

Abstract
In the final stages of the herpes simplex virus 1 (HSV-1) life cycle, a viral nucleocapsid buds into a vesicle of trans-Golgi network (TGN)/endosome origin, acquiring an envelope and an outer vesicular membrane. The virus-containing vesicle then traffics to the plasma membrane where it fuses, exposing a mature virion. Although the process of directed egress has been studied in polarized epithelial cell lines, less work has been done in nonpolarized cell types. In this report, we describe a study of HSV-1 egress as it occurs in nonpolarized cells. The examination of infected Vero cells by electron, confocal, and total internal reflection fluorescence (TIRF) microscopy revealed that HSV-1 was released at specific pocket-like areas of the plasma membrane that were found along the substrate-adherent surface and cell-cell-adherent contacts. Both the membrane composition and cytoskeletal structure of egress sites were found to be modified by infection. The plasma membrane at virion release sites was heavily enriched in viral glycoproteins. Small glycoprotein patches formed early in infection, and virus became associated with these areas as they expanded. Glycoprotein-rich areas formed independently from virion trafficking as confirmed by the use of a UL25 mutant with a defect in capsid nuclear egress. The depolymerization of the cytoskeleton indicated that microtubules were important for the trafficking of virions and glycoproteins to release sites. In addition, the actin cytoskeleton was found to be necessary for maintaining the integrity of egress sites. When actin was depolymerized, the glycoprotein concentrations dispersed across the membrane, as did the surface-associated virus. Lastly, viral glycoprotein E appeared to function in a different manner in nonpolarized cells compared to previous studies of egress in polarized epithelial cells; the total amount of virus released at egress sites was slightly increased in infected Vero cells when gE was absent. However, gE was important for egress site formation, as Vero cells infected with gE deletion mutants formed glycoprotein patches that were significantly reduced in size. The results of this study are interpreted to indicate that the egress of HSV-1 in Vero cells is directed to virally induced, specialized egress sites that form along specific areas of the cell membrane.
INTRODUCTION
To ensure efficient transmission and replication, many viruses have developed mechanisms for directed spread (reviewed by Mothes et al. [37]). As infection moves from cell to cell, it is more advantageous for a virus to be released at locations adjacent to sites of entry than to travel to entry sites after egress. Although many viruses share common pathways in the process of directed spread, each virus is unique in its approach (11, 20, 27, 43, 44, 57, 67).
Herpes simplex virus 1 (HSV-1) is a double-stranded DNA (dsDNA) virus with a 152-kbp genome. The DNA is enclosed in 3 layers: an inner icosahedral capsid, a composite of numerous proteins that form the tegument, and a host-derived envelope incorporating the viral glycoproteins. During the process of virion formation, capsids are assembled in the nucleus and then enveloped in the cytoplasm (17, 32). In the course of envelopment, the capsid encounters viral glycoproteins on a vesicle that is believed to be derived from the trans-Golgi network (TGN) (30, 48, 54, 58). Tegument proteins bind to the capsid and to the glycoprotein tails. Negative curvature is induced, and the capsid buds into the vesicle, acquiring an envelope and an outer vesicular membrane. This virus-containing vesicle then moves to the plasma membrane, where it fuses and releases a mature enveloped virion. Most released virions remain cell associated until very late in infection (40).
In a natural setting, HSV-1 infects epithelial cells of mucosal surfaces and then moves on to establish latent infections in the sensory neurons that innervate these areas. Mucosal tissues consist of highly polarized epithelial cells in the apical layers of the epidermis as well as less differentiated cells in the basal region (34, 46). It has been suggested that infection spreads from cell to cell in different directions depending on a cell's location in a tissue layer and the cell's differentiation status (35, 42, 67). The virus is released at cell junctions in highly polarized epithelial cells (cells with tight junctions and high transepithelial resistance [TER]) (21), allowing rapid lateral spread through the monolayer and the formation of a lesion or cold sore. Directed egress in less polarized cells (i.e., no tight junctions, low TER) of the epidermis has not been extensively examined, although previous electron microscopy studies suggest that virus is randomly released (21). In neurons, newly formed virions traffic down axon microtubules for release at synaptic terminals and at virally induced varicosities (4, 6). Infection can then move swiftly through neural networks and back down to epithelial surfaces where replication is reinitiated in an area that is advantageous for host-to-host spread. In both polarized epithelial cells and neurons, the directed spread of the virus is dependent upon viral glycoprotein E (gE) (2, 7, 8, 21, 28, 29, 60, 61, 64).
gE is a transmembrane protein that forms a dimer with gI (18, 19). Early during infection, gE colocalizes with TGN46 in the TGN, while at late times gE moves to cell junctions in polarized epithelial cells (9, 30). Although gE is not essential for HSV-1 growth in culture, virus mutants lacking gE are severely limited in spread in vivo. The inhibition appears to occur after envelopment, as mutants produce the same amount of infectious virus as wild-type (WT) HSV-1 (2, 7, 8, 29, 61). In highly differentiated, polarized epithelial cells, virus lacking gE becomes randomly released rather than specifically directed to cell junctions, and infection spreads cell to cell much less efficiently (21, 64). In addition, progeny virus is unable to be transferred from infected epithelial cells to neurons, even though ΔgE mutants are competent for neuron entry (29, 61). Furthermore, in neurons infected with a ΔgE mutant, newly formed progeny virus cannot efficiently enter into and traffic down axons to egress sites (28, 51, 60). Infection is, therefore, much more limited.
Egress has not been extensively studied in less polarized epithelial cells, such as those located in the basal layers of the epidermis. It is important to examine the process of viral release in a variety of cell types to fully understand the mechanism of HSV-1 egress. In the study described here, we examined the process of HSV egress as it occurs in Vero cells, a nonpolarized epithelial cell line that lacks tight junctions and has a low TER. It was observed that progeny virions were released at specific sites on the plasma membrane that concentrated along the substrate-adherent surface of the cell as well as at cell-cell contact areas despite the lack of mature junctional complexes at these locations. Electron microscopy (EM), confocal microscopy, and total internal reflection fluorescence (TIRF) microscopy were employed to characterize the formation and expansion of egress sites along the substrate-adherent surface of infected cells.
MATERIALS AND METHODS
Cells and viruses.
Vero African Green Monkey kidney cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco) with 10% fetal bovine serum (FBS; Atlanta Biologicals) on 75-cm2 Nunc EasYFlasks. Cells were incubated at 37°C in 7% CO2 and passaged every 4 to 6 days with a medium change at day 2. Viruses used were the green fluorescent protein (GFP)-tagged VP26 (K26GFP) mutant developed by Prashant Desai (5), KOS-HSV-1, and the KUL25NS mutant (ΔUL25) developed by Fred Homa (31). Both the K26GFP mutant and WT KOS HSV were passaged on Vero cells. gE mutants, whose development is described below, were passaged in a similar manner on Vero cells. ΔUL25 mutant stocks were made on the 8-1 complementing cell line (31).
Antibodies.
gB (DL16) and gD (DL11) monoclonal antibodies were a kind gift from Gary Cohen and Roselyn Eisenberg. Each was used at a 1:1,500 dilution in 1% bovine serum albumin (BSA). The gE polyclonal antibody used in Western blotting was kindly provided by Harvey Friedman. It was used at a 1:6,000 dilution. 8F5 monoclonal antibody (56) was used to label major capsid protein VP5 at a 1:1,500 dilution. Commercially available antibodies included monoclonal anti-α-tubulin (T6074; 1:1,000; Sigma) and monoclonal anti-vinculin (V-9131; 1:600; Sigma). Actin was labeled with Texas Red-X phalloidin (T7471; Invitrogen) at 1:500 in phosphate-buffered saline (PBS). Rhodamine-labeled wheat germ agglutinin (WGA; RL-1022) was obtained from Vector Laboratories, Burlingame CA, and diluted to 2.5 μg/ml in PBS. Alexa Fluor 594 goat anti-mouse IgG (A11005; Invitrogen) and Alexa Fluor 488 goat anti-mouse IgG (A11001; Invitrogen) were the secondary antibodies used throughout. They were used at a 1:1,000 dilution.
Infection protocol.
