

Abstract
Interferon (IFN) regulatory factors (IRFs) are a family of transcription factors involved in regulating type I IFN genes and other genes participating in the early antiviral host response. To better understand the mechanisms involved in virus-induced central nervous system (CNS) inflammation, we studied the influence of IRF1, -3, -7, and -9 on the transcriptional activity of key genes encoding antiviral host factors in the CNS of mice infected with lymphocytic choriomeningitis virus (LCMV). A key finding is that neither IRF3 nor IRF7 is absolutely required for induction of a type I IFN response in the LCMV-infected CNS, whereas concurrent elimination of both factors markedly reduces the virus-induced host response. This is unlike the situation in the periphery, where deficiency of IRF7 almost eliminates the LCMV-induced production of the type I IFNs. This difference is seemingly related to the local environment, as peripheral production of type I IFNs is severely reduced in intracerebrally (i.c.) infected IRF7-deficient mice, which undergo a combined infection of the CNS and peripheral organs, such as spleen and lymph nodes. Interestingly, despite the redundancy of IRF7 in initiating the type I IFN response in the CNS, the response is not abolished in IFN-β-deficient mice, as might have been expected. Collectively, these data demonstrate that the early type I IFN response to LCMV infection in the CNS is controlled by a concerted action of IRF3 and -7. Consequently this work provides strong evidence for differential regulation of the type I IFN response in the CNS versus the periphery during viral infection.
INTRODUCTION
Type I interferons (IFNs) (predominantly IFN-α/β) are among the earliest cytokines to be produced in the innate host response to a viral infection. The initiation of this response relies on the sensing of viral invasion by different types of cellular pathogen-recognizing receptors (PRRs) (26, 38). They include several of the Toll-like receptors (TLRs), together with an increasing number of cytosolic receptors detecting nucleic acid sequences indicative of viral presence within the host cell (24, 26, 27, 30, 38, 41, 49, 56, 59). Upon engagement of the relevant PRRs, several transcription factors, including interferon regulatory factors (IRFs), activator protein 1 (AP-1), and NF-κB, are activated and translocated into the nucleus to induce the expression of proinflammatory cytokines and type I IFNs. Induction of a broad range of type I IFNs and IFN-stimulated genes (ISGs) involves several members of the IRF family. In fibroblasts and epithelial cells, the production of type I IFNs is biphasic, and in the earliest response, phosphorylation of IRF3 leads to the production of small amounts of IFN-β and IFN-α4 (34, 51). The secreted IFNs act in a paracrine/autocrine manner with locally expressed type I IFN receptors (IFNARs), and this interaction causes several important secondary response molecules, including IRF7, to become upregulated in the type I IFN-exposed cells. As a result of IRF7 induction, the expression of all type I IFN species is facilitated, and the full-range type I IFN response is triggered (20, 52). In plasmacytoid dendritic cells (pDCs), the sequence of events is slightly different from that described above, in part because IRF7 is constitutively expressed at a high level in these cells. For this reason, pDCs respond very efficiently, even at the earliest sensing of viral infection, and large amounts of IFN-α are rapidly produced and released from the cells (3, 47).
Other IRFs also play a role in regulating the early type I IFN response. IRF1 may be involved in CpG-B induced IFN-β production (40) and has recently been found to play a crucial role in virus-induced, IPS-1-dependent signaling from peroxisomes (13). In addition, upon binding of type I IFN to its receptors, IRF9 associates with phosphorylated STAT1/STAT2 dimers to form the canonical IFN-activated transcription factor ISGF3. ISGF3, as well as IRF3 and IRF7, may bind to IFN response elements (ISREs) present in the promoters of ISGs and activate the transcription of these genes, which limit viral replication through multiple mechanisms (48, 53).
Originally, IFNs were described based on their capacity to induce and activate a number of proteins that inhibit various steps in the viral life cycle (54, 55). Protein kinase R and 2,5-oligoadenylate synthase (2,5-OAS) are classical examples of this type of molecule, which directly interferes with viral replication in type I IFN-exposed cells (2, 15, 22, 28). However, type I IFNs also profoundly impact the capacity of responding cells to produce a variety of cytokines and chemokines, e.g., CXCL10, which are essential for activation of endothelial cells and recruitment of innate effector cells, e.g., NK cells and monocytes, to the site of infection. Thus, the induction of a potent type I IFN response seems to represent a cornerstone in the early innate response to viral infection. However, many details regarding the early regulation of the antiviral host response and the precise role of type I IFNs in this context are still unknown, and interestingly, as more and more information regarding different viral infections in different organ sites is becoming available, the number of variations on the overall theme keeps increasing.
In this report, we have focused on studying the roles of key IRFs in controlling the early type I interferon-dependent response to infection of the central nervous system (CNS) with the prototypic arenavirus, lymphocytic choriomeningitis virus (LCMV). Understanding the regulation of the type I IFN host response in the virus-infected CNS is particularly important, as appropriate balancing of the inflammatory process in this sensitive organ is of the utmost importance with regard to the clinical outcome of infection and the long-term well-being of the host (45). Furthermore, the absence of pDCs and other cells with high constitutive expression of IRF7 from the neural parenchyma (12) makes it likely that early events in this organ are different from what is observed in most other organ sites.
Following intracerebral (i.c.) infection of the CNS, LCMV induces an innate response that is most clearly expressed by day 3 postinfection (p.i.) (1, 39). In addition to this early, nonspecific host response, the i.c. infection also triggers the adaptive immune system and the expansion of virus-specific CD8+ T cells in the spleen and, to a lesser degree, cervical lymph nodes, and upon their differentiation into mature effector cells, many are recruited to the infected CNS (14). A key factor in this recruitment seems to be the local production of the chemokine CXCL10, which interacts with the chemokine receptor CXCR3 on the activated CD8 T cells (6, 7). Upon contact between incoming CD8 T cells and virus-infected cells in different parts of the CNS, large amounts of type II IFN are released, further increasing the local production of important chemoattractants, including CXCL10 (8). The whole process culminates in a severe CD8+ T-cell-mediated inflammatory reaction in essential parts of the CNS, cerebral edema, and death 7 to 9 days after virus inoculation (8, 35). Using IFNAR-deficient (IFNAR−/−) mice, we have previously shown that type I IFNs are essential in activating certain aspects of the local antiviral host response, including the early production of CXCL10 (8). However, it is not known how the early type I IFN response is regulated within the LCMV-infected CNS, nor has the cellular source(s) been described.
MATERIALS AND METHODS
Mice.
