL response. Thus, because these responses persist during chronic infection, there appears to be a situation where the optimal balance for HIV-1 is a tradeoff of maintaining RC for incomplete evasion of CTLs (driving CTL persistence). This intermediate scenario represents “partial escape.”
To investigate whether this phenomenon occurs for epitopes presented by other HLA types, the evolution of HIV-1 from persons with chronic infection was examined after ex vivo passaging in the absence of CTL selection to observe whether chronically CTL-targeted epitopes would “revert” as a reflection of reduced RC due to CTL pressure in vivo. HIV-1 cultures were established from four participants with chronic untreated HIV-1 infection and viremia of at least 5 × 103 log RNA copies per ml plasma (range, 7,500 to 19,000) who were enrolled in a University of California, Los Angeles, Institutional Review Board-approved study. None had the HLA B*13, B*27, or B*57 haplotype. The average duration of infection of these subjects was 14.3 years (median, 15; range, 6 to 21), with an average blood CD4+ T lymphocyte count of 395 cells/ml (range, 284 to 594).
HIV-1-specific targeting was defined by standard gamma interferon (IFN-γ) enzyme-linked immunosorbent spot assays (ELISpot) using 15-mer peptides overlapping by 11 amino acids and spanning the entire HIV-1 consensus subtype B sequence proteome (NIH AIDS Research and Reference Reagent Program), as previously described (5, 31, 32). Recognized epitope regions were defined as singly targeted peptides or the region of overlap between targeted consecutive overlapping peptides; in most cases, a likely minimal epitope could be inferred from the HLA type of the subject and known epitopes reported in the Los Alamos National Laboratory HIV Immunology Database (Table 1). The median number of epitope regions targeted by these four participants was 9.5, with a range of 9 to 17 (Fig. 1). The most highly targeted protein was Pol (median, 5.5 epitope regions; range, 2 to 8), followed by Gag (median, 3.5 epitope regions; range, 2 to 7).
Table 1.
Statistical analysis of amino acid changes within mapped CTL epitope regions
| Subject no. (HLA haplotype) | Epitope (aa range)a | HLA restriction | Amino acid substitutionsa | Fisher's exact test resultb | dN/dSc | Reversion to consensusd |
|---|---|---|---|---|---|---|
| 00017 (A02, 03; B44, 51; C04, 14) | Integrase (26–36) | B51 | L28I | P = 0.0277 | ns | No |
| 00019 (A02, 02; B44, 50; C06, 16) | Protease (69–83) | A02 | I77V | P = 0.0294 | 8.0 | Yes |
| Rev (17–31) | A02 | F21S | P = 0.0062 | ND | No | |
| Vpr (53–67) | A02 | S63I | P = 0.0297 | 6.4 | Yes | |
| 00025 (A02, 03; B15, 40; C02, 03) | p17 (9–23) | B40 | E11G | P = 0.0031 | ns | Yes |
| p17 (9–23) | B40 | K12E | P = 0.0031 | ns | Yes | |
| p17 (21–35) | A03 | R26K | P = 0.0288 | ns | Yes | |
| p17 (21–35) | A03 | Q28K | P = 0.0032 | ns | Yes | |
| p17 (21–35) | A03 | R30K | P = 0.0001 | ns | Yes | |
| p17 (21–35) | A03 | L34I | P = 0.0031 | ns | Yes | |
| p17 (49–63) | A02 | I61L | P = 0.0031 | ns | Yes | |
| p17 (73–83) | A02 | R76K | P < 0.00001 | ns | No | |
| p17 (73–83) | A02 | I82V | P < 0.00001 | 5.7 | No | |
| p17 (81–95) | A02 | R91K | P < 0.00001 | ns | No | |
| p17 (81–95) | A02 | V94I | P < 0.00001 | ns | No | |
| p17 (81–95) | A02 | R95K | P < 0.00001 | ns | Yes | |
| p24 (76–90) | A02, B40 | L83V | P < 0.00001 | ns | No | |
| p24 (76–90) | A02, B40 | P87H | P = 0.0006 | 1.5 | Yes | |
| p24 (136–146) | B15 | M136L | P = 0.0067 | 1.5 | Yes | |
| Protease (69–83) | A02 | Y69H | P < 0.00001 | ns | Yes | |
| Protease (69–83) | A02 | K70N | P < 0.00001 | ns | No | |
| Protease (69–83) | A02 | I77V | P < 0.00001 | ns | Yes | |
| RT (282–304) | B15 | I293V | P = 0.0001 | ND | No | |
| 00026 (A01, 02; B08, 44; C05, 07) | RT (82–92) | Unknown | K83R | P < 0.00001 | ns | Yes |
| Integrase (262–276) | B15, B42 | X263R | ns | 4.6 | Yes | |
| Integrase (278–288) | Unknown | X281V | ns | 4.6 | Yes |
Numbering according to the HXB2 reference sequence. aa, amino acid; X, any amino acid.
