

Abstract
Functional analysis of Bifidobacterium genes is essential for understanding host-Bifidobacterium interactions with beneficial effects on human health; however, the lack of an effective targeted gene inactivation system in bifidobacteria has prevented the development of functional genomics in this bacterium. Here, we report the development of a markerless gene deletion system involving a double crossover in Bifidobacterium longum. Incompatible plasmid vectors were used to facilitate a second crossover step. The conditional replication vector pBS423-ΔrepA, which lacks the plasmid replication gene repA, was integrated into the target gene by a first crossover event. Subsequently, the replicative plasmid pTBR101-CM, which harbors repA, was introduced into this integrant to facilitate the second crossover step and subsequent elimination of the excised conditional replication vector from the cells by plasmid incompatibility. The proposed system was confirmed to work as expected in B. longum 105-A using the chromosomal full-length β-galactosidase gene as a target. Markerless gene deletion was tested using the aga gene, which encodes α-galactosidase, whose substrates include raffinose. Almost all the pTBR101-CM-transformed strains became double-crossover recombinants after subculture, and 4 out of the 270 double-crossover recombinants had lost the ability to assimilate raffinose. Genotype analysis of these strains revealed markerless gene deletion of aga. Carbohydrate assimilation analysis and α-galactosidase activity measurement were conducted using both the representative mutant and a plasmid-based aga-complemented strain. These functional analyses revealed that aga is the only gene encoding a functional α-galactosidase enzyme in B. longum 105-A.
INTRODUCTION
Bifidobacteria are generally known as beneficial microbes that produce health-promoting effects by interacting with their hosts (20, 23). Some bifidobacteria are commercially used as probiotics in yogurt products and as pharmaceutical agents. To better understand bifidobacteria, the genome sequences of several probiotic bifidobacterial species, including Bifidobacterium longum NCC2705 (46), B. longum DJO10A (26), B. longum subsp. infantis ATCC 15697 (47), Bifidobacterium breve UCC2003 (34), and Bifidobacterium animalis subsp. lactis DSM 10140 and Bl-04 (4), have been analyzed. Although knowledge of the genome sequences enables the prediction of bifidobacterial gene function, the clarification of the detailed mechanisms of interactions is not possible with genomic information alone. The most effective approach for directly analyzing gene function is targeted gene mutagenesis. However, this approach is still not easily applicable to studies of bifidobacteria (14), despite recent progress (15, 32–34, 41).
Currently, Escherichia coli-Bifidobacterium shuttle vectors based on cryptic plasmids have been developed, but they have relatively low transformation frequencies (14). As for gene mutagenesis systems, the construction of vector-insertion mutants by single crossover and gene disruption mutants by double crossover through allelic exchange between drug resistance genes and target genes has been reported (15, 32–34, 41). Markerless gene deletion, which leaves no exogenous DNA in mutated alleles, has not yet been achieved in bifidobacteria. Markerless gene deletion has several advantages: (i) it is stable, (ii) it avoids polar effects on the expression of adjacent genes, and (iii) it enables multiple gene deletions in a genome.
Markerless gene deletion using the double-crossover method has been achieved in various bacteria (19, 28, 31, 35, 38). Generally, a low frequency of the second crossover event is an obstacle to successful gene deletion, and strategies for efficiently selecting second-crossover recombinants are required. Conditional replication vectors such as thermosensitive replication vectors (7, 17, 35, 45) and counterselectable marker genes, e.g., sacB (31, 35, 38) and pheS, encoding the phenylalanyl-tRNA synthase α-subunit (5, 50), have been used to overcome this problem in other bacteria. A similar system should be applicable in bifidobacteria, but reliable conditional replication vectors and suitable counterselectable marker genes are currently unavailable.
To achieve markerless gene deletion in the bifidobacterial genome, a strategy based on conditional plasmid replication was conceived (Fig. 1). The plasmid for the first crossover functions as a conditional replication vector as a result of the deletion of the gene encoding the replication protein RepA. This plasmid can only replicate in bifidobacterial cells when RepA is provided in trans. When this plasmid is integrated into the target gene, supplying RepA is expected to induce the conditional replication of the integrated plasmid. This will result in severe growth defects (17), presumably due to the interference with chromosome replication as replication starts from not only the chromosome's origin of replication but also the origin in the integrated plasmid. Under these conditions, only the cells in which a second crossover has occurred, excising the integrated plasmid from the chromosome and leaving only the chromosomal replication origin, can survive. To supply RepA, a second plasmid harboring the repA gene is introduced into first-crossover integrants. Second-crossover recombinants that lost the first plasmid can be efficiently isolated through selection with appropriate antibiotics, as the second plasmid is incompatible with the conditional replication vector. In this study, the proposed strategy was tested to introduce markerless gene deletion in the B. longum 105-A chromosome using the aga gene, which encodes α-galactosidase (α-Gal), as a target.
Fig 1.

Outline of markerless gene deletion system in Bifidobacterium longum 105-A. GeneA represents a model target gene for markerless gene deletion. ΔGeneA lacks the internal region of GeneA and is cloned in the conditional replication vector (first plasmid), which lacks the repA gene encoding the plasmid replication initiation protein RepA (indicated as ΔrepA). The region absent from ΔGeneA is identified by a cross. Theoretically, two types of GeneA alleles can be generated by the first crossover, and ΔGeneA deletion mutants and wild-type revertants can arise in the second-crossover recombinants. Here, only the simplified scheme for the generation of ΔGeneA deletion mutants was described to represent the concept. In the first crossover, the first plasmid harboring ΔGeneA was introduced into the host strain B. longum 105-A. Here, the 5′ region of ΔGeneA is used as a homologous region for the first crossover. The first-crossover integrant harbors both the GeneA and ΔGeneA alleles in its chromosome. The region derived from the first plasmid harboring ΔGeneA in the chromosome of the first-crossover integrants is identified by a dashed line. For transformation of the RepA+ plasmid, the introduction of the repA-harboring second plasmid (RepA+ plasmid) into the first-crossover integrant results in the supply of the plasmid replication protein RepA to the cells. During the second crossover and excision, the integrated first plasmid is expected to be excised with GeneA from the chromosome as a result of the second crossover event facilitated by RepA. In exclusion, the excised first plasmid with GeneA has been excluded due to the incompatibility between two plasmids. Objective ΔGeneA deletion mutants can be found among the second-crossover recombinants. AntAr, resistance gene for antibiotic A; AntBr, resistance gene for antibiotic B.