

Abstract
Bananas are among the most widely consumed foods in the world. In Uganda, the country with the second largest banana production in the world, bananas are the most important staple food. The objective of this study was to analyze banana-associated microorganisms and to select efficient antagonists against fungal pathogens which are responsible for substantial yield losses. We studied the structure and function of microbial communities (endosphere, rhizosphere, and soil) obtained from three different traditional farms in Uganda by cultivation-independent (PCR-SSCP fingerprints of 16S rRNA/ITS genes, pyrosequencing of enterobacterial 16S rRNA gene fragments, quantitative PCR, fluorescence in situ hybridization coupled with confocal laser scanning microscopy, and PCR-based detection of broad-host-range plasmids and sulfonamide resistance genes) and cultivation-dependent methods. The results showed microhabitat-specific microbial communities that were significant across sites and treatments. Furthermore, all microhabitats contained a high number and broad spectrum of indigenous antagonists toward identified fungal pathogens. While bacterial antagonists were found to be enriched in banana plants, fungal antagonists were less abundant and mainly found in soil. The banana stem endosphere was the habitat with the highest bacterial counts (up to 109 gene copy numbers g−1). Here, enterics were found to be enhanced in abundance and diversity; they provided one-third of the bacteria and were identified by pyrosequencing with 14 genera, including not only potential human (Escherichia, Klebsiella, Salmonella, and Yersinia spp.) and plant (Pectobacterium spp.) pathogens but also disease-suppressive bacteria (Serratia spp.). The dominant role of enterics can be explained by the permanent nature and vegetative propagation of banana and the amendments of human, as well as animal, manure in these traditional cultivations.
INTRODUCTION
Bananas and plantains grown in the tropic and subtropic regions are among the important crops worldwide (39). They are versatile, e.g., bananas and plantains are staple foods and fruit for a population of over 100 million people in the Great Lake region of Africa covering Uganda, Tanzania, Kenya, Burundi, and parts of Democratic Republic of Congo (15). Uganda is the second largest banana-producing country in the world. Production is harvested solely from small-scale systems characterized by a high (4 to 22 per farm) cultivar diversity (19). Of particular importance is the Musa sp. strain AAA-EAHB, the East African Highland cultivar group with its center of diversity in this region. Uganda produces more than 10% of the global bananas, but these are locally consumed or sold within Uganda or neighboring countries fresh green or ripe or as banana beer, wine, and rum (4). Banana farmers face numerous problems in maintaining production, including low soil fertility and pest and disease pressure (19). Fusarium wilt caused by the soil fungus Fusarium oxysporum Schlecht f. sp. cubense (E.F.Sm) Synder & Hassen (FOC) is one of the most destructive diseases of bananas in Uganda (44). FOC represents a complex of eight phylogenetic lineages; its ability to cause disease on all or some of the current race differential cultivars has evolved convergently in the taxon (18, 44). Furthermore, in recent years, results obtained by molecular tools have changed our understanding of fungal diseases. There are emerging fungal threats to plant and ecosystem health, which are often caused by multipathogen complexes (11, 16). Here, diverse saprophytic or opportunistic microorganisms are involved in the expression of symptoms of these diseases; this fact should be considered in disease control (9). Cultivar substitution for the important susceptible cultivars (Bogoya [Gros Michel—AAA]), (Kayinja [Pisang awak—ABB], Sukali Ndizi [Kamaramasenge subgroup—AAB], and Kisubi [Ney Poovan—AB]) is one proposed approach to reducing losses to Fusarium wilt in Uganda, although consumer preference limits this approach (19). Since Fusarium clamydospores persist in the soil for decades, no cultural or agronomic practices that are useful for the growth of susceptible cultivars on infested soils have been identified (44).
Biocontrol using indigenous, disease-suppressive microorganisms provides promising perspectives for sustainable plant protection (9, 29, 46). Plants, similar to other eukaryotes, harbor a highly diverse microbiome with specific functions and traits; novel molecular techniques have enhanced our knowledge about its structure and function (32). For example, bacterial endophytes residing within plants without causing disease can efficiently support plant growth promotion and/or antagonism toward phytopathogens (31). The endosphere was shown to be an important bioresource of plant beneficial microbes (9, 38). In addition to microorganisms with symbiotic or parasitic plant-microbe interactions, the endosphere can be a reservoir for potentially human pathogenic bacteria such as human enteric pathogens (7, 12). Recent outbreaks highlight important deficiencies in our understanding of the ecology of enteric pathogens outside of their human and animal hosts (43).
The banana plant, which is the largest herbaceous flowering plant, forms unique microhabitats for endophytic microorganisms (39). Their main upright stem is succulent with more than 90% water. This very juicy pseudostem is a cylinder of leaf-petiole sheaths arising from a fleshy rhizome or corm (40). Shoots sprout from the corm replacing the pseudostems, which flower and bear a bunch of fruits, maintaining the original mat indefinitely. These shoots can also be detached and used to plant new fields. The rhizome is an interesting but largely unexplored habitat for endophytic microorganisms. Bananas are propagated through tissue cultures (39). For the application of biological control agents, tissue cultures or tissue-cultured banana plantlets are interesting targets. Biological control of Fusarium wilt mainly based on nonpathogenic endophytic Fusarium oxysporum has been reported (17, 36), although these are not yet commercially available. We sought here to analyze the structure and function of banana-associated microbial communities in the pseudostem (endosphere) and root (rhizosphere) compared to bulk soil and to identify indigenous antifungal antagonists.
MATERIALS AND METHODS
Experimental design and sampling.
Three banana farms representative of the smallholder cropping system in Central Uganda were selected. All were characterized by their high cultivar diversity, including intercropping with a diversity of crops (tomato, pumpkin, papaya, and beans) and combined with animal husbandry (cows, pigs, and chickens). In addition, each of the three selected sites showed unique characteristics: field site 1 was exposed to sun and manual cropping practices; field site 2 was comparable to field 1, except that chemical herbicides (Weedmaster herbicide; BASF) had been used; and field site 3 had a diversity of associated trees. The latter was divided into parts a (agroforest, shaded) and b (without trees, nonshaded). In April 2010, the soil, root (rhizosphere), and interior of the pseudostem (endosphere) were sampled from each field site: four independent composite samples from one plant and three microenvironments were taken from each site. The samples were collected in sterile plastic bags and stored under cooled conditions over 6 days until preparation in the laboratory described in Berg et al. (6, 8). Briefly, for the isolation of banana-associated microorganisms, 5 g of rhizosphere and soil was resuspended with 45 ml of sterile 0.85% NaCl solution (saline) and shaken for 3 min, and the supernatant was decanted into 50-ml tubes. Endosphere samples were washed with 45 ml of sterile saline, and the pieces were transferred to sterile WhirlPaks (Nasco, Fort Atkinson, WI) and homogenized with mortar and pestle after adding 10 ml of sterile saline. The same supernatants were used for all cultivation and cultivation-independent investigations.
Isolation and characterization of fungal pathogens.