Wild-type C57BL/6 (WT) mice were purchased from Taconic M&B (Ry, Denmark). IFNAR (IFNR1)-, IFN-β-, and IRF7-deficient mice on a C57BL/6 background were the progeny of breeder pairs maintained at the Panum Institute, University of Copenhagen; IFNAR- and IRF7-deficient mice both originated in the animal facility of R. Zinkernagel, Universitätspital, Zürich, Switzerland, while the IFN-β-deficient mice originally came from T. Leandersson, Lund University, Sweden. IRF1-deficient mice came from the Jackson Laboratory, Bar Harbor, ME. IRF9-deficient mice were bred at University Libre de Bruxelles, Gosselies, Belgium, and IRF3-deficient mice came from Karolinska Institute, Stockholm, Sweden. IRF3/7 double-knockout (KO) (IRF3/7−/−) mice were either bred at the Technical University Munich (single IRF KO mice have been described by Honda et al. [18] and were originally obtained from the Riken Bioresource Center, Japan, with permission from T. Taniguchi) or came from University Libre de Bruxelles, Gosselies, Belgium. All mice from outside sources were allowed to acclimatize to the local environment for at least a week before entering into experiments; by that time, the animals were typically 7 to 10 weeks old. The animals were housed under specific-pathogen-free conditions, validated by testing of sentinels for unwanted infections according to Federation of European Laboratory Animal Science Association standards; no such infections were detected. Female mice were used in most experiments, but when both sexes were used, no gender effect was observed. Experiments were conducted in accordance with Danish national guidelines regarding animal experiments as approved by the Danish Animal Experiments Inspectorate, Ministry of Justice.
Virus infection.
Mice were infected i.c. with a virus dose of 103 50% lethal doses (LD50) (∼200 PFU) of LCMV Traub. LCMV is a noncytolytic virus that causes little if any disease in immunodeficient mice (10, 21). However, intracerebral inoculation of LCMV leads to infection of the CNS, and in adult immunocompetent mice, the result is a severe CD8+ T cell-mediated meningoencephalitis to which the animals succumb around day 8 or 9 p.i (9).
Isolation of total RNA for quantitative PCR (Q-PCR).
Brains from deeply anesthetized and exsanguinated mice were immediately removed, snap-frozen in liquid nitrogen, and stored in a liquid nitrogen freezer. Total RNA was extracted from the homogenized brains by use of an RNeasy midi kit (Qiagen, Hilden, Germany).
Detection of mRNA in the brain by quantitative PCR.
mRNA (1 μg) was reverse transcribed to cDNA using a RevertAid First Strand cDNA synthesis kit (MBT Fermentas). For the Q-PCR reaction, Brilliant SYBRGreen QPCR Mastermix was used according to the manufacturer's instructions (Stratagene, AH Diagnostics). Alternatively, Brilliant II QPCR Mastermix and hexachlorofluorescein (HEX)- or 6-carboxyfluorescein (FAM)-conjugated probes were used. The sequences of the primers and probes are shown in Table 1; as IFN-α covers more than 20 isoforms, primers and probes were designed using a previously published consensus sequence (23). Target gene expression was normalized against the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or porphobilinogen (PBDG) housekeeping gene.
Table 1.
Primers and probes used in the study
| Gene | Primer or probe | Sequence (5′–3′) |
|---|---|---|
| CXCL10 | Forward primer | CGA TGA CGG GCC AGT GAG AAT G |
| Reverse primer | TCA ACA CGT GGG CAG GAT AGG CT | |
| 2′,5′-OAS | Forward primer | CTT TGA TGT CCT GGG TCA TGT |
| Reverse primer | CTC CGT GAA GCA GGT AGA G | |
| IRF3 | Forward primer | TGG GCA GCA CAG CTG ACA TGA |
| Reverse primer | GCC CAT TGC CCA GCC CTT | |
| IRF7 | Forward primer | GCC TTG GGT TCC TGG ATG TGA |
| Reverse primer | TGG GGC CAT GGG GCT GTA | |
| IFN-α | Forward primer | TGC AAC CCT CCT AGA CTC ATT CT |
| Reverse primer | CCA GCA GGG CGT CTT CCT | |
| Probe | FAM–CTG CAT CAG ACA GCC TTG CAG GTC ATT-BHQ-1a | |
| IFN-β | Forward primer | TGA ATG GAA AGA TCA ACC TCA CCT A |
| Reverse primer | CTC TTC TGC ATC TTC TCC GTC A | |
| Probe | FAM–AGG GCG GAC TTC AAG ATC CCT ATG GA-BHQ-1 | |
| GAPDH | Forward primer | CAA TGT GTC CGT CGT GGA |
| Reverse primer | GAT GCC TGC TTC ACC ACC | |
| PBGD | Forward primer | GTG AGT GTG TTG CAC GAT C |
| Reverse primer | GGG TCA TCT TCT GGA CCA T | |
| Probe | HEX–CTC TGC TTC GCT GCA TTG CTG-BHQ-1 |
BHQ, black hole quencher.
The Q-PCR program used in an Mx3000P Real-Time QPCR instrument was as follows: denaturation (95°C for 10 min) and 40 cycles of denaturation (95°C for 30 s), annealing (58 to 60°C for 60 s), and extension (72°C for 30 s). Each reaction was run in duplicate or triplicate plus a control without reverse transcriptase and a control without template.
The results were analyzed using Mx3000P system software. The relative expression ratio (R) in each sample was calculated by a mathematical model based on the amplification efficiency (46): R = EtargetΔCP (control−sample)/EreferenceΔCP (control−sample). An amplification efficiency (E) of 100% corresponds to a doubling of the PCR product per cycle. E is calculated from the slope of a standard curve based on a 10-fold titration of each primer used (E = 10−1/slope). Thus, Etarget corresponds to the target gene primers and Ereference to the housekeeping gene primer (GAPDH or PBDG). In this paper, WT brains infected i.c. 3 or 7 days earlier with LCMV were used as standard-curve templates. ΔCP(control − sample) refers to the difference in threshold cycles (CT) between day 0 (control) and day 3, 5, or 7 p.i. (sample). CT reflects the number of cycles it takes to reach a point in which the fluorescent signal is first recorded as statistically significant above background (46).
Quantitative PCRs for ISGs.
To evaluate the expression of ISGs, we used an RT2 profiler PCR array system kit from SABiosciences (Frederick, MD) dedicated to the analysis of ISGs (PAMM 016). The preparation of cDNA, the running of the assays, and the analysis of the results were all performed according to the manufacturer's instructions.
In situ hybridization.