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. All chemicals were of analytical grade and were commercially available. Cultures of B. longum 105-A (29) were routinely grown in half-strength (1/2) MRS broth containing glucose as the fermentable carbohydrate source (Becton, Dickinson and Co., Franklin Lakes, NJ) (22) and supplemented with 0.02% (wt/vol) l-cysteine and 0.34% (wt/vol) sodium ascorbate (1/2 MRSCS). Carbohydrate assimilation by B. longum was examined in semisynthetic medium (SSM) containing raffinose, melibiose, or glucose as the sole carbohydrate source (20 g/liter) (39). The SSM, which did not contain l-tryptophan and l-asparagine, was autoclaved at 110°C for 10 min, and then its pH was adjusted to pH 6.8 with NaOH. Bacterial growth was monitored by measuring the optical density at 660 nm (OD660) using a Mini Photo 518R (Taitec, Saitama, Japan) or UV-1800 (Shimadzu, Kyoto, Japan) spectrophotometer. When necessary, the following antibiotics were added to both the broth and the agar medium: ampicillin (100 μg/ml), chloramphenicol (Cm; 2.5 μg/ml), rifampin (Rif; 0.1 μg/ml), and spectinomycin (Sp; 75 μg/ml). Bifidobacterial strains were cultured at 37°C under anaerobic conditions in an anaerobic chamber (N2:CO2:H2 at 80:10:10; Coy Laboratory Products, Inc., Grass Lake, MI). Cultures of E. coli were grown aerobically in Luria-Bertani (LB) medium or SOC medium as described previously (44).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Escherichia coli | ||
| DH5α | F− ϕ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ− thi-1 gyrA96 relA1 | Invitrogen |
| HMS174 | F− λ− recA1 IN(rrnD-rrnE)1 rph-1 rpoB331(Rifr) hsdR19 | 8 |
| JM109 | recA1 endA1 gyrA96 thi hsdR17(rK− mK+) e14(mcrA) supE44 relA1 Δ(lac-proAB) F′[traD36 proAB+ lacIq lacZΔM15] | TaKaRa Bio Inc. |
| Bifidobacterium longum | ||
| 105-A | Human fecal isolate, transformation host strain | 29 |
| 105-A βgal::pBS423-ΔrepAβgal | First-crossover integrant of pBS423-ΔrepAβgal into chromosomal β-Gal gene locus | This study |
| 105-A aga::pBS423-ΔrepAΔaga | First-crossover integrant of pBS423-ΔrepAΔaga into chromosomal aga locus | This study |
| 105-A Δaga | aga deletion mutant of 105-A | This study |
| 105-A Δaga/pAga135 | Plasmid-based aga complementation strain of 105-A Δaga by introduction of pAga135 | This study |
| 105-A Δaga/pKKT427 | Empty vector (pKKT427)-introduced derivative of 105-A Δaga | This study |
| BK25 | Human fecal isolate harboring cryptic plasmid pTB4 | 13 |
| Plasmids | ||
| pAga135 | 6.4 kbp, pKKT427 derivative containing aga and 135-bp upstream region including putative aga promoter, Spr | This study |
| pBS423 | 4.9 kbp, E. coli-Bifidobacterium shuttle vector, pMB1 ori pTB4 ori repA+ Spr | This study |
| pBS423-ΔrepA | 4.4 kbp, pTB4 ori ΔrepA Spr | This study |
| pBS423-ΔrepAΔaga | 6.2 kbp, derivative of pBS423-ΔrepA containing integrated fragment of 5′ and 3′ regions of aga, Spr | This study |
| pBS423-ΔrepAβgal | 9.0 kbp, derivative of pBS423-ΔrepA containing the β-Gal gene, Spr | This study |
| pBR322 | 4.4 kbp, E. coli cloning vector, rop Tcr Apr | TaKaRa Bio Inc. |
| pBRASTA101 | 5.0 kbp, E. coli-Bifidobacterium shuttle vector, pMB1 ori pTB6 ori Spr | 48 |
| pC194 | 2.9 kbp, small R plasmid found in Staphylococcus aureus, Cmr | 18 |
| pGEM-T Easy | 3.0 kbp, TA cloning vector, Apr | Promega Corp. |
| pGEM-T-aga | 7.9 kbp, 4.9 kbp of B. longum 105-A genomic DNA fragment containing aga cloned in pGEM-T Easy | This study |
| pKKT427 | 3.9 kbp, E. coli-Bifidobacterium shuttle vector, ColE1 ori pTB6 ori Spr | 51 |
| pTANS19 | 1.9 kbp, E. coli cloning vector, pMB1 ori Spr | This study |
| pTB4 | 2.9 kbp, cryptic plasmid in B. longum BK25 | This study |
| pTBR101 | 7.3 kbp, E. coli-Bifidobacterium shuttle vector, pMB1 ori pTB4 ori repA+ Apr effective only in E. coli | This study |
| pTBR101-CM | 8.4 kbp, Cmr gene cloned in pTBR101, pMB1 ori pTB4 ori repA+ Apr effective only in E. coli | This study |
Apr, ampicillin resistance; Cmr, chloramphenicol resistance; Spr, spectinomycin resistance; Tcr, tetracycline resistance.
DNA manipulations.
The oligonucleotide primers used in this study are listed in Table S1 in the supplemental material. Reagents used in molecular biological procedures were purchased from TaKaRa Bio, Inc. (Otsu, Japan), unless otherwise indicated. Chromosomal DNA was purified from cultured bifidobacterial cells using a previously described method (13) with one modification: prior to the extraction of chromosomal DNA using Isoplant II (Nippon Gene Co., Ltd., Tokyo, Japan), the cells were treated with 30 mg/ml lysozyme dissolved in a 25% (wt/vol) solution of sucrose at 37°C for 1 h. Molecular cloning in E. coli was conducted as described elsewhere (44). Plasmid DNA from E. coli was prepared using a QIAprep miniprep kit (Qiagen, Hilden, Germany). Plasmids were purified from B. longum cells as described previously (27). PCR was performed using Primestar HS DNA polymerase with GC buffer (TaKaRa Bio) according to the manufacturer's instructions. Amplified DNA fragments were purified using a MinElute PCR purification kit (Qiagen) or a MinElute gel extraction kit (Qiagen). Agarose gel electrophoresis was conducted as described previously (13). Nucleotide sequences were determined by the dideoxy method at TaKaRa Bio and Operon Biotechnologies, Inc. (Tokyo, Japan). BLAST programs were used for sequence similarity analysis (1). Southern hybridization analysis was conducted as described previously (13). BamHI-digested chromosomal DNA (4 μg) from each strain was analyzed. The 3′ region of aga (1.1 kbp) was amplified from pBS423-ΔrepAΔaga with the primer pair aga13f/aga8r and used as a probe.
Electrotransformation of B. longum strains.
Electrotransformation of B. longum was performed using a previously described method, with some modifications (3). In brief, B. longum 105-A and first-crossover integrants were cultured anaerobically until the OD660 reached 0.5 to 0.6 in 1/2 MRSCS medium and 1/2 MRSCS-Sp medium, respectively, both of which were supplemented with 50 mM sucrose. Cells were then harvested by centrifugation at 32,300 × g at 4°C for 15 min and washed twice with ice-cold buffered sucrose (1 mM ammonium citrate buffer containing 50 mM sucrose, pH 6.0) and resuspended in the buffered sucrose. Electroporation was conducted using Gene Pulser (Bio-Rad Laboratories, Hercules, CA) with settings of 12 kV/cm, 25 μF, and 200 Ω. After an electric pulse, the cells were cultured anaerobically in 1/2 MRSCS supplemented with 50 mM sucrose for 3 h and spread onto 1/2 MRSCS agar medium (without sucrose supplementation) containing antibiotics corresponding to the plasmid used. The plates were incubated anaerobically at 37°C for 48 h. Sucrose in the 1/2 MRSCS medium and the buffered sucrose has been reported to improve the transformation efficiency in bifidobacteria (3).
Construction of incompatible plasmids for markerless gene deletion.
Two incompatible plasmids, pBS423-ΔrepA and pTBR101-CM, were constructed for markerless gene deletion. All of the plasmids named below were constructed by the introduction of ligated products into E. coli HMS174 (8) and subsequent selection on LB agar medium containing appropriate antibiotics. To construct pBS423-ΔrepA, a conditional replication vector for the first crossover event, the Sp resistance (Spr) gene [Sp adenyltransferase AAD(9)] from pBRASTA101 (48) was first ligated with a pMB1 ori-containing fragment from pUC18. The resulting plasmid was named pTANS19. The cryptic plasmid pTB4 from B. longum BK25 (13) was digested with BamHI and ligated into pTANS19. The resulting plasmid, E. coli-Bifidobacterium shuttle vector pBS423 (Fig. 2A), was digested with HincII to remove repA. HincII-digested pBS423 was separated into 4.4- and 0.5-kbp fragments (the latter contained a part of repA), and the 4.4-kbp fragment was self ligated. The resulting plasmid was named pBS423-ΔrepA (RepA−, pTB4 ori+, Spr) (Fig. 2B) and was shown not to replicate in B. longum 105-A.
