

Abstract
Because productive infection by parvoviruses requires cell division and is enhanced by oncogenic transformation, some parvoviruses may have potential utility in killing cancer cells. To identify the parvovirus(es) with the optimal oncolytic effect against human glioblastomas, we screened 12 parvoviruses at a high multiplicity of infection (MOI). MVMi, MVMc, MVM-G17, tumor virus X (TVX), canine parvovirus (CPV), porcine parvovirus (PPV), rat parvovirus 1A (RPV1A), and H-3 were relatively ineffective. The four viruses with the greatest oncolytic activity, LuIII, H-1, MVMp, and MVM-G52, were tested for the ability, at a low MOI, to progressively infect the culture over time, causing cell death at a rate higher than that of cell proliferation. LuIII alone was effective in all five human glioblastomas tested. H-1 progressively infected only two of five; MVMp and MVM-G52 were ineffective in all five. To investigate the underlying mechanism of LuIII's phenotype, we used recombinant parvoviruses with the LuIII capsid replacing the MVMp capsid or with molecular alteration of the P4 promoter. The LuIII capsid enhanced efficient replication and oncolysis in MO59J gliomas cells; other gliomas tested required the entire LuIII genome to exhibit enhanced infection. LuIII selectively infected glioma cells over normal glial cells in vitro. In mouse models, human glioblastoma xenografts were selectively infected by LuIII when administered intratumorally; LuIII reduced tumor growth by 75%. LuIII also had the capacity to selectively infect subcutaneous or intracranial gliomas after intravenous inoculation. Intravenous or intracranial LuIII caused no adverse effects. Intracranial LuIII caused no infection of mature mouse neurons or glia in vivo but showed a modest infection of developing neurons.
INTRODUCTION
Each year there are over 14,000 new cases of malignant glioma in the United States, and these are associated with high mortality; glioblastoma multiforme (GBM), the most common malignant glioma, has a median survival of only 12 to 15 months, despite currently available surgical, radiotherapeutic, and chemotherapeutic treatments (48). Among the novel therapeutic approaches being investigated for GBM is the use of viral agents to selectively infect tumor cells (51, 56). According to the nature and design of the agent, tumor cell infection may have a number of therapeutic consequences, including direct cell lysis, expression of prodrug convertases to target chemotherapy to the tumor, stimulation of antitumor immunity, or expression of proteins to alter the tumor microenvironment. In the case of replication-competent oncolytic viruses, replication of the viral agent within infected cells allows progeny virus spread to neighboring tumor cells. A number of different viruses are being studied in the context of targeting cancer cells, including glioma; these include herpesvirus, adenovirus, reovirus, Newcastle disease virus, measles virus, vesicular stomatitis virus, and poliovirus (8, 29, 30, 34, 37, 51, 56).
Some autonomous parvoviruses of the genus Parvovirus (e.g., minute virus of mice [MVM], H-1, and LuIII) show an innately oncoselective replication and toxicity (21, 42). Infection is dependent on spontaneous cell transition through S phase (14), and therefore, nondividing cells such as mature neurons are not susceptible, an important safety feature when targeting glioma in the brain. Additionally, protein expression, DNA replication, and overall propagation of these viruses are generally favored in oncogenically transformed cells, whether transformed by naturally occurring mutations or experimentally transformed by oncogenic viruses, chemical mutation, or sequential transduction with specific oncogenes (9, 36, 55). In contrast to the Erythrovirus genus (e.g., parvovirus B19), there is no human disease associated with any member of the Parvovirus genus (16). Parvovirus-mediated oncosuppression has been observed in vivo for a number of tumor types (43), including GBM, against which H-1 parvovirus has shown particular promise in rat models (22). Recently, based on this and other promising preclinical studies (15, 26, 31, 43), a clinical phase I trial of H-1 against GBM has been initiated (clinical trials.gov NCT01301430). In addition, we and others (44, 53) have previously studied in vitro infection characteristics of murine parvovirus strains MVMi and MVMp. However, we are not aware of any broad survey of Parvovirus genus members against human GBMs to identify those viruses with the greatest natural propensity toward infection and oncolysis in GBMs or other cancers.
Here, we tested 12 wild-type parvoviruses and 2 recombinant viruses in a series of in vitro and in vivo experiments to identify which virus was most effective in infecting, replicating in, and killing human glioblastoma cells with minimal infection of normal brain cells. Among these 14 parvoviruses, LuIII, a virus originally isolated from a human tumor (24), showed the greatest ability to infect and kill a broad range of human glioma cells. We examined the underlying mechanism of LuIII efficacy and show that efficient multiple-cycle infection, as measured by a virus expansion assay, correlates with progressive GBM cell loss after infection at a low multiplicity of infection (MOI). We also addressed the mechanism of LuIII tropism for glioma by generating recombinant parvoviruses containing the LuIII capsid together with the NS1 protein of MVM. In vivo, direct injection of LuIII specifically infected intracranial U87 tumors and delayed growth of subcutaneous U87 tumors. Intravenous injection of LuIII specifically targeted intracranial or subcutaneous U87 tumors and spared normal tissue, including brain, liver, and spleen. Normal mice infected intravenously or intracranially with LuIII showed no symptomatic infection. We conclude that LuIII has a favorable efficacy and safety profile for treatment of human glioblastoma and therefore merits further development as a potential therapeutic agent for this disease.
MATERIALS AND METHODS
Viral stocks.
Parvoviral stocks were propagated in simian virus 40 (SV40)-transformed human kidney cell line 324K or HeLa cells and purified away from empty capsids over iodixanol gradients, and genome titers were established by Southern blotting and hybridization as described previously (19), to a radiolabeled oligonucleotide (5′-AAC TTT CCA TTT AAT GAC TGT ACC AAC AA-3′) whose sequence within the NS1 helicase region is highly conserved among parvoviruses. MVMp, the prototype strain of minute virus of mice, was recovered from plasmid pdbMVp (10). MVMi, the immunosuppressive strain, was recovered from plasmid pMVMi (20). MVM-G52, MVMc, and MVM-G17 are additional mutant strains of MVM (5). LuIII was derived from plasmid pLuIII-NdeI, described earlier (11). The H-1, H-3, tumor virus X (TVX), canine parvovirus (CPV), porcine parvovirus (PPV), and rat parvovirus 1A (RPV1A) stocks were propagated, and their titers were determined as for the rest. MVM-LuCap, described earlier (36), is a recombinant MVMp with the capsid gene (VP2) of LuIII substituted for the capsid gene of MVMp. MVM-LuCap/mP4 was constructed by cloning a mutated P4 promoter (mP4) from a recombinant MVMp/mP4 plasmid into MVM-LuCap. This promoter mutation, also termed “4-between,” is described in the text and elsewhere (35). Because there is no single cell type in which one can grow all the viruses tested in the present work, we cannot generate a monotonic PFU assay for all of these parvoviruses. Instead, to maintain a means of quantitative assessment of parvovirus MOI, we used a common measure of genomes/cell (g/c). Parvovirus stocks were kindly provided by Peter Tattersall (Yale University).
Cell culture.
Human glioma U87, U373, U118, MO59J, and A172, human kidney 324K, and mouse glioma GL261 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) with 10% fetal bovine serum (FBS), penicillin, streptomycin, nonessential amino acids, and 20 mM HEPES buffer. rU87 is U87 engineered to express red fluorescent protein (RFP) (34). Mouse brain cultures were propagated in neurobasal medium (Invitrogen) with B-27 serum-free supplement (Invitrogen). Human glial cell cultures were isolated through explant cultures and tested for immunoreactivity to glial fibrillary acidic protein (GFAP) (52). Human cell preparation and use were approved by the Yale University Human Investigation Committee. All cells were kept in a humidified atmosphere containing 5% CO2 at 37°C.
Cell growth assays.
Twenty-four-well plates were seeded with 1 × 104 cells per well and incubated overnight, and then cells were infected in triplicate with parvovirus at a high or a low MOI: 30,000 or 1,000 genomes per cell, respectively. Cell density in triplicate wells was measured by counting cells per field with the 10× objective of an Olympus Optical IX71 microscope with a SPOT-RT camera (Diagnostic Instruments, Sterling Heights, MI). The high-MOI experiment was performed twice, and the data shown are representative. Cell doubling time was calculated by assessing cell density as described above in uninfected wells on day 0 and day 2 and using the formula DT = t × ln2/ln(Nt/N0), where DT is doubling time, t is time interval between measurements (48 h), N0 is initial number of cells/high-power field (hpf), and Nt is number of cells/hpf at time t, as described elsewhere (41).