Cells were plated on glass coverslips in 6-well plates at 3 × 105 cells/well. Twenty-four h later, cells were infected on ice (MOI of 10 unless otherwise stated) in 1% FBS-DMEM for 1 h. Cells then were warmed for 1 h at 37°C, after which they were acid washed (40 mM citric acid, 10 mM KCl, 135 mM NaCl, pH 3) for 1 min to inactivate extracellular virus (3). Acid was removed with 3 rinses in warm 1% FBS-DMEM, and cells were returned to 37°C. Cells were fixed at noted times after infection with 4% paraformaldehyde (PFA) for 10 min. Those that required antibody or WGA staining were permeabilized with 0.2% Triton X-100 and incubated in blocking buffer (10% goat serum and 1% BSA in PBS) for 1 h at room temperature.
Electron microscopy.
At 12 h postinfection (hpi), coverslip-grown Vero cells infected as described above were fixed in 2.5% glutaraldehyde for 12 h. Samples then were postfixed for 30 min in 1% (wt/vol) osmium tetroxide, dehydrated in increasing concentrations of ethanol, and infiltrated with EPON-812 (Electron Microscopy Sciences, Inc.). Embedding was achieved by inverting epoxy-filled BEEM capsules onto the coverslips. To remove BEEM capsules with the embedded samples from coverslips, samples were transferred between liquid nitrogen and boiling water, causing the glass coverslip to separate from the resin. Ultrathin sections (70 to 80 nm) prepared on a Leica Ultracut UCT ultramicrotome using a Diatome diamond knife were collected on 200-mesh copper grids and contrast stained with lead citrate and uranyl acetate as described previously (39). Images were recorded on film using a Philips 400T transmission electron microscope operated at 80,000 eV.
Immunofluorescence microscopy.
TIRF images were obtained on an inverted IX70 Olympus microscope with a 1.45-numeric-aperture (NA) (oil) Plan Apochromat 60× TIRF objective lens that was fitted with a Ludl modular automation controller (Ludl Electronic Products) and operated by MetaMorph software (Invitrogen). Images were acquired with a charge-coupled device camera (Retiga Exi; Qimaging). Confocal images were taken on a Zeiss LSM 510 scanning confocal microscope with the use of a Plan Apochromat 100×/1.4-NA oil immersion lens. Samples were excited with an argon laser and a 543-nm HeNe laser with photomultiplier tube image detection.
Nocodazole/cytochalasin B treatment.
At 4, 8, or 11.5 hpi, Vero cells were treated with either 1.7 μg/ml cytochalasin B (C6762; Sigma), 10 μg/ml nocodazole (M1404; Sigma), or an equal volume of dimethyl sulfoxide (DMSO) (BP231; Fisher) dissolved in 1% FBS-DMEM. Both drugs were previously solubilized in DMSO. At 12 hpi, cells were fixed in 4% paraformaldehyde for 10 min at room temperature and processed for immunofluorescence microscopy. In a second experimental design, cells that were treated with cytochalasin B at 11.5 hpi were rinsed at 12 hpi with 2 rinses in PBS and 2 rinses in warm 1% DMEM. Incubation was continued at 37°C until 12.75 hpi. At this time, cells were fixed with 4% paraformaldehyde and processed for immunofluorescence microscopy.
Image analysis.
TIRF images of 20 to 30 cells per sample set were analyzed with ImageJ software. The pixel area was calculated for both the total cell area and the patch areas as determined by WGA staining. Only glycoprotein patches larger than 300 pixels in area were used for quantifications. Virion density was determined by transferring the cell outlines as determined on combined channel images onto the green channel pictures. Background was subtracted from these pictures by using the rolling-ball algorithm (53) and setting the radius to 50 pixels. Integrated density was then calculated and divided by the area in question to get the virion density. P values were calculated using the t test for 2 samples assuming equal variances. Two-tail results were used for the verification of statistical significance.
Construction of recombinant HSV gE mutants.
A bacterial artificial chromosome (BAC) containing the HSV-1 KOS strain genome was used to generate recombinant viruses. The detailed protocol has been described previously (1). A gE null mutant (gEΔ) was made by replacing the gE start codon ATG with stop codon TAA. The gE cytoplasmic tail deletion mutant (gEΔCT), generated by adding two copies of stop codon (TAATAA) between residues W446 and R447 right after the transmembrane domain, was previously reported (14). gEΔCT rescue virus was generated by removing the stop codons. Correct clones were verified by HindIII digestion, PCR analysis, and DNA sequencing of the corresponding region. The resulting BAC plasmids were purified and then transfected into Vero cells with Lipofectamine 2000. After the appearance of cytopathic effects (3 to 4 days), transfected cells were harvested and used to infect new Vero cell monolayers to produce a viral stock.
Analysis of gE mutant expression and virion incorporation.
Vero cells were infected with wild-type HSV or gE mutants at a multiplicity of infection (MOI) of 5. To analyze gE expression, the medium was collected at 16 to 20 h postinfection, cleared at 2,500 rpm for 10 min, and then centrifuged at 26,000 rpm for 1 h at 4°C in a Beckman SW41 rotor through a 30% (wt/vol) sucrose cushion (1.7 ml). The virus-containing pellets were dissolved in 100 μl 1× SDS-PAGE sample buffer, and an approximately equal amount of virus normalized to VP5 was loaded into an SDS-PAGE gel for Western blot analyses. The infected cell lysates were also analyzed for the expression levels of gE and its mutants. Blots were probed with α-gE polyclonal antibody (see above) at a 1:6,000 dilution.
Determination of viral output.
Very little virus (less than 1 virion per 3 infected cells) was released from the cell surface into the supernatant by 12 hpi. Therefore, titer was determined for cell-associated virus only unless stated otherwise. To determine the amount of infectious, cell-associated virus, infected cells were harvested in PBS with the aid of a cell scraper. Samples were frozen and thawed twice and placed in a bath sonicator for 5 s. The titer was then determined by limiting dilution assay with a 4% agar overlay. On day 3, plaques were detected by a 5-h incubation in 0.5 mg/ml thiazolyl blue tetrazolium bromide stain diluted in medium (M2128; Sigma).
RESULTS
Virus egress occurs at specific sites on adherent surfaces of Vero cells.
To study the process of HSV-1 egress in Vero cells, electron microscopy was used to observe the pattern of progeny virus association with the cell surface. Glass-grown Vero cells were infected with KOS HSV-1 at an MOI of 10, and at 12 h postinfection cells were fixed on coverslips and processed for thin sectioning. At this time point, progeny virions that were not transferred to nearby cells were found to be associated with the parental cell surface with few virions released into the media. Micrographs showed that at 12 hpi, the majority of virions were observed at specific areas on the cell surface, rather than in a randomly dispersed release pattern (Fig. 1A to D). Virus-containing regions were located at cell-cell contact sites and at areas along the adherent cell surface. There were approximately 3-fold more virions at these locations than on the nonadherent upper cell surface, although this is likely an underestimation, since some virions along the upper cell surfaces are expected to be noninfectious parental virus particles that did not enter the cell. At both the substrate-adherent surface and at cell-cell contact site egress locations, additional membrane was present allowing a curvature in the membrane at the site and the creation of a pocket-like structure (Fig. 1A to C). This was not the case in uninfected cell samples; the adherent cell membrane of mock-infected Vero cells was tightly apposed to the coverslip surface (Fig. 1E and F). Although many virions were observed exterior to the plasma membrane in infected cells, none were observed near the interior side of the membrane (Fig. 1A to D). The few virions seen on other membrane surfaces were often in areas adjacent to cell-cell contacts (Fig. 1D, bracket). Similar results were obtained whether the cells were in a confluent monolayer or if they were less densely plated; the movement of virions to the substrate-adherent surface was not induced by a lack of available cell-cell-adherent surfaces.
Fig 1.