Fig 2.

Structures of the plasmids used in markerless gene deletion. Genes are identified by open arrows, except for the repA gene, which is identified by black arrows. Representative restriction sites in the plasmids are shown. DNA regions in plasmids pBS423 (A), pTBR101 (C), and pTBR101-CM (D) are shown in the surrounding circle (dashed lines) of each plasmid and are divided according to the source of the DNA region. Names of derived genetic materials are shown on the surrounding circle. (A) The E. coli-Bifidobacterium shuttle vector pBS423 was constructed by cloning the cryptic plasmid pTB4 from B. longum BK25 into the BamHI site of pTANS19. (B) The conditional replication vector pBS423-ΔrepA was constructed by the self-circularization of the 4.4-kbp HincII fragment of pBS423. Because pBS423-ΔrepA lacks the repA gene, which encodes the replication initiation protein RepA, it cannot replicate in B. longum 105-A. (D) To construct pTBR101-CM, which is incompatible with pBS423-ΔrepA, a chloramphenicol resistance gene (Cmr) from pC194 (cryptic plasmid of Staphylococcus aureus) was cloned into the HindIII site of pTBR101 (C), which consists of pTB4 from B. longum BK25 and pBR322. Spr, spectinomycin resistance gene; Apr, ampicillin resistance gene.
To construct pTBR101-CM, a repA-positive plasmid for facilitation of the second crossover step, bifidobacterial plasmid pTB4 was purified from B. longum BK25, digested with BamHI, and ligated into BamHI-digested pBR322. The resulting plasmid, E. coli-Bifidobacterium shuttle vector pTBR101 (Fig. 2C), was digested with HindIII. The HindIII linker fragment was ligated to the MspI-Sau3AI-digested DNA fragment from pC194 containing the Cm resistance (Cmr) gene (18). The HindIII-digested fragment containing the Cmr gene was ligated into HindIII-digested pTBR101, and the resulting plasmid was named pTBR101-CM (RepA+, pTB4 ori+, Cmr) (Fig. 2D).
Construction of vectors for the first crossover.
Two first-crossover vectors were constructed for different objectives, pBS423-ΔrepAβgal for validation of the integration and efficient excision/exclusion of the vector at the β-galactosidase (β-Gal) gene locus and pBS423-ΔrepAΔaga for the introduction of the markerless gene deletion in the aga locus.
For the construction of pBS423-ΔrepAβgal, the full-length β-Gal gene of B. longum 105-A, which shows 99% nucleotide identity to BLLJ_1486 in B. longum subsp. longum JCM 1217T (15), was used as the target gene. A 4.6-kbp fragment containing the full-length β-Gal gene was amplified by PCR, using B. longum 105-A chromosomal DNA as a template and the lacZ Fw/lacZ Rv primer pair. The amplified product was digested with PstI and ligated into PstI-digested pBS423-ΔrepA. Ligated products were introduced into E. coli JM109 by electroporation. Transformants were selected on LB agar containing Sp and supplemented with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactoside; 40 μg/ml) and IPTG (isopropyl-1-thio-β-d-galactopyranoside; 0.1 mM). Colonies that showed blue color, indicating the successful cloning of the β-Gal gene, were isolated.
For the construction of pBS423-ΔrepAΔaga, the 5′ and 3′ regions of aga, but lacking the internal region of aga containing the predicted α-Gal catalytic site (Fig. 3A), were used for the construction. A 4.9-kbp fragment containing the full-length aga gene was amplified by PCR using B. longum 105-A chromosomal DNA as the template and the aga1f/aga2r oligonucleotide primer pair. The amplified fragment was purified and cloned into the TA cloning vector pGEM-T Easy (Promega Corp., Madison, WI). The resulting plasmid was named pGEM-T-aga. The 5′ and 3′ regions of the cloned fragment (0.9 and 1.1 kbp, respectively) were separately amplified by PCR using pGEM-T-aga as the template and aga11f/aga12r and aga13f/aga8r as the respective primer pairs. These fragments were fused together by performing a second amplification, covering the 25-bp overlapping region inserted at the 5′ terminus of the primer aga13f, with the aga11f/aga8r primer pair. The amplified 2.0-kbp fragment was digested with PstI and ligated into PstI-digested pBS423-ΔrepA. The ligated products were introduced into E. coli JM109, and transformants were selected on LB-Sp agar medium.
Fig 3.

(A) Schematic of the markerless gene deletion procedures conducted in B. longum 105-A. The first crossover event is shown in the upper panel, and the second crossover event is in the lower panel. The first crossover between the target gene (aga) and the conditional replication vector pBS423-ΔrepAΔaga, harboring the Δaga allele, which lacks the internal region of aga (C in the chromosome structure), can take place with homologous region A or B, yielding two types of first-crossover integrants, designated a and b. Primers used to check the genotypes of first-crossover integrants are shown as open arrows, with names above the predicted structure of the target locus of each first-crossover integrant. The expected lengths of the amplicons generated using these primers are indicated by dashed lines below the predicted structures. The second crossover event, facilitated by the supply of RepA by the plasmid pTBR101-CM, can take place with homologous region A or B in the first-crossover integrants, designated a and b. The second crossover event yielded two types of strains, aga deletion mutants and wild-type revertants. Predicted restriction sites for BamHI and the predicted lengths of the restriction fragments are indicated in the structure of the aga locus for each strain. (B to D) Genotype analyses of the pBS423-ΔrepAΔaga transformants to confirm integration of pBS423-ΔrepAΔaga into the aga locus. Amplified fragments produced using the aga11f/aga8r primer pair (B), the aga11f/TB4-1 primer pair (C), and the SpR4/aga8r primer pair (D) were subjected to agarose gel electrophoresis. A 1-kb DNA ladder (New England BioLabs, Inc., Ipswich, MA) was used as a molecular mass marker (lane M). The sizes of representative marker fragments are shown to the left of each panel. Each lane shows amplified DNA generated from a DNA template: lanes 1 to 8, candidate first-crossover integrants; lane P, pBS423-ΔrepAΔaga (positive control for amplification); lane W, B. longum 105-A (wild-type); and lane N, no template DNA (negative control for amplification). The sizes of the amplified fragments are shown to the right of each panel. (E) Southern hybridization analysis of markerless aga deletion. Samples of BamHI-digested chromosomal DNA (4 μg) were electrophoresed in a 0.7% agarose gel. Each lane corresponds to a DNA sample from a Bifidobacterium strain: lane 1, B. longum 105-A; lane 2, 105-A Δaga; lane 3, wild-type revertant (after the second crossover). Electrophoresed total DNA was transferred to a nylon membrane. The 3′ region of aga (region B in panel A) was used as a probe. Hybridization signals were detected after exposure for 30 min. The sizes of the hybridized fragments are indicated to the left of the panel.
Construction of the first-crossover integrants.