Virus expansion assays.
Cells were seeded at 2,500 cells per 6-mm-diameter spot on Teflon-coated “spot slides” (Cell-Line Associates, Inc., Newfield, NJ) and infected in duplicate the next day with purified viral stocks at 1,000 genomes per cell. Spot slide cultures were fixed at 24 or 72 hours postinfection (hpi) in 2.5% paraformaldehyde, permeabilized in 0.1% Triton X-100, and stained with monoclonal murine antibody against NS1 (54), and primary antibodies were detected with Texas Red-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). Nuclei were visualized by addition of DAPI (4′,6-diamidino-2-phenylindole). To assess the percentage of DAPI-stained nuclei expressing NS1, images were acquired with a Nikon OptiPhot epifluorescence microscope fitted with a Kodak digital camera driven by MDS 290 software.
Oncolysis assay.
Ten thousand cells were seeded per well in 24-well dishes and infected the next day with parvovirus at the indicated genomes per cell or mock infected. Cultures were incubated for 7 days, at which point dead cells were rinsed away, and any cells remaining attached to the surface were fixed and stained with Leishman's solution.
EtHD staining.
To label dead cells, ethidium homodimer (EtHD) was added to infected or uninfected control cultures at a final concentration of 4 μM. Triplicate wells for each condition were incubated with EtHD for 30 min at 37°C before assessment of cell number and EtHD+ (red fluorescent) cell number in multiple high-magnification fields.
Immunofluorescence.
Cultured cells or histology sections were fixed in 4% paraformaldehyde (20 min); rinsed with phosphate-buffered saline (PBS); permeabilized by being washed 5 times for 15 min each in PBS with 0.1% l-lysine, 1% bovine serum albumin (BSA), 0.4% Triton-X; blocked in washing buffer plus 2% normal horse serum (NHS); and exposed to primary antibody in blocking solution. Primary antibody was rabbit polyclonal antibody directed either against viral NS proteins (4) or against the LuIII coat (raised against purified LuIII capsids by Pocono Farms, Canadensis, PA). Cells were rinsed in PBS three times for 15 min each, exposed to donkey anti-rabbit antibody conjugated to Alexa Fluor (488 nm) (Invitrogen, Carlsbad, CA) in PBS with 0.4% Triton-X, and washed in PBS. Phase-contrast and fluorescent images were captured with an Olympus Optical (Tokyo, Japan) IX71 fluorescence microscope with a SPOT-RT camera (Diagnostic Instruments, Sterling Heights, MI).
Animal procedures.
Animal experiments were approved by and performed in accordance with the institutional guidelines of the Yale University Institutional Animal Care and Use Committee. Immunodeficient homozygous CB17-SCID mice 7 to 8 weeks of age were obtained from Taconic Farms (Germantown, NY). Bilateral subcutaneous flank tumors were established by injection of approximately 7 × 105 U373 cells mixed with 1.5 × 105 rU87 cells, in 50 μl sterile PBS. To establish intracranial tumors, mice were anesthetized by intraperitoneal injection of a combination of ketamine and xylazine (100 and 10 mg/kg of body weight, respectively) and then stereotactically injected with 20,000 rU87 cells in 1 μl of PBS into the striatum (2 mm lateral and 0.4 mm rostral to the bregma at a 3-mm depth). Virus was injected either via tail vein, by direct injection into subcutaneous tumors, or stereotactically into intracranial tumors, as described in the text. Subcutaneous tumor dimensions were measured by caliper, and tumor volume (V) was estimated by the formula for the volume of an ellipsoid of length a and uniform width b, i.e., V = 4/3 π(a/2)(b/2)(b/2). For histology, animals were killed with a pentobarbital overdose and perfused transcardially with 4% paraformaldehyde in PBS.
Statistical analysis.
Comparisons were made with 2-tailed unpaired t tests, using KaleidaGraph software v3.6 (Synergy Software). Ratios were analyzed after logarithmic conversion of data [x′ = log(x + 1)] as recommended for statistical analysis of ratios (45).
RESULTS
High-MOI screening of 12 parvoviruses for the ability to suppress human glioblastomas U87 and U373.
In order to identify the parvovirus(es) with the greatest oncolytic potential in human glioblastoma, we assembled a large and diverse 12-member panel of wild-type Parvovirus genus members. Strains of the mouse virus minute virus of mice (MVM) tested included the prototype strain (MVMp), the immunosuppressive strain (MVMi), MVM-G52, MVM-G17, and MVMc. The natural host of LuIII parvovirus, originally isolated as a contaminant of a human tumor (24), remains unknown. H-1 and H-3 (also known as Kilham rat virus) are rat parvoviruses more closely related to one another than to either MVM or LuIII (5). Other parvoviruses tested included porcine parvovirus (PPV), canine parvovirus (CPV), rat parvovirus 1A (RPV1A), and tumor virus X (TVX) (also with an unknown host [24]). Human glioblastomas U87 and U373 were seeded at a low density and infected the next day at a high MOI (30,000 genomes per cell). To assess the ability of tumor cells to proliferate, cell density was measured at 0, 1, 3, 5, and 7 days postinfection (dpi). Whereas other viruses that can infect glioma, for instance, vesicular stomatitis virus, may show a relatively low particle/infectivity ratio of 5 to 50 particles/PFU (3, 47), parvoviruses are relatively inefficient in binding, internalization, and nuclear trafficking (12), and the particle-to-infectivity ratio can be on the order of 500 to 1,000 particles/PFU (38). In parallel, single infected cells generate a very high number of parvovirus progeny particles. To cast a broad net to identify parvoviruses that show any promise of oncolytic targeting, we started with a high-MOI experiment.
Results for human glioblastoma U87 are shown in Fig. 1A. By 7 dpi, mock-infected U87 grew to well over 600% of the original cell density. LuIII was unique among all 12 parvoviruses in its ability to prevent U87 cell proliferation after high-MOI infection. Not only was cell proliferation blocked, but cell density in LuIII-infected cultures at 7 dpi was significantly reduced to 14% of its original value (P < 0.0001), indicating that only LuIII was able to kill the majority of cells after high-MOI infection. Cells remaining attached to the culture dish were rare in LuIII-infected cultures 7 dpi, as shown by the micrograph in Fig. 1B (strong oncosuppression). In contrast, cells infected with parvoviruses MVMp, MVMi, MVM-G52, MVM-G17, MVMc, and H-1 showed cell proliferation, although at a reduced rate relative to that of mock-infected cells, reaching between 300 and 500% of their original density by 7 dpi (Fig. 1A). A representative micrograph for these infections is seen in Fig. 1B (moderate oncosuppression). There was little to no effect on cell growth after infection by RPV1A, TVX, CPV, PPV, and H-3 (Fig. 1A), demonstrating an inability of these viruses to significantly impede U87 cell growth, even after high-MOI infection (Fig. 1B, negligible oncosuppression).
Fig 1.

Screening 12 parvoviruses against human glioblastoma U87. (A) U87 cells were infected with a panel of 12 parvoviruses at a high multiplicity of infection (MOI) (30,000 genomes per cell) or mock infected. Cell density was assessed 0, 1, 3, 5, and 7 dpi by microscopy. G52, MVM-G52; G17, MVM-G17. Infection phenotypes were grouped as described in the text by the magnitude of the oncosuppressive effect at this high MOI, demonstrating negligible (dotted lines), moderate (dashed lines), or strong (solid lines) oncosuppression. Bars, standard errors of the means. (B) Representative micrographs from 7 dpi are shown.
We next tested a different human glioma to determine if the results from U87 generalized to other gliomas. High-MOI screening results for human glioblastoma U373 are shown in Fig. 2. Mock-infected cells grew to over 600% of their original density by 5 dpi. Infection of U373 by five parvoviruses (LuIII, MVMp, MVM-G52, H-1, and H-3) at a high MOI was able to significantly reduce cell density below its original value by 7 dpi (Fig. 2A, P < 0.005 for all); a representative micrograph from 7 dpi is shown in Fig. 2B (strong oncosuppression). MVMc and MVM-G17 were not able to prevent or reverse cell growth; however, U373 infected by these viruses grew at a lower rate than did mock-infected cells (Fig. 2B, moderate oncosuppression). In contrast, U373 infected with MVMi, CPV, PPV, TVX, or RPV1A grew at a rate comparable to that of mock-infected cells (Fig. 2B, negligible oncosuppression). With the exception of MVMc, MVM-G17, and MVMi, the experiments with all the other viruses shown in Fig. 1 and 2 were repeated, independently, and yielded highly similar results; in both instances, LuIII, H-1, MVMp, and MVM-G52 were identified as the top overall performers.