P value for Fisher's exact test. ns, not significant (P value > 0.05).
dN/dS, ratio of the rate (d) of nonsynonymous replacements (N) to the rate of synonymous replacements (S). ns, not significantly greater than 1; ND, not determined.
Determined by comparison to the Los Alamos National Laboratory HIV Sequence Database 2004 consensus B sequence.
Fig 1.
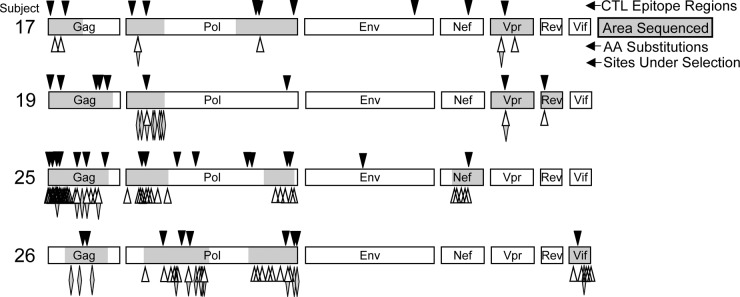
Summary of CTL targeting and associated amino acid changes. The black arrowheads mark the locations of CTL responses within the HIV-1 coding regions for each subject as determined by IFN-γ ELISpot mapping of CD8+ T lymphocytes using a consensus subtype B peptide library (no responses were seen against Vpu or Tat). The regions shaded in gray were sequenced before and after ex vivo passaging (see Fig. 2 for the exact sequence regions). Amino acid sites that changed with passaging are marked with open arrowheads. Amino acid sites that displayed evidence of positive selection with passaging are marked with shaded diamonds (see Tables 1 and 2 for the exact amino acid positions).
Virus was recovered from peripheral blood mononuclear cells by expanding CD4+ T lymphocytes using a CD3/CD8-bispecific antibody (29, 30, 33, 35). Recovered viruses were passaged weekly in freshly expanded CD4+ T lymphocytes from multiple, non-HLA-matched, healthy HIV-1-uninfected donors for 10 to 14 weeks (median, 11). All cells were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum, l-glutamine, penicillin, streptomycin, and recombinant human interleukin-2 at 50 U/ml (NIH AIDS Research and Reference Reagent Program). Sequencing of HIV-1 was performed on genomic DNA from the infected cell cultures using standard PCR cycling conditions and primer pairs from a previously described HIV-1 subtype B primer set; baseline sequences were obtained by plasma reverse transcription-PCR (RT-PCR) (4). Multiple PCR products were cloned using the TOPO TA kit (Invitrogen). Approximately 10 individual clones were sequenced (median, 9) for each epitope region (Fig. 1) pre- and postpassaging. All sequences were aligned with the Los Alamos National Laboratory HIV Sequence Database consensus sequences for subtype B using BioEdit and then analyzed for amino acid changes occurring after passaging.
Across all subjects, 26 amino acid changes within CTL-targeted epitope regions were noted between the consensus sequences of baseline and passaged viruses (Table 1). One participant (subject 25) had the greatest numbers of both amino acid changes and CTL responses (Fig. 1). Sequence changes were evaluated for statistical significance by Fisher's exact test as well as analyzed for selection pressure using the PAML software program and the likelihood method of Nielsen and Yang (36, 37). Of the 26 changes, 24 were statistically significant and 7 showed evidence of significant positive selective pressure by the ratio of the nonsynonymous mutation rate to the synonymous mutation rate (dN/dS > 1). A total of 17 of the 26 substitutions (65%) were changes from nonconsensus to the subtype B consensus amino acid, suggesting optimization of viral RC after passaging ex vivo.
