

Abstract
Staphylococcus aureus strains producing the bacteriophage-encoded staphylococcal enterotoxin A (SEA) were divided into two groups, high- and low-SEA-producing strains, based on the amount of SEA produced. After growth under favorable conditions in batch cultures, 10 of the 21 strains tested produced more than 1,000 ng/ml SEA, and 9 strains produced less than 10 ng/ml SEA; two enterotoxigenic strains, MRSA252 and Newman, produced intermediate levels of SEA (around 450 ng/ml). The differences in the production of SEA were found to be associated with the expression level of sea and whether the strains hosted the sea1 or sea2 version. Furthermore, differences in nucleotide sequence in the Siphoviridae phage region showed two clonal lineages of the high-SEA-producing strains. One of these lines was correlated with the capacity for a massive increase in SEA levels by prophage induction as demonstrated using mitomycin C (MC). This was also confirmed by the occurrence of additional sea expression, presumed to be initiated by a latent phage promoter located upstream of the endogenous sea promoter. Remarkably, the SEA level was increased