Fig 2.

Screening 12 parvoviruses against glioblastoma U373. (A) U373 cells were infected with a panel of 12 parvoviruses at a high MOI (30,000 genomes per cell) or mock infected. Cell density was assessed 0, 1, 3, 5, and 7 dpi by microscopy. G52, MVM-G52; G17, MVM-G17. Infection phenotypes were grouped as described in the text by the magnitude of the oncosuppressive effect at this high MOI, demonstrating negligible (dotted lines), moderate (dashed lines), or strong (solid lines) oncosuppression. Bars, standard errors of the means. (B) Representative micrographs from 7 dpi are shown.
Progressive infection and oncolysis after low-MOI infection of five human glioblastomas.
In the high-MOI screen of 12 parvoviruses against two human GBM lines, only four viruses were able to achieve strong oncosuppression in at least one line and at least moderate oncosuppression in the other line: LuIII, H-1, MVMp, and MVM-G52. We therefore studied these four viruses in low-MOI experiments designed to be more sensitive to differences in viral efficiency. Furthermore, to better determine the effective host range of these parvoviruses across a broader variety of human glioblastomas, we used five different human glioblastoma derivatives, each with a different set of gene mutations; in addition to U87 and U373, we tested human glioblastomas A172, U118, and MO59J.
When susceptible cells are infected by parvovirus at a low MOI, only a minority of cells are infected. If the rate of viral replication is sufficient in these cells, progeny virus will go on to infect an ever-increasing proportion of the proliferating glioma cells over time; here, we use the term “progressive” for this phenotype. At a lower rate of viral replication, the percentage of cells infected may stay constant or decrease over time as the cells proliferate, resulting in a “balanced” or “restrictive” infection, respectively (46). We hypothesized that the infection phenotype for any particular parvovirus-cancer cell pair should correlate with the ability to kill a GBM culture after low-MOI infection. The in vitro expansion assay mimics, and is designed to predict, the relative ability of a virus to progressively infect a tumor in vivo that is only partially infected after initial exposure to virus. We infected all five human GBM lines at a low MOI (1,000 genomes/cell) with MVMp, MVM-G52, LuIII, and H-1; inocula were washed off and replaced with fresh medium after 2 h of infection. Parallel slides were fixed 24 hpi or 72 hpi and immunostained with polyclonal antibody against viral NS1 protein, a protein with a high level of homology in different parvoviruses. The percentage of cells infected for each virus at both time points is shown in Fig. 3A. At 24 hpi, only a minority of cells (below 15%) were infected in all cases. At 72 hpi, the percentage of cells infected by LuIII was significantly greater than the percentage of cells infected at 24 hpi in all five glioblastomas (P < 0.05 for all), indicating a progressive infection phenotype for LuIII in all five lines. By this criterion, H-1 progressively infected U373 and MO59J but showed only balanced or restrictive infection in the other three lines. In contrast, MVMp and MVM-G52 did not show progressive infection in any of these cell lines.
Fig 3.

Low multiplicity of infection of five human glioblastomas: virus expansion and cell growth. (A) Expansion assay. Five human glioblastoma lines were infected at a low MOI (1,000 genomes per cell) with LuIII, H-1, MVMp, or MVM-G52 (G52). The percentage of cells expressing viral NS1 was assessed at 24 and 72 hpi. Here, infection is characterized as progressive when the percentage of cells infected 72 hpi is greater than the percentage infected 24 hpi. Progressively infecting viruses are shown by solid lines (others are shown by dotted lines). (B) Cell growth with low-MOI parvovirus. The growth of five human glioblastomas was assessed at a low MOI (1,000 genomes per cell) with LuIII, H-1, MVMp, or MVM-G52 (G52) or after mock infection. Cell density at 0, 1, 3, 5, and 7 dpi was assessed by microscopy. Viruses able to suppress or reverse cell growth are shown by solid lines (others are shown by dotted lines).
The capacity for progressive infection theoretically translates to the ability to infect the entire culture over time, and thus, a low-MOI parvovirus infection should be able to arrest and reverse the proliferation of infected cells by means of an ever-increasing rate of virus progeny production and cell lysis. To test this prediction, we infected five different GBMs at the same low MOI used above (1,000 genomes per cell, seen in Fig. 3A to be sufficient to infect only a minority of cells initially [<15%]) and cell density was measured every other day until 7 dpi (Fig. 3B). Consistent with the results of the expansion assay, LuIII arrested and reversed cell proliferation in all five cell lines when infection was initiated at a low MOI. Also consistent with our prediction, H-1 arrested and reversed cell growth only in those cell lines in which it had demonstrated progressive infection: U373 and MO59J (compare Fig. 3A and Fig. 3B). Neither MVMp nor MVM-G52 reduced growth of any glioblastoma cell line, consistent with their inability to progressively infect these lines.
Mechanisms underlying LuIII infection of GBM.
We found LuIII to be the most broadly effective oncolytic agent against human GBM, as it could progressively infect all five GBM lines tested. In contrast, the next best parvovirus, H-1, progressively infected and consequently reversed cell growth only in U373 and MO59J. We examined the relative contributions of the two major LuIII proteins, the nonstructural NS1 and the capsid protein VP2, in GBM oncolysis. We used a recombinant chimeric parvovirus, MVM-LuCap, based on MVMp, in which the MVM VP2 capsid gene is replaced by its LuIII VP2 homolog (Fig. 4A).
Fig 4.
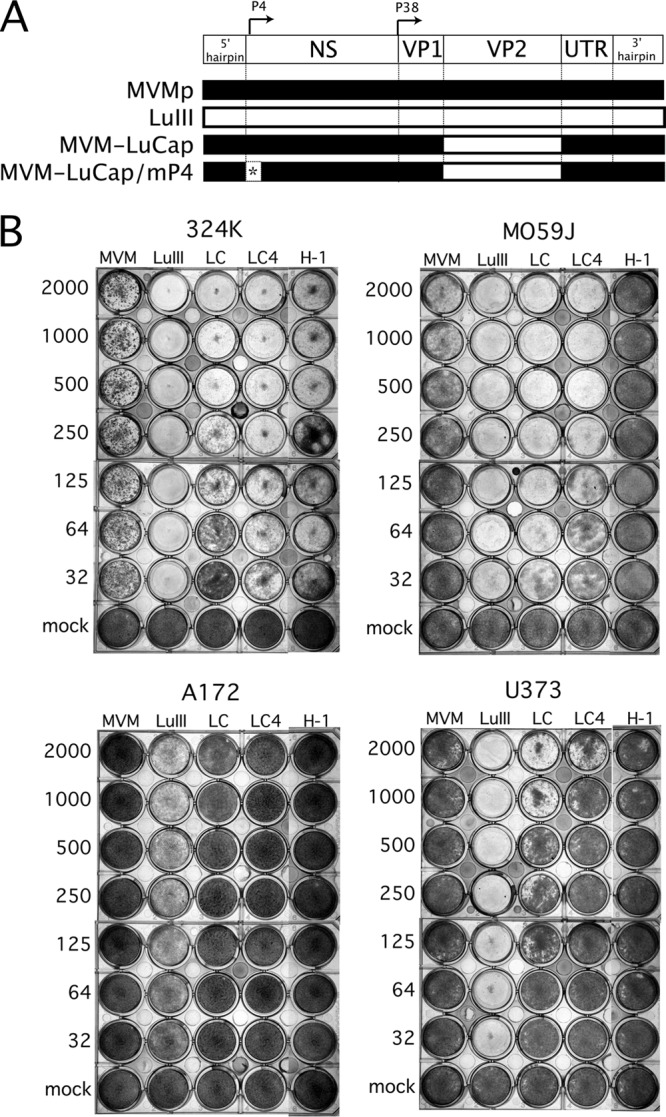
Influence of parvoviral genotype on GBM tropism and killing. (A) Genetic map indicating the design of chimeric MVM-LuCap and MVM-LuCap/mP4. P4 and P38 promoters are shown; the “4-between” P4 mutation (mP4) is indicated by an asterisk. (B) Oncolysis assay. Serially diluted inocula of five different parvoviruses were tested for killing against broadly susceptible control cells 324K and against three human glioblastoma lines—M0595, U373, and A172. Subconfluent cells were infected at the indicated number of genomes per cell and were stained for viable cells 7 dpi with Leishman's solution. A dark well is indicative of a healthy GBM monolayer; wells that appear white or clear are ones in which the GBM cells have been killed by the respective parvovirus. LC, MVM-LuCap; LC4′, MVM-LuCap/mP4.