Material from heavily infected banana pseudostem with characteristic symptoms of Fusarium wilt was collected near field site 3. For the isolation of fungi, slices of infected banana pseudostem were applied on synthetic nutrient agar (SNA) medium containing 1 g of KH2PO4, 1 g of KNO3, 0.5 g of MgSO4·7H2O, 0.5 g of KCl, 0.2 g of glucose, 0.2 g of sucrose, 0.6 ml of 1 N NaOH, and 22 g of agar per liter of demineralized water (pH 6.5) (34). After autoclaving for 20 min, several antibiotics were added (10 mg of chlorotetracycline liter−1, 50 mg of dihydrostreptomycin sulfate liter−1, and 100 mg of penicillin G liter−1), followed by incubation at 20°C for 3 to 5 days. Fungal cultures were isolated and purified on potato dextrose agar (PDA) plates. For identification, ITS genes were amplified with the primer pair ITS1f-ITS4r (47) in 20 μl. The PCR was performed as follows: 94°C for 5 min, followed by 36 cycles of 94°C for 30 s, 54°C for 35 s, and 72°C for 40 s, with a 72°C final extension step for 10 min. The PCR product was purified using a Wizard SV gel and PCR Clean-Up system (Promega, Mannheim, Germany). The samples were sequenced, and the ITS sequences were aligned with sequences of the NCBI sequence databases using the BLASTn algorithm according to the method of Altschul et al. (1). Fungal isolates were routinely grown on SNA medium and stored at −70°C in liquid storage medium with 60% glycerol.
Isolation of banana-associated bacteria and fungi.
Solutions obtained from soil and banana tissues were serially diluted and plated onto R2A (Difco, Detroit, MI) medium, King's B medium, MacConkey medium (Roth, Karlsruhe, Germany), SNA medium (34), and TSM according to the method of Smith et al. (41). The Trichoderma-specific medium (TSM) contained 1.0 g of Ca(NO3)2·4H2O, 0.26 g of KNO3, 0.26 g of MgSO4·7H2O, 0.12 g of KH2PO4, 1.0 g of CaCl2 · 2H2O 0.05 g of citric acid, 2.0 g of sucrose, and 20.0 g of agar per liter of distilled water. After autoclaving for 20 min, 1.0 ml of Igepal Ca-630, 50 mg of tetracycline liter−1, 50 mg of captan liter−1, and 5.0 mg of vinclozolin liter−1 were added. The numbers of CFU were determined after 5 days of incubation at 20°C.
For further characterization, a total of 384 bacterial strains per habitat (eight strains per replicate and medium) were randomly selected and subcultured on nutrient agar (NA); 586 fungal colonies from SNA medium (171 from the rhizosphere, 235 from the endosphere, and 180 from soil) and 59 Trichodermaisolates from TSM from soil were picked and subcultured on PDA. Bacterial and fungal isolates were encoded by using a combination of numbers and letters: (i) microenvironment (R = rhizosphere, E = endosphere, and S = soil), (ii) sampled field (1 to 4; 3 = 3a, 4 = 3b), (iii) cultivation medium (R2A, MC = MacConkey, KB = King's B, SNA, and TSM), (iv) replicate (1 to 4), and (v) number of isolate (bacteria 1 to 8, fungi 1 to 25). The isolates were purified and stored at −70°C, the bacterial cultures were stored in a liquid nutrient broth (NB) with glycerol (70% [vol/vol]), and the fungi were stored in a liquid storage medium. The medium for the fungi contained the following sterile compounds: 60 ml of glycerol, 20 ml of glucose (50% [wt/vol]), 10 ml of peptone (20% [wt/vol]), and 10 ml of yeast extract (10% [wt/vol]).
Screening and characterization of antagonistic microorganisms.
Bacterial and fungal isolates were screened for their antagonistic activity toward the fungal pathogens Colletotrichum musae (Berk. Et Curt.) ARX, Fusarium chlamydosporum Wollenw. & Reinking, and F. oxysporum Schlecht. f. sp. cubense. We used a dual-culture in vitro assay on Waksman agar, containing 10.0 g of tryptone, 10.0 g of glucose, 5.0 g NaCl, 3.0 g of yeast extract, and 20.0 g of agar per liter of distilled water. Zones of inhibition were measured after 5 days of incubation at 20°C according to the method of Berg et al. (6). All strains were tested in two independent replicates.
Identification of antagonistic microorganisms.
DNA from antagonistic isolates was prepared according to the protocol of Berg et al. (6). Amplified rRNA gene restriction analysis (ARDRA) using the restriction endonuclease HhaI (MP Biomedicals, Solon, OH) was applied to group isolates at the genus level. Isolates displaying similar ARDRA patterns were further analyzed using BOX-PCR genomic fingerprinting. BOX-PCR fingerprints were performed using the BOX_A1R primer (5′-CTA CGG CAA GGC GAC GCT GAC G-3′) as described by Rademaker and De Bruijn (37). The PCR conditions were used as specified by Berg et al. (8), and PCR products were separated by gel electrophoresis on 1.5% agarose gels. Antagonists with either individual ARDRA patterns or different BOX patterns (cutoff level, 80% similarity) were identified by partial 16S rRNA gene sequence analysis according to the method of Berg et al. (6). The PCR product was sequenced using an Applied Biosystems 31301 genetic analyzer sequencer (Data Collection v3.0, Sequencing Analysis v5.2; Applied Biosystems, Foster City, CA) at the sequencing core facility ZMF, Medical University of Graz, Graz, Austria. The obtained sequences were aligned with reference gene sequences from GenBank using the BLAST algorithm. The sequences obtained were submitted to the EMBL nucleotide sequence database.
Total community DNA isolation.
Total community DNA was extracted using a FastDNA spin kit for soil (MP Biomedicals) according to the manufacturer's protocol and used for fingerprinting of the 16S rRNA/ITS genes, quantitative PCR, and the amplicon sequencing approach for enterics.
Microbial fingerprints by SSCP analysis of the 16S rRNA/ITS genes (PCR-SSCP).
Fingerprinting of microbial communities by single-stranded conformational polymorphism (SSCP) was carried out as described by Schwieger and Tebbe (42). Bacterial 16S rRNA gene sequences were amplified by PCR using the bacterial primer pair Unibac-II-515f (5′-GTG CCA GCA GCC GC-3′) and Unibac-II-927rP (5′-CCC GTC AAT TYM TTT GAG TT-3′) according to the method of Zachow et al. (49). The PCR was performed by using a total volume of 60 μl containing 1× Taq&Go PCR Mastermix (MP Biomedicals, Eschwege, Germany), 1.5 mM MgCl2, 0.2 μM concentrations of each primer, and 1 μl of template DNA (95°C for 5 min, followed by 32 cycles of 95°C for 20 s, 54°C for 15 s, and 72°C for 30 s, with a final elongation at 72°C for 10 min). For the analysis of the Pseudomonas community, a nested PCR was performed. In the first PCR, Pseudomonas was selectively amplified with the primers F311Ps (5′-CTG GTC TGA GAG GAT GAT CAG T-3′) and 1459rPsP (5′-AAT CAC TCC GTG GTA AAC GT-3′) (33); this was followed by a second PCR with the primer pair Unibac-II-515f and Unibac-II-927rP. The reaction mixture for the first PCR (20 μl) was composed of 1× Taq&Go PCR Mastermix, 2.25 mM MgCl2, 0.5 mg of bovine serum albumin ml−1, 1.5% dimethyl sulfoxide, 0.2 μM concentrations of each primer, and 1 μl of template DNA (94°C for 7 min, followed by 30 cycles of 94°C for 45 s, 56°C for 2 min, and 72°C for 2 min, with a final elongation at 72°C for 10 min). Fungal communities were analyzed using the internal transcribed spacer region (ITS) primer pairs ITS1f/ITS4r and ITS1f/ITS2 (47) by a two-step PCR. In a first reaction mixture of 20 μl, 1× Taq&Go PCR Mastermix, 0.5 μM concentrations of each primer, 2.5 mM MgCl2, and about 20 ng of template were combined. The second PCR was performed in a 60-μl reaction mixture that was the same as that used for the first PCR. As a template, 1 μl from the primary PCR was used. Prior to separation using the TGGE Maxi system (Biometra, Göttingen, Germany) at 400 V and 26°C, the obtained amplicons were treated with λ-exonuclease (New England Biolabs, Ipswich, MA). Silver staining was used for the the routine detection of DNA bands in SSCP gels (5). Dominant bands were excised from SSCP gels as described previously (42). Gel-extracted DNA was stored 3 to 5 days at 4°C, purified with Wizard SV Gel and PCR Clean-Up System (Promega, Mannheim, Germany), and reamplified with a final primer pair. Samples were sequenced and for phylogenetic analysis and the identification of related sequences; the BLAST algorithm according to the method of Altschul et al. (1) was used.