Mice were deeply anesthetized with tribromoethanol (Sigma-Aldrich, Denmark) and decapitated. The brains were rapidly dissected and frozen on dry ice. Subsequently, the brains were cut into serial 30-μm cryostat sections, mounted on RNase-free Super Frost Plus glass slides (Hounisen, Denmark), and stored in sealed boxes at −80°C. The detection of IFN-β mRNA was performed in accordance with the method of Lambertsen et al. (31) using a mixture of two alkaline phosphatase (AP)-labeled DNA probes (5′-GTTGATGGAGAGGGCTGTGGTGGAGAAG-3′ and 5′-ATTCACTACCAGTCCCAGAGTCCGCCTC-3′) (4 pmol/ml; designed by use of Oligo-design software version 6.0 and fabricated by DNA Technology A/S [Aarhus, Denmark]) that were complementary to bases 62 to 89 and 638 to 655, respectively, of murine IFN-β mRNA (NM 010510). The hybridization signal, consisting of a purple reaction product, was developed for 72 h. The specificity of the hybridization was documented by showing that (i) hybridization with individual probes yielded a signal fainter than but otherwise similar to hybridization with the probe mixture, (ii) 100-fold excess of unlabeled probes was able to outperform the AP-labeled probes, (iii) the hybridization signal was abolished in RNase A (Pharmacia Biotech)-digested sections, and (iv) incubation in hybridization buffer without probe yielded no signal. The overall suitability of the tissue for hybridization was ensured by hybridization for GAPDH mRNA (31).
Combined in situ hybridization and immunohistochemistry.
After development of the in situ hybridization signal using a standard procedure (35), the sections were rinsed in Tris-buffered saline (TBS) buffer containing 1% Triton and then incubated with monoclonal Alexa 488-conjugated mouse anti-glial fibrillary acidic protein (GFAP) IgG1 (5 μg/ml; A21294; Invitrogen, Denmark), Alexa 488-conjugated mouse IgG1 isotype control (5 μg/ml; MG120; Caltag Laboratories), monoclonal rat anti-CD3 (2.5 μg/ml; MCA500A488; Serotec, Germany) plus Alexa 594-conjugated donkey-anti-rat IgG (10 μg/ml; A21209; Invitrogen), or rat IgG2a isotype control (2.5 μg/ml; 26D11204; Nordic BioSite ApS, Denmark) plus Alexa 594-conjugated donkey-anti-rat IgG (10 μg/ml) diluted in TBS buffer plus 10% bovine serum. After a final rinse in TBS, sections were counterstained with DAPI (4′,6-diamidino-2-phenylindole) nuclear staining. Bright-field pictures of the in situ hybridization signal were inverted before the green channel or red channel was selected and merged with the channels showing the fluorescence signals by using Photoshop software (Adobe Photoshop CS4 Extended, version 11.0.2; Adobe Systems Inc.).
Statistical analysis.
Quantitative results were compared using the Mann-Whitney U test. A P value of <0.05 was considered evidence of statistical significance.
RESULTS
The role of IRFs in regulating the early type I IFN response in the LCMV-infected CNS.
The IRFs, and in particular IRF3 and IRF7, are generally assumed to play a key role in activating the type I IFN transcriptional response to viral infection (19). To investigate the role of IRFs in regulating the early LCMV-induced type I IFN response, we infected IRF1−/−, IRF3−/−, IRF7−/−, IRF9−/−, and WT mice with LCMV i.c., and 3 days later, we measured the level of mRNAs for 2,5-OAS and CXCL10 in the CNS. The expression levels of these two ISGs were used as surrogate markers for the induction of type I IFN, because we had previously observed that the level of expression of type I IFNs in the LCMV-infected CNS borders on the level of detection (8); this approach is validated by our observation that there is no virus-induced increase in the expression of either of these genes in LCMV-infected IFNAR−/− mice (8).
As can be seen in Fig. 1, lack of IRF1, which has recently been found to play a crucial role in regulating RNA virus-induced, IPS-dependent signaling from peroxisomes (13), did not impact the expression of either CXCL10 or 2,5-OAS in the LCMV-infected CNS. In contrast, absence of either IRF3 or IRF7 slightly reduced the LCMV-induced increase in expression of CXCL10 and 2,5-OAS (Fig. 1). However, in both cases, the reductions were small (∼2- to 3-fold) and not statistically significant in every experiment. Interestingly, deficiency of IRF9 differentially affected the expression of the tested ISGs. Thus, while the expression of mRNA for 2,5-OAS was markedly reduced in these mice compared to that in matched WT mice, CXCL10 was similarly expressed in the LCMV-infected CNS whether or not IRF9 was present (Fig. 1).
Fig 1.

Roles of IRF1, IRF3, IRF7, and IRF9 in regulating the early expression of CXCL10 and 2,5-OAS. Using Q-PCR, the levels of mRNA for CXCL10 and 2,5-OAS were determined in the CNS of IRF1−/− (IRF1), IRF3−/− (IRF3), IRF7−/− (IRF7), IRF9−/− (IRF9), and WT mice infected i.c. with LCMV 3 days earlier. The mRNA levels in the brains of sham-inoculated WT mice were used as set points (R = 1). The points represent individual mice, and group averages are indicated by horizontal lines. The asterisks indicate statistically significant differences between knockout and WT mice.
As IRF3 or IRF7 normally plays the major role in regulating the type I IFN response to viral infection, the above-mentioned results were somewhat surprising. Therefore, to examine whether these factors showed reciprocal redundancy, we next investigated the expression of CXCL10 and 2,5-OAS in IRF3/7−/− mice. Unlike the situation in singly deficient mice, the expression of both genes was consistently found to be reduced (by about a factor of 10) in doubly deficient mice (Fig. 2), indicating that IRF3 and IRF7 acted in concert in regard to the early activation of the type I IFN response.
Fig 2.
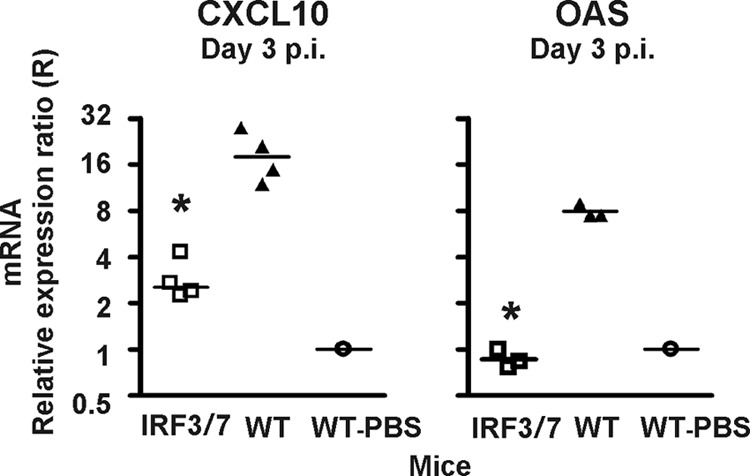
Absence of both transcription factors IRF3 and IRF7 markedly reduces the expression of CXCL10 and 2,5-OAS in the LCMV-infected CNS. Using Q-PCR, the levels of mRNA for CXCL10 were determined in the CNS of IRF3/IRF7−/− (IRF3/7) and WT mice infected i.c. with LCMV 3 days earlier. The mRNA level in the brain of a sham-inoculated mouse was used as a set point (R = 1). The points represent individual mice, and group averages are indicated by horizontal lines. The asterisks indicate statistically significant differences between knockout and WT mice.