The first-crossover integrants were constructed by the introduction of the first-crossover vectors, either pBS423-ΔrepAβgal or pBS423-ΔrepAΔaga, to B. longum 105-A by electroporation. Transformants were selected on 1/2 MRSCS-Sp agar medium. Integration of the first-crossover vector into an objective gene locus was verified by genomic PCR using following primers: pBS423-ΔrepAβgal integrants SpR1/SpR3 and bgal1/UC18pBS423 (see Fig. S1A in the supplemental material) for the detection of the Spr gene and pBS423-ΔrepAβgal integration sequence, respectively, and pBS423-ΔrepAΔaga integrants aga11f/aga8r, aga11f/TB4-1, and aga8r/SpR4 (Fig. 3A) for the detection of the pBS423-ΔrepAΔaga integration sequence. The validated strains were used as the first-crossover integrant of each vector.
Triggering of the second crossover by the introduction of pTBR101-CM.
pTBR101-CM was introduced into the first-crossover integrant, either 105-A βgal::pBS423-ΔrepAβgal or 105-A aga::pBS423-ΔrepAΔaga, by electroporation to facilitate the second crossover step through excision of integrated pBS423-ΔrepA-derived first-crossover vectors. Transformants were selected on 1/2 MRSCS-Cm agar medium as the Cm-resistant colonies, and they were anaerobically subcultured overnight in 1/2 MRSCS-Cm medium. Subcultures were conducted twice. Excised plasmid was expected to be eliminated from the cells due to plasmid incompatibility during the subcultures. Second-crossover recombinants were selected by using an Sp-sensitive (Sps) phenotype as an indicator of the elimination of the excised plasmid. This was tested by a replica plating method using 1/2 MRSCS-Cm and 1/2 MRSCS-CmSp agar media. The excision of pBS423-ΔrepA-derived first-crossover vectors was verified by genomic PCR using the following primer pairs: for pBS423-ΔrepAβgal, SpR1/SpR3 and bgal1/UC18pBS423; for pBS423-ΔrepAΔaga, aga11f/aga8r, aga11f/TB4-1, and aga8r/SpR4. In principle, the second crossover event in 105-A βgal::pBS423-ΔrepAβgal yields original wild-type 105-A strain with respect to the β-Gal gene allele, and that in 105-A aga::pBS423-ΔrepAΔaga yields either wild-type revertant or a mutant with respect to the aga allele. To check if the recombination rates actually change when pTBR101-CM is introduced into the cell, control experiments were conducted in both of the first-crossover integrants without adding pTBR101-CM in the reaction mixture for the electroporation step (mock transformation experiment) for comparison. In this case, Cm was not added to the culture media.
Construction of the markerless aga-defective mutants.
The second-crossover recombinants from 105-A aga::pBS423-ΔrepAΔaga were cultured in SSM supplemented with raffinose as the sole carbohydrate source using 96-well microtiter plates. Strains that showed no growth were selected as candidates for aga deletion. The genotypes of the candidates were checked by aga-specific PCR, which was conducted using chromosomal DNA as the template and the aga11f/aga8r primer pair. Subsequently, one candidate was cultured anaerobically overnight in 1/2 MRSCS-Rif broth at 37°C to cure pTBR101-CM, which was propagated in the cells. The culture was streaked onto 1/2 MRSCS agar medium. Plasmid-cured strains were selected as Cm-sensitive (Cms) colonies by replica plating using 1/2 MRSCS and 1/2 MRSCS-Cm agar media. After curing, detailed genotype analysis of the candidate aga deletion strain was conducted by Southern hybridization and the sequencing of the aga deletion region. The aga deletion mutant was named 105-A Δaga.
Construction of the aga-complemented strain.
The upstream region of aga containing the putative promoter was determined from the draft genome sequence of B. longum 105-A (H. Yoshikawa, S. Fukiya, and A. Yokota, unpublished results) and a previous report (21). A 2.5-kbp DNA fragment containing aga and an upstream region (135 bp) was amplified by PCR using pGEM-T-aga as the template and the aga20f/aga21r primer pair. The products were digested with SalI and ligated into SalI-digested pKKT427 (51) (Spr). The ligated products were introduced into E. coli DH5α, and transformants were selected on LB-Sp agar medium. The resulting aga complementation plasmid was named pAga135. Purified pAga135 was subsequently introduced into 105-A Δaga by electroporation, and an Sp-resistant strain was isolated on 1/2 MRSCS-Sp agar medium. The propagation of pAga135 was confirmed by agarose gel electrophoresis of the purified plasmid. The strain, named 105-A Δaga/pAga135, was used as a plasmid-based aga-complemented strain. An empty vector-introduced strain, 105-A Δaga/pKKT427, was similarly constructed through the introduction of the shuttle vector pKKT427 into 105-A Δaga.
Measurement of α-Gal activity.
Strains 105-A, 105-A Δaga, 105-A Δaga/pAga135, and 105-A Δaga/pKKT427 were cultured overnight in 40 ml of 1/2 MRSCS (Sp was added when necessary). Cells were harvested by centrifugation at 19,000 × g at 4°C for 15 min and washed with saline. The cells were resuspended in SSM containing 20 g/liter raffinose and incubated for 6 h under anaerobic conditions at 37°C to induce α-Gal expression. The cells were harvested by centrifugation, washed twice with ice-cold 50 mM potassium phosphate buffer (pH 6.0) containing 0.015% (wt/vol) dithiothreitol, and disrupted three times using a French pressure cell (Ohtake Works, Tokyo, Japan) at 125 MPa. Cell debris was removed by centrifugation. The resulting supernatant was used as a crude extract. Protein concentration was determined by the Bradford method using a Bio-Rad protein assay kit (Bio-Rad Laboratories). The measurement of α-Gal activity was performed according to a previously described method (36) in a 300-μl reaction mixture containing 50 mM potassium phosphate buffer (pH 6.0), 5 mM p-nitrophenyl-α-d-galactopyranoside (25026-81; Nacalai Tesque, Inc., Kyoto, Japan) (as a substrate), and 60 μl of crude extract, diluted appropriately in 50 mM potassium phosphate buffer (pH 6.0) containing 0.5 mg/ml bovine serum albumin. The enzyme reaction was started by adding the crude extract to the reaction mixture followed by incubation at 35°C for 5 or 10 min. The reaction was stopped by adding 600 μl of 1 M Na2CO3. A blank reaction mixture was prepared by adding 600 μl of 1 M Na2CO3 prior to the addition of the crude extract. The release of p-nitrophenol was monitored in a 1-cm-wide cuvette by measuring absorbance at 400 nm with a DU800 spectrophotometer (Beckman Coulter Inc., Fullerton, CA). The absorbance value for the blank was subtracted from that for the samples. A molar extinction coefficient of 5,560 M−1 cm−1 for p-nitrophenol was used in the calculation. The specific activity was expressed as μmol of p-nitrophenol per min per mg protein.
Nucleotide sequence accession numbers.
Nucleotide sequences of the plasmids constructed were deposited into the DDBJ/EMBL/GenBank databases (accession no. AB687999 for pBS423, including information for the deleted region in pBS423-ΔrepA, and AB688000 for pTBR101-CM).
RESULTS
Validation of the proposed system at β-Gal gene locus in B. longum 105-A.
In the proposed system, the plasmid pBS423-ΔrepA (RepA− pTB4 ori+ Spr), which lacks the gene encoding the replication protein RepA, was used as the conditional replication vector. A second plasmid, pTBR101-CM (RepA+ pTB4 ori+ Cmr), which was incompatible with pBS423-ΔrepA, was used to supply RepA. Uncertainty as to the presence of sufficient recombination activity in B. longum 105-A led us to first examine whether integration and excision events readily occur using the full-length β-Gal gene as the target.