Tag-encoded pyrosequencing analysis.
To analyze the taxonomic composition of the bacterial communities in the soil, rhizospheres, and endospheres of banana plants, bar-coded PCR amplicons were sequenced with the Roche 454 GS FLX Titanium platform (11). A first PCR was performed with the specific primers for enterics Entero-F234 and Entero-R1423 according to the method of Binh et al. (10). A nested PCR were carried out as described by Heuer et al. (23) using the primer pair F984 and R1378 modified for multiplex 454 sequencing according to the specification of Roche. A preparative gel (1.5% 0.5× Tris-borate-EDTA) was used to purify the amplicons with the Wizard SV Gel and PCR Clean-Up system (Promega). Composite samples containing equimolar aliquots of PCR products from all four replicates per habitat were used for sequencing. Pyrosequencing was accomplished by LGC Genomics (Berlin, Germany).
Raw sequence data were denoised and quality filtered using the default parameter of the open-source software QIIME (13). Resulting reads were trimmed by length (≥440 bp) and analyzed by using the BLAT pipeline integrated into the web interface SnoWMAn version 1.8 (https://epona.genome.tugraz.at/snowman/). The NCBI database was used as a reference database. Taxonomic assignment to family and genus levels was accomplished by using the integrated RDB classifier with an 80% confidence threshold. Rarefaction curves of normalized read counts were obtained by applying the Aligner and the furthest-neighbor approach of the complete linkage clustering and rarefaction tools of the RDP-pyrosequencing pipeline. Phylotype clusters were built at a 5% genetic distance, corresponding to the level of the genus. Shannon indices were also calculated as a measure for diversity.
Quantitative PCR.
Enterobacteriaceae were quantified by determining the number of 16S rRNA genes specifically amplified using the primers DG74f and RW01r (22). The 16S rRNA gene of Serratia plymuthica HRO-C48 served as a reference fragment. First, the fragment was ligated into the vector pGEM-T (Promega) and transformed into E. coli DH5α. The insert was amplified from the vector with M13 and M13r standard primers and used for the generation of standard curves. Amplification from total extracted DNA from soil, rhizosphere, and endosphere was carried out with the Rotor-Gene 6000 system (Corbett, Mortlake, Australia). Reaction components per 10 μl included 1 μl of diluted DNA extracts (1:500), 5.0 μl of 2× Kapa SYBR Fast Mastermix (Kapa Biosystems, Woburn, MA), and 2.5 pmol of each primer. To factor the inhibitory effects of coextracted compounds from environmental samples, PCRs performed for standard curves were amended with 1 μl of 1:500-diluted DNA-free matrices of respective samples. Quantitative PCRs were carried out as follows: an initial denaturation at 95°C for 10 min, followed by 40 cycles of 92°C for 30 s, 52°C for 30 s, and 72°C for 60 s. All reactions were prepared in duplicates in each of the two independently repeated runs. Calculated gene counts were normalized for each sample based on the DNA concentration determined spectrometrically by the NanoDrop 2000c system (Thermo Scientific, Wilmington, MA).
Fluorescence in situ hybridization coupled with confocal laser scanning microscopy (FISH-CLSM).
Thin sections of banana endorhiza and endosphere were fixed in 3:1 4% paraformaldehyde solution–phosphate-buffered saline (PBS) for 6 h at 4°C and then washed three times in ice-cold PBS before storage at −20°C in 1:1 ice-cold PBS–ice-cold 96% ethanol. For the FISH staining, the sections were briefly washed in PBS and placed on poly-l-lysine-coated glass slides. The samples were dried at 43°C for ∼10 min and then incubated with 10 μl of 0.5 mg of lysozyme (Sigma-Aldrich, Steinheim, Germany) ml−1 for 10 min at room temperature to increase the bacterial cell wall permeability to the FISH probes. The samples were then rinsed with ice-cold PBS, dehydrated sequentially with three ethanol solutions (50, 70, and 96%, 3 min each time), and then quickly rinsed with ice-cold PBS. All hybridizations were performed at 43°C for 2 h in a buffer containing 0.9 M NaCl, 0.02 M Tris-HCl, 0.01% sodium dodecyl sulfate, 30% ultrapure formamide (Invitrogen), and 5.0 to 8.3 ng of each FISH probe μl−1 (pH 8). The following FISH probes were applied: NONEUB-Cy5a, NONEUB-Cy3a, NONEUB-ATTO448 (45), EUB338 (2), EUB338IIm, EUB338III (14), ALF968 (28), BET42a, GAM42a, GAM42a competitor (30), and EnterbactD (35). Due to their nonoverlapping fluorescent spectra, Cy3-, Cy5-, and ATTO488-labeled probes could be applied simultaneously. The hybridization buffer was immediately removed, and the samples were incubated for 10 to 15 min with prewarmed (45°C) washing buffer (0.02 M Tris-HCl, 102 mM NaCl, and 5 mM EDTA [pH 8]). The washing buffer was removed, and the samples were rinsed with ice-cold water. The sections were finally dried with soft compressed air, immediately mounted with ProLong Gold antifade reagent (Molecular Probes, Eugene, OR), and stored at 4°C until investigation by CLSM.
CLSM was performed with a Leica TCS SPE confocal microscope (Leica Microsystems, Mannheim, Germany). The fluorescent dyes ATTO488, Cy3, and Cy5 labeling the FISH probes were sequentially excited with 488-, 532-, and 635-nm laser beams, respectively; the emitted light was detected in the ranges of 500 to 540 nm, 550 to 610 nm, and 650 to 700 nm, respectively. The setting of the photomultiplier (gain and offset, corresponding to brightness and contrast, respectively) were individually optimized for every channel and every field of view, in order to improve the signal/noise ratio. Confocal stacks were acquired with a Leica ACS APO 40× oil CS objective lens (NA, 1.15) and a Leica ACS APO 63× oil CS objective lens (NA, 1.30) by applying a z-step of 0.25 to 0.5 μm. Three-dimensional models based on spots and isosurfaces were generated with the software Imaris 7.0 (Bitplane, Zurich, Switzerland).
PCR-based detection of broad-host-range plasmids and sulfonamide resistance genes.
Primers targeting the IncP-1 (3), IncN, and IncQ plasmids (20), as well as the sul1 and sul2 genes (25, 48), were amplified from DNA directly extracted from soils. The PCR amplicons were analyzed by gel electrophoresis and subsequent Southern blot hybridization, as previously described by Götz et al. (20).