Finally, since IκΒ kinase ε (IKKε), together with TANK-binding kinase 1 (TBK1), targets serine residues that are important for full transcriptional activation of IRF3 and -7 in most cell types (16), we examined whether the expression of 2,5-OAS and CXCL10 was reduced in mice lacking IKKε. However, similar levels of expression of both ISGs were found in the infected mice irrespective of their genotype (see Fig. S1 in the supplemental material), indicating that IKKε is not essential to the activation of IRF3 and/or -7 in the LCMV-infected CNS. This is in accordance with the findings of McWhirter et al. and Panne et al., whose in vitro studies have shown that TBK1 alone may suffice for phosphorylation of IRF3 in vitro (36, 43).
Failure to demonstrate a major role of IRF7 in regulating the early type I IFN response in the CNS.
The above-mentioned results could indicate that activation of either IRF3 or IRF7 individually would suffice for the induction of a nearly complete type I IFN response in the LCMV-infected CNS. In contrast, IRF7 is believed to represent the master regulator of the type I IFN transcriptional response in mice undergoing systemic viral infection (20, 32). In order to ascertain that the observed tissue-related difference in the regulation of the type I IFN response is real, the following experiments were conducted.
First, we analyzed whether absence of IRF7 might impact the kinetics of ISG activation; could it be that analysis on day 3 was simply too late to detect an important early role of IRF7 in the initiation of the type I IFN response in the CNS? To investigate this point, WT and IRF7−/− mice were infected i.c. with LCMV, and 1 and 2 days after infection, their brains were processed for evaluation of ISG activation. As can be seen in Fig. 3, we observed low-grade upregulation of 2,5-OAS in WT mice already on day 1 after infection, whereas it was not evident in IRF7−/− mice. At 2 days after infection, the pattern observed resembled that previously found on day 3 after infection (Fig. 1), i.e., substantial upregulation of 2,5-OAS was evident in both groups of mice; however, as previously noted, the induced response was slightly reduced (2- to 3-fold) in IRF7−/− mice compared to matched WT mice. Regarding CXCL10, no virus-induced increase in gene expression was detected in either group of mice on day 1 after infection, and on day 2 after infection, as on day 3, similar levels of expression were observed regardless of the host genotype. Taken together, these results strongly suggested that absence of IRF7, if anything, only slightly delayed the induction of the type I IFN response to LCMV in the CNS.
Fig 3.

Absence of IRF7 does not impact the kinetics of the early type I IFN-dependent response. Using Q-PCR, the levels of mRNA for CXCL10 and 2,5-OAS were determined in the CNS of IRF7−/− (IRF7) and WT mice infected i.c. with LCMV 1 and 2 days earlier. The mRNA levels in the brains of matched sham-inoculated WT mice were used as set points (R = 1). The points represent individual mice, and group averages are indicated by horizontal lines.
Nevertheless, as this conclusion hinges on the analysis of only two ISGs, and potentially, different ISGs might be differentially affected by absence of IRF7, we proceeded by analyzing the expression of a wider range of ISGs in the CNS of LCMV-infected IRF7−/− mice, comparing expression levels to those in similarly infected WT mice. As can be seen in Fig. 4, nearly all of the ISGs upregulated in the CNS of LCMV-infected WT mice were also upregulated in the CNS of IRF7−/− mice (29 versus 25). Moreover, a direct comparison of expression levels in infected WT and IRF7−/− mice revealed very few genes not expressed to about the same level in both mouse strains (Fig. 4, bottom; for the complete data set, see Table S1 in the supplemental material). Consequently, in view of the range of ISGs tested in the latter experimental setup, we find it reasonable to conclude that there is no mandatory requirement for IRF7 in the transcriptional regulation of the early type I IFN response in the LCMV-infected CNS.
Fig 4.
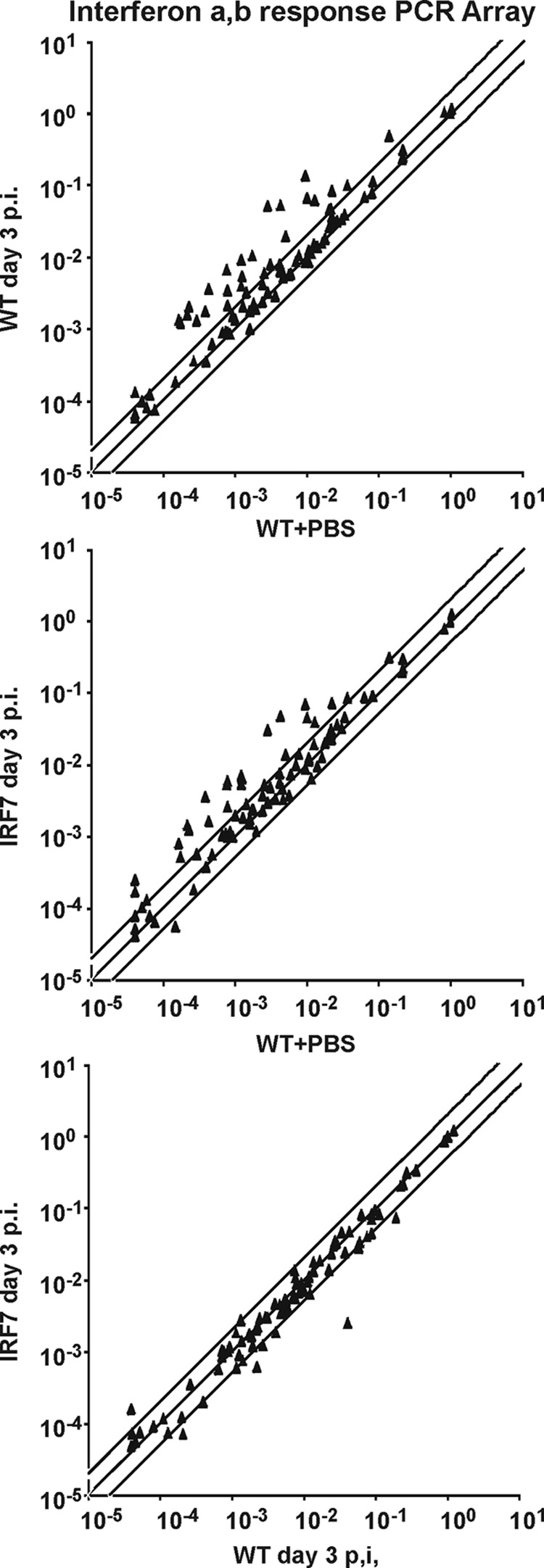
Expression of IFN-regulated genes in the LCMV-infected CNS is independent of IRF7. mRNA was extracted from the CNS of IRF7−/− (IRF7) and WT mice injected with LCMV 3 days earlier; the brains from WT mice injected with PBS served as a reference. The expression of 86 classical IFN-stimulated genes was analyzed by Q-PCR. The results represent averages of 2 or 3 mice per group. (Top) Gene expression in infected versus sham-inoculated WT mice. (Middle) Gene expression in infected IRF7−/− mice compared to sham-inoculated WT mice. (Bottom) Gene expression in infected IRF7−/− mice compared to infected WT mice. The lines represent equal expression and 2-fold up- or downregulation.