The parvoviral P4 promoter drives NS gene expression in parvoviruses. Into the P4 promoter of MVM-LuCap, we engineered a mutation which consists of a single-base-pair deletion, thus generating MVM-LuCap/mP4 (Fig. 4A). This mutation reduces P4 promoter affinity for cellular parvoviral initiation factor (PIF) (which regulates genomic replication), thereby facilitating interactions of cellular transcription factors with the promoter's overlapping CRE (cyclic AMP response element), which is a Ras-responsive (oncoselective) component of the P4 promoter (7, 40). The P4 promoter mutation enhanced the oncoselectivity of MVMp in SV40-transformed human fibroblast line 324K (35); MVM-LuCap/mP4 had not been previously tested. These two recombinant viruses, together with wild-type viruses MVMp, LuIII, and H-1, were tested for oncolytic potency against parvovirus-permissive 324K cells, as well as three human glioblastomas, MO59J, U373, and A172. Serial dilutions of virus were added to subconfluent cells at multiplicities of infection ranging between 32 and 2,000 viral genomes per cell (g/c) and allowed to incubate for 7 days, at which time any remaining attached cells were stained (Fig. 4B).
Control cultures of 324K cells grew to confluence. In contrast, all five viruses effectively prevented 324K cells from growing to confluence, even at the lower MOI, consistent with an ability of these viruses to progressively infect these cultures after initially infecting only a small minority of cells. Although all viruses were effective, LuIII had the greatest potency, followed by MVM-LuCap/mP4, H-1, MVM-LuCap, and then MVMp. MVM-LuCap/mP4 showed an advantage over parental MVM-LuCap in 324K cells (compare at 64 and 32 g/c), consistent with the enhancement that this mutation confers on MVMp in these cells (35).
In MO59J, LuIII was considerably better than MVMp and H-1 at glioma suppression, particularly at a low MOI. Importantly, MVMp-LuCap was as effective as LuIII, and both viruses were superior to MVMp, indicating that the LuIII VP2 capsid gene alone is sufficient to confer on MVM a robust expansion and killing phenotype in these cells. In contrast to our findings in 324K cells, however, in MO59J the P4 promoter mutation was detrimental rather than beneficial to MVM-LuCap oncolysis, as MVM-LuCap/mP4 showed less oncolysis than did MVM-LuCap. In both U373 and A172 glioma cells, LuIII was substantially more effective than each of the other viruses, including the MVMp-LuCap and MVMp-LuCap/mP4 recombinant viruses, indicating that in these cell types, as distinct from 324K and MO59J, the LuIII capsid gene in MVM-LuCap is not sufficient to allow robust expansion and killing. Still, in U373, MVM-LuCap does show some advantage over the parental MVMp (Fig. 4B; compare at 2,000 g/c). Thus, although the LuIII capsid enhances infection in two of three gliomas tested, the complete LuIII genome, including both the NS and VP genes, confers the broadest overall glioma-killing phenotype for parvoviruses. Furthermore, in contrast to findings in 324K cells, the P4 promoter mutation was detrimental to infection and oncolysis in at least two GBMs (MO59J and U373).
To corroborate the view that LuIII infection resulted in cell death, rather than simply repression of mitosis, we infected GBM cells in culture with LuIII (10,000 genomes/cell) and, at 72 and 96 hpi, stained parallel cultures with ethidium homodimer (EtHD), a dye that selectively labels dead cells with a fluorescent red color (Fig. 5A to D). Between 72 and 96 hpi, cell density increased in uninfected cultures but decreased by over 35% in infected cultures (P < 0.01, data not shown), consistent with an ongoing oncolytic effect. Further evidence of an oncolytic mechanism of LuIII oncosuppression comes from the finding that cell death rate, as measured by EtHD staining, was over 5-fold higher in infected cultures than in uninfected cultures (P < 0.0001) (Fig. 5E). The percentage of dead cells is an underestimate, as soon after dying, cells detach from the culture substrate and float into the medium, leaving cellular debris on the culture substrate.
Fig 5.
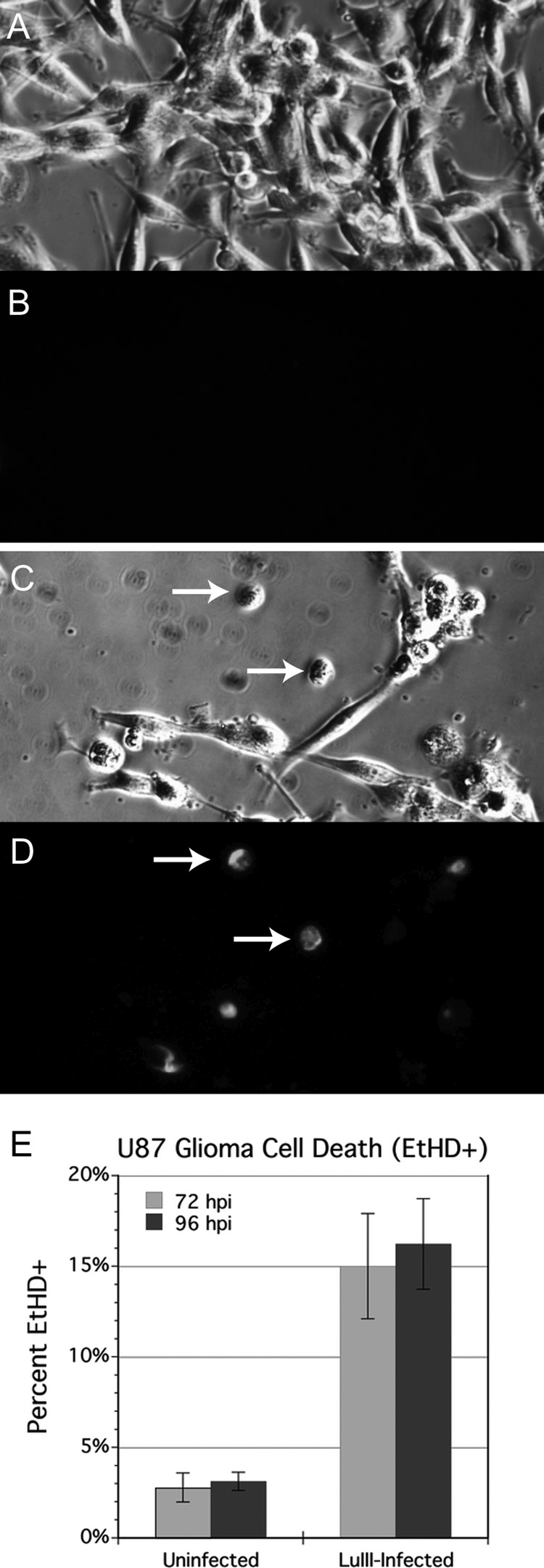
Oncolytic effect of LuIII on U87 glioma. Cultures were uninfected (A and B) or infected with LuIII at 10,000 genomes per cell (C and D). Cell density and ethidium homodimer (EtHD) staining for dead cells (red fluorescence) were assessed in parallel wells at 72 and 96 hpi (shown). Incidence of cell death as measured by EtHD staining was at least 5 times greater in LuIII-infected cultures (E).
LuIII is highly selective for glioblastoma versus normal glia in vitro irrespective of cell proliferation rate.