Spr transformants carrying pBS423-ΔrepAβgal were obtained with an efficiency of 50 CFU/μg DNA. Genomic PCR analysis of the isolated transformants indicated that pBS423-ΔrepAβgal was successfully integrated into the chromosomal β-Gal gene locus as a result of the first crossover (see Fig. S1B and C in the supplemental material). The introduction of pTBR101-CM into the first-crossover pBS423-ΔrepAβgal integrants yielded transformants (7.5 × 102 CFU/μg DNA) on 1/2 MRSCS-Cm agar. After the subsequent culture of pTBR101-CM transformants in 1/2 MRSCS-Cm medium, 100% of the colonies tested were Sps (Table 2). In contrast, only 0.125% of the mock transformants without using pTBR101-CM showed a Sps phenotype after the culture in 1/2 MRSCS medium. Therefore, it was found that the introduction of pTBR101-CM dramatically increased the frequency of the appearance of the second-crossover recombinants.
Table 2.
Frequency of detection of Sps colonies during the culture of first-crossover integrantsa
| Addition of pTBR101-CM to reaction mixture for electroporation | No. of tested colonies (A) | No. of Sps second-crossover recombinants (B) | Detection frequency (%) |
|---|---|---|---|
| 105-A βgal:: pBS423-ΔrepAβgal | |||
| − | 2,396 | 3 | 0.125 |
| + | 3,568 | 3,568 | 100 |
| 105-A aga:: pBS423-ΔrepAΔaga | |||
| − | 963 | 1 | 0.104 |
| + | 1,645 | 1,643 | 99.9 |
The reaction mixtures were supplemented with pTBR101-CM for electroporation (+) or left unsupplemented (−). Detection frequency was determined by the following equation: (number of Sps second-crossover recombinants/number of tested colonies) × 100.
The lack of amplification in genomic PCR analysis of selected Sps strains (see Fig. S1D and E in the supplemental material) indicated the successful excision of pBS423-ΔrepAβgal from the chromosome by the second crossover. To confirm the elimination of the excised pBS423-ΔrepA by using incompatibility, plasmids in the second-crossover recombinants were extracted and introduced into E. coli DH5α. Many transformants derived from pTBR101-CM appeared in the LB-Cm agar. In contrast, none of the transformants derived from pBS423-ΔrepA were detected in LB-Sp agar. These results indicate that the excised pBS423-ΔrepA was eliminated from cells because of plasmid incompatibility. Taken together, the proposed system was verified to function as expected.
Markerless deletion of aga in B. longum 105-A.
This system was next applied for markerless gene deletion into the aga gene, which putatively encodes α-Gal (Fig. 3A). The α-Gal-encoding gene was selected as the target because α-Gal hydrolyzes raffinose, which is known to increase bifidobacterial populations in the intestinal tract of rats and humans (9, 10). As shown in Table 3, three genes annotated as α-Gal were found in the draft genome sequence of B. longum 105-A (Yoshikawa et al., unpublished). The contribution of each gene to the α-Gal activity in B. longum 105-A cells was not known. Among these genes, aga and galA3 are conserved in the genomes of B. longum NCC2705 and B. longum DJO10A (Table 3). Furthermore, the molecular mass of the deduced aga polypeptide was similar to those of purified α-Gals from other bifidobacteria (approximately 80 kDa as a monomer) (16, 24, 49). Therefore, aga was inferred to contribute to α-Gal activity and raffinose assimilation in B. longum 105-A.
Table 3.
Annotated α-galactosidase genes in B. longum 105-A draft genome and their distribution in B. longum strains NCC2705 and DJO10A
| Annotated α-Gala gene in 105-A |
B. longum strain |
||||||
|---|---|---|---|---|---|---|---|
| 105-A |
NCC2705 |
DJO10A |
|||||
| Length of gene (bp) | Calculated molecular massb (kDa) | Nucleotide sequence identityc (%) | Annotated α-Gala gene | Nucleotide sequence identityc (%) | Annotated α-Gala gene | Nucleotide sequence identityc (%) | |
| aga (galA1) | 2,307 | 83.3 | 100 | BL_1518 (aga) | 98.1 | BLD_1483 (galA1) | 99.0 |
| galA2 | 1,863 | 68.1 | 100 | NDd | NDd | BLD_1496 (galA2) | 99.8 |
| galA3 | 1,410 | 52.4 | 100 | BL_0177 | 98.7 | BLD_1538 (galA3) | 99.5 |
α-Gal, α-galactosidase.
The molecular mass of the deduced polypeptide was calculated.
Nucleotide sequence identity to the corresponding α-Gal gene in B. longum 105-A.
ND, not determined because the galA2 homolog was absent from B. longum NCC2705.
Fragments of both the 5′ and 3′ regions of aga (each approximately 1 kbp in length), which lacked the sequence for the putative active site of the protein, were used as the homologous regions to construct the conditional replication vector pBS423-ΔrepAΔaga. An outline of experimental procedures is illustrated in Fig. 3A. When pBS423-ΔrepAΔaga was introduced into B. longum 105-A by electroporation, Spr transformants were formed with an efficiency of 2.2 × 102 CFU/μg DNA. After single-colony isolation, strains harboring pBS423-ΔrepAΔaga integrated into the chromosomal aga locus were selected by aga-specific PCR (primer pair aga11f/aga8r). The amplicons of both wild-type (3.4 kbp) and mutant (2.0 kbp) aga alleles were identified in Spr transformants (Fig. 3B). To identify integration patterns, PCR was conducted using different primer pairs. Amplicons of 3.8 kbp (primer pair aga11f/TB4-1; Fig. 3C) and 2.2 kbp (primer pair aga8r/SpR4; Fig. 3D) were detected in the same transformants. These results demonstrated that the selected strains were first-crossover integrants of pBS423-ΔrepAΔaga (105-A aga::pBS423-ΔrepAΔaga) in which recombination had taken place in the 3′ region of the aga locus (region B) (Fig. 3A).
To construct the markerless aga deletion mutants, pTBR101-CM was introduced into 105-A aga::pBS423-ΔrepAΔaga, and Cmr transformants were formed with an average efficiency of 3.9 × 102 CFU/μg DNA. Randomly selected Cmr transformants (288 colonies) were subcultured in 1/2 MRSCS-Cm broth to select for second-crossover recombinants. The subcultures were checked for Sps and the inability to assimilate raffinose by the replica method. The results indicated a high frequency of the appearance of the second-crossover recombinants, as 270 of the 288 subcultured strains were Sps (94%). In this case, 4 of the 270 Sps strains (1.5%) lacked raffinose assimilation ability and were thus considered candidate aga deletion mutants. Genotype analysis of all of these candidates by aga-specific PCR supported their inability to grow on raffinose (data not shown). In addition, the same aga-specific PCR analysis of raffinose-assimilating Sps strains indicated that these strains were wild-type aga allele revertants (data not shown). In a separate experiment, the usefulness of pTBR101-CM was also confirmed at the aga locus. As shown in Table 2 when pTBR101-CM was not introduced into the integrant, the ratio of Sps colonies among the tested colonies was found to be dramatically decreased, indicating the facilitation of the second crossover step by the plasmid. One of the four candidate strains was selected and used to cure pTBR101-CM, which remained in the cells. Rif was employed as a curing agent because it was previously used to cure plasmids in E. coli (6, 30). After Rif treatment, 84% of the colonies grown on 1/2 MRSCS agar plates were Cms. The curing of the plasmid was also confirmed by the introduction of the extracted plasmid fraction from a Cms strain into E. coli, which yielded no Cmr E. coli transformants. These results indicated that pTBR101-CM was successfully removed from the candidate strain by Rif treatment. The deletion of aga from the pTBR101-CM-cured candidate strain was further confirmed by Southern hybridization (Fig. 3E) and sequence analysis. These results demonstrated successful markerless aga gene deletion in B. longum 105-A. This mutant was named 105-A Δaga.