Differential role of IRF7 in regulating the early type I IFN response in the periphery versus the CNS.
The above-mentioned findings clearly do not conform to recent observations pointing to an absolutely pivotal role of the IRF7 signaling pathway regarding the type I IFN response in the blood of mice systemically infected with LCMV (32). This could indicate that signaling pathways different from those relevant in the periphery were involved in transmitting the sensing of LCMV in the CNS. To study this possibility, we harvested the sera together with the spleens and brains of IRF7−/− and WT mice infected i.c. 3 days earlier. Since about 90% of the virus inoculum given i.c. ends up in the circulation (37), i.c.-infected mice are also subject to a systemic infection, and we could therefore directly compare the roles of IRF7 in regulating the type I IFN response in the periphery versus the CNS of the same animals.
Confirming published data (32), no IFN-α was detectable in the sera of IRF7−/− mice (by use of enzyme-linked immunosorbent assay [ELISA]), while this type of IFN could readily be demonstrated in the sera of 4/4 infected WT mice (Fig. 5A). Consistent with this result, we found a reduction in the level of expression of mRNA for type I IFNs, in particular IFN-α, in the spleens of infected IRF7−/− mice compared to WT mice. In contrast, low but seemingly identical expression levels of mRNA for IFN-β were detected in the CNS of IRF7−/− and WT mice (no IFN-α expression could be detected with certainty in the CNS) (Fig. 5B and C).
Fig 5.

Differential regulation of type I IFN in the periphery versus the CNS of LCMV-infected mice. WT and IRF7−/− (IRF7) mice were infected i.c. with LCMV, and 3 days later, blood was drawn for analysis of serum IFN-α. Subsequently, the mice were sacrificed, and spleens and brains were removed for analysis of IFN-α and -β expression. (A) Serum IFN-α (IFNa) levels 3 days after i.c. infection. The points represent individual mice, and the group average is indicated by the horizontal line. The dotted line indicates the limit of detection; ND, not detectable. The asterisk indicates a statistically significant difference between knockout and WT mice. (B) IFN-β (IFNb) expression in spleens and brains 3 days after i.c. infection; averages ± standard deviations (SD) of 3 or 4 mice/group. (C) IFN-α (IFNa) expression in spleens and brains 3 days after i.c. infection; averages ± SD of 3 or 4 mice/group.
Role of IFN-β in regulating the early type I IFN response in the LCMV-infected CNS.
The above-mentioned results strongly indicate that different signaling pathways are involved in the sensing of LCMV in the periphery versus the CNS. This could be explained if different cell types contributed to the sensing of the infection in different organ sites, leading to the production of different types of type I IFN. Since robust production of IFN-α tends to rely heavily on IRF7 (20, 32), a predominant production of IFN-β by various resident cells of the CNS might explain the observed difference regarding the importance of IRF7 in the CNS versus the periphery, where pDCs and other hematopoietic cells appear to play a dominant role (25, 32).
To evaluate the participation of IFN-β in the early type I IFN response in the CNS of LCMV-infected mice, we compared IFN-β−/− mice to IFNAR−/− and WT mice with regard to the expression of CXCL10 and 2,5-OAS in the CNS on day 3 after LCMV infection (Fig. 6). While the expression of both CXCL10 and 2,5-OAS was found to be lower in IFN-β−/− mice than in WT mice, the reduction did not recapitulate that observed in IFNAR−/− mice, indicating that type I IFNs, in addition to IFN-β, contribute significantly to the early IFN response in the LCMV-infected CNS.
Fig 6.
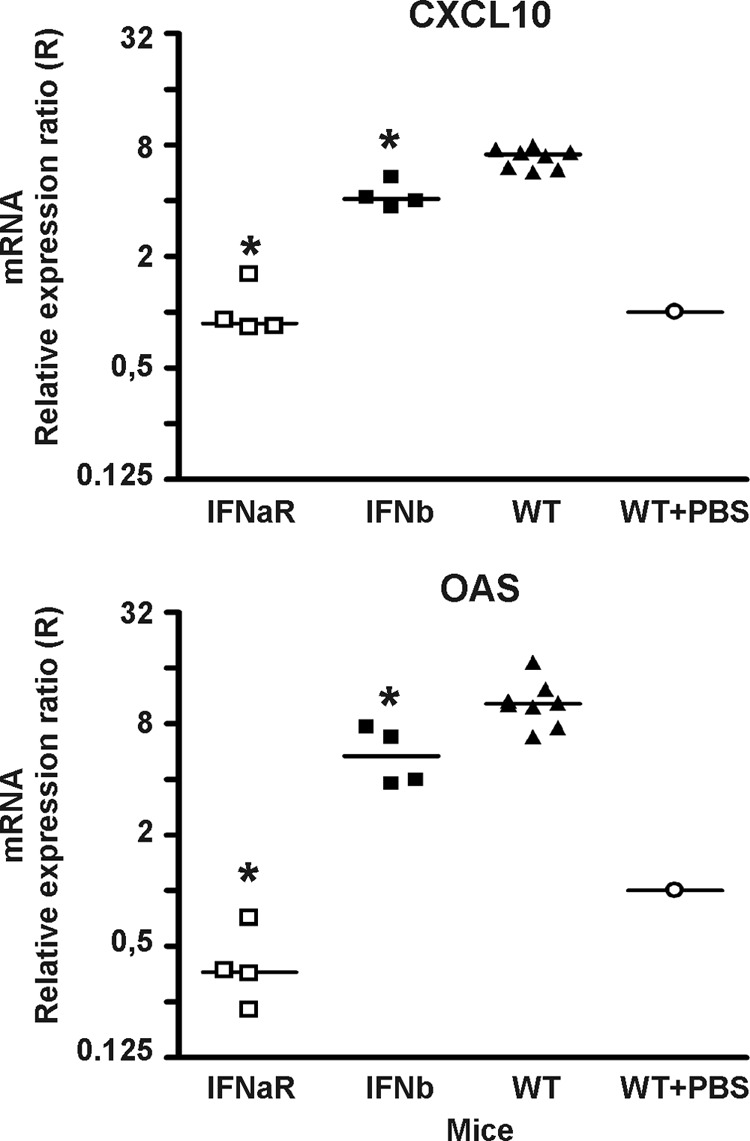
Robust early CXCL10 and 2,5-OAS expression requires both IFN-α and -β. Using Q-PCR, the levels of mRNA for CXCL10 and 2,5-OAS were determined in the CNS of IFN-β−/− (IFNb), IFNAR−/− (IFNaR), and WT mice infected i.c. with LCMV 3 days earlier. The mRNA level in the brain of a sham-inoculated mouse was used as a set point (R = 1). The points represent individual mice, and group averages are indicated by horizontal lines. The asterisks indicate statistically significant differences between knockout and WT mice.