Given the broad and potent efficacy of LuIII in our panel of human glioblastomas, we sought to gauge how selective LuIII infection is for tumor cells over normal cells. Parvovirus infection is S phase dependent and therefore should be highly selective for tumors in the mature brain, which contains primarily nondividing neurons as well as rarely dividing glial cells. Cell proliferation rate is a potential confounder when analyzing the oncoselectivity of a virus whose growth is enhanced by factors expressed in S phase. Due to dysregulation of the cell cycle, tumor cells typically proliferate at a rate higher than that of their corresponding cell of origin. To control for cell proliferation rate, we examined the infectivity of LuIII in human glia, human glioma U87, and U87 in which the cell proliferation rate was diminished by growing the cells in 1% fetal bovine serum (FBS). In addition to testing the infectivity of LuIII at 24 hpi (MOI, 8,000 genomes/cell; Fig. 6A and C), the doubling time of each of these three cultures was determined, as described in Materials and Methods, in parallel uninfected wells (Fig. 6B). As expected, the doubling time of U87 grown in 10% serum (29 ± 6.6 h) was faster than that of human glia grown in 10% serum (50 ± 3.5 h). However, culturing U87 in a reduced serum concentration (1%) prolonged the doubling time to a value (57 ± 11 h) even greater than that of glia. The infectivity of LuIII 24 hpi (percent staining for NS1) was 3.6% (±0.4%) for human glia. In U87 grown in 10% serum, infectivity was 49.9% (±5.7%), a 13.8-fold-greater infectivity than that in glia. In U87 grown in 1% serum, LuIII infectivity was 45% (±3.1%), which is statistically equivalent to infectivity in U87 grown in 10% serum and 12.5-fold greater than that observed in glia.
Fig 6.

LuIII is highly selective for glioblastoma versus normal glia in vitro irrespective of cell proliferation rate. Human glial cultures (hGlia) and human glioblastoma (U87) were infected with LuIII at 8,000 genomes per cell. In parallel cultures, U87 were grown in 1% FBS, to increase the cell doubling time. Cell doubling times (B) were determined in parallel wells. At 24 hpi, cells were fixed and immunostained for viral NS protein to determine infectivity rates (A). Representative micrographs are shown in panel C.
Intratumoral LuIII reduces the growth of subcutaneous glioblastomas.
To assess the oncolytic efficacy of LuIII in vivo, tumors were established in 10 SCID mice, using a 5:1 mixture of U373 and rU87 cells, and mice were evenly divided into a treatment group and a control group; some mice received two tumor cell injections. rU87 glioblastoma cells were engineered to express red fluorescent protein (RFP) (34). To allow us to measure tumor size on an ongoing basis, a subcutaneous tumor site was used. Twelve tumors were generated that reached a minimum 2-mm diameter. When a tumor in the treatment group reached a 2-mm diameter, 1 × 1010 genomes of LuIII were injected directly into the treatment group tumors (n = 7), and on day 4, treated tumors were given an equivalent second dose of LuIII. Five tumors were untreated controls and were considered to reach day zero when they were 2 mm in diameter. Tumor dimensions were measured every other day, and the volume was calculated; results are shown in Fig. 7A. LuIII reduced tumor growth substantially, achieving statistical significance (P < 0.05 by unpaired t test) by 9 dpi and continuing thereafter. Over the 21 days of the experiment postinoculation, untreated tumors grew 59-fold on average, whereas LuIII-treated tumors grew 14.4-fold, a 4-fold difference. The experiment was halted after 21 dpi because some of the tumors had reached a maximum allowable size (1 cm3), per animal use regulations. In a parallel experiment, LuIII-treated subcutaneous rU87 tumors were harvested between 12 and 14 dpi and immunostained for viral protein NS1. Many red tumor cells showed NS1 immunoreactivity, consistent with ongoing replication of LuIII (Fig. 7B). Virus-infected cells were found in scattered regions of the tumor. No LuIII immunostaining was found in normal tissue around the tumor.
Fig 7.
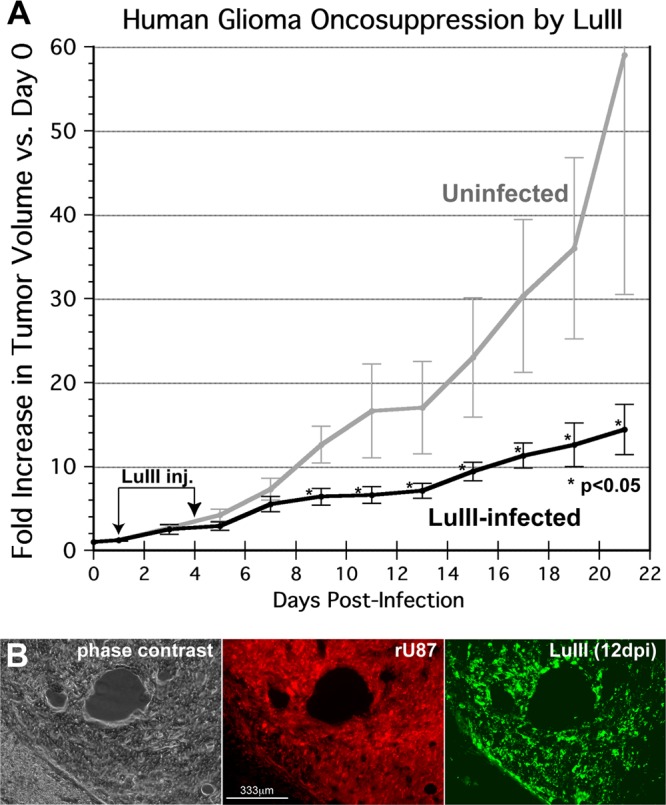
Intratumoral LuIII inhibits growth of subcutaneous human GBM tumors. (A) Tumor volume. LuIII parvovirus (1 × 1010 genomes) was injected in 7 tumors, and 5 tumors were left uninfected. Tumor dimensions were measured daily by caliper; the average of tumor volumes relative to day 0 is shown. Bars, standard errors of the means. (B) Histology, 12 dpi. Phase-contrast, red filter (red fluorescent rU87 tumor cells), and green filter (immunostaining for viral NS and VP proteins) micrographs of a treated tumor at 12 dpi are shown.
Intracranial rU87 tumors are selectively infected by LuIII after intratumoral injection.
Intracranial rU87 tumors were established in the striatum of SCID mice (n = 5) and directly injected on day 20 with 1010 genomes of LuIII in a volume of 1 μl. Histological examination of two brains immunostained for viral protein at 6 dpi and one brain at 9 dpi revealed selective infection of all tumors, infecting approximately 40 to 80% of each tumor (Fig. 8A, B, and C). Normal brain cells outside the region of the tumor showed no sign of infection (Fig. 8D and E), as was the case for liver and spleen (Fig. 8F, G, H, and I) as assessed by NS immunostaining.
Fig 8.

rU87 tumors are selectively infected in vivo by LuIII. Intracranial tumors were stereotactically injected directly with 1 × 1010 genomes of LuIII. Panel A (phase contrast) shows the tumor and normal surrounding tissue; panels B (red fluorescent tumor) and C (green immunostaining for NS1) are from a representative mouse sacrificed 6 dpi. The virus infects only the tumor; no infection is seen in normal brain tissue. Enlargements in panels D and E show the specificity of LuIII infection. Liver (F) and spleen (H) from the mouse shown in panel A show no immunostaining for virus (G and I, respectively). Four days after intravenous administration of LuIII in SCID mice, subcutaneous tumors (phase contrast [J] and red fluorescent tumor [K]) show substantial infection by immunofluorescence (L); intracranial tumors (M and N) were also partly infected 3 days after intravenous LuIII (O).
Intravenous LuIII targets glioma implanted into the brain or subcutaneously.
After unilateral intracranial and bilateral subcutaneous rU87 tumors were established in SCID mice, 1011 genomes of LuIII were injected intravenously (tail vein) in a volume of 100 μl. Histological examination with NS1 immunostaining 6 days after virus inoculation revealed multiple sites of infection in both subcutaneous (Fig. 8J, K, and L) and intracranial (Fig. 8M, N, and O) tumors but not in surrounding normal tissue. Two of three mice showed virus infection of both subcutaneous tumors and brain tumors after intravenous inoculation; one mouse showed no immunostaining for NS1 in its brain tumor. In some cases, infection was widespread in a significant region of the tumor (Fig. 8L); in other tumors, the level of infection was less substantial. These data suggest that LuIII has the potential ability both to target peripheral tumors and to cross the blood-brain barrier to infect cerebral tumors. Based on NS1 immunocytochemistry, there was no evidence for infection in normal brain tissue, liver, or spleen after intravenous virus application.
LuIII is minimally pathogenic.
To test virus safety in the brain, seven normal adult Swiss Webster mice were injected intracranially with 5 × 109 genomes of LuIII. Weight loss in all animals was minimal (7% or less) and transient, and all animals gained weight and were asymptomatic over the course of a month. Histological examination of brain, liver, and spleen in two randomly selected mice sacrificed 5 weeks postinfection revealed no evidence of viral protein expression (Fig. 9A and B). No obvious cytopathic effects were seen.