Location of progeny virions in HSV-1-infected Vero cells. (A to D) Thin-section electron micrographs of infected Vero cells fixed and processed on coverslips at 12 hpi. Note that the majority of virions are released in pockets along the adherent cell surface (arrowheads) and at cell-cell contact points (arrows), with some virus above cell-cell contacts (bracket). Panel B is an enlargement of the left arrowhead in panel A; panel C is an enlargement of the right arrowhead in panel A. (E and F) Electron micrographs of mock-infected Vero cells. Note that, unlike infected cells, the cell membrane is closely apposed to the coverslip edge. In panels A to F the coverslip interface is marked by the thin line near the bottom surface of the cell. (G) Consecutive Z-stack confocal images of a representative glass coverslip-grown Vero cell infected with VP26-GFP mutant virus. Sections begin at the coverslip and go up by 0.25-μm increments. Asterisks mark the two sections above the coverslip where the majority of virions can be seen. Other sections have far fewer virions. The nucleus (containing VP26-GFP-labeled capsid assembly areas) is outlined. Arrows indicate virus released along a cell-cell contact. Images were obtained with a Philips 400T transmission electron microscope (A to F) and a Zeiss LSM 510 confocal microscope with an inverted 100× lens (G).
Confocal microscopy was utilized to observe the release of virions in larger numbers of infected Vero cells. Cells grown on glass coverslips were infected with a GFP-tagged VP26 mutant (K26GFP) and fixed at 12 hpi. Confocal Z-stack images showed that GFP-labeled virions were concentrated along the adherent surfaces of cells. A representative series is shown in Fig. 1G. Most virions outside the nucleus were detected at the two planes closest to the coverslip (those marked by an asterisk). The majority of virions that were visible above the two planes were located at cell-cell contact points (Fig. 1G, arrows in slices 4 to 6). The large GFP-containing areas located in the nucleus are capsid assembly compartments. In the following sections we further characterize the egress sites that form along the coverslip-adherent cell surface.
Viral egress sites are enriched in glycoproteins.
Due to a high cytoplasmic signal of viral proteins, egress sites along the adherent cell surface were best visualized using total internal reflection fluorescence (TIRF) microscopy. This method allowed the area of excitation to be restricted to a small plane above the coverslip. A standard TIRF field is 70 to 300 nm above the coverslip. The angle of the laser can be adjusted to obtain the desired excitation field within this range (50). In the following studies, the laser was set at the maximum angle to allow the excitation of the largest area possible, which is 300 nm above the coverslip.
Using TIRF microscopy, we found that GFP-labeled virions clustered at specific sites along the adherent cell surface in a manner similar to that observed with EM (Fig. 2). In addition, infection induced the recruitment of glycoproteins to egress locations. Wheat germ agglutinin (WGA) binds to N-acetylglucosamine and N-acetylneuraminic acid (sialic acid) residues on glycoproteins, whether viral or cellular in origin (45). When infected Vero cells were fixed, permeabilized, and stained with rhodamine-labeled WGA, it was observed that viral release sites were stained much more strongly than the surrounding cell membrane (Fig. 2A and E), indicating that the regions where cell-associated viral particles were observed had a much greater concentration of glycoproteins. Such focal concentrations were absent from uninfected cells, where the WGA stain was light and diffuse (Fig. 2D). In fact, WGA staining outside the patches was much brighter in infected cells than in uninfected cells, suggesting a greater amount of surface glycoprotein expression overall. There were often several viral egress patches per infected cell, yet cells with a single large patch were also seen. Patches could be expansive and were generally peripheral, as shown in Fig. 2B.
Fig 2.

Glycoprotein enrichment at coverslip-adherent surface egress sites. (A) TIRF micrographs of a VP26-GFP HSV-1-infected Vero cell at 12 hpi fixed, permeabilized, and stained with rhodamine-conjugated WGA (Rh-WGA) to mark glycosylated proteins. As in EM pictures, GFP-labeled virions were found to cluster at specific sites along the cell surface. Note that glycosylated proteins accumulate at these sites. A magnified image of the box is shown in panel E. (B) TIRF micrographs of a VP26-GFP HSV-infected Vero cell at 12 hpi that had been treated with α-gB (DL16) antibody. (C) TIRF micrographs of a VP26-GFP HSV-infected Vero cell treated with α-gD antibody (DL11). Staining indicates that viral glycoproteins accumulate at egress sites. Arrowheads mark two egress sites where the pocket-like structure of the sites is apparent. The holes in the donuts are areas where the membrane has extended beyond the 300-nm laser excitation range. Virus is rarely visible in the holes, indicating that virions are closely bound to the cell membrane. (D) Mock-infected Vero cell permeabilized and stained with WGA shown at that same exposure as that for the infected cell in panel A. The cell edge is outlined as determined by increasing the brightness of the image until the edge was visible. The size bar in panel C is also relevant for panels A to D. (F) HSV-1-infected Vero cell stained with both rhodamine-WGA and α-gD antibody. Note that there is complete colocalization between the two. Alexa 594-conjugated secondary antibodies were used in panels B, C, and F.
Viral glycoproteins were found to be a component of WGA-stained patches. Infected Vero cells were fixed at 12 hpi and stained with α-gB, α-gD, α-gH, or α-gE monoclonal antibody. Staining for gB and gD are shown in Fig. 2B and C, while gH and gE staining is not shown. All viral glycoproteins were found in greater amounts in the regions where cell-associated virus was concentrated compared to the surrounding membrane. Viral glycoprotein staining in infected cells entirely overlapped with WGA staining (Fig. 2F). Viral proteins appear to compose a large portion of the glycoprotein patches, but cellular proteins also may be recruited. Since both viral glycoprotein staining and WGA staining similarly define the size and shape of egress locations, patches were labeled using rhodamine-WGA in the following figures. Patches were observed in all cells at 12 hpi that were labeled with viral glycoprotein antibodies, suggesting that the majority of infected cells form these structures (data not shown). Many glycoprotein-stained patches resembled a donut in shape (Fig. 2C, arrowheads). This phenomenon was due to the pocket-like structure of these sites. The holes in the donuts were the result of the membrane rising above the 300-nm excitation limit in the center of the patches.
Concentrations of virions and glycoproteins along the adherent surface were not specific to Vero cells but seemed to be associated with a nonpolarized state. Similar adherent surface glycoprotein patches were observed in infected HeLa cells (a nonpolarized cell type), but glycoproteins appeared to accumulate only at cell-cell junctions in polarized HEC-1A cells (data not shown).
Glycoprotein patches form independently from trafficking virions.
Vero cells infected at an MOI of 10 showed neither glycoprotein staining nor VP26-GFP virus signal at 4 hpi (Fig. 3A, top). At 6 h postinfection, small glycoprotein patches were detectable, although virions were rarely seen associating with these areas (Fig. 3A, middle). By 8 hpi, glycoprotein patches were more widespread and low levels of virus were consistently detected associating with the patches (Fig. 3A, bottom). This observation indicates that release sites were determined early in infection. Glycoproteins arrived first, modifying the composition of the egress site membrane. Virions then associated with these membranes at later time points.
Fig 3.