As a more global measurement of whether the sequence evolution after ex vivo passaging reflected selective pressure, the diversity of pre- and postpassaging sequences was assessed using SENDBS with the HKY model and 500 bootstrap replicates (23). Sequence diversity tended to decrease after passaging, although this reached statistical significance only for four regions (Fig. 2). Overall, these results suggested the evolution of HIV-1 toward an optimized fittest sequence in the absence of CTL pressure, therefore resulting in a loss of diversity.
Fig 2.
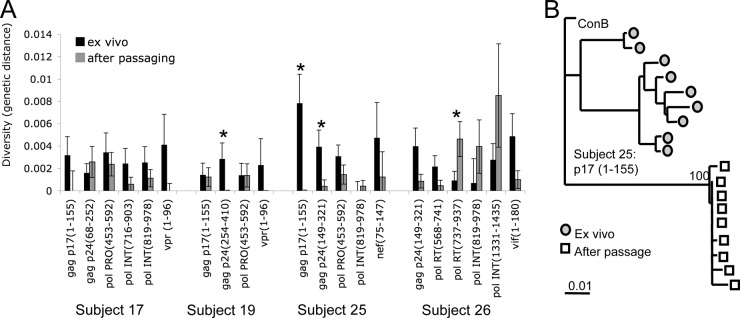
Changes in sequence diversity with passaging. (A) The average diversity of the initial ex vivo (black bars) and after passaging (gray bars) virus sequences was calculated and evaluated by 500 bootstrap replicates to give a mean and standard error. Significant differences in pre- and postpassaging values are marked with an asterisk (t test; P < 0.05). (B) A representative neighbor-joining phylogenetic tree for subject 25 p17 Gag (amino acids 1 to 155) shows that the passaged sequences have less diversity and form an independent cluster with 100% bootstrap support.
Several amino acids flanking the epitope regions also demonstrated change after passaging. Because changes in neighboring amino acids can affect epitope processing (10, 14, 28, 38) or compensate for a loss of RC from epitope escape mutations (19), the analysis was broadened to examine available sequences flanking the epitope regions, and at least 30 nucleotides (nt) and as much as 750 nt of flanking sequence were examined. Maximum likelihood and neighbor-joining phylogenetic trees (with 500 bootstrap replicates) were constructed using PHYLIP 3.64 (11) to determine the relationship between the initial and passaged viruses. For each subject, the passaged viruses clustered with strong bootstrap support for 13 of the 21 regions and did not intermingle with the initial sequences, indicating significantly directed evolution (Fig. 2B).
Next, the 13 regions demonstrating directed evolution were examined for differential selective pressure between the initial ex vivo and passaged viruses using the SelectionLRT program of HyPhy (25). Four regions demonstrated significantly changed dN/dS ratios, indicating that the ex vivo viruses were under substantially different selective pressure than the passaged viruses. The dN/dS ratios for the ex vivo clusters were <1 in all cases (mean, 0.41; range, 0.15 to 0.66), and the ratios along the branches separating the 2 groups were significantly higher in all cases (mean, 3.29; range, 0.77 to 7.30). These results indicated that purifying selection dominated in vivo and that evolution ex vivo without CTL pressure was dominated by positive selection.
A site-by-site analysis for selection revealed 24 sites under positive selection during ex vivo passaging (Table 2). Seven sites were within mapped targeted epitope regions for the subjects, 5 corresponded to known HLA-associated polymorphisms for HLA types of the subjects (provided by Simon Mallal; data not shown), and the remaining 12 did not fall within mapped epitope regions or correspond to known HLA-associated polymorphisms. The observation of positively selected sites in regions outside our defined epitope regions and the overall changes in diversity described above were consistent with escape or compensatory mutations outside the epitope regions or other escape mutations within epitopes that were missed in our CTL mapping. Overall, the data suggested that CTLs shape the evolution of protein regions via multiple changes within or flanking targeted epitopes through direct escape mutations and/or compensatory mutations.
Table 2.