Fig 9.
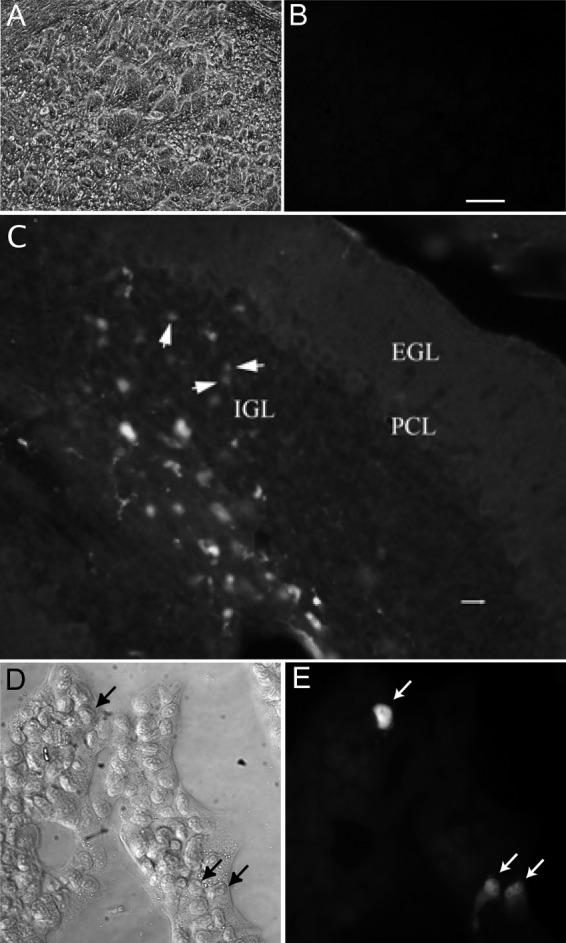
LuIII infection of murine brain and glioma. (A and B) LuIII was injected directly into the brain (striatum) of normal adult Swiss Webster mice. Immunostaining showed no evidence of virus infection in a set of serial sections through the injection sites. (A and B) Phase-contrast (A) and green fluorescence (B) images are shown; bar, 150 μm. (C) Eight-day-old Swiss Webster pups were injected in the cerebellum with LuIII. Infected cells were found in the internal granule cell layer (IGL), as detected with LuIII NS immunostaining. The micrograph shown is 6 days postinoculation. Bar, 14 μm. EGL, external granule cell layer; PCL, Purkinje cell layer. (D and E) Murine glioma was infected with LuIII (8,000 genomes/cell) and immunostained for viral NS1 protein 24 hpi. Phase-contrast (D) and immunofluorescence (E) images are shown.
Five normal adult Swiss Webster mice were injected via tail vein with 1010 genomes of LuIII in a volume of 100 μl. All mice survived to beyond 30 days (at which point they were euthanized) and exhibited no signs of illness throughout other than transient and minimal (<6%) weight loss. Histological examination of brain, liver, and spleen in two randomly selected mice sacrificed 7 weeks postinfection revealed no evidence of viral protein expression (data not shown), and all organs showed a normal appearance.
To test further the safety of LuIII, we used severely immunocompromised SCID mice. Six adult SCID mice were injected via tail vein with 1 × 1010 genomes of LuIII in a volume of 100 μl. All mice survived to beyond 4 weeks (at which point they were euthanized), exhibiting no symptoms and nothing more than transient and minimal (<7%) weight loss. That these mice remained healthy in the absence of a substantive T or B cell response underlines the high safety level of the virus. No adult SCID and Swiss Webster mice showed any obvious sign of deteriorating health, and no neurological symptoms were detected.
In adult mice, we found no evidence of infection of neurons even after direct injection of LuIII into the brain. As parvoviruses are dependent on cell division for productive infection, this raises the possibility that developing brain cells during the period of mitosis might be infected. To test the hypothesis that LuIII might infect developing neurons, we used neonatal mice at postnatal day 8, a time when cerebellar granule cells are still undergoing mitosis. Eleven Swiss Webster neonates were injected intracranially with 1 × 109 genomes of LuIII, eight into the cerebellum and three into the cortex. In cortex or other rostral regions of the brain, we found no infected cells in neonatal brain at 4 dpi, 9 dpi, or 30 dpi. However, we found a small number of infected cells in the cerebellum of three mice, one euthanized 4 dpi and two mice sacrificed at 6 dpi (Fig. 9C). These data suggest that postmitotic brain cells in the adult show no infection by LuIII but that developing murine brain cells undergoing cell division may be infected by LuIII.
Given this evidence that LuIII can and does infect murine cells, we also tested a murine glioma line, GL261, for LuIII infectivity. At an MOI of 8,000 genomes/cell, less than 0.1% of mouse normal glia were infected after 24 hpi (not shown). In contrast, 1.5% of mouse GL261 glioma cells were infected (Fig. 9D and E) during the same interval, a level of infectivity more than 10-fold greater. These data suggest that LuIII has a preference for mouse glioma over normal mouse glia. Similar concentrations of virus in human glioma generated a level of infection over an order of magnitude greater than that in mouse glioma, as shown above, consistent with a preference of LuIII for human glioma over mouse glioma. Similarly, we found lower levels of LuIII infectivity in other mouse glioma and melanoma cell lines relative to their human counterparts (data not shown).
DISCUSSION
Although a wide variety of parvovirus genus members have been identified, studies of parvoviral growth in human glioblastomas appear to have been limited to MVM (44, 53) and H-1 (1, 26). In our hands, the majority of parvoviruses that we tested showed only weak or insubstantial effectiveness in infection and destruction of human glioma; these included MVMi, MVMc, MVM-G17, TVX, CPV, PPV, RPV1A, and H-3. Our high-MOI screen identified LuIII, H-1, MVMp, and MVM-G52 as having the greatest potential. For any parvovirus-cell type pair, our characterization of in vitro infection as progressive depends on whether the infected proportion of the cultured cancer cells increases over time due to efficient production of infectious progeny viruses. Of all parvoviruses that we tested, only LuIII infection was progressive in five of five human glioblastomas infected at a low virus multiplicity, resulting in complete death of all gliomas in vitro. LuIII was particularly effective in U373 and MO59J; low-MOI LuIII (32 genomes per cell), infecting only a small fraction of the cells initially, progressively infected and killed all glioma cells.
Mechanism of LuIII action.
Our experiments with MVM-LuCap compared to MVMp show that the LuIII capsid plays a role in facilitating infection of human GBM, at least in the case of two gliomas, MO59J and U373. This is consistent with previous studies showing the critical importance of parvoviral capsids at determining host cell range, evidently operating at a postentry, pre-gene-expression step early in the viral life cycle (6, 12, 36). The role of the autonomous parvoviral capsid in governing species and tissue tropism is thus analogous to the key role that the capsid of parvovirus adeno-associated virus (AAV) plays in defining its tissue tropism (2). Although the LuIII capsid enhanced fitness of MVMp in human glioma MO59J, the entire LuIII genome, including untranslated regions and nonstructural protein genes, was required for a robust oncolysis of U373 and A172 gliomas. The major nonstructural protein, NS1, has multiple vital functions, including genome replication and packaging. NS1 also plays a critical role in cytotoxicity; in the absence of infection, NS1 expression alone leads to cell death, particularly in transformed cells (33). Our data showing that wild-type LuIII has greater cytolytic actions in glioma than does recombinant LuIII with the MVM NS1 protein (MVM-LuCap) suggest that the LuIII NS1 protein might be sufficiently different to confer a cytolytic advantage. Parvoviruses can induce apoptosis of glioblastomas, mediated via cathepsins, even in cells resistant to chemotherapeutic drug-induced, caspase-mediated apoptosis (15). The natural host of LuIII, isolated from a human tumor, is unknown; however, it has an affinity for replicating in transformed human tissues over rodent tissues (25), a property which may make it more suited to cytolytic infection of human tumors than the mouse virus MVMp or the rat virus H-1.
Parvoviral oncoselectivity.