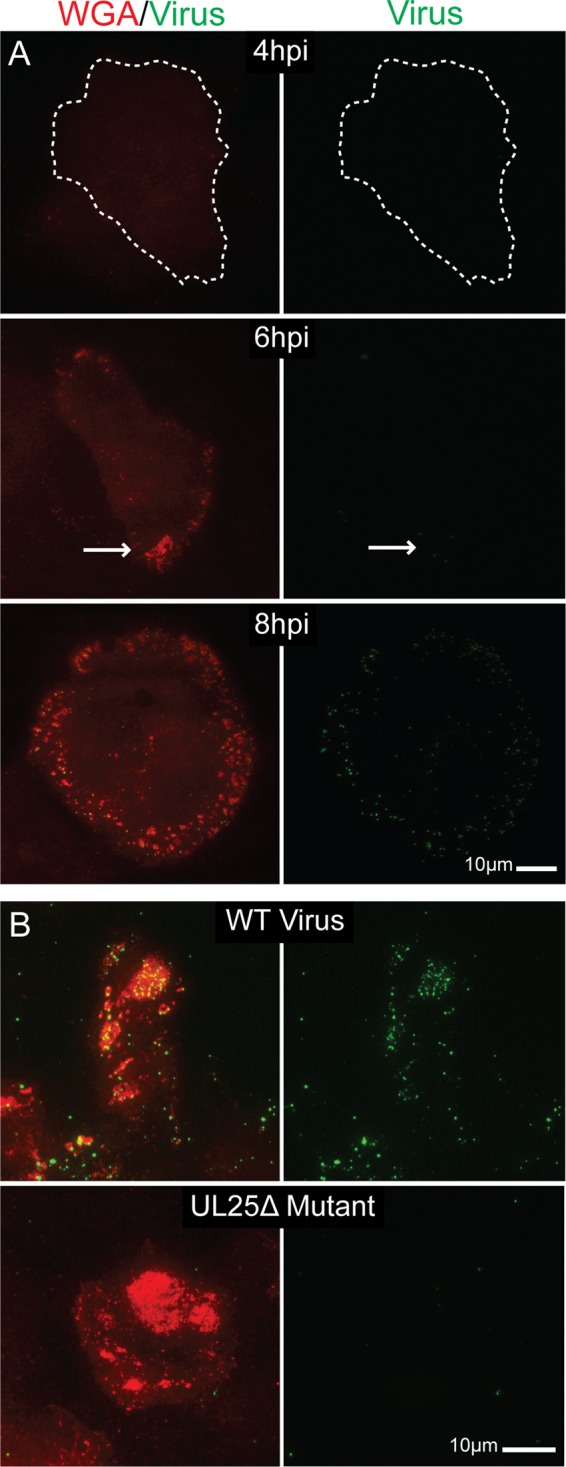
TIRF micrographs showing formation of glycoprotein patch sites uncoupled from virus egress. (A) Vero cells infected with VP26-GFP HSV-1 were fixed at 4, 6, and 8 hpi. Patch glycoproteins were labeled with rhodamine-WGA. All images were taken at the same exposure. In the 4-hpi image, the edge of the adherent surface of the cell (as determined by increasing the brightness of the image) is outlined. Note that neither glycoprotein patches nor virus are visible at 4 hpi. By 6 hpi, glycoprotein patches are beginning to form (arrow) on the adherent surface, but virus still is not evident. At 8 hpi, there is pronounced glycoprotein staining on the cell surface and virions are beginning to accumulate in these areas. Images indicate that patches form before viral egress. (B) Vero cells infected with wild-type KOS HSV-1 (top) or UL25Δ mutant HSV (bottom) were fixed at 12 hpi. In a UL25Δ infection, capsids are retained in the nucleus. Egress site glycoproteins were labeled with WGA. Virus was labeled with α-VP5 major capsid protein antibody and Alexa 488-labeled secondary antibody. Infections suggest that adherent surface glycoprotein patches form independently of viral release.
To further explore the differential trafficking between virions and glycoproteins, Vero cells were infected with a ΔUL25 mutant (KUL25NS). The gene product of UL25 is needed for the packaging of DNA into the capsid; when it is absent, DNA is not packaged and capsids do not exit the nucleus. However, protein expression continues normally (31). If glycoprotein movement to the cell surface was induced by virus envelopment and trafficking, then we would expect to see a drop in the size of glycoprotein patches in cells infected with the ΔUL25 virus compared to the WT. If the glycoprotein concentrations observed were actually glycoproteins on virion envelopes, then we would expect to see no glycoproteins on the surface with the ΔUL25 mutant.
To test this idea, Vero cells were infected with either WT KOS-HSV-1 or the ΔUL25 mutant for 12 h. Cells were then fixed, permeabilized, and stained with WGA. Capsids were labeled with α-VP5 monoclonal antibody and Alexa 488-labeled secondary antibody. In the KOS-infected cells, virus and glycoproteins were found to colocalize at release site patches as expected (Fig. 3B, top). In ΔUL25 mutant-infected cells, glycoproteins formed normal patches even though capsids were not present (Fig. 3B, bottom). In addition, the patches were approximately equal in size to those formed in the infection with wild-type UL25 gene product (data not shown). These results provide evidence that patch glycoproteins are in the cell membrane of infected Vero cells, and glycoproteins traffic to and accumulate at viral egress sites independently of virions.
Release sites are depleted of cytoskeletal elements.
It has been previously shown that the cellular cytoskeleton is altered during HSV-1 infection. The microtubule organizing center (MTOC) is disrupted and microtubules become sparse and disorganized (23, 26, 62). Although peripheral anti-alpha-tubulin-stained microtubules could be seen in our samples, results showed that these microtubules did not colocalize with virions at peripheral egress sites (Fig. 4A). Actin stress fibers are also largely depolymerized during infection (16, 41, 59, 63). When infected Vero cells were stained for actin with Texas Red-labeled phalloidin, few stress fibers were visible but the actin cortex (the unbundled, highly branched actin lining the cell membrane) could still be seen in most cases (Fig. 4B). Contrary to our expectations, the actin cortex was depleted at the viral egress sites. It is unlikely that the actin holes observed were due to limitations of the TIRF laser; depleted areas were still apparent when the entire membrane was within range of the laser. In addition, the majority of areas where actin was cleared contained a glycoprotein patch whose edges closely followed the line of actin depletion even if few virions were visible (data not shown). A similar result was observed with focal adhesions (Fig. 4C). Although patches often formed shapes that mimicked large focal adhesions, focal adhesions were not present at the viral egress sites; they were depleted in areas where the patches formed. Areas of microtubule, actin, and focal adhesion depletion were rarely detected in mock-infected cells (Fig. 4D to F). They appeared only after HSV infection.
Fig 4.

Location of cytoskeletal elements in relation to viral egress sites on the coverslip-adherent surface of HSV-1-infected Vero cells as shown by TIRF microscopy. (A to C) VP26-GFP HSV-infected Vero cells stained with α-tubulin antibody to label microtubules (A), Texas Red-phalloidin (TR-Phalloidin) to stain actin (B), or α-vinculin antibody to mark focal adhesions (C). Note that areas where virions accumulate are depleted of all three cytoskeletal elements. The lined areas mark representative egress sites where the depletion has occurred. The starred area in panel B is an adjacent uninfected cell. (D to F) Mock-infected cells stained with α-tubulin antibody (D), Texas Red-phalloidin (E), or α-vinculin antibody (F). Note that large areas of cytoskeletal depletion are rarely seen in uninfected cells. Alexa 594-conjugated goat anti-mouse secondary antibody was used in panels A, C, D, and F. The size marker in panel F is relevant for all panels.
Both actin and microtubules contribute to glycoprotein patch formation and virion trafficking to release sites.
Although viral egress sites were depleted of cytoskeletal elements, it was likely that microtubules, actin, or both were involved in virus trafficking. The expected size and density of virus-containing vesicles along with the nonrandom release of virions suggested that a structural element was assisting their movement and delivery to their destination. The possible role of microtubules and actin was investigated by depolymerizing their filaments at increasing intervals after infection. VP26-GFP HSV-1-infected cells were treated at 4 hpi (before patches were visible and infectious virus detectable), 8 hpi (patches were expanding, a few virions could be seen), or 11.5 hpi (patches were large and contained many virus particles). Treatment included either 10 μg/ml nocodazole to depolymerize microtubules, 1.7 μg/ml cytochalasin B to depolymerize actin, or 3.3 μl/ml DMSO as a control. Each drug concentration used was the minimal amount needed to completely depolymerize the cytoskeleton. Treated cells were fixed at 12 hpi and stained with rhodamine-WGA to define the patch outlines. Resulting TIRF images (Fig. 5A to C) were then analyzed as described in Materials and Methods.
Fig 5.

Role of microtubules and actin in virus/glycoprotein trafficking and patch formation. Cells were infected with VP26-GFP HSV-1 and fixed at 12 hpi. (A) TIRF micrograph of DMSO-treated sample stained with rhodamine-WGA to mark glycoprotein patches. (B) TIRF micrographs of infected cells treated for the noted intervals during infection with 10 μg/ml nocodazole (Noc). Cells were stained with WGA. Note that patches in the 4- to 12-hpi treatment sample are smaller, with fewer virions than the control. (C) TIRF micrographs of infected cells treated for the noted periods with 1.7 μg/ml cytochalasin B (CyB). Patches were labeled with WGA. Note that treatment has disrupted glycoprotein patch structures. (D) Graph depicts the percentage of cell membrane that is covered in glycoprotein-rich patches. Numbers were obtained by dividing the sum of the patch areas (that were above the 300-pixel cutoff) by the total area of the cellular adherent membrane visible in TIRF micrographs. (E) Graph depicts the amount of virus on the cellular adherent membrane visible by TIRF microscopy. Twenty-five cells per sample set were analyzed. Error bars are for the standard errors of the means. P values of treated samples compared to DMSO control samples are labeled the following: *, <0.05; **, <0.005; and ***, <0.0005.