Functional analyses of aga using B. longum 105-A Δaga.
To clarify the function of aga, the complementation of aga in 105-A Δaga was conducted using the plasmid pAga135, which harbors the aga open reading frame and a 135-bp upstream region containing the putative aga promoter. A transformant, 105-A Δaga/pAga135, was used in the functional analysis, together with the wild-type 105-A strain, 105-A Δaga, and the empty vector-introduced derivative 105-A Δaga/pKKT427.
Carbohydrate assimilation analysis using SSM revealed that 105-A Δaga was unable to grow after cultivation for 24 h on either melibiose or raffinose, two substrates for α-Gal (Fig. 4). In contrast, Δaga/pAga135 showed growth levels comparable to those of 105-A on all tested carbohydrates. These results indicated that aga is the gene responsible for the assimilation of melibiose and raffinose.
Fig 4.
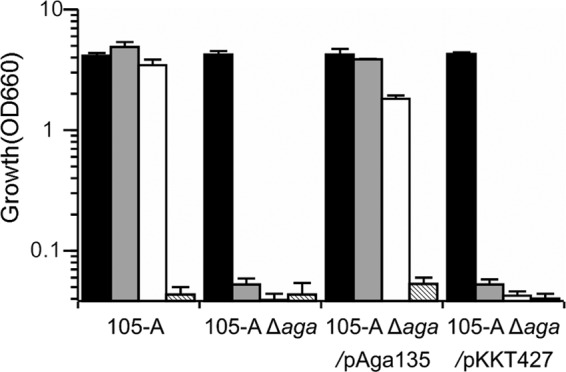
Growth profiles of B. longum strains 105-A, 105-A Δaga, 105-A Δaga/pAga135, and 105-A Δaga/pKKT427. Bacteria were cultured anaerobically at 37°C for 24 h in semisynthetic medium containing glucose (filled bars), raffinose (gray bars), or melibiose (open bars) as the sole carbohydrate source. Striped bars indicate cultures without the addition of a carbohydrate source. Data are shown as means ± standard errors of three independent experiments.
α-Gal activity was next measured in cells incubated for 6 h in SSM containing raffinose as the sole carbohydrate. As expected, α-Gal activity was not detected in 105-A Δaga (Table 4). α-Gal activity was successfully restored in Δaga/pAga135 but not in 105-A Δaga/pKKT427 (Table 4). Taken together, these results clearly identify aga as the only functional α-Gal gene necessary for the assimilation of α-galactosides such as melibiose and raffinose in B. longum 105-A. These results also suggest that the other annotated α-Gal genes in the B. longum 105-A genome, galA2 and galA3 (Table 3), make little contribution, if any, to α-Gal activity under the conditions used in this study.
Table 4.
α-Galactosidase activities of B. longum 105-A and its derivatives
| Strain | Sp acta |
|---|---|
| 105-A | 4.44 ± 0.36 |
| 105-A Δaga | Not detected |
| 105-A Δaga/pAga135 | 1.30 ± 0.09 |
| 105-A Δaga/pKKT427 | Not detected |
The activities were measured after induction by raffinose as described in Materials and Methods. Results are mean values of α-galactosidase specific activity [μmol min−1 (mg protein)−1] with standard errors as determined in duplicate on three biological replicates.
DISCUSSION
In this study, a markerless gene deletion system was successfully developed using a double-crossover method in B. longum 105-A. Despite the emerging importance of bifidobacteria in human health, genetic manipulation systems for improving our understanding of these bacteria, especially efficient methods for targeted gene mutagenesis, have not yet been established. Several B. breve UCC2003 gene insertion mutants (32–34, 41), in which a nonreplicating vector containing internal DNA fragments of the target gene was integrated into the target gene by a single-crossover method, were recently reported. A mutant of B. longum NCC2705 has also been generated (15). In this mutant, gene disruption was achieved by a double-crossover method in which a region homologous to the target gene along with an inserted antibiotic resistance gene was cloned into a general E. coli vector, and allelic exchange was performed. These methods represent important progress in the functional analysis of bifidobacterial genes, although exogenous sequences (vector sequences and/or selection marker genes) were left in the mutated allele by both methods. In contrast, the proposed method is a markerless deletion method that does not leave any exogenous sequences in the mutated allele.
The following factors are considered to be the characteristics of the developed system.
(i) Facilitation of the second crossover step.
A low-frequency second crossover event is generally the bottleneck in gene mutagenesis based on the double-crossover system. In this study, a conditional replication vector lacking repA [pBS423-ΔrepA (Spr)] for integration into the chromosome (first crossover event) and a second plasmid to supply RepA (pTBR101-CM) were used to facilitate the second crossover step. As shown in Table 2, pTBR101-CM apparently increased the second-crossover frequency. Still, one can also consider the possibility that the introduced pTBR101-CM served simply as a strong selective pressure to the first-crossover integrants, allowing the preferential growth of preexisting spontaneous second-crossover recombinants after the electrotransformation step. However, this may not be the case, as there seemed to be no contamination of the spontaneous second-crossover recombinants in the competent cells (Sp was included in the medium for preparation of the competent cells). Though not shown in the results, colony PCR analysis of the pTBR101-CM transformants clearly indicated that the excision of the integrated plasmid from the chromosome had already occurred in the transformed colonies before subcultures. Taking these findings together, the supply of RepA was judged to increase the second-crossover frequency, which led to the facilitation of the second crossover step.
(ii) Exclusion of excised vector by plasmid incompatibility.
Plasmid incompatibility has been used as a method to eliminate unnecessary plasmids from cells during bacterial gene mutagenesis by double-crossover recombination (11, 25, 37, 43). This system was applied to markerless gene deletion. The use of two incompatible vectors, pBS423-ΔrepA and pTBR101-CM, enabled the excised vector pBS423-ΔrepA (containing a wild-type or mutated allele) to be effectively excluded from cells. In principle, either plasmid could be excluded. However, in this case, retaining pTBR101-CM was the only option, because pBS423-ΔrepA cannot replicate on its own. The Sps plus Cmr combination ensured the acquisition of the desired recombinants.
(iii) Successful curing of pTBR101-CM by Rif.
The remaining pTBR101-CM in cells was easily cured by Rif treatment, as has been reported for E. coli (6, 30). Successful curing of the replicative vector in the markerless gene deletion mutant means that multiple gene deletions are possible using the developed system. Multiple gene mutations have not been introduced into bifidobacteria and are theoretically limited by mutagenesis methods that leave antibiotic resistance markers, such as Sp (29), tetracycline (2), and erythromycin (42), in the mutated allele, owing to the limited availability of these markers in bifidobacteria.
Temperature-sensitive vectors harboring a temperature-sensitive rep gene have been commonly used for targeted gene mutagenesis in other bacteria (7, 17, 35, 45). However, no reliable temperature-sensitive vector is currently available for use in bifidobacteria. In addition, mutagenic effects of heat shock stress on the genome sequence are a concern, because heat shock-induced GroEL/GroES chaperone is known to protect error-prone DNA polymerases IV (Pol IV) and Pol V from degradation in E. coli (12). Homologs of the chaperone and the DNA polymerases are also found in the genome sequences of B. longum 105-A and other bifidobacteria.