Statistics.
All data (CFU, percentages of antagonists, and quantitative PCR) were analyzed for significance using analysis of variance (ANOVA) and Tukey's post hoc test (P > 0.05) and studied by two-factor ANOVA by using Statistical Product and Service Solutions for Windows (release 9.0.1; SPSS, Inc., Chicago, IL). SSCP- and BOX-PCR-generated fingerprints were evaluated using the GelCompar II version 5.1 software package (Applied Maths, Kortrijk, Belgium). A similarity matrix was constructed using Pearson's correlation coefficients (r), and cluster analysis was done by using the unweighted pair group method with average linkages (UPGMA). The resulting clusters of samples were examined for statistical significance by applying the permutation test with 10,000 random permutations of the single samples (26).
Nucleotide sequence accession numbers.
The accession numbers for sequences submitted to the EMBL nucleotide sequence database are HE588041 to HE588079 for the bacterial isolates and HE586686 to HE586734 for the fungal isolates.
RESULTS
Abundances of banana-associated microbial communities.
The CFU counts on R2A (bacteria), King's B (Pseudomonas), MacConkey agar (enterics), and SNA (fungi) determined are summarized in Fig. S1 in the supplemental material. For bacteria, CFU counts were in the range of 7.7 to 9.4 log10 CFU per g fresh weight (g fw−1). Similar numbers were calculated for enterics grown on MacConkey agar: endosphere samples contained the highest abundances with 8.8 (±0.1) log10 CFU g fw−1, followed by the rizosphere with 7.1 (±0.3) log10 CFU g fw−1 and soil with 6.0 (±0.6) log10 CFU g fw−1. Fungal counts were in the range of 5.4 to 6.2 log10 CFU g fw−1. The CFU counts determined for the different field sites were rather similar. Statistically significant differences were found, however, between the microenvironments: the endosphere with log10 9.4 ± 0.1 CFU g fw−1 showed the highest abundances for all microbial groups compared to the rhizosphere (log10 8.4 ± 0.3 CFU g fw−1) and soil (log10 7.7 ± 0.3 CFU g fw−1).
To verify the high numbers calculated for enterics on MacConkey agar, a cultivation-independent measurement was included: enterics were monitored by quantitative PCR using specific primers (Fig. 1). Using this technique, we confirmed the high abundances of enterics: the endosphere with log10 7.9 ± 0.2 gene copy numbers g fw−1 was statistically higher compared to the rhizosphere (log10 6.8 ± 0.4 gene copy numbers g fw−1) and soil (log10 6.3 ± 0.8 gene copy numbers g fw−1).
Fig 1.
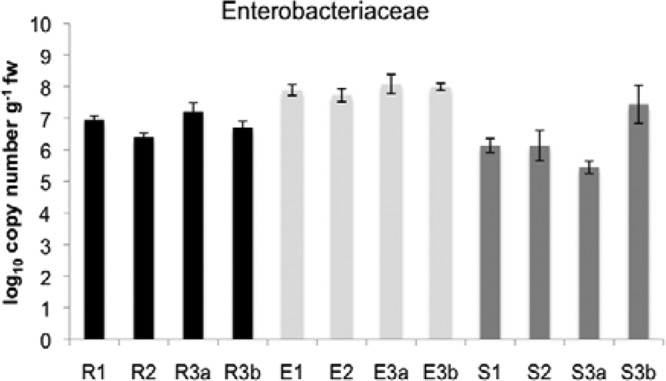
16S rRNA copy numbers per gram of fresh weight (g fw−1) determined by Enterobacteriaceae-specific quantitative PCR. Log10 values are shown for the rhizosphere (R), the endosphere (E), and soil (S) for the four investigated field sites: field 1, no herbicide treatment, no agro-forest; field 2, intensive herbicide treatment, no agro-forest; field 3a, no herbicide treatment, agro-forest; and field 3b, no herbicide treatment, no agro-forest. Error bars indicate confidence intervals at P = 0.005.
Molecular fingerprinting of banana-associated microbial communities.
SSCP analysis of 16S rRNA/ITS genes amplified from DNA obtained from the rhizosphere, endosphere, and surrounding bulk soil yielded an impressive number of bands representing bacteria and fungi for all samples (see, for example, Fig. 2 for the endosphere samples). According to cluster analyses and statistical evaluation, the composition of the bacterial and especially of the Pseudomonas community showed microenvironment-specific effects. Site-specific effects were partly detected between the fields, especially for the Pseudomonas-specific community but not in general (Fig. 3). Soil communities in the investigated fields showed higher and statistically significant differences than the plant-associated communities. Bacterial communities in the endosphere were characterized by the highest degree of heterogeneity. From these endophytic communities several dominant bands were identified as Flavobacteria, Sphingobacteria, Alphaproteobacteria (Brevundimonas), and Betaproteobacteria (Delftia, Herbaspirillum, Azoarcus, Acidovorax, and Diaphorobacter), as well as Gammaproteobacteria (Buttiauxella, Pseudomonas, and Serratia) (see Fig. 2 and Table S1 in the supplemental material). The high level of diversity and heterogeneity of the endosphere communities was confirmed by the analyses of the fungal communities. They were microenvironment specific but not statistically significantly different among the field sites (Fig. 2). Sequencing of characteristic bands resulted in identification of putative members of Basidiomycota: Geastrum corollinum, Pluteus albostipitatus, Agaricus bisporus, Hyphodontia nothofagi, Coprinus fissolanatus, Bullera oryzae, and the yeast Cryptococcus zeae (see Table S1 in the supplemental material), although their identification level was in part very low.
Fig 2.
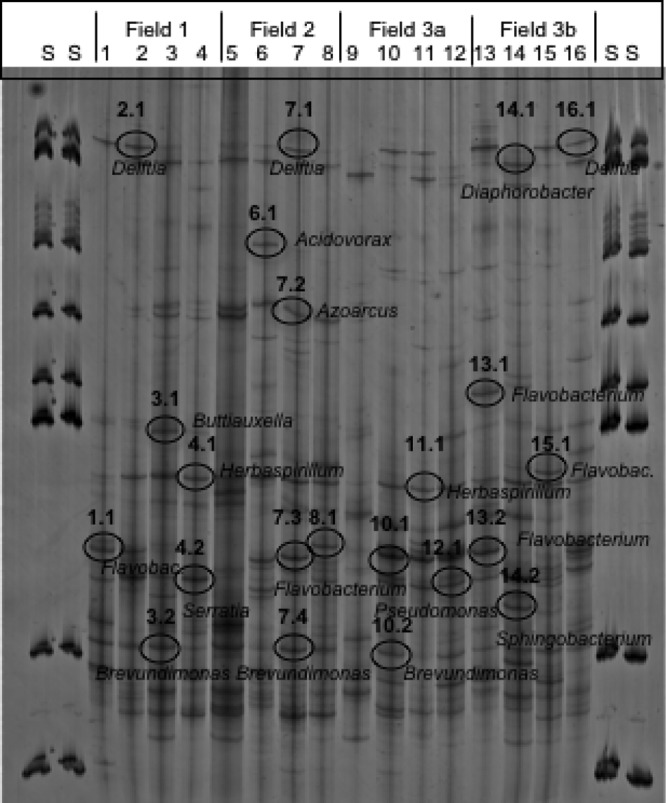
Molecular fingerprints of 16S rRNA gene fragments amplified from endosphere samples of all four field sites and separated by SSCP. Four lanes of each variant represent independent replicates. Lanes S, 1-kb ladder. Marked bands were sequenced and compared to those in the GenBank database using the BLAST algorithm. Detailed results are presented in Table S1 in the supplemental material.