Topological and cellular localization of type I IFN expression in the LCMV-infected CNS.
Having established a central role for local type I IFN production in the activation of the innate response to LCMV infection of the CNS, it was of interest to determine which cell types were involved in synthesizing type I IFN. Since IFN-β appears to be more strongly expressed in the LCMV-infected CNS than other type I IFNs (8), we decided to focus on expression of this cytokine. First, we performed in situ hybridization on brain sections to study the topology of IFN-β expression as a function of time after virus inoculation. Overall, IFN-β mRNA was detected in cells scattered in the meningeal membrane, around the injection site and in the ependymal cells lining the lateral ventricles on day 3 (Fig. 7A, C, and E) and to an even greater degree on day 7 (Fig. 7B, D, and F) after virus inoculation. In general, the level of IFN-β mRNA expression was low, but it was clearly increased compared to the level of IFN-β mRNA induced by PBS injection (data not shown). Importantly, the controls were all devoid of signal confirming the specificity of the in situ hybridization reaction. Furthermore, GAPDH mRNA was expressed at normal high levels (data not shown), documenting the appropriate quality of the tissue for use with in situ hybridization.
Fig 7.
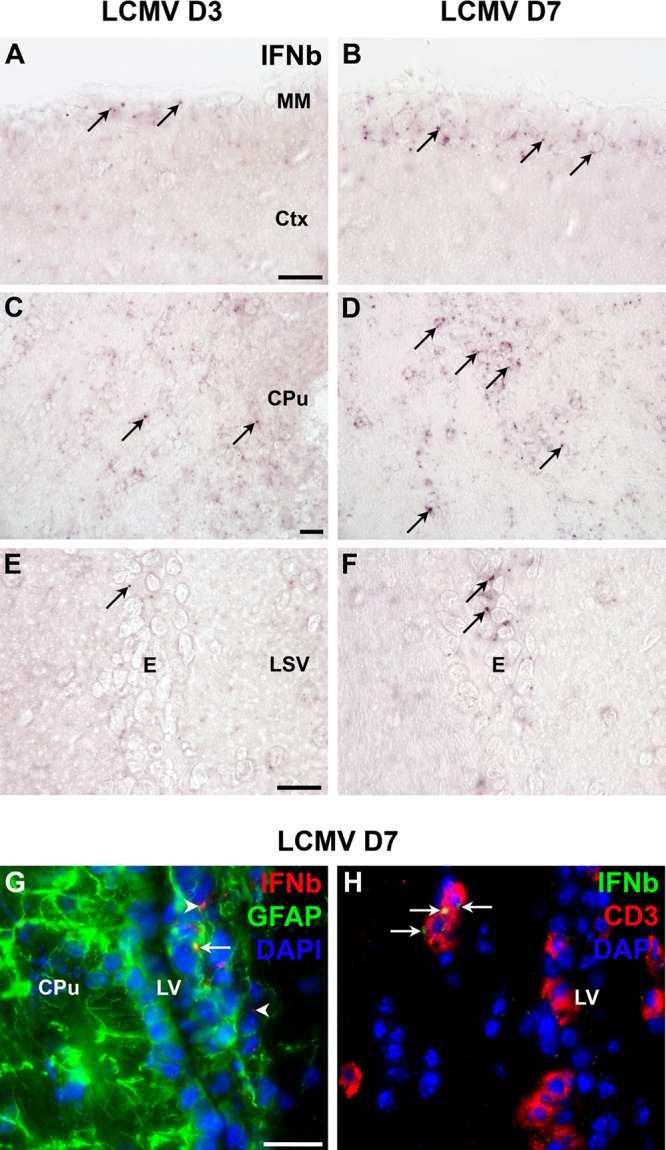
In situ hybridization for IFN-β mRNA in LCMV-injected CNS of C57BL/6 mice. (A to F) LCMV-induced expression of IFN-β (IFNb) mRNA in meningeal membrane cells (A and B), in parenchymal cells close to the injection site (C and D), and in ependymal cells (E and F) at day 3 (A, C, and E) and at day 7 (B, D, and F) after i.c. infection with LCMV. (G and H) Combined in situ hybridization and immunohistochemistry for IFN-β mRNA and GFAP (G) or CD3 (H) showing IFN-β expression in both GFAP+ (G, arrow) and GFAP− (G, arrowheads) cells, as well as in CD3+ cells (H, arrows) infiltrating the brain parenchyma adjacent to the lateral ventricle 7 days after i.c. LCMV infection. CPu, caudoputamen; Ctx, cerebral cortex; E, ependymal cell layer; LSV, lateral septal nucleus, ventral part; LV, lateral ventricle; MM, meningeal membrane. Scale bars, 20 μm.
Next, despite the low level of single-cell expression, we attempted to determine which cell types expressed IFN-β mRNA. Combined in situ hybridization for IFN-β mRNA and immunostaining for GFAP protein on brain tissue from mice infected i.c. 7 days earlier suggested that the IFN-β mRNA was expressed by GFAP+ cells, which could be either ependymal cells or astrocytes, since both cell types were visualized by the GFAP staining, as well as GFAP− cells (Fig. 7G). In line with the latter observation, combined in situ hybridization for IFN-β mRNA and immunostaining for CD3 protein revealed that IFN-β mRNA was also expressed by CD3+ cells in the brains of mice infected i.c. 7 days earlier (Fig. 7H).
DISCUSSION
Early protection of the host against viral infection relies on early detection of incoming virus followed by swift and efficient activation of the innate immune system. It has long been known that the type I IFN response represents one of the earliest responses to viral invasion and that type I IFNs may act through direct inhibition of viral replication in exposed cells, as well as through the induction of inflammation and recruitment of antiviral effector cells (55). Overall, this makes type I IFNs key mediators in the early antiviral host response. In regard to LCMV infection of the CNS, we have previously found that this infection causes an early increase in the expression of 2,5-OAS and CXCL10. Moreover, using IFNAR−/− mice, we showed that this upregulation is dependent on type I IFNs. However, as we could not with certainty detect type I IFN mRNA in the CNS during the early phase of infection, the possibility that the local effects primarily reflected the penetration of systemically produced type I IFNs from the circulation could not be formally excluded.