Theoretically, after the introduction of gene mutations by the double-crossover method, the frequencies of the appearance of the wild-type and the mutant genotypes should be the same. However, it was found that the number of aga deletion mutants was far lower than that of revertants (4 versus 266). The reason for the low frequency of gene deletion mutants is unclear, but it is possible that there was a difference in recombination efficiency between the homologous regions A and B used in aga deletion (Fig. 3A). Therefore, the recombination frequency was checked using plasmids consisting of homologous region A or B in pBS423-ΔrepA. These plasmids were introduced into B. longum 105-A, and the appearance of first-crossover integrants was examined. However, no significant difference between homologous regions A and B was observed (data not shown). These results suggest that a difference in recombination frequency between these homologous regions was not a cause of the low occurrence of gene deletion mutants. An unexpected selective disadvantage associated with the aga deletion might be involved in this tendency. Further comparisons using other genes are needed to address this point.
aga was shown to be the sole gene responsible for α-Gal activity in B. longum 105-A. Successful complementation in both the growth on melibiose and the α-Gal enzyme activity by introduction of pAga135 into α-Gal-deficient E. coli JW4080 (melA mutant) also supported the observation (data not shown). Although gene deletion mutants for the other annotated α-Gal genes (galA2 and galA3) were not constructed, it seemed that these genes are not expressed or do not encode proteins with α-Gal activity. The nucleotide similarity between aga and galA2 or galA3 was calculated to be 47 or 48%, respectively. The molecular sizes of the predicted polypeptides encoded by these genes were smaller than that of aga (Table 3) and those of purified α-Gals in other bifidobacteria (approximately 80 kDa) (16, 24, 49), suggesting that these genes are unlikely to encode proteins with α-Gal activity.
The applicability of the developed system to other bifidobacterial species is unclear. It may depend on the host range of the plasmids pBS423-ΔrepA and pTBR101-CM, both of which were derived from a cryptic plasmid, pTB4, that is harbored by B. longum BK25 (13). Another factor will be the transformation efficiency of a given bifidobacterial strain. The average transformation efficiency of an E. coli-Bifidobacterium shuttle vector pBS423 into B. longum 105-A was 1.1 × 105 CFU/μg DNA, whereas in the case of aga deletion, the transformation efficiency of pBS423-ΔrepAΔaga for the construction of the first-crossover integrants decreased to 2.2 × 102 CFU/μg DNA. The average transformation efficiency of the replicative pTBR101-CM vector into the first-crossover integrants was 3.9 × 102 CFU/μg DNA. According to our calculation from previously reported data on host/vector systems, the median transformation efficiency in bifidobacteria was up to 103 CFU/μg DNA (14). It therefore appears that the application of the developed system to other bifidobacteria will be limited to the strains that have relatively high transformation efficiency, at least more than 104 CFU/μg DNA with the replicative vector pBS423. In fact, preliminary trials for the deletion of aga in B. longum subsp. longum JCM 1217T, B. longum subsp. infantis JCM 1222T, and B. breve JCM 1192T were not successful, probably due to the low transformation efficiencies as well as appearance of false-positive transformants. The optimization of transformation conditions and antibiotic concentrations for transformant selection will allow markerless gene deletion in other strains. The methylation of the vectors using bifidobacterial modification enzymes is one way to increase the transformation efficiency by avoiding the cleavage of introduced DNA by restriction systems in bifidobacteria (33, 51).
The markerless gene deletion method reported here can be used for functional gene analysis as well as for other applications. For example, this method may have advantages in the construction of food-grade host strains, as the use of antibiotic resistance genes has not been approved for the construction of food-grade hosts (40, 45). Moreover, the method may be used to insert exogenous genes into the bifidobacterial chromosome. It is anticipated that therapeutic proteins and hormones for medical treatments can be stably expressed in genetically engineered bifidobacteria (14).
In conclusion, a rational system for markerless gene deletion was successfully developed in B. longum 105-A. Although the applicability of this system to other bifidobacteria remains to be demonstrated and there is much room for improvement, we believe that this technique will accelerate functional gene analysis in bifidobacteria.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Masayuki Okuyama (Research Faculty of Agriculture, Hokkaido University) for instruction on α-galactosidase activity measurement.
This study was supported in part by a Grant-in-Aid for Young Scientists (B) from the Japan Society for the Promotion of Science (no. 23780072 to S.F.), by a Grant-in-Aid for Regional R&D Proposal-Based Program Grant from Northern Advancement Center for Science & Technology of Hokkaido Japan (no. T-3-5 to S.F.), and by a Ph.D. Student Research Support Program Grant from Hokkaido University Clark Memorial Foundation to Y.H. We also thank the Research Center, Nippon Beet Sugar Mfg. Co., Ltd. (Obihiro, Japan), for financial support.
Footnotes
Published ahead of print 11 May 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 2.Álvarez-Martín P, Belén Flórez A, Margolles A, del Solar G, Mayo B. 2008. Improved cloning vectors for bifidobacteria, based on the Bifidobacterium catenulatum pBC1 replicon. Appl. Environ. Microbiol. 74:4656–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Argnani A, Leer RJ, van Luijk N, Pouwels PH. 1996. A convenient and reproducible method to genetically transform bacteria of the genus Bifidobacterium. Microbiology 142:109–114 [DOI] [PubMed] [Google Scholar]
- 4.Barrangou R, et al. 2009. Comparison of the complete genome sequences of Bifidobacterium animalis subsp. lactis DSM 10140 and Bl-04. J. Bacteriol. 191:4144–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett AR, et al. 2008. Genetic tools for allelic replacement in Burkholderia species. Appl. Environ. Microbiol. 74:4498–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bazzicalupo P, Tocchini-Valentini GP. 1972. Curing of an Escherichia coli episome by rifampicin (acridine orange/F+/F−/Hfr/lac). Proc. Natl. Acad. Sci. U. S. A. 69:298–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biswas I, Gruss A, Ehrlich SD, Maguin E. 1993. High-efficiency gene inactivation and replacement system for gram-positive bacteria. J. Bacteriol. 175:3628–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell JL, Richardson CC, Studier FW. 1978. Genetic recombination and complementation between bacteriophage T7 and cloned fragments of T7 DNA. Proc. Natl. Acad. Sci. U. S. A. 75:2276–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dinoto A, et al. 2006. Population dynamics of Bifidobacterium species in human feces during raffinose administration monitored by fluorescence in situ hybridization-flow cytometry. Appl. Environ. Microbiol. 72:7739–7747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dinoto A, et al. 2006. Modulation of rat cecal microbiota by administration of raffinose and encapsulated Bifidobacterium breve. Appl. Environ. Microbiol. 72:784–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fedorova ND, Highlander SK. 1997. Generation of targeted nonpolar gene insertions and operon fusions in Pasteurella haemolytica and creation of a strain that produces and secretes inactive leukotoxin. Infect. Immun. 65:2593–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster PL. 2007. Stress-induced mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 42:373–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukiya S, Sugiyama T, Kano Y, Yokota A. 2010. Characterization of an insertion sequence-like element, ISBlo15, identified in a size-increased cryptic plasmid pBK283 in Bifidobacterium longum BK28. J. Biosci. Bioeng. 110:141–146 [DOI] [PubMed] [Google Scholar]