Amino acids undergoing positive selection during passaging
| Subject no. | Protein | Amino acid changea | dN/dSb | P valuec |
|---|---|---|---|---|
| 00017 | Protease | X37A** | 51.5 | <0.01 |
| Vpr | X20L | 7.8 | ns | |
| 00019 | Protease | T37S** | 8.0 | <0.01 |
| Protease | E48G | 8.0 | <0.01 | |
| Protease | I77V* | 8.0 | <0.01 | |
| Vpr | S63I* | 6.4 | ns | |
| 00025 | Gag p17 | I82V* | 5.7 | ns |
| Gag p24 | P87H* | 1.5 | ns | |
| Gag p24 | X102 | 1.5 | ns | |
| Gag p24 | M136L* | 1.5 | ns | |
| Gag p24 | T171V | 1.5 | ns | |
| 00026 | Gag p24 | I91V | 3.4 | ns |
| Gag p24 | X120N** | 3.4 | ns | |
| Gag p24 | X171V | 3.4 | ns | |
| RT | D192N | 4.1 | 0.05 | |
| RT | V245T** | 4.1 | 0.05 | |
| RT | T286X | 4.1 | 0.05 | |
| RT | D324P | 4.1 | 0.05 | |
| RT | I329X** | 4.1 | 0.05 | |
| RT | Q334N | 4.1 | 0.05 | |
| Integrase | D232X | 4.6 | ns | |
| Integrase | V281X* | 4.6 | ns | |
| Integrase | R263G* | 4.6 | ns | |
| Vif | X147Y | 1.4 | ns |
Numbering according to the HXB2 reference sequence. X, any amino acid; *, within a CTL epitope determined by ELISpot mapping; **, although not within a CTL epitope as determined by ELISpot mapping, these mutations have been reported as associated with HLA alleles present in the subject's haplotype.
dN/dS, ratio of the rate (d) of nonsynonymous replacements (N) to the rate of synonymous replacements (S).
ns, not significant.
Finally, in order to determine whether the changes observed during adaptation in culture had a direct effect on viral fitness, the replication capacities of baseline and passaged viruses were compared. The GagPol coding regions from baseline and passaged samples were amplified and cloned in bulk into an NL4-3-based proviral clone, each with a different reporter gene, and then used to make recombinant reporter virus as previously described (1) (subject 19 was not tested because no remaining baseline sample was available.). The gagpol region was selected since this region contained most of the observed changes and is most likely to have a direct effect on the virus's ability to replicate in vitro. Reporter viruses were sequenced to confirm that they contained the expected polymorphisms and substitutions and that they were phylogenetically indistinguishable from the previously sequenced quasispecies at the respective time points. Paired baseline and passaged viruses were cocultured at low multiplicities of infection (MOIs), and each reporter copy number, normalized to the β-actin copy number, was measured by quantitative PCR (qPCR) on days 1, 3, and 5 as previously described (2). Replication rates, expressed as increases in log10 copies (normalized)/day, were compared (Fig. 3); for each subject, the passaged sample had a higher replication rate than the baseline sample, with the differences for subjects 17 and 26 being highly statistically significant at P values of 0.002 and 0.0004, respectively. This demonstrates that the sequence changes in GagPol observed after passage resulted in an increased replication capacity and implies that the mutations associated with CTL selective pressure at baseline indeed result in a diminished in vivo fitness.
Fig 3.

Comparison of the replication capacities of the ex vivo and passaged viruses. Recombinant NL4-3-based reporter viruses carrying the GagPol coding region from baseline and passaged viruses from subjects 17, 25, and 26 were created. Paired baseline and passaged samples, each with a different reporter, were grown simultaneously at low MOIs in coculture, and viral copy number was measured by qPCR on days 1, 3, and 5. The mean and standard deviation of the slope of the growth curve of each sample (log10 copies/day) were calculated based on duplicate infections. P values for the differences between baseline and passaged virus replication rates are shown above each group of paired samples.
Because CTL responses depend on antigenic stimulation, they decay when fully escaped. Thus, the observed stability of CTL responses during chronic infection demonstrates the lack of complete escape (16), which, in contrast, is seen frequently during acute infection (6, 14, 24), suggesting that the earliest escape mutations have low fitness costs that allow complete escape with little fitness cost. In contrast, however, escape mutations in immunodominant epitopes targeted during chronic infection by persons with the protective HLA types B*13, B*27, and B*57 are not associated with the loss of CTL responses against those epitopes (7, 15, 19, 21, 26, 27). In these cases, the high fitness costs for escape likely limit options to epitope variants that are either still recognized well enough to drive CTL proliferation or associated with such severe loss in RC that competing levels of the index virus persist. Thus, this appears to represent “partial escape,” where HIV-1 does not appear to have the capability to generate an epitope mutant in which the loss of RC is completely outweighed by the evasion of CTLs targeting that epitope. This situation leads to a net loss of fitness through a combination of residual CTL recognition and a loss of RC and is believed to be a major mediator of the protective effects of these HLA types.