It was found that the depolymerization of the microtubules during infection caused a significant reduction in the percentage of cell membrane covered by glycoprotein patches. Greater effects were observed at the earlier time point addition (Fig. 5B and D). In addition, depolymerization at early time points caused a reduction in the total number of virions on the visible cell membrane at the adherent surface (Fig. 5B and E). No significant effect was observed when nocodazole was added later in infection after patches had formed and many virions were present. The determination of virus titers showed that there was no statistically significant effect on titer throughout several experimental repetitions (data not shown), although small reductions in infectious virus are difficult to measure. The results are interpreted to indicate that the lack of microtubules impedes the trafficking of glycoproteins to the cell surface. The decrease in virus at the cell membrane is also likely due to inhibited transport to the surface, although the depolymerization of microtubules could also be hindering the envelopment of virus.
Surprisingly, it was observed that disrupting the actin cytoskeleton decreased the number and size of egress patches at all time points tested (Fig. 5C and D). This effect was more pronounced when the cytoskeleton was depolymerized early in infection, but it was also seen when actin was depolymerized after patches had already formed. As actin was disrupted, the patches that had formed were dispersed into very small glycoprotein aggregations with the occasional associated virion. The result that previously formed patches disassemble when filamentous actin is removed is interpreted to indicate that actin filaments are maintaining the structure of egress sites.
Treatment with cytochalasin B early in infection decreased the amount of virus on the cell surface (Fig. 5E). This suggested that actin is involved in trafficking of virus to egress locations as well as maintaining patch integrity. There was no statistically significant difference in titers of cell-associated or supernatant virus for all treatments, so dispersing the patches after formation likely did not induce the release of the surface virions into the supernatant (data not shown). The observed decrease in detectable virus on the membrane when cells were treated from 11.5 to 12 hpi was probably due to the diffusion of virions onto the top membrane, where they could not be detected using TIRF microscopy. Actin could be assisting the trafficking of virions from the cytoplasm to the cell surface, or it could be assisting newly secreted virus to move to patch locations through actin surfing. We conclude that actin contributes to both virus trafficking to egress sites and to the maintenance of viral and possibly cellular components at these sites.
The structure of egress sites can reform after disruption.
During the process of secretion in secretory cell types, actin is locally depolymerized in the cortex, allowing the passage of a vesicle. Existing studies have found that the depolymerized areas are often not much larger than the secretory vesicle, and the cytoskeleton generally reforms after exocytosis has occurred (13, 47). One explanation that we considered for the existence of actin holes at viral egress sites was that actin depolymerized to allow the exocytosis of the virus, and the holes remained due to a global block in actin repolymerization. We also considered the possibility that the actin cortex as a whole is lost during infection (likely through a block in actin polymerization) and viral release is directed to areas where this has occurred.
To test these ideas, Vero cells with observable patches (Fig. 6A) were treated with 1.7 μg/ml cytochalasin B for 30 min. Actin was depolymerized as expected, as shown in Fig. 6B. The drug was then rinsed out and infection continued for another 45 min. Samples were stained with either WGA or phalloidin. The results showed that the actin cortex was able to reform after depolymerization; there was no apparent block in actin filament polymerization or elongation (Fig. 6C). In addition, the viral egress sites were able to reform after disruption; patches consisting of dense glycoproteins and GFP-labeled virus were visible after the 45-min recovery period (Fig. 6C). It is unclear whether the dispersed virus/glycoproteins reformed into patches or the visible patches were simply new virus released since the restoration of the actin cortex. In either case, we conclude that actin was able to polymerize normally and infected cells were able to create new egress sites late in infection. The clearance of actin at these sites appears to be locally rather than globally induced.
Fig 6.
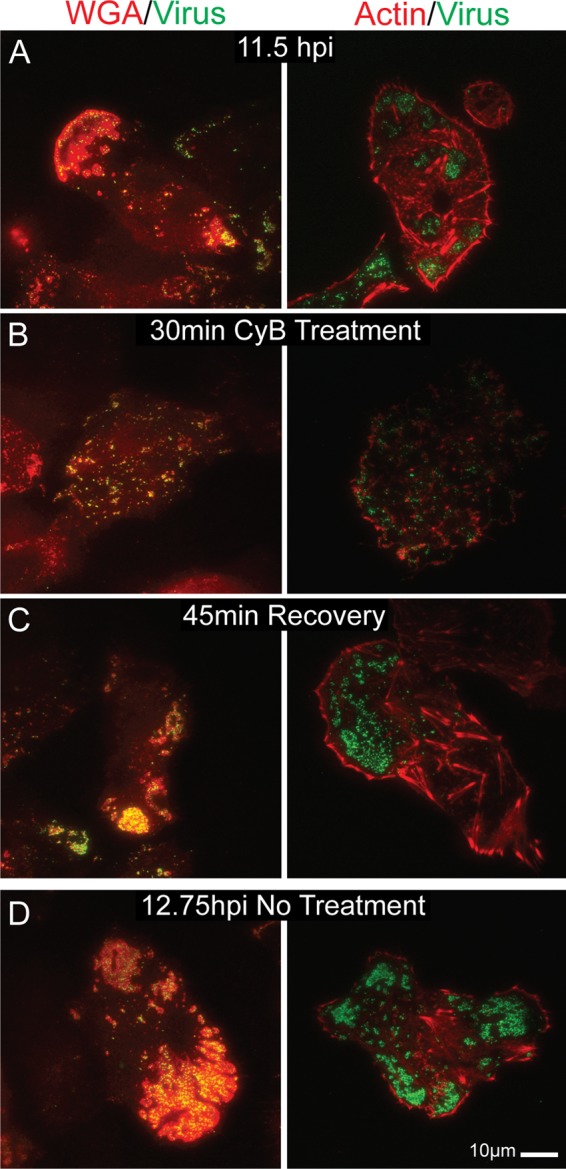
TIRF micrographs showing egress site reformation after actin depolymerization-induced disruption. VP26-GFP-infected Vero cells at 11.5 hpi (A) were treated with 1.7 μg/ml cytochalasin B for 30 min (B). (C) Toxin was then rinsed out and the infection continued for another 45 min. Sample cells were fixed at each step and stained with Texas Red-phalloidin or rhodamine-WGA. (D) Untreated control. Note that both the actin cortex and viral egress sites were able to reorganize after disruption. There is no global block in actin polymerization.
Glycoprotein E is necessary for glycoprotein patch formation but not trafficking of virus to release sites.
Directed egress in polarized cells such as keratinocytes and neurons is dependent upon viral glycoprotein E (gE). gE null mutants produce normal levels of progeny virus, but the trafficking of virions to egress sites is disrupted (8, 21, 29, 51, 60). We therefore wanted to test the role of this protein in HSV-1-infected nonpolarized cells. To test the function of gE in the trafficking of viral components to the coverslip-adherent cell surface, we ultilized ΔgE, gEΔCT, and gEΔCT rescue mutants. Using a bacterial artificial chromosome system, the start codon was replaced with a stop codon in the gE gene sequence. To create the gE cytoplasmic tail deletion mutant, two stop codons were added at residue 446 directly after the transmembrane region (14). The expression of gE was rescued by removing these stop codons.
The mutants were verified by harvesting the supernatant and cell lysates of infected Vero cells and staining for the appropriate protein (Fig. 7). The results showed that staining for gE was seen in the WT and rescue infection but was absent from the cell lysates and virions of the gE deletion mutants. The gE cytoplasmic tail deletion protein was incorporated into the virion at a much lower level and was less stable than its full-length counterpart. In cell lysate samples, the gE band was shifted compared to that of virions due to the presence of large amounts of immature gE (Fig. 7).
Fig 7.