LuIII is very selective for human glioma compared to normal human glia. Even when the proliferation rates of normal glia and glioma were matched, LuIII infectivity was more than 12-fold greater in glioma. The selectivity of LuIII for glioma therefore derives only in part from the parvoviral dependence on S phase (14). Additional oncoselective features of parvoviruses include elements within the viral P4 promoter which drive NS gene expression, including a cyclic AMP-responsive element (CRE) (40). The P4 mutation that we employed was designed to enhance oncoselectivity, particularly in ras-upregulated cells, by allowing greater access of CRE to its cognate cellular transcription factors (7, 35). This mutation enhanced MVMp infection of 324K cells (35) and also enhanced infection by MVMp-LuCap in 324K cells. In striking contrast, it did not enhance infection in any of three glioma lines tested and was even detrimental in two lines. The P4 mutation employed here, therefore, can exert either a positive or a negative impact on viral propagation efficiency, depending on the host cell.
Safety in the brain.
As neurons in the central nervous system (CNS) show little capacity for replication or regeneration, it is important that any virus that targets brain tumors show minimal infection of normal brain cells. Glia in the brain occasionally divide, and yet we found that even in glia proliferating in culture, LuIII infectivity is greatly reduced relative to glioma. Direct injection of LuIII either into a glioma within the brain or directly into the brain did not result in detectable infection of normal cells of any type. Intravenous or direct inoculation of high-MOI LuIII showed no obvious infection outside the implanted gliomas. Even infections of immunocompromised SCID mice, which have a deficient T and B cell response to infection, caused no adverse effects or lethality. Although LuIII shows a general preference for human over murine glioma, the virus did infect mouse glioma and dividing mouse brain cells, and thus, this species preference is not an absolute restriction. LuIII is also reported not to infect embryonic human tissue (24). Consideration of the safety of LuIII in humans will benefit from studies of species that are more closely related, particularly nonhuman primates.
Although we found no sign of infection in the adult brain, we did find some infection by LuIII in the developing cerebellum of neonatal mice. We purposefully injected the virus into the cerebellum at postnatal day 8 as granule cells are still undergoing mitosis, in contrast to the rest of the brain, where mitosis has already been completed. Infected cerebellar cells were found in the internal granule cell layer; as granule cells divide in the external layer and then migrate past the Purkinje cells into the internal granule cell layer, our data suggest that the cells may have been infected during replication in one region of the cerebellum and then the infected cell migrated into a nearby region. We found no sign of infection in the adult cerebellum.
Parvoviral targeting of glioblastoma in vivo.
LuIII specifically targets human GBM in vivo; after intratumoral or intravenous administration, infection was detected selectively in U87 tumors in SCID mice. This is consistent with studies of H-1 specifically targeting U87 in athymic RNU rats (22). LuIII substantially depressed the growth of subcutaneous U87 tumors in mice in the present study. Repeated administration of LuIII may further enhance its efficacy, as it does for other parvoviruses (22).
Other parvoviruses such as H-1 can initiate an antitumor immune response by the adaptive immune system (22, 31). An antitumor immunogenicity would enhance LuIII's oncolytic potential. That LuIII's progressive infection phenotype may directly kill more tumor cells than does that of H-1 may also enhance the ability of LuIII to evoke an immune response against the infected tumor. Supporting evidence for this view comes from the findings that a cytotoxic treatment in vivo that killed melanocytes stimulated, in a dose-dependent manner, an effective antimelanoma immune response (13).
LuIII versus H-1 and MVM.
Of the parvoviruses previously considered to show oncolytic potential, H-1 has received the most attention for a role as an antiglioma agent and is currently in human clinical trials (clinical trials.gov NCT01301430), this being primarily due to studies from the Division of Tumor Virology at the German Cancer Research Center in Heidelberg. Our data suggest that LuIII also merits consideration for clinical trials. In our hands, LuIII was superior to H-1 in targeting and killing a broader range of human gliomas, and at a lower MOI. Where low levels of LuIII killed all human gliomas tested, H-1 was successful in only two of five gliomas, suggesting that H-1 may have a more restricted range among human gliomas. The efficacy of H-1 was previously studied in two rat GBM lines, a human glioma line (U343MG), and several short-term cultures of primary human gliomas (15, 26). Killing of rat glioma RG2 by H-1, a rat parvovirus, was highly efficient. In contrast, H-1 infection of the human glioma line U343MG, and 3 of 4 primary human gliomas, when performed at an MOI 50-fold higher than that for the rat glioma, slowed but did not prevent continued cell proliferation through 6 dpi; only 1 of 4 human gliomas showed a reduction in cell number (26). The continued growth of most human gliomas even with high-MOI H-1 infection suggests inefficient and nonprogressive viral replication. Overall, the results of previous studies are consistent with our own in that H-1 demonstrated significant variability among human gliomas in its killing efficiency, sometimes able to reverse cell proliferation and sometimes not.
Although MVMp is efficient in targeting rodent tumors (17, 23, 28, 50), its effectiveness is reduced in human tumor cells (18), and this property may be dependent on the coat protein. When the MVMp capsid is replaced with that of LuIII, virus growth is dramatically enhanced in in human glioblastoma (present study) and in human fibroblasts transformed by hTERT, Ras, and SV40 large T antigen (36). The superiority of LuIII over MVM-LuCap in glioma further suggests that the NS of LuIII is likewise superior to that of MVM in human glioma.
The five glioma lines used here were selected for their high degree of genetic heterogeneity; they vary, for example, in p53, epidermal growth factor (EGF) receptor, mdr, DNA PK repair, p14, p16, interferon (IFN), and transforming growth factor beta (TGF-β) status. They share some mutation of PTEN (27, 32). The ability of LuIII to progressively infect and kill a diverse array of gliomas suggests that the efficient infection and cytolytic action of LuIII generalize to a variety of different human glioblastoma genotypes, an advantage in targeting tumor cells which are genetically heterogeneous not only among separate tumors (39) but even within different cells in a single tumor (49).
In sum, given the broad range of efficient LuIII replication among human gliomas, the absence of neurotoxicity of LuIII, and the ability to selectively target gliomas in vivo after systemic administration, we conclude from our studies that LuIII is an important viral candidate meriting further consideration for therapy of malignant glioblastoma and that both the LuIII capsid and NS1 nonstructural protein are critical for maximal effectiveness.
ACKNOWLEDGMENTS
We are indebted to Peter Tattersall for kindly providing many parvovirus stocks, for sharing lab resources, and for many helpful suggestions. We thank G. Wollmann, M. Robek, S. Ahmadi, and W. Andres for manuscript suggestions and Y. Yang, J. Davis, and V. Rogulin for technical assistance.
Grant support was provided by NIH RO1 CA 029303, NIH RO1 CA161048, and NIH RO1 CA 124737.