While escape from CTLs otherwise has usually been considered a dichotomous process, our results suggest that this is not the case and suggest that escape during chronic infection likely falls along a continuum of partial escape for common nonprotective HLA types as well. Once HIV-1 is removed from the selective pressure of CTLs in vivo, targeted regions show amino acid substitutions and a loss of diversity that indicate optimization of fitness through reversion of escape mutations. The majority of substitutions correspond to consensus amino acids for subtype B, further supporting evolution toward the presumed fittest sequence. Some of these substitutions do not match the consensus sequence, suggesting either that the consensus residue represents a common escape mutation shared by the majority of persons (17, 20) or that the optimal amino acid is context dependent. Formal fitness assessments demonstrate that the observed sequence changes result in fitness gains, confirming that the evolution of sequences ex vivo reflects selection of fitter variants, in agreement with predictions that fitter members rapidly overgrow quasispecies (8). Thus, it appears that partial escape commonly imposes a fitness cost that contributes to CTL antiviral effects in chronic infection in the same manner as that shown for immunodominant responses mediated by HLA types associated with superior immune control.
Although we observed consistent trends that strongly suggest the phenomenon of partial escape, larger sample sizes with more subjects will be required to define the frequency of this process. More data also will be required to explore the pathways for epitope escape and factors determining the degree of fitness loss; in the context of escapes with high fitness costs for protective HLA types, there are examples of changes in the T cell receptor-binding residues (7, 19, 21) and HLA-binding anchor residue mutations (9, 26, 27), as well as flanking sequence mutation (10). Finally, a caveat to these studies is that fitness costs of sequence polymorphisms in vitro may differ from those in vivo, particularly for nonstructural proteins, e.g., Nef (34). Still, our results provide support for the phenomenon of partial escape and its contribution to the antiviral activity of CTLs.
Nucleotide sequence accession numbers.
GenBank accession numbers are as follows: subject 17 sequences, JQ927572 to JQ927633 and JQ928097 to JQ928119; subject 19 sequences, JQ927634 to JQ927693; subject 25 sequences, JQ927694 to JQ927779; and subject 26 sequences, JQ927780 to JQ927865 and JQ928120 to JQ928139.
ACKNOWLEDGMENTS
We thank our study subjects for their participation and Simon Mallal for providing data on HLA-associated sequence polymorphisms in HIV-1.
This work was supported by NIH/NIAID grant AI043203 (O.O.Y.).
Mirablle Dagarag is currently a senior research scientist at Diagnostic Products Corporation. Basim Khan was a medical student at the David Geffen School of Medicine at the University of California, Los Angeles.