Fig 3.
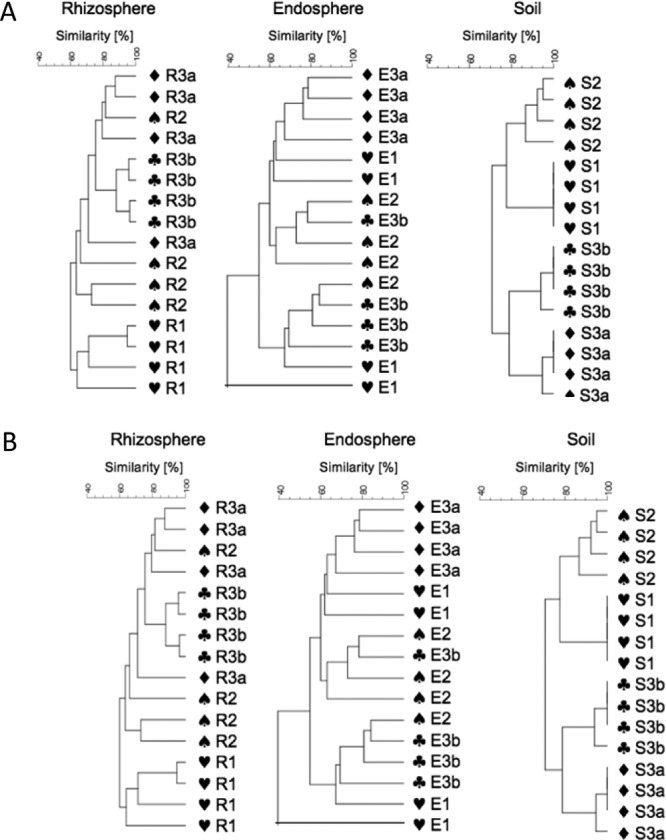
Clustering of the SSCP profiles of Pseudomonas communities (A) and fungal communities (B) obtained from the rhizosphere (R), endosphere (E), and soil (S) for the four investigated field sites: field 1, no herbicide treatment, no agro-forest; field 2, intensive herbicide treatment, no agro-forest; field 3a, no herbicide treatment, agro-forest; and field 3b, no herbicide treatment, no agro-forest. The dendrograms were generated from the SSCP community profiles and clustered by the UPGMA method using average linkages with GelCompar II.
Pyrosequencing of enteric 16S rRNA gene fragments.
All samples from the representative field site 3 characterized by a high microbial diversity were studied by a pyrosequencing-based analysis of partial 16S rRNA gene sequences specific for enterics of all quality sequences, 83.0% could be classified below the domain level. To determine rarefaction curves, operational taxonomic units (OTU) were identified at sequence divergences of 5% (genus level) (see Fig. S2 in the supplemental material). Furthermore, Shannon indices of diversity (H′) at a 95% similarity level were calculated: they were lower in all plant microhabitats, with H′ = 0.55 for the rhizosphere (shaded), H′ = 0.40 for the second rhizosphere sample (nonshaded), and H′ = 0.55 for the endosphere, than in bulk soil (H′ = 2.99). Most of the determined enteric genera were found in all microenvironments (Table 1): Citrobacter, Enterobacter, Escherichia, Klebsiella, Pantoea, Raoultella, Salmonella, and Serratia. The genus Yersinia was exclusively found in the endosphere, and Pectobacterium was exclusively found in soil. Interestingly, the proportion of genera varied greatly especially between the plant-associated microenvironments and soil. Enterobacter was predominant in plant-associated habitats (31.9 to 44.8%), and Pantoea was predominant in soil (23.5%). The composition of enteric bacteria in the endosphere differed strongly from the rhizosphere and soil. Here, in addition to Enterobacter (31.9%), we found that Pantoea (14.0%), Raoultella (12.3%), Klebsiella (11.4%), and Serratia (11.3%) were the most important genera. For the rhizosphere, an impact of agro-forest was found for the Serratia community, which was greater in nonshaded areas (23.0%) than in shaded areas (12.1%). In contrast to genera well known for beneficial plant-microbe interaction, such as Enterobacter and Serratia, potential human pathogenic were found in general only in low abundances: Escherichia (1.1 to 1.4% in all microenvironments), Salmonella (1.1 to 1.3% in endosphere and soil), and Yersinia (1.3% in only endosphere).
Table 1.
Enteric communities in three different microenvironments of field site 3
| Genus | Relative % of sequencesa |
|||
|---|---|---|---|---|
| Rhizo 3a | Rhizo 3b | Endo 3b | Soil 3b | |
| Buttiauxella | 1.6 | 0.0 | 0.0 | 2.6 |
| Citrobacter | 5.4 | 2.6 | 3.9 | 7.0 |
| Enterobacter | 44.8 | 44.6 | 31.9 | 15.5 |
| Erwinia | 1.5 | 0.0 | 0.0 | 4.9 |
| Escherichia | 1.4 | 0.0 | 1.1 | 1.3 |
| Klebsiella | 9.5 | 5.7 | 11.5 | 12.2 |
| Kluyvera | 1.7 | 2.1 | 3.8 | 1.4 |
| Pantoea | 14.5 | 13.2 | 14.0 | 23.5 |
| Pectobacterium | 0.0 | 0.0 | 0.0 | 5.0 |
| Raoultella | 2.4 | 3.0 | 12.3 | 2.7 |
| Salmonella | 0.0 | 0.0 | 1.1 | 1.3 |
| Serratia | 12.1 | 23.0 | 11.3 | 18.3 |
| Yersinia | 0.0 | 0.0 | 1.3 | 0.0 |
| Others | 5.1 | 5.9 | 7.7 | 4.4 |
The percentage of major genera was determined by pyrosequencing 16S rRNA amplicons from metagenomic DNA extracted from the rhizosphere (Rhizo 3a, no herbicide treatment, agro-forest; Rhizo 3b, no herbicide treatment), the endosphere (Endo 3b), and soil (Soil 3b). The identification of the closest strain based on the 16S rRNA sequence similarity was achieved using the online analysis tool SnoWMAn 1.8. Phylogenetic groups accounting for ≤1% of all quality sequences are summarized in the artificial group “Others.”
PCR-based detection of plasmid replicon and sulfonamide resistance genes.
The PCR-based detection of the broad-host-range plasmids belonging to the IncQ, IncP-1α, IncP-1β, IncP-1ε, and the IncN group in soil DNA (S1 to S16) revealed that plasmid replicon sequences that are typically associated with manure-fertilized soils were not detectable in the soils from all four sites. However, the sul1 and the sul2 genes could be detected both in samples S1 and S2. In addition, the sul2 was also detected in samples S9 and S14.
FISH-CLSM of the endorhiza and endospheres of banana plants.
In both endophytic microenvironments, a large number of alpha-, beta-, and gammaproteobacteria were detected. Using specific probes for enterics, bacteria of this group also were visualized: they form large colonies in close interaction with other groups (Fig. 4). In the endosphere, enterics constitute up to 30% of the visible bacterial cells.