- 14.Fukiya S, Suzuki T, Kano Y, Yokota A. 2011. Current status of Bifidobacterium gene manipulation technologies, p 33–51 In Sonomoto K, Yokota A. (ed), Lactic acid bacteria and bifidobacteria: current progress in advanced research. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 15.Fukuda S, et al. 2011. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469:543–547 [DOI] [PubMed] [Google Scholar]
- 16.Goulas T, Goulas A, Tzortzis G, Gibson GR. 2009. A novel α-galactosidase from Bifidobacterium bifidum with transgalactosylating properties: gene molecular cloning and heterologous expression. Appl. Microbiol. Biotechnol. 82:471–477 [DOI] [PubMed] [Google Scholar]
- 17.Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR. 1989. New method for generating deletions and gene replacements in Escherichia coli. J. Bacteriol. 171:4617–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horinouchi S, Weisblum B. 1982. Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol. 150:815–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ichimura M, et al. 2010. Efficient electrotransformation of Bacteroides fragilis. Appl. Environ. Microbiol. 76:3325–3332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleerebezem M, Vaughan EE. 2009. Probiotic and gut lactobacilli and bifidobacteria: molecular approaches to study diversity and activity. Annu. Rev. Microbiol. 63:269–290 [DOI] [PubMed] [Google Scholar]
- 21.Klijn A, et al. 2006. Construction of a reporter vector for the analysis of Bifidobacterium longum promoters. Appl. Environ. Microbiol. 72:7401–7405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurdi P, Kawanishi K, Mizutani K, Yokota A. 2006. Mechanism of growth inhibition by free bile acids in lactobacilli and bifidobacteria. J. Bacteriol. 188:1979–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leahy SC, Higgins DG, Fitzgerald GF, van Sinderen D. 2005. Getting better with bifidobacteria. J. Appl. Microbiol. 98:1303–1315 [DOI] [PubMed] [Google Scholar]
- 24.Leder S, Hartmeier W, Marx SP. 1999. α-Galactosidase of Bifidobacterium adolescentis DSM 20083. Curr. Microbiol. 38:101–106 [DOI] [PubMed] [Google Scholar]
- 25.Lee CA, Saier MH., Jr 1983. Use of cloned mtl genes of Escherichia coli to introduce mtl deletion mutations into the chromosome. J. Bacteriol. 153:685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JH, et al. 2008. Comparative genomic analysis of the gut bacterium Bifidobacterium longum reveals loci susceptible to deletion during pure culture growth. BMC Genomics 9:247 doi: 10.1186/1471-2164-9-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JH, O'Sullivan DJ. 2006. Sequence analysis of two cryptic plasmids from Bifidobacterium longum DJO10A and construction of a shuttle cloning vector. Appl. Environ. Microbiol. 72:527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leenhouts K, et al. 1996. A general system for generating unlabelled gene replacements in bacterial chromosomes. Mol. Gen. Genet. 253:217–224 [DOI] [PubMed] [Google Scholar]
- 29.Matsumura H, Takeuchi A, Kano Y. 1997. Construction of Escherichia coli-Bifidobacterium longum shuttle vector transforming B. longum 105-A and 108-A. Biosci. Biotechnol. Biochem. 61:1211–1212 [DOI] [PubMed] [Google Scholar]
- 30.Messing J, Staudenbauer WL, Hofschneider PH. 1972. Inhibition of minicircular DNA replication in Escherichia coli 15 by rifampicin. Nat. New Biol. 238:202–203 [DOI] [PubMed] [Google Scholar]
- 31.Mizoguchi H, Tanaka-Masuda K, Mori H. 2007. A simple method for multiple modification of the Escherichia coli K-12 chromosome. Biosci. Biotechnol. Biochem. 71:2905–2911 [DOI] [PubMed] [Google Scholar]
- 32.O'Connell Motherway M, et al. 2008. Characterization of ApuB, an extracellular type II amylopullulanase from Bifidobacterium breve UCC2003. Appl. Environ. Microbiol. 74:6271–6279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Connell Motherway M, O'Driscoll J, Fitzgerald GF, van Sinderen D. 2009. Overcoming the restriction barrier to plasmid transformation and targeted mutagenesis in Bifidobacterium breve UCC2003. Microb. Biotechnol. 2:321–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Connell Motherway M, et al. 2011. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proc. Natl. Acad. Sci. U. S. A. 108:11217–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okibe N, Suzuki N, Inui M, Yukawa H. 2011. Efficient markerless gene replacement in Corynebacterium glutamicum using a new temperature-sensitive plasmid. J. Microbiol. Methods 85:155–163 [DOI] [PubMed] [Google Scholar]
- 36.Okuyama M, et al. 2009. Catalytic mechanism of retaining α-galactosidase belonging to glycoside hydrolase family 97. J. Mol. Biol. 392:1232–1241 [DOI] [PubMed] [Google Scholar]
- 37.Ostroff RM, Vasil ML. 1987. Identification of a new phospholipase C activity by analysis of an insertional mutation in the hemolytic phospholipase C structural gene of Pseudomonas aeruginosa. J. Bacteriol. 169:4597–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pavelka MS, Jr, Jacobs WR., Jr 1999. Comparison of the construction of unmarked deletion mutations in Mycobacterium smegmatis, Mycobacterium bovis Bacillus Calmette-Guérin, and Mycobacterium tuberculosis H37Rv by allelic exchange. J. Bacteriol. 181:4780–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perrin S, Warchol M, Grill JP, Schneider F. 2001. Fermentations of fructo-oligosaccharides and their components by Bifidobacterium infantis ATCC 15697 on batch culture in semi-synthetic medium. J. Appl. Microbiol. 90:859–865 [DOI] [PubMed] [Google Scholar]
- 40.Peterbauer C, Maischberger T, Haltrich D. 2011. Food-grade gene expression in lactic acid bacteria. Biotechnol. J. 6:1147–1161 [DOI] [PubMed] [Google Scholar]
- 41.Pokusaeva K, et al. 2011. Cellodextrin utilization by Bifidobacterium breve UCC2003. Appl. Environ. Microbiol. 77:1681–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossi M, Brigidi P, Gonzalez Vara y Rodriguez A, Matteuzzi D. 1996. Characterization of the plasmid pMB1 from Bifidobacterium longum and its use for shuttle vector construction. Res. Microbiol. 147:133–143 [DOI] [PubMed] [Google Scholar]
- 43.Ruvkun GB, Ausubel FM. 1981. A general method for site-directed mutagenesis in prokaryotes. Nature 289:85–88 [DOI] [PubMed] [Google Scholar]
- 44.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 45.Sasaki Y, Ito Y, Sasaki T. 2004. thyA as a selection marker in construction of food-grade host-vector and integration systems for Streptococcus thermophilus. Appl. Environ. Microbiol. 70:1858–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schell MA, et al. 2002. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc. Natl. Acad. Sci. U. S. A. 99:14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sela DA, et al. 2008. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. U. S. A. 105:18964–18969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanaka K, Samura K, Kano Y. 2005. Structural and functional analysis of pTB6 from Bifidobacterium longum. Biosci. Biotechnol. Biochem. 69:422–425 [DOI] [PubMed] [Google Scholar]
- 49.Xiao M, Tanaka K, Qian XM, Yamamoto K, Kumagai H. 2000. High-yield production and characterization of α-galactosidase from Bifidobacterium breve grown on raffinose. Biotechnol. Lett. 22:747–751 [Google Scholar]
- 50.Xie Z, Okinaga T, Qi F, Zhang Z, Merritt J. 2011. Cloning-independent and counterselectable markerless mutagenesis system in Streptococcus mutans. Appl. Environ. Microbiol. 77:8025–8033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yasui K, et al. 2009. Improvement of bacterial transformation efficiency using plasmid artificial modification. Nucleic Acids Res. 37:e3 doi: 10.1093/nar/gkn884 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.