Western blot analysis of gE expression in deletion and rescue mutants. Cell lysates and supernatants of wild-type HSV- and gE mutant-infected Vero cells were collected at 18 hpi. Samples were run on an SDS-polyacrylamide gel, transferred, and blotted with α-gE antibody. The gE antibody used is against the extracellular domain of gE (25). α-VP5 was used as a loading control. Note that gE is expressed normally in the WT and rescue samples (gEΔCT.R) but not in the deletion mutant-infected samples. The arrowhead points to a nonspecific band. Truncated gE is indicated by a star.
To test the role of gE in the creation of release sites and the egress of virions at these sites, Vero cells grown on glass coverslips were infected with ΔgE, gEΔCT, and gEΔCT rescue mutants. A low MOI of 0.3 was used to ensure infections arose from a single virion. To compare virus production, titer samples were collected at 10 hpi, a time at which each infected cell was producing virus but second-generation infected cells were not yet doing so. In this way, the effect of the gE deletions on the formation of infectious virus could be determined separately from effects on cell-to-cell spread. At 12 hpi, infected Vero cells were fixed with 4% PFA and treated with α-VP5 antibody (labeling major capsid protein) and Alexa 488-conjugated secondary antibody. Samples were then stained with rhodamine-WGA to label glycoprotein patches and viewed using TIRF microscopy. Twenty cells per infection sample were measured as described in Materials and Methods for both patch coverage and density of virions on the cell surface.
The results showed that the lack of gE or the gE cytoplasmic tail caused a reduction in the percentage of cell surface covered in glycoprotein patches compared to that of the rescue virus (Fig. 8A and B). However, while there was an inhibition in glycoprotein patch formation, the number of virions on the cell surface of the ΔgE mutants increased slightly (Fig. 8A and C) for unknown reasons. There was no statistical difference in titers, indicating that the production of progeny infectious virus was not increased (Fig. 8D). The results are interpreted to indicate that gE is important for the expansion of glycoprotein patches at egress locations, but unlike in polarized epithelial cells, gE does not direct virions to these sites. The gE protein appears to play different roles in the spread of infection in polarized and nonpolarized epithelial cells.
Fig 8.

Effect of gE and gE cytoplasmic tail deletions on patch formation and progeny virion trafficking. (A) TIRF images of Vero cells infected with the ΔgE mutant, the gEΔCT mutant, or the gEΔCT rescue virus at an MOI of 0.3 for 12 h. Cells were fixed and treated with rhodamine-WGA and α-VP5 antibody with Alexa 488-conjugated secondary antibody. Note that patches are smaller in size in the gE deletion mutant-infected cells, but the virion number in those patches is increased at 12 hpi. (B) Quantitative determination of the percentage of adherent cell membrane covered by glycoprotein patches and (C) the amount of virions on the total adherent membrane. Quantifications support the visual data. (D) Infected cells were harvested at 10 hpi, and the titer was determined by limiting-dilution plaque assay. Note that gE deletions had little effect on the production of progeny virus titer despite the observation that the number of virions on the adherent surface was increased. P values of deletion mutants compared to rescue virus are labeled the following: *, <0.05; **, <0.005; and ***, <0.0005. Bars represent standard errors of the means.
DISCUSSION
Within the mucosal tissues where cold sore lesions form, there are both highly differentiated and less differentiated epithelial cells. While there has been some study of directed egress in polarized epithelial cells (cells with tight junctions, compositionally distinct membranes, and high TER), egress in nonpolarized cell types has not been explored as extensively. Here, we report the results of a study in which the release of HSV virions in nonpolarized Vero cells was investigated. The major findings are summarized as follows. (i) HSV-1 egress in Vero cells is a directed process that occurs at adherent surfaces. (ii) Both the membrane composition and cytoskeletal structure of egress sites are modified during infection. These modifications begin before virus is released, and they proceed even when capsid trafficking is blocked. (iii) Trafficking of both virus and membrane components is dependent on microtubules and, to a lesser extent, actin. (iv) Actin determines the structure of egress locations; when actin is depolymerized the sites disperse. (v) Viral glycoprotein E aides in the formation of egress sites. These findings are discussed in greater detail below.
Virus is directed to adherent surfaces rather than cell-cell junctional proteins in Vero cells.
The observation that the majority of newly secreted virus associated with the cell membrane at specific sites along cell-cell and cell-coverslip contact areas indicates that the sorting of virus in nonpolarized cells is a directed rather than random process. Previous reports have shown a random, dispersed association of virions with the surface of HEp-2 nonpolarized cells at 17 hpi (21). We observed directed egress in Vero cells between 8 and 15 hpi. By 17 hpi, Vero cells were rounded and virus covered most cell surfaces (data not shown). It is possible that HEp-2 cells exhibit the directed sorting of virions at earlier time points as well. Although virus titers in Vero cells peak around 24 hpi, we ended our studies at 12 h because infection was observed to transfer to adjacent noninfected cells before 12 hpi (data not shown). The investigation of viral release during this 12-h time period thus gives the most information about viral egress during cell-to-cell spread. Although it has been hypothesized that the release of virions at cell-contacting lateral surfaces in polarized epithelial cells is determined by the presence of cell-cell junctional complexes (9), our results suggest that other elements are directing egress in nonpolarized cells. Vero cells do not express E-cadherin, and exogenously expressed nectin-1 does not traffic to egress sites (our unpublished observations). The state of being adherent to a substrate or cell may, therefore, be sufficient to direct egress in some cell types, including Vero and HeLa cells. This may be due to the directionality of the cytoskeleton or to cellular proteins expressed along adherent surfaces.
Infection induces the creation of specialized egress sites.
In the course of infection, the adherent surfaces of Vero cells were modified to create discernible viral egress sites. First, pocket-like membrane invaginations formed at egress locations that were visible by both EM and TIRF microscopy. In contrast, the plasma membrane of uninfected cells was tightly apposed to the coverslip. Second, the cell membrane at egress sites was extremely rich in glycoproteins. Comparable concentrations were never observed in uninfected cells. Lastly, cytoskeletal structures were modified at release sites with the depletion of microtubules, actin, and focal adhesions. Overall, the release of HSV-1 at egress sites shares similarities with the formation of virological synapses by retroviruses. Virological synapses form at areas of adhesion between infected and noninfected cells. Viral and cellular proteins are recruited to synapse sites, exocytosis is polarized toward these areas, and virions are released between the two cells (22). The observation that HSV-1 egress sites formed on adherent surfaces of Vero cells is a reasonable finding; in vivo the majority of adherent surfaces are those in contact with adjacent cells. In addition, viral glycoproteins concentrated at egress sites, and the lack of patches on nonadherent surfaces, suggests that secretion was polarized toward these structures. Future work will determine whether HSV-1 egress patches function as virological synapses and assist the cell-to-cell spread of infection.
The cellular cytoskeleton assists egress in several possible ways.
Although viral release sites were depleted of cytoskeletal elements, both microtubules and, to a lesser extent, actin were found to be important for the movement of viral components to egress locations. Microtubules are known to function in the long-distance movement of secretory vesicles, while actin has been reported to serve as both a barrier and an aide to secretion.
It is possible that actin surfing is playing a role in virus/glycoprotein patch formation. Virus may be released elsewhere on the cell surface and then surf through the movement of actin filaments to the sites where patches form. We observed that when actin motility was inhibited with blebbistatin, patch size was decreased (not shown). Whether this is due to inhibition in surfing or a block at a step previous to viral release will be explored in future studies.
In addition to assisting virus/glycoprotein trafficking, actin was found to play a structural role in the maintenance of HSV egress sites; when the actin cortex was depolymerized with cytochalasin B, glycoprotein patches and associated virions dispersed across the cell surface (Fig. 5 and 6). The maintenance of virions at the observed sites suggests an interaction with a protein or lipid within the glycoprotein-rich areas. The putative protein could be maintained at egress sites by actin in two ways. The virion-interacting protein could be bounded by an actin fence (24, 36); that is, the protein or lipid could be unable to move past the peripheral cortical actin and its membrane-associated proteins. It is also possible that the protein is directly/indirectly bound to the actin cytoskeleton. The depletion of actin we observed at release sites makes the former a likely possibility, although there may be low levels of actin at these sites that are not detectable by phalloidin. Future work should be able to characterize the proposed association between virus and cell surface protein and what role it plays in cell-to-cell spread.