Fig 4.

Occurrence of Enterobacteriaceae in the endosphere and endorhiza of banana plants. FISH with the probes EnterbactD-ATTO488, ALF968-Cy5, and EUBMIX-Cy3 of banana thin sections and CLSM showed colonization of the endosphere (A, B, and C) and endorhiza (D). (A and B) Violet, Enterobacteriaceae; yellow, Alphaproteobacteria; red, other bacteria. Panel B shows the thre-dimensional computer reconstruction of panel A, using Imaris7.0. (C) Violet, Enterobacteriaceae (arrows); yellow, Alphaproteobacteria; red, other bacteria or plant structures (long, parallel bars). (D) Violet, Enterobacteriaceae (arrows) or plant cell walls; yellow, Alphaproteobacteria; red: other bacteria. Scale bars, 20 μm.
Antagonistic potential of the microbial community toward Fusarium wilt.
In a first step, different fungal strains were isolated from the infected tissues. Those with the typical Fusarium morphology were identified as Fusarium chlamydosporum Wollenw. & Reinking and F. oxysporum Schlecht. f. sp. cubense. A third species, which is known as pathogen causing Colletotrichum speckle on banana plants, was identified as Colletotrichum musae (Berk. Et Curt.) ARX. Considering what is currently known about multipathogen diseases, all three isolates were included in antagonism studies.
To evaluate the indigenous antagonistic potential within the microbial communities, 1,152 bacterial and 586 fungal isolates randomly selected were screened by dual testing regarding their in vitro antagonistic activity toward the fungal pathogens. In general, the highest proportion of bacterial strains with antagonistic properties was found in the endosphere (9.4%), followed by the rhizosphere (6.5%) and soil (4.4%). The source of fungal antagonists showed a contrasting picture: 15.0% of the soil fungi displayed antagonistic properties, followed by fungi from the rhizosphere (12.3%) and endosphere (2.6%).
To assess the diversity of bacterial antagonists, isolates with an activity toward at least two of the potentially pathogenic fungi (37 isolates) were characterized genotypically, and a representative selection of 17 strains was identified by partial 16S rRNA gene sequencing (see Table S2 in the supplemental material). Using restriction fragment length polymorphism of the 16S rRNA (i.e., ARDRA), the antagonistic isolates could be clustered into five groups: (i) Burkholderia, (ii) Serratia, (iii) Pseudomonas, (iv) Bacillus 1, and (v) Bacillus 2. Altogether, 11 different species with antagonistic properties were identified: Burkholderia cepacia, B. multivorans, B. plantarii, Pseudomonas cichorii, P. fluorescens, P. putida, P. cichorii, P. palleroniana, Serratia marcescens, Bacillus indicus, and B. weihenstephanensis. To analyze the genotypic diversity within the taxonomic groups at the population level, the BOX-PCR patterns of the whole bacterial genome were used. The Burkholderia and Pseudomonas clusters displayed an especially high genotypic diversity. Interestingly, the Serratia isolates from different microhabitats (soil and endosphere) and field sites (2, 3a, and 3b) showed identical fingerprints. The contrasting diversity using BOX fingerprints is illustrated for seven Burkholderia and Serratia isolates (Fig. 5).
Fig 5.
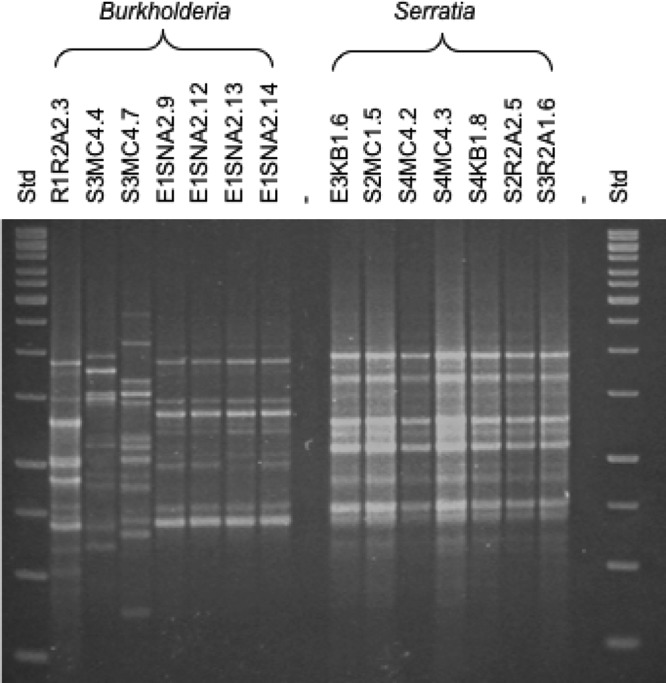
BOX patterns of bacterial antagonists of Burkholderia species and Serratia marcescens isolates obtained from the rhizosphere (R), endosphere (E), and soil (S) of the investigated field sites.
Of 36 fungal isolates with antagonistic properties grouped according their ARDRA patterns, as well as their morphology, 26 morphogroups were found that were represented by the genera Fusarium, Penicillium, Paecilomyces, Mortierella, Bionectria, Hypocrea, and Faurelina. Twelve different species were identified, among them a high diversity of Penicillium species (P. brevicompactum, P. chrysogenum, P. coprobium, P. janthinellum, and P. phialosporum), well-known species with pathogenic properties (Fusarium oxysporum and F. solani), and only a few species known for their antagonistic activity (Paecilomyces lilacinus, P. marquandii, and Hypocrea lixi/Trichoderma harzianum). The results of the detailed characterization of the 36 fungal isolates are presented in Table S3 in the supplemental material.
DISCUSSION
In our study we found microhabitat-specific microbial communities and functional guilds in bananas, which were independent from the site and different managements (intercropping, agro-forest, and herbicides). Several unexpected findings—including the unique microbiology in the banana endosphere, the dominant role of Enterobacteriaceae in all banana-associated microhabitats investigated, and Fusarium wilt, which was found as disease complex—are discussed in detail.
The endosphere (pseudostem) of bananas was identified as an extraordinary microhabitat for bacteria because here we found (i) the highest bacterial abundances up to 109 16S rRNA CFU g fw−1, (ii) the highest number of enterics up to 109 16S rRNA gene copy numbers g fw−1 (using quantitative PCR), and (iii) the highest proportion of bacterial antagonists over fungal pathogens. The microhabitats of plants differ not only in their abiotic parameters but also in their microbial communities (7, 49). Due to the fact that plant roots exude an enormous range of potentially valuable small-molecular-weight compounds into the rhizosphere (29, 32), this plant-associated microhabitat contains the main microbiome of plants in general. In contrast, the above-ground parts are less colonized; nevertheless, they harbor a diverse and specifically adapted microbial community (27). For bananas, we obtained higher CFU counts in the endosphere than in the rhizosphere. Unfortunately, little is known about the chemical composition within the very juicy pseudostem of banana plants. In addition to polyphenols and antioxidants, several bioactive, antibacterial ingredients were identified (40). Regardless, we found the endosphere of bananas to be a comfortable environment for microorganisms.