In this study, we uncovered a marked difference in the way in which the detection of the same virus is transmitted in the CNS compared to the periphery. Thus, while absence of IRF7 very substantially reduces the systemic type I IFN response to LCMV and markedly affects the course of the visceral infection (32), little or no reduction was observed in the LCMV-infected CNS. From this observation, it may immediately be concluded that the response measured in the CNS predominantly, if not exclusively, reflects the local production of type I IFNs and not a passage of these cytokines across the blood-brain barrier. Regarding the difference in the functional importance of IRF7, this is likely to reflect the fact that different cell types are involved as the primary producers of type I IFN in the two situations. Thus, no cells with high constitutive expression of IRF7 can be demonstrated in the CNS (12). In contrast, previously published data suggest that there is a relatively high constitutive expression of IRF3 by certain resident cells of the CNS (42), and this may explain why the otherwise canonical upregulation of IRF7 is not essential in this organ unless IRF3 is also lacking. In the periphery, on the other hand, pDCs and other hematopoietic cells constitutively expressing IRF7 are prominent and play a pivotal role in producing type I IFNs, particularly IFN-α, as a first line of antiviral defense (25, 32). The nearly normal type I IFN response in the CNS of IRF7−/− mice therefore suggests that inside the CNS, non-pDCs would dominate as producers of type I IFNs. Consistent with this hypothesis, we found that most cells taking part in the early type I IFN response in the LCMV-infected CNS are the parenchymal CNS cells next to the injection site and the cells forming the meningeal and ependymal linings; this is not surprising and is in full agreement with the distribution of virus-infected cells at this time (11, 17). The type I IFN response increases over time (8, 50), and with time, GFAP+ astrocytes and recruited CD3+ cells were also found to produce type I IFN, which is in agreement with previous findings showing that astrocytes (44) and murine T cells (29) are able to produce IFN-β. Other IFN-β-producing cell candidates are neurons (12) and macrophages (33, 57). At this later time point, we also observed a few very intensely stained cells, which could be pDCs recruited to the site of infection as a result of ongoing inflammation.
Because IRF7 is required for robust production of IFN-α in the periphery (20, 32, 52), the finding that deficiency of this transcription factor only marginally impacted the type I IFN-related response in the CNS led us to suspect that IFN-β would dominate as the functionally most important type I IFN produced inside the LCMV-infected CNS. As mentioned above, directly measuring type I IFNs in the LCMV-infected CNS is very difficult, but a comparison of the levels of expression of CXCL10 and 2,5-OAS in mice lacking only IFN-β to those in mice lacking all type I IFN signaling (due to lack of IFNAR) revealed that IFN-β could not be the only type I IFN to be produced in the LCMV-infected CNS, nor does it appear to be essential for virus-induced upregulation of other type I IFNs in the organ. The reason for this negative result may lie in the fact that IFN-α4 can also be produced in the absence of IRF7 (34) and that this type I IFN may suffice for triggering of a nearly normal early response in the CNS.
Finally, we found that while the expression of 2,5-OAS was markedly reduced in IRF9−/− mice, IRF9 was redundant with regard to LCMV-induced upregulation of CXCL10 in the CNS. This finding points to a dichotomy in the regulation of ISGs in the LCMV-infected CNS. Thus, while the expression of 2,5-OAS seems to be regulated by the canonical IFN-activated transcription factor ISGF3, composed of STAT1, STAT2, and IRF9, alternative ISRE-binding factors may suffice for type I IFN-dependent upregulation of CXCL10. The latter interpretation is supported by preliminary data indicating that CXCL10 upregulation also occurs in the absence of STAT1 (J. E. Christensen and J. P. Thomsen, unpublished results). We do not know the reasons for this differential regulation of ISGs in the LCMV-infected CNS, but it is possible that different cell types with different requirements regarding IFNAR signaling may dominate in expression of different ISGs in the CNS.
In summary, our studies revealed that the signaling pathways involved in translating the sensing of a viral infection into an early type I IFN-associated response may be differentially wired in the CNS than in the periphery, probably as a reflection of organ-related differences in the cell types that dominate early aspects of this process. These results improve our understanding regarding the immune privilege of the CNS and underscore the cell-type-related complexity of IFN induction. Hopefully, greater insight into the transcriptional regulation of the earliest inflammatory response to a viral invasion may eventually further the development of novel strategies for prevention of an overzealous host response in the early phase of a viral infection, such as the “cytokine storm” associated with highly pathogenic influenza virus infection (4, 5, 58).
Supplementary Material
ACKNOWLEDGMENTS
The technical assistance of Benjamin Jensen, Peter Rasmussen, and Deanna Bardenfleth is gratefully acknowledged.
This work was supported in part by the Danish Medical Research Council, The Novo Nordisk Foundation, the Lundbeck Foundation, and a Manufacturer Vilhelm Pedersen and Wife's Grant. Stanislas Goriely is a research fellow at the FRS-FNRS (Belgium).
Footnotes
Published ahead of print 18 April 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Asensio VC, Campbell IL. 1997. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J. Virol. 71:7832–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baglioni C, Minks MA, Maroney PA. 1978. Interferon action may be mediated by activation of a nuclease by pppA2′p5′A2′p5′A. Nature 273:684–687 [DOI] [PubMed] [Google Scholar]
- 3. Barchet W, et al. 2002. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J. Exp. Med. 195:507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baskin CR, et al. 2009. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. U. S. A. 106:3455–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheung CY, et al. 2002. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360:1831–1837 [DOI] [PubMed] [Google Scholar]
- 6. Christensen JE, de Lemos C, Moos T, Christensen JP, Thomsen AR. 2006. CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J. Immunol. 176:4235–4243 [DOI] [PubMed] [Google Scholar]
- 7. Christensen JE, et al. 2004. Efficient T-cell surveillance of the CNS requires expression of the CXC chemokine receptor 3. J. Neurosci. 24:4849–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Christensen JE, et al. 2009. Fulminant lymphocytic choriomeningitis virus-induced inflammation of the CNS involves a cytokine-chemokine-cytokine-chemokine cascade. J. Immunol. 182:1079–1087 [DOI] [PubMed] [Google Scholar]
- 9. Christensen JP, Marker O, Thomsen AR. 1994. The role of CD4+ T cells in cell-mediated immunity to LCMV: studies in MHC class I and class II deficient mice. Scand. J. Immunol. 40:373–382 [DOI] [PubMed] [Google Scholar]
- 10. Christoffersen PJ, Volkert M, Rygaard J. 1976. Immunological unresponsiveness of nude mice to LCM virus infection. Acta Pathol. Microbiol. Scand. C 84C:520–523 [DOI] [PubMed] [Google Scholar]
- 11. Cole GA, Gilden DH, Monjan AA, Nathanson N. 1971. Lymphocytic choriomeningitis virus: pathogenesis of acute central nervous system disease. Fed. Proc. 30:1831–1841 [PubMed] [Google Scholar]