Glycoprotein E is important for the formation of egress site glycoprotein patches.
gE has been reported to direct the trafficking of progeny virions to cell junctions in epithelial cells and synapses in neurons (4, 6, 21). Surprisingly, the deletion of gE in a Vero cell infection did not decrease the amount of virus associating with bottom surface egress locations; in fact, the number was increased. At this time, we do not have a theory for why this occurs. The absence of gE did decrease the amount of glycoproteins at these sites, resulting in much smaller egress patches. Therefore, gE appears to be involved in egress site formation in nonpolarized cells, but it does not function in a manner similar to that in polarized cell infections.
We considered the prospect that the observed decrease in patch size was due to the loss of gE stabilization of the surface glycoprotein aggregations. Previous reports have indicated that gE interacts with multiple glycoproteins and can stabilize these associations (10, 49). Although a slight dispersal is perhaps visible in ΔgE- and gEΔCT-infected cells (Fig. 8), it is not definitively more dispersed than it is in the rescue virus images. If the lack of gE did result in the disruption of glycoprotein associations but this dispersal was not visible, then those dispersed glycoproteins would have to have been removed from the surface, i.e., endocytosed. We think it unlikely that slightly disrupted (complete disruption would lead to no patches) glycoprotein aggregations would be endocytosed at the egress sites, and minimally disrupted aggregations would be unlikely to diffuse past the actin fence to areas where endocytosis would be more likely to occur. In the event that glycoproteins are held at the egress sites by linkage to low levels of actin, one could argue that gE is involved in this linkage. However, previous studies have not been able to observe a link between gE and the cytoskeleton; when the cytoskeleton is isolated from detergent-disrupted cells, gE remains in the soluble portion (9).
A small drop in patch size would be expected when a viral glycoprotein is deleted. The absence of gE as the cause of the observed decrease suggests that there is more gE in patches than the other cellular and viral glycoproteins combined. There does not appear to be more gE in egress patches than gD as shown by immunofluorescence (not shown); however, the difference may be hidden by different antibody affinities.
It is also possible that gE directs the trafficking of glycoprotein-containing vesicles to the cell surface. Previous works suggested that viral glycoprotein-containing vesicles can traffic independently to egress locations (9, 51, 52, 55, 65). In some HSV-1-infected neurons, capsids are shipped down the axon in unenveloped form (33, 52, 66), and glycoproteins are transported on separate vesicles. When gE is absent, the trafficking of both capsids and glycoprotein-containing vesicles into axons is greatly reduced (28, 51, 60). This indicates that gE can direct the movement of viral proteins by direct or indirect means. gE's targeting of vesicle transport may be limited to glycoproteins in nonpolarized cell types.
Hypothesized function of glycoprotein patches.
There are several roles that glycoprotein patches could play in an epithelial cell infection. Viral glycoproteins concentrated in the cell membrane may be able to recruit entry receptors on adjacent uninfected cells. This may lead to a more efficient transfer of virions to a target cell. gE in concert with gI is known to function as an Fc receptor (12, 15, 19, 38), and the observed reduction in patch size with the ΔgE mutants suggests that there are large amounts of gE protein in the patches. It is possible that gE at the egress sites can function to protect progeny virus from attack from the humoral immune system. There is also the prospect that proteins within the patch membrane are able to assist with the fusion of the outgoing virus-containing vesicle with the cell surface. Previous reports have concluded that Vero cells infected with a ΔgE mutant are inhibited in cell-cell spread and cell-neuron spread (2, 29). In one study it was found that the transfer of virus from infected cells to neurons is almost completely blocked in a gE null infection. However, gE null virus added to axons directly has no trouble entering and traveling to the neuron cell body to initiate infection (29, 61). Assuming the viral egress sites that form along cell-glass contact surfaces function similarly to those that form between cells, the presence of a virion may not be sufficient to induce infection in an adjacent cell; the buildup of cellular and/or viral proteins at these locations may be necessary for cell-cell spread. Since free virus is able to enter both epithelial cells and neurons without difficulty, it is likely not obligatory for glycoproteins on an infected cell to induce the creation of a specialized entry site on an adjacent noninfected cell. However, since the majority of progeny virions remain attached to the host cell surface until very late in infection, it is quite possible that proteins recruited to egress sites are necessary for viral detachment. It is our hope that future studies in this area will clarify the role that the recruited glycoproteins play in the cell-to-cell spread of HSV-1 infection.
ACKNOWLEDGMENTS
We are extremely grateful to Alan (Rick) Horwitz for the use of his TIRF microscope. We also thank Jessica Zareno for her generous time and technical support. In addition, we acknowledge the procedural support provided by Stacey Guillot of the Advanced Microscopy Facility at the University of Virginia. We are indebted to Gary Cohen, Roselyn Eisenberg, and Harvey Friedman for the kind gifts of their anti-HSV glycoprotein antibodies. Likewise, we thank Fred Homa and Prashant Desai for providing HSV-1 mutants. Furthermore, we are grateful to Amy Bouton and Keena Thomas for their generosity in advice and reagents. Lastly, we thank Jesse Seamon for computer access and technical support.
We are grateful to David Castle, Dan Engel, Rick Horwitz, Dean Kedes, Ulrike Lorenz, Tom Parsons, Dorothy Schafer, Judy White, and John Wills for helpful advice on experimental design.
This work was supported by NIAID training grant AI007046, NIH grant AI041644 awarded to J.C.B., and NIAID grant AI071286 awarded to J.W.W.
Footnotes
Published ahead of print 24 April 2012
REFERENCES
- 1. Baird NL, Starkey JL, Hughes DJ, Wills JW. 2010. Myristylation and palmitylation of HSV-1 UL11 are not essential for its function. Virology 397:80–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balan P, et al. 1994. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins gG, gE, gI or the putative gJ. J. Gen. Virol. 75:1245–1258 [DOI] [PubMed] [Google Scholar]
- 3. Brunetti CR, et al. 1995. Role of mannose-6-phosphate receptors in herpes simplex virus entry into cells and cell-to-cell transmission. J. Virol. 69:3517–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Regge N, et al. 2006. Alpha-herpesvirus glycoprotein D interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J. Cell Biol. 174:267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Desai P, Person S. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL. 2008. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 18:35–51 [DOI] [PubMed] [Google Scholar]
- 7. Dingwell KS, et al. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 68:834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dingwell KS, Doering LC, Johnson DC. 1995. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J. Virol. 69:7087–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dingwell KS, Johnson DC. 1998. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 72:8933–8942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Favoreel HW, Nauwynck HJ, Van Oostveldt P, Mettenleiter TC, Pensaert MB. 1997. Antibody-induced and cytoskeleton-mediated redistribution and shedding of viral glycoproteins, expressed on pseudorabies virus-infected cells. J. Virol. 71:8254–8261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Felts RL, et al. 2010. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc. Natl. Acad. Sci. U. S. A. 107:13336–13341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frank I, Friedman HM. 1989. A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. J. Virol. 63:4479–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giner D, et al. 2005. Real-time dynamics of the F-actin cytoskeleton during secretion from chromaffin cells. J. Cell Sci. 118:2871–2880 [DOI] [PubMed] [Google Scholar]
- 14. Han J, Chadha P, Meckes DG, Jr, Baird NL, Wills JW. 2011. Interaction and interdependent packaging of tegument protein UL11 and glycoprotein e of herpes simplex virus. J. Virol. 85:9437–9446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanke T, Graham FL, Lulitanond V, Johnson DC. 1990. Herpes simplex virus IgG Fc receptors induced using recombinant adenovirus vectors expressing glycoproteins E and I. Virology 177:437–444 [DOI] [PubMed] [Google Scholar]
- 16. Heeg U, Dienes HP, Muller S, Falke D. 1986. Involvement of actin-containing microfilaments in HSV-induced cytopathology and the influence of inhibitors of glycosylation. Arch. Virol. 91:257–270 [DOI] [PubMed] [Google Scholar]