Using all methods, members of Enterobacteriaceae were identified as a major component in the bacterial community of bananas. Pseudomonas is the classical plant-associated genus; Pseudomonas strains are well known for their ability to colonize plants and to assume a beneficial plant-microbe interaction via growth promotion, stress reduction, and antagonism toward phytopathogens (21, 29, 46). They were also identified as one of the main components and important disease-suppressive bacteria in suppressive soils analyzed by PhyloChip-based metagenomics (32). By cultivation, we found an abundant Pseudomonas community in bananas; they provide one cluster of antagonists, but they were less abundant than enterics (data not shown). The high abundance of enterics can be explained by amendments of both human and animal manure in this traditional field sites. This manure is often the only nutrient supply for this extensively managed agricultural ecosystem. Fertilization of arable soils with animal manure has recently shown to be a source of antibiotic resistance genes and plasmids in soil bacteria (24). The plasmids targeted in the present study are typically associated with piggery manure (10). The low abundance of plasmids might point to the absence of selective pressure by antibiotics used typically in farms. Furthermore, on all field sites, bananas had been cultivated for a long time. In these traditional farms, which were managed by only one family but over many generations, banana mats are permanent through the corm with a succession of vegetative suckers. The corm and older stems are the reservoir for the endophytic microorganisms of the new shoot. Although enterics were well-known members of the human and animal microbiomes, recent outbreaks of food-borne diseases changed our understanding of enterics and plant hosts. Indeed, many enterics are good colonizers of plant tissues and multiply in the rhizosphere (12, 43). The mechanisms responsible for colonization of the rhizosphere and antagonistic activity against plant pathogens are similar to those responsible for the colonization of human organs and tissues and for pathogenicity (7). Escherichia, Klebsiella, Salmonella, and Yersinia spp. were abundant in the endospheres of bananas. However, we did not investigate banana fruits, which is an important aspect also regarding food safety. Although our data provide evidence for the occurrence of enteric pathogens in the endosphere, very little information on their virulence relative to the one of their clinical counterparts is available.
In addition to Fusarium oxysporum f. sp. cubense, which is a well-known pathogen of bananas and causes serious wilting symptoms (18, 44), F. chlamydosporum and C. musae have been identified in disease symptoms. All three strains were included in bacterial (but not in fungal) antagonism studies because it has been shown that opportunistic pathogens often play a role in symptom expression (17, 18). However, which role they play and how they interact with each other has yet to be investigated. All microhabitats contained a high number and a broad spectrum of indigenous bacterial and fungal isolates with antagonistic activity toward the fungal pathogens of banana tested here. Several new antagonists were identified, e.g., Bacillus indicus, Pseudomonas palleroniana, Faurelina elongata, and Penicillium spp. Unfortunately, a high number of selected strains belong to potential plant pathogens (Burkholderia plantarii, Pseudomonas cichorii, F. solani, and F. oxysporum) or potential human pathogens (Burkholderia cepacia, B. multivorans, and Serratia marcescens). Further tests are needed to assess their pathogenic capacity. Treatment with antagonistic bacteria, fungi, or mixed consortia is a promising objective to control Fusarium wilt in bananas, and allochthonous strains are often more effective than indigenous strains (50). They can be applied already in tissue cultures or banana plantlets and have to be adapted to the specific conditions in the banana pseudostem.
There are emerging fungal threats to plant and ecosystem health and, in addition, fungal diseases are caused by multipathogen complexes (16). Microbial biodiversity, balanced plant-microbe interactions, and the promotion of beneficial bacteria are key points to avoiding pathogenic interactions. Ecology-based selection and careful use of biocontrol agents is one possibility to stabilize agricultural ecosystems. Our study showed that bananas are a propitious habitat for microbial communities of bacteria with functions that are important for plant health. Moreover, due to the high abundance and diversity of enterics, these bacteria also have significant implications with regard to human health issues.
Supplementary Material
ACKNOWLEDGMENTS
We thank Birgit Lukesch and Massimiliano Cardinale (Graz, Austria) for valuable assistance in microscopy, Ellen Krögerrecklenfort (Braunschweig, Germany) for technical assistance, and Christin Zachow and Martina Köberl (Graz, Austria) for generous support.
This study was supported by the Federal Ministry of Finance of the Republic of Austria through the Austrian Development Agency.
Footnotes
Published ahead of print 4 May 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein data-base search programs. Nucleic Acids Res. 25: 3389– 3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amann RI, et al. 1990. Combination of 16S rRNA targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56: 1919– 1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahl MI, Burmolle M, Meisner A, Hansen LH, Sørensen SJ. 2009. All IncP-1 plasmid subgroups, including the novel epsilon subgroup, are prevalent in the influent of a Danish wastewater treatment plant. Plasmid 62: 134– 139 [DOI] [PubMed] [Google Scholar]
- 4.Bagamba F, et al. 2006. Awareness of banana bacterial wilt control in Uganda. 1. Farmers' perspective. Afr. Crop Sci. J. 14: 157– 164 [Google Scholar]
- 5.Bassam BJ, Caetano-Anolles G, Gresshoff PM. 1991. Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal. Biochem. 80: 81– 84 [DOI] [PubMed] [Google Scholar]
- 6.Berg G, et al. 2002. Plant-dependent genotypic and phenotypic diversity of antagonistic rhizobacteria isolated from different Verticillium host plants. Appl. Environ. Microbiol. 68: 3328– 3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berg G, Eberl L, Hartmann A. 2005. The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ. Microbiol. 7: 1673– 1685 [DOI] [PubMed] [Google Scholar]
- 8.Berg G, et al. 2006. The rhizosphere effect on bacteria antagonistic toward the pathogenic fungus Verticillium differs depending on plant species and site. FEMS Microbiol. Ecol. 56: 250– 261 [DOI] [PubMed] [Google Scholar]
- 9.Berg G. 2009. Plant-microbe interactions promoting plant growth and health: perspectives for controlled use of microorganisms in agriculture. J. Appl. Microbiol. Biotechnol. 84: 11– 18 [DOI] [PubMed] [Google Scholar]
- 10.Binh CTT, Heuer H, Gomes NCM, Kaupenjohann M, Smalla K. 2010. Similar bacterial community structure and high abundances of sulfonamide resistance genes in field-scale manures, p 141– 166 Nova Science Publishers, Hauppauge, NY [Google Scholar]
- 11.Binladen J, et al. 2007. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One 2: 1– 9 doi:10.1371/journal.pone.0000197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brandl MT. 2006. Fitness of human enteric pathogens on plants and implications for food safety. Annu. Rev. Phytopathol. 44: 367– 392 [DOI] [PubMed] [Google Scholar]