Footnotes
Published ahead of print 2 May 2012
REFERENCES
- 1. Ali A, Jamieson BD, Yang OO. 2003. Half-genome human immunodeficiency virus type 1 constructs for rapid production of reporter viruses. J. Virol. Methods 110:137–142 [DOI] [PubMed] [Google Scholar]
- 2. Ali A, Yang OO. 2006. A novel small reporter gene and HIV-1 fitness assay. J. Virol. Methods 133:41–47 [DOI] [PubMed] [Google Scholar]
- 3. Allen TM, et al. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 79:13239–13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Altfeld M, et al. 2002. HIV-1 superinfection despite broad CD8+ T-cell responses containing replication of the primary virus. Nature 420:434–439 [DOI] [PubMed] [Google Scholar]
- 5. Balamurugan A, et al. 2008. Primary human immunodeficiency virus type 1 (HIV-1) infection during HIV-1 Gag vaccination. J. Virol. 82:2784–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Borrow P, et al. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3:205–211 [DOI] [PubMed] [Google Scholar]
- 7. Boutwell CL, Rowley CF, Essex M. 2009. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J. Virol. 83:2460–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coffin JM. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267:483–489 [DOI] [PubMed] [Google Scholar]
- 9. Crawford H, et al. 2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 81:8346–8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Draenert R, et al. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J. Exp. Med. 199:905–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Felsenstein J. 1989. PHYLIP—phylogeny inference package (version 3.2). Cladistics 5:164–166 [Google Scholar]
- 12. Goulder PJ, et al. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3:212–217 [DOI] [PubMed] [Google Scholar]
- 13. Jamieson BD, et al. 2003. Epitope escape mutation and decay of human immunodeficiency virus type 1-specific CTL responses. J. Immunol. 171:5372–5379 [DOI] [PubMed] [Google Scholar]
- 14. Jones NA, et al. 2004. Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J. Exp. Med. 200:1243–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kelleher AD, et al. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koibuchi T, et al. 2005. Limited sequence evolution within persistently targeted CD8 epitopes in chronic human immunodeficiency virus type 1 infection. J. Virol. 79:8171–8181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leslie A, et al. 2005. Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA. J. Exp. Med. 201:891–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Y, et al. 2006. Selection on the human immunodeficiency virus type 1 proteome following primary infection. J. Virol. 80:9519–9529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martinez-Picado J, et al. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80:3617–3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matthews PC, et al. 2009. HLA footprints on human immunodeficiency virus type 1 are associated with interclade polymorphisms and intraclade phylogenetic clustering. J. Virol. 83:4605–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miura T, et al. 2009. HLA-associated viral mutations are common in human immunodeficiency virus type 1 elite controllers. J. Virol. 83:3407–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moore CB, et al. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439–1443 [DOI] [PubMed] [Google Scholar]
- 23. Nei M, Jin L. 1989. Variances of the average numbers of nucleotide substitutions within and between populations. Mol. Biol. Evol. 6:290–300 [DOI] [PubMed] [Google Scholar]
- 24. Phillips RE, et al. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354:453–459 [DOI] [PubMed] [Google Scholar]
- 25. Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679 [DOI] [PubMed] [Google Scholar]
- 26. Prado JG, et al. 2009. Functional consequences of human immunodeficiency virus escape from an HLA-B*13-restricted CD8+ T-cell epitope in p1 Gag protein. J. Virol. 83:1018–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schneidewind A, et al. 2008. Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. J. Virol. 82:5594–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Troyer RM, et al. 2009. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 5:e1000365 doi:10.1371/journal.ppat.1000365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong JT, Colvin RB. 1987. Bi-specific monoclonal antibodies: selective binding and complement fixation to cells that express two different surface antigens. J. Immunol. 139:1369–1374 [PubMed] [Google Scholar]
- 30. Wong JT, Colvin RB. 1991. Selective reduction and proliferation of the CD4+ and CD8+ T cell subsets with bispecific monoclonal antibodies: evidence for inter-T cell-mediated cytolysis. Clin. Immunol. Immunopathol. 58:236–250 [DOI] [PubMed] [Google Scholar]
- 31. Yang OO, et al. 2005. Human immunodeficiency virus type 1 clade B superinfection: evidence for differential immune containment of distinct clade B strains. J. Virol. 79:860–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang OO, Daar ES, Ng HL, Shih R, Jamieson BD. 2011. Increasing CTL targeting of conserved sequences during early HIV-1 infection is correlated to decreasing viremia. AIDS Res. Hum. Retroviruses 27:391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang OO, et al. 1997. Suppression of human immunodeficiency virus type 1 replication by CD8+ cells: evidence for HLA class I-restricted triggering of cytolytic and noncytolytic mechanisms. J. Virol. 71:3120–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang OO, et al. 2003. Determinant of HIV-1 mutational escape from cytotoxic T lymphocytes. J. Exp. Med. 197:1365–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang OO, et al. 1997. Lysis of HIV-1-infected cells and inhibition of viral replication by universal receptor T cells. Proc. Natl. Acad. Sci. U. S. A. 94:11478–11483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13:555–556 [DOI] [PubMed] [Google Scholar]
- 37. Yang Z, Nielsen R, Goldman N, Pedersen AM. 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yokomaku Y, et al. 2004. Impaired processing and presentation of cytotoxic-T-lymphocyte (CTL) epitopes are major escape mechanisms from CTL immune pressure in human immunodeficiency virus type 1 infection. J. Virol. 78:1324–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]