- 17. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 18. Johnson DC, Feenstra V. 1987. Identification of a novel herpes simplex virus type 1-induced glycoprotein which complexes with gE and binds immunoglobulin. J. Virol. 61:2208–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson DC, Frame MC, Ligas MW, Cross AM, Stow ND. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 62:1347–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson DC, Huber MT. 2002. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 76:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson DC, Webb M, Wisner TW, Brunetti C. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 75:821–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jolly C, Sattentau QJ. 2004. Retroviral spread by induction of virological synapses. Traffic 5:643–650 [DOI] [PubMed] [Google Scholar]
- 23. Kotsakis A, Pomeranz LE, Blouin A, Blaho JA. 2001. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J. Virol. 75:8697–8711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kusumi A, et al. 2005. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 34:351–378 [DOI] [PubMed] [Google Scholar]
- 25. Lin X, Lubinski JM, Friedman HM. 2004. Immunization strategies to block the herpes simplex virus type 1 immunoglobulin G Fc receptor. J. Virol. 78:2562–2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu M, Schmidt EE, Halford WP. 2010. ICP0 dismantles microtubule networks in herpes simplex virus-infected cells. PLoS One 5:e10975 doi:10.1371/journal.pone.0010975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Llewellyn GN, Hogue IB, Grover JR, Ono A. 2010. Nucleocapsid promotes localization of HIV-1 gag to uropods that participate in virological synapses between T cells. PLoS Pathog. 6:e1001167 doi:10.1371/journal.ppat.1001167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McGraw HM, Awasthi S, Wojcechowskyj JA, Friedman HM. 2009. Anterograde spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. J. Virol. 83:8315–8326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McGraw HM, Friedman HM. 2009. Herpes simplex virus type 1 glycoprotein E mediates retrograde spread from epithelial cells to neurites. J. Virol. 83:4791–4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McMillan TN, Johnson DC. 2001. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 75:1928–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McNab AR, et al. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 72:1060–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mettenleiter TC, Klupp BG, Granzow H. 2006. Herpesvirus assembly: a tale of two membranes. Curr. Opin. Microbiol. 9:423–429 [DOI] [PubMed] [Google Scholar]
- 33. Miranda-Saksena M, et al. 2009. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J. Virol. 83:3187–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morita K, et al. 1998. Subcellular distribution of tight junction-associated proteins (occludin, ZO-1, ZO-2) in rodent skin. J. Investig. Dermatol. 110:862–866 [DOI] [PubMed] [Google Scholar]
- 35. Moriyama K, Imayama S, Mohri S, Kurata T, Mori R. 1992. Localization of herpes simplex virus type 1 in sebaceous glands of mice. Arch. Virol. 123:13–27 [DOI] [PubMed] [Google Scholar]
- 36. Morone N, et al. 2006. Three-dimensional reconstruction of the membrane skeleton at the plasma membrane interface by electron tomography. J. Cell Biol. 174:851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mothes W, Sherer NM, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J. Virol. 84:8360–8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagashunmugam T, et al. 1998. In vivo immune evasion mediated by the herpes simplex virus type 1 immunoglobulin G Fc receptor. J. Virol. 72:5351–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Newcomb WW, Brown JC. 2009. Time-dependent transformation of the herpesvirus tegument. J. Virol. 83:8082–8089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Newcomb WW, Brown JC. 2010. Structure and capsid association of the herpesvirus large tegument protein UL36. J. Virol. 84:9408–9414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Norrild B, Lehto VP, Virtanen I. 1986. Organization of cytoskeleton elements during herpes simplex virus type 1 infection of human fibroblasts: an immunofluorescence study. J. Gen. Virol. 67:97–105 [DOI] [PubMed] [Google Scholar]
- 42. Ohara PT, Chin MS, LaVail JH. 2000. The spread of herpes simplex virus type 1 from trigeminal neurons to the murine cornea: an immunoelectron microscopy study. J. Virol. 74:4776–4786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Owens RJ, Dubay JW, Hunter E, Compans RW. 1991. Human immunodeficiency virus envelope protein determines the site of virus release in polarized epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 88:3987–3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pais-Correia AM, et al. 2010. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 16:83–89 [DOI] [PubMed] [Google Scholar]
- 45. Peters BP, Ebisu S, Goldstein IJ, Flashner M. 1979. Interaction of wheat germ agglutinin with sialic acid. Biochemistry 18:5505–5511 [DOI] [PubMed] [Google Scholar]
- 46. Pummi K, et al. 2001. Epidermal tight junctions: ZO-1 and occludin are expressed in mature, developing, and affected skin and in vitro differentiating keratinocytes. J. Investig. Dermatol. 117:1050–1058 [DOI] [PubMed] [Google Scholar]
- 47. Rak GD, Mace EM, Banerjee PP, Svitkina T, Orange JS. 2011. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol. 9:e1001151 doi:10.1371/journal.pbio.1001151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Remillard-Labrosse G, Mihai C, Duron J, Guay G, Lippe R. 2009. Protein kinase D-dependent trafficking of the large herpes simplex virus type 1 capsids from the TGN to plasma membrane. Traffic 10:1074–1083 [DOI] [PubMed] [Google Scholar]
- 49. Rizvi SM, Raghavan M. 2003. Responses of herpes simplex virus type 1-infected cells to the presence of extracellular antibodies: gE-dependent glycoprotein capping and enhancement in cell-to-cell spread. J. Virol. 77:701–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sarkar A, Robertson RB, Fernandez JM. 2004. Simultaneous atomic force microscope and fluorescence measurements of protein unfolding using a calibrated evanescent wave. Proc. Natl. Acad. Sci. U. S. A. 101:12882–12886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Snyder A, Polcicova K, Johnson DC. 2008. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 82:10613–10624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Snyder A, Wisner TW, Johnson DC. 2006. Herpes simplex virus capsids are transported in neuronal axons without an envelope containing the viral glycoproteins. J. Virol. 80:11165–11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sternberg S. 1983. Biomedical image processing. IEEE Computer 16:22–34 [Google Scholar]
- 54. Sugimoto K, et al. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tomishima MJ, Enquist LW. 2001. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. J. Cell Biol. 154:741–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Trus BL, Newcomb WW, Booy FP, Brown JC, Steven AC. 1992. Distinct monoclonal antibodies separately label the hexons or the pentons of herpes simplex virus capsid. Proc. Natl. Acad. Sci. U. S. A. 89:11508–11512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tucker SP, Compans RW. 1993. Virus infection of polarized epithelial cells. Adv. Virus Res. 42:187–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Turcotte S, Letellier J, Lippe R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 79:8847–8860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Van Minnebruggen G, Favoreel HW, Jacobs L, Nauwynck HJ. 2003. Pseudorabies virus US3 protein kinase mediates actin stress fiber breakdown. J. Virol. 77:9074–9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang F, et al. 2005. Herpes simplex virus type 1 glycoprotein e is required for axonal localization of capsid, tegument, and membrane glycoproteins. J. Virol. 79:13362–13372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang F, et al. 2010. Herpes simplex virus type 2 glycoprotein E is required for efficient virus spread from epithelial cells to neurons and for targeting viral proteins from the neuron cell body into axons. Virology 405:269–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ward PL, Avitabile E, Campadelli-Fiume G, Roizman B. 1998. Conservation of the architecture of the Golgi apparatus related to a differential organization of microtubules in polykaryocytes induced by syn mutants of herpes simplex virus 1. Virology 241:189–199 [DOI] [PubMed] [Google Scholar]
- 63. Winkler M, Dawson GJ, Elizan TS, Berl S. 1982. Distribution of actin and myosin in a rat neuronal cell line infected with herpes simplex virus. Arch. Virol. 72:95–103 [DOI] [PubMed] [Google Scholar]
- 64. Wisner T, Brunetti C, Dingwell K, Johnson DC. 2000. The extracellular domain of herpes simplex virus gE is sufficient for accumulation at cell junctions but not for cell-to-cell spread. J. Virol. 74:2278–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wisner TW, Johnson DC. 2004. Redistribution of cellular and herpes simplex virus proteins from the trans-Golgi network to cell junctions without enveloped capsids. J. Virol. 78:11519–11535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wisner TW, Sugimoto K, Howard PW, Kawaguchi Y, Johnson DC. 2011. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J. Virol. 85:5919–5928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zurzolo C, et al. 1992. Opposite polarity of virus budding and of viral envelope glycoprotein distribution in epithelial cells derived from different tissues. J. Cell Biol. 117:551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]