- 13.Caporaso JG, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7: 335– 336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daims H, Bühl A, Amann R, Schleifer K- H, Wagner M. 1999. The domain-specific probe EUB388 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22: 434– 444 [DOI] [PubMed] [Google Scholar]
- 15.Eledu CA, Karamura EB, Tushemereirwe WK. 2010. Agroecological distribution of banana systems in the Great Lakes Region. Afr. Crop Sci. J. 12: 33– 42 [Google Scholar]
- 16.Fisher MC, et al. 2012. Emerging fungal threats to animal, plant and ecosystem health. Nature 484: 186– 194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsyth LM, Smith LJ, Aitken EA. 2006. Identification and characterization of non-pathogenic Fusarium oxysporum capable of increasing and decreasing Fusarium wilt severity. Mycol. Res. 110: 929– 935 [DOI] [PubMed] [Google Scholar]
- 18.Fourie G, Steenkamp ET, Ploetz RC, Gordon TR, Viljoen A. 2011. Current status of the taxonomic position of Fusarium oxysporum formae specialis cubense within the Fusarium oxysporum complex. Infect. Genet. Evol. 11: 533– 542 [DOI] [PubMed] [Google Scholar]
- 19.Gold CS, Kiggundu A, Abera AMK, Karamura D. 2002. Diversity, distribution, and farmer preference of Musa cultivars in Uganda. Exp. Agric. 38: 39– 50 [Google Scholar]
- 20.Götz A, et al. 1996. Detection and characterization of broad-host-range plasmids in environmental bacteria by PCR. Appl. Environ. Microbiol. 62: 2621– 2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haas D, Défago G. 2005. Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol. 3: 307– 319 [DOI] [PubMed] [Google Scholar]
- 22.Handschur M, Pinar G, Gallist B, Lubitz W, Haslberger AG. 2005. Culture free DGGE and cloning based monitoring of changes in bacterial communities of salad due to processing. Food Chem. Toxicol. 43: 1595– 1605 [DOI] [PubMed] [Google Scholar]
- 23.Heuer H, Kresek M, Baker P, Smalla K, Wellington EMH. 1997. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 63: 3233– 3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heuer H, et al. 2012. IncP-1ε plasmids are important vectors of antibiotic resistance genes in agricultural systems: diversification driven by class 1 integron gene cassettes. Front. Microbiol. doi:10.3389/fmicb.2012.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerrn MB, Klemmensen T, Frimodt-Møller N, Espersen F. 2002. Susceptibility of Danish Escherichia coli strains isolated from urinary tract infections and bacteraemia, and distribution of sul genes conferring sulphonamide resistance. J. Antimicrob. Chemother. 50: 513– 516 [DOI] [PubMed] [Google Scholar]
- 26.Kropf S, Heuer H, Grüning M, Smalla K. 2004. Significance test for comparing complex microbial community fingerprints using pairwise similarity measures. J. Microbiol. Methods 57: 187– 195 [DOI] [PubMed] [Google Scholar]
- 27.Lindow SE, Brandl MT. 2003. Microbiology of the phyllosphere. Appl. Environ. Microbiol. 69: 1875– 1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loy A, Maixner F, Wagner M, Horn M. 2007. probeBase: an online resource for rRNA-targeted oligonucleotide probes: new features 2007. Nucleic Acids Res. 35: D800– D804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lugtenberg B, Kamilova F. 2009. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 63: 541– 556 [DOI] [PubMed] [Google Scholar]
- 30.Manz W, Amann R, Ludwig W, Wagner M, Schleifer K-H. 1992. Phylogenetic oligodeoxynucleotide probes for the major subclasses of Proteobacteria: problems and solutions. Syst. Appl. Microbiol. 62: 4504– 4513 [Google Scholar]
- 31.Mei C, Flinn BS. 2010. The use of beneficial microbial endophytes for plant biomass and stress tolerance improvement. Recent Pat. Biotechnol. 1: 81– 95 [DOI] [PubMed] [Google Scholar]
- 32.Mendes R, et al. 2011. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 27: 1097– 1100 [DOI] [PubMed] [Google Scholar]
- 33.Milling A, Lembke A, Schönfeld J, Smalla K. 2004. Survival and activity of the Ralstonia solanacearum antagonist Pseudomonas chlororaphis 24-4 in the rhizosphere of tomato and its impact on the indigenous bacterial community. IOBC/WPRS Bull. 27: 177– 186 [Google Scholar]
- 34.Nirenberg HI. 1976. Untersuchungen über die morphologische und biologische Differenzierung in der Fusarium-Sektion Liseola. Mitt. Biol. Bundesanst. Land-Forstwirtsch. Berl.-Dahl. 169: 1– 117 [Google Scholar]
- 35.Ootsubo M, et al. 2002. Oligonucleotide probe for detecting Enterobacteriaceae by in situ hybridization. J. Appl. Microbiol. 93: 60– 68 [DOI] [PubMed] [Google Scholar]
- 36.Paparu P, et al. 2008. Screenhouse and field persistence of nonpathogenic endophytic Fusarium oxysporum in Musa tissue culture plants. Microb. Ecol. 55: 561– 568 [DOI] [PubMed] [Google Scholar]
- 37.Rademaker JLW, De Bruijn FJ. 1997. Characterization and classification of microbes by REP-PCR genomic fingerprinting and computer-assisted pattern analysis, p 151– 171 In Caetano-Anolle G, Gresshoff PM. (ed), DNA markers: protocols, applications, and overviews. Wiley, New York, NY [Google Scholar]
- 38.Reinhold-Hurek B, Hurek T. 2011. Living inside plants: bacterial endophytes. Curr. Opin. Plant Biol. 14: 435– 443 [DOI] [PubMed] [Google Scholar]
- 39.Robinson JC, Saúco VG. 2010. Bananas and plantains, 2nd ed CAB International, Oxford, United Kingdom: [Google Scholar]
- 40.Saravanan K, Aradhya SM. 2011. Polyphenols of pseudostem of different banana cultivars and their antioxidant activities. J. Agric. Food Chem. 59: 3613– 3623 [DOI] [PubMed] [Google Scholar]
- 41.Schwieger F, Tebbe CC. 1998. A new approach to utilize PCR-single-strand-conformation polymorphism for 16S rRNA gene-based microbial community analysis. Appl. Environ. Microbiol. 64: 4870– 4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith VL, Wilcox WF, Harman GE. 1990. Potential for biological control of Phytophthora root and crown rots of apple by Trichoderma and Gliocladium spp. Phythopathology 80: 880– 885 [Google Scholar]
- 43.Teplitski M, Warriner K, Bartz J, Schneider KR. 2011. Untangling metabolic and communication networks: interactions of enterics with phytobacteria and their implications in produce safety. Trends Microbiol. 19: 121– 127 [DOI] [PubMed] [Google Scholar]
- 44.Tushemereirwe WK, Kangire A, Kubiriba J, Nakyanzi M, Gold CS. 2004. Diseases threatening banana diversity in Uganda. Afr. Crop Sci. J. 12: 19– 26 [Google Scholar]
- 45.Wallner G, Amann R, Beisker W. 1993. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry 14: 136– 143 [DOI] [PubMed] [Google Scholar]
- 46.Weller DM. 2007. Pseudomonas biocontrol agents of soilborne pathogens: looking back over 30 years. Phytopathology 97: 250– 256 [DOI] [PubMed] [Google Scholar]
- 47.White TJ, Bruns TD, Lee SB, Taylor JW. 1990. PCR protocols: a guide to methods and applications: amplification and direct sequencing of fungal rRNA genes for phylogenetics, p 315– 322 Academic Press, New York, NY [Google Scholar]
- 48.Wu S, Dalsgaard A, Hammerum AM, Porsbo LJ, Jensen LB. 2010. Prevalence and characterization of plasmids carrying sulfonamide resistance genes among Escherichia coli from pigs, pig carcasses, and human. Acta Vet. Scand. 52: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zachow C, Tilcher R, Berg G. 2008. Sugar beet-associated bacterial and fungal communities show a high indigenous antagonistic potential against plant pathogens. Microb. Ecol. 55: 119– 129 [DOI] [PubMed] [Google Scholar]
- 50.Zachow C, Fatehi J, Cardinale M, Tilcher R, Berg G. 2010. Strain-specific colonization pattern of Rhizoctonia antagonists in the root system of sugar beet. FEMS Microbiol. Ecol. 74: 124– 135 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.