Footnotes
Published ahead of print 2 May 2012
REFERENCES
- 1. Abschuetz A, et al. 2006. Oncolytic murine autonomous parvovirus, a candidate vector for glioma gene therapy, is innocuous to normal and immunocompetent mouse glial cells. Cell Tissue Res. 325:423–436 [DOI] [PubMed] [Google Scholar]
- 2. Arruda VR, Xiao W. 2007. It's all about the clothing: capsid domination in the adeno-associated viral vector world. J. Thromb. Haemost. 5:12–15 [DOI] [PubMed] [Google Scholar]
- 3. Arstila P. 1976. Quantitative studies on adsorption, elution, and haemagglutination of vesicular stomatitis virus. Arch. Virol. 51:51–58 [DOI] [PubMed] [Google Scholar]
- 4. Ball-Goodrich LJ, Tattersall P. 1992. Two amino acid substitutions within the capsid are coordinately required for acquisition of fibrotropism by the lymphotropic strain of minute virus of mice. J. Virol. 66:3415–3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Besselsen DG, Romero MJ, Wagner AM, Henderson KS, Livingston RS. 2006. Identification of novel murine parvovirus strains by epidemiological analysis of naturally infected mice. J. Gen. Virol. 87:1543–1556 [DOI] [PubMed] [Google Scholar]
- 6. Brownstein DG, et al. 1992. The pathogenesis of infection with minute virus of mice depends on expression of the small nonstructural protein NS2 and on the genotype of the allotropic determinants VP1 and VP2. J. Virol. 66:3118–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burnett E, Tattersall P. 2003. Reverse genetic system for the analysis of parvovirus telomeres reveals interactions between transcription factor binding sites in the hairpin stem. J. Virol. 77:8650–8660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Candolfi M, et al. 2010. Gene therapy-mediated delivery of targeted cytotoxins for glioma therapeutics. Proc. Natl. Acad. Sci. U. S. A. 107:20021–20026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cornelis JJ, et al. 1988. Transformation of human fibroblasts by ionizing radiation, a chemical carcinogen, or simian virus 40 correlates with an increase in susceptibility to the autonomous parvoviruses H-1 virus and minute virus of mice. J. Virol. 62:1679–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cotmore SF, Tattersall P. 1992. In vivo resolution of circular plasmids containing concatemer junction fragments from minute virus of mice DNA and their subsequent replication as linear molecules. J. Virol. 66:420–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cotmore SF, Tattersall P. 2005. Genome packaging sense is controlled by the efficiency of the nick site in the right-end replication origin of parvoviruses minute virus of mice and LuIII. J. Virol. 79:2287–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cotmore SF, Tattersall P. 2007. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 70:183–232 [DOI] [PubMed] [Google Scholar]
- 13. Daniels GA, et al. 2004. A simple method to cure established tumors by inflammatory killing of normal cells. Nat. Biotechnol. 22:1125–1132 [DOI] [PubMed] [Google Scholar]
- 14. Deleu L, Pujol A, Faisst S, Rommelaere J. 1999. Activation of promoter P4 of the autonomous parvovirus minute virus of mice at early S phase is required for productive infection. J. Virol. 73:3877–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Piazza M, et al. 2007. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J. Virol. 81:4186–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dupont F. 2003. Risk assessment of the use of autonomous parvovirus-based vectors. Curr. Gene Ther. 3:567–582 [DOI] [PubMed] [Google Scholar]
- 17. Dupont F, et al. 2000. Tumor-selective gene transduction and cell killing with an oncotropic autonomous parvovirus-based vector. Gene Ther. 7:790–796 [DOI] [PubMed] [Google Scholar]
- 18. Enderlin M, et al. 2009. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. 16:149–160 [DOI] [PubMed] [Google Scholar]
- 19. Farr GA, Tattersall P. 2004. A conserved leucine that constricts the pore through the capsid fivefold cylinder plays a central role in parvoviral infection. Virology 323:243–256 [DOI] [PubMed] [Google Scholar]
- 20. Gardiner EM, Tattersall P. 1988. Evidence that developmentally regulated control of gene expression by a parvoviral allotropic determinant is particle mediated. J. Virol. 62:1713–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geletneky K, Herrero y Calle M, Rommelaere J, Schlehofer JR. 2005. Oncolytic potential of rodent parvoviruses for cancer therapy in humans: a brief review. J. Vet. Med. B Infect. Dis. Vet. Public Health 52:327–330 [DOI] [PubMed] [Google Scholar]
- 22. Geletneky K, et al. 2010. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol. 12:804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giese NA, et al. 2002. Suppression of metastatic hemangiosarcoma by a parvovirus MVMp vector transducing the IP-10 chemokine into immunocompetent mice. Cancer Gene Ther. 9:432–442 [DOI] [PubMed] [Google Scholar]
- 24. Hallauer C, Kronauer G, Siegl G. 1971. Parvoviruses as contaminants of permanent human cell lines. I. Virus isolation from 1960–1970. Arch. Gesamte Virusforsch. 35:80–90 [DOI] [PubMed] [Google Scholar]
- 25. Hallauer C, Siegl G, Kronauer G. 1972. Parvoviruses as contaminants of permanent human cell lines. 3. Biological properties of the isolated viruses. Arch. Gesamte Virusforsch. 38:366–382 [DOI] [PubMed] [Google Scholar]
- 26. Herrero y Calle M, et al. 2004. Parvovirus H-1 infection of human glioma cells leads to complete viral replication and efficient cell killing. Int. J. Cancer 109:76–84 [DOI] [PubMed] [Google Scholar]
- 27. Ishii N, et al. 1999. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 9:469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Legrand C, Mousset S, Salome N, Rommelaere J. 1992. Cooperation of oncogenes in cell transformation and sensitization to killing by the parvovirus minute virus of mice. J. Gen. Virol. 73:2003–2009 [DOI] [PubMed] [Google Scholar]
- 29. Lichty BD, Power AT, Stojdl DF, Bell JC. 2004. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol. Med. 10:210–216 [DOI] [PubMed] [Google Scholar]
- 30. Markert JM, et al. 2000. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 7:867–874 [DOI] [PubMed] [Google Scholar]
- 31. Moehler MH, et al. 2005. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum. Gene Ther. 16:996–1005 [DOI] [PubMed] [Google Scholar]
- 32. Nister M, et al. 1988. Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-alpha and their receptors in human malignant glioma cell lines. Cancer Res. 48:3910–3918 [PubMed] [Google Scholar]
- 33. Op De Beeck A, et al. 2001. NS1- and minute virus of mice-induced cell cycle arrest: involvement of p53 and p21(cip1). J. Virol. 75:11071–11078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ozduman K, Wollmann G, Piepmeier JM, van den Pol AN. 2008. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J. Neurosci. 28:1882–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paglino J, Burnett E, Tattersall P. 2007. Exploring the contribution of distal P4 promoter elements to the oncoselectivity of minute virus of mice. Virology 361:174–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paglino J, Tattersall P. 2011. The parvoviral capsid controls an intracellular phase of infection essential for efficient killing of stepwise-transformed human fibroblasts. Virology 416:32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paglino JC, van den Pol AN. 2011. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: rare resistance is overcome by blocking interferon pathways. J. Virol. 85:9346–9358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paradiso PR. 1981. Infectious process of the parvovirus H-1: correlation of protein content, particle density, and viral infectivity. J. Virol. 39:800–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parsons DW, et al. 2008. An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perros M, et al. 1995. Upstream CREs participate in the basal activity of minute virus of mice promoter P4 and in its stimulation in ras-transformed cells. J. Virol. 69:5506–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ramasamy R, et al. 2012. Basic fibroblast growth factor modulates cell cycle of human umbilical cord-derived mesenchymal stem cells. Cell Prolif. 45:132–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rommelaere J, Cornelis JJ. 1991. Antineoplastic activity of parvoviruses. J. Virol. Methods 33:233–251 [DOI] [PubMed] [Google Scholar]
- 43. Rommelaere J, et al. 2010. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 21:185–195 [DOI] [PubMed] [Google Scholar]
- 44. Rubio MP, Guerra S, Almendral JM. 2001. Genome replication and postencapsidation functions mapping to the nonstructural gene restrict the host range of a murine parvovirus in human cells. J. Virol. 75:11573–11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ryder EF, Robakiewicz P. 2001. Statistics for the molecular biologist: group comparisons. Curr. Protoc. Mol. Biol. 67:A.3I.1–A.3I.22 doi:10.1002/0471142727.mba03is43 [DOI] [PubMed] [Google Scholar]
- 46. Tattersall P, Bratton J. 1983. Reciprocal productive and restrictive virus-cell interactions of immunosuppressive and prototype strains of minute virus of mice. J. Virol. 46:944–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thompson KA, Yin J. 2010. Population dynamics of an RNA virus and its defective interfering particles in passage cultures. Virol. J. 7:257 doi:10.1186/1743-422X-7-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wen PY, Kesari S. 2008. Malignant gliomas in adults. N. Engl. J. Med. 359:492–507 [DOI] [PubMed] [Google Scholar]
- 49. Westphal M, Lamszus K. 2011. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat. Rev. Neurosci. 12:495–508 [DOI] [PubMed] [Google Scholar]
- 50. Wetzel K, et al. 2001. Transduction of human MCP-3 by a parvoviral vector induces leukocyte infiltration and reduces growth of human cervical carcinoma cell xenografts. J. Gene Med. 3:326–337 [DOI] [PubMed] [Google Scholar]
- 51. Wollmann G, Ozduman K, van den Pol AN. 2012. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J. 18:69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. 2010. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J. Virol. 84:1563–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wollmann G, Tattersall P, van den Pol AN. 2005. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J. Virol. 79:6005–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yeung DE, et al. 1991. Monoclonal antibodies to the major nonstructural nuclear protein of minute virus of mice. Virology 181:35–45 [DOI] [PubMed] [Google Scholar]
- 55. Zeicher M, et al. 2003. Oncoselective parvoviral vector-mediated gene therapy of cancer. Oncol. Res. 13:437–444 [DOI] [PubMed] [Google Scholar]
- 56. Zemp FJ, Corredor JC, Lun X, Muruve DA, Forsyth PA. 2010. Oncolytic viruses as experimental treatments for malignant gliomas: using a scourge to treat a devil. Cytokine Growth Factor Rev. 21:103–117 [DOI] [PubMed] [Google Scholar]