- 12. Delhaye S, et al. 2006. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. U. S. A. 103:7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dixit E, et al. 2010. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141:668–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doherty PC, Allan JE, Lynch F, Ceredig R. 1990. Dissection of an inflammatory process induced by CD8+ T cells. Immunol. Today 11:55–59 [DOI] [PubMed] [Google Scholar]
- 15. Durbin RK, Mertz SE, Koromilas AE, Durbin JE. 2002. PKR protection against intranasal vesicular stomatitis virus infection is mouse strain dependent. Viral Immunol. 15:41–51 [DOI] [PubMed] [Google Scholar]
- 16. Fitzgerald KA, et al. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496 [DOI] [PubMed] [Google Scholar]
- 17. Gilden DH, Cole GA, Monjan AA, Nathanson N. 1972. Immunopathogenesis of acute central nervous system disease produced by lymphocytic choriomeningitis virus. I. Cyclophosphamide-mediated induction by the virus-carrier state in adult mice. J. Exp. Med. 135:860–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Honda K, et al. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434:1035–1040 [DOI] [PubMed] [Google Scholar]
- 19. Honda K, Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6:644–658 [DOI] [PubMed] [Google Scholar]
- 20. Honda K, et al. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777 [DOI] [PubMed] [Google Scholar]
- 21. Hotchin J, Weigand H. 1961. The effects of pretreatment with X-rays on the pathogenesis of lymphocytic choriomeningitis in mice. I. Host survival, virus multiplication and leukocytosis. J. Immunol. 87:675–681 [PubMed] [Google Scholar]
- 22. Hovanessian AG, Brown RE, Kerr IM. 1977. Synthesis of low molecular weight inhibitor of protein synthesis with enzyme from interferon-treated cells. Nature 268:537–540 [DOI] [PubMed] [Google Scholar]
- 23. Hughes TK, Jr, Chin R, Tyring SK, Rady PL. 1994. Distinction of mouse interferon-alpha subtypes by polymerase chain reaction utilizing consensus primers and type-specific oligonucleotide probes. J. Interferon Res. 14:117–120 [DOI] [PubMed] [Google Scholar]
- 24. Ishii KJ, et al. 2006. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 7:40–48 [DOI] [PubMed] [Google Scholar]
- 25. Jung A, et al. 2008. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J. Virol. 82:196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137 [DOI] [PubMed] [Google Scholar]
- 27. Kawai T, et al. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988 [DOI] [PubMed] [Google Scholar]
- 28. Kerr IM, Brown RE, Hovanessian AG. 1977. Nature of inhibitor of cell-free protein synthesis formed in response to interferon and double-stranded RNA. Nature 268:540–542 [DOI] [PubMed] [Google Scholar]
- 29. Klimpel GR, Infante AJ, Patterson J, Hess CB, Asuncion M. 1990. Virus-induced interferon alpha/beta (IFN-alpha/beta) production by T cells and by Th1 and Th2 helper T cell clones: a study of the immunoregulatory actions of IFN-gamma versus IFN-alpha/beta on functions of different T cell populations. Cell Immunol. 128:603–618 [DOI] [PubMed] [Google Scholar]
- 30. Kumar H, et al. 2006. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 203:1795–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lambertsen KL, Gregersen R, Drojdahl N, Owens T, Finsen B. 2001. A specific and sensitive method for visualization of tumor necrosis factor in the murine central nervous system. Brain Res. Brain Res. Protoc. 7:175–191 [DOI] [PubMed] [Google Scholar]
- 32. Lang PA, et al. 2009. Hematopoietic cell-derived interferon controls viral replication and virus-induced disease. Blood 113:1045–1052 [DOI] [PubMed] [Google Scholar]
- 33. Ma Y, et al. 1994. Outer surface lipoproteins of Borrelia burgdorferi stimulate nitric oxide production by the cytokine-inducible pathway. Infect. Immun. 62:3663–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marie I, Durbin JE, Levy DE. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17:6660–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matullo CM, O'Regan KJ, Hensley H, Curtis M, Rall GF. 2010. Lymphocytic choriomeningitis virus-induced mortality in mice is triggered by edema and brain herniation. J. Virol. 84:312–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McWhirter SM, et al. 2004. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 101:233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mims CA. 1960. Intracerebral injections and the growth of viruses in the mouse brain. Br. J. Exp. Pathol. 41:52–59 [PMC free article] [PubMed] [Google Scholar]
- 38. Mogensen TH, Paludan SR. 2005. Reading the viral signature by Toll-like receptors and other pattern recognition receptors. J. Mol. Med. 83:180–192 [DOI] [PubMed] [Google Scholar]
- 39. Nansen A, Marker O, Bartholdy C, Thomsen AR. 2000. CCR2+ and CCR5+ CD8+ T cells increase during viral infection and migrate to sites of infection. Eur. J. Immunol. 30:1797–1806 [DOI] [PubMed] [Google Scholar]
- 40. Negishi H, et al. 2006. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc. Natl. Acad. Sci. U. S. A. 103:15136–15141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. 2005. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J. Exp. Med. 202:1333–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ousman SS, Wang J, Campbell IL. 2005. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J. Virol. 79:7514–7527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Panne D, McWhirter SM, Maniatis T, Harrison SC. 2007. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 282:22816–22822 [DOI] [PubMed] [Google Scholar]
- 44. Park C, et al. 2006. TLR3-mediated signal induces proinflammatory cytokine and chemokine gene expression in astrocytes: differential signaling mechanisms of TLR3-induced IP-10 and IL-8 gene expression. Glia 53:248–256 [DOI] [PubMed] [Google Scholar]
- 45. Paul S, Ricour C, Sommereyns C, Sorgeloos F, Michiels T. 2007. Type I interferon response in the central nervous system. Biochimie 89:770–778 [DOI] [PubMed] [Google Scholar]
- 46. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prakash A, Smith E, Lee CK, Levy DE. 2005. Tissue-specific positive feedback requirements for production of type I interferon following virus infection. J. Biol. Chem. 280:18651–18657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rasmussen SB, Reinert LS, Paludan SR. 2009. Innate recognition of intracellular pathogens: detection and activation of the first line of defense. APMIS 117:323–337 [DOI] [PubMed] [Google Scholar]
- 49. Sabbah A, et al. 2009. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 10:1073–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sandberg K, Eloranta ML, Campbell IL. 1994. Expression of alpha/beta interferons (IFN-alpha/beta) and their relationship to IFN-alpha/beta-induced genes in lymphocytic choriomeningitis. J. Virol. 68:7358–7366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sato M, et al. 1998. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 441:106–110 [DOI] [PubMed] [Google Scholar]
- 52. Sato M, et al. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548 [DOI] [PubMed] [Google Scholar]
- 53. Schmid S, Mordstein M, Kochs G, Garcia-Sastre A, Tenoever BR. 2010. Transcription factor redundancy ensures induction of the antiviral state. J. Biol. Chem. 285:42013–42022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227–264 [DOI] [PubMed] [Google Scholar]
- 55. Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381 [DOI] [PubMed] [Google Scholar]
- 56. Takeuchi O, Akira S. 2001. Toll-like receptors; their physiological role and signal transduction system. Int. Immunopharmacol. 1:625–635 [DOI] [PubMed] [Google Scholar]
- 57. Toshchakov V, et al. 2002. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 3:392–398 [DOI] [PubMed] [Google Scholar]
- 58. Tumpey TM, et al. 2005. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 79:14933–14944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Unterholzner L, et al. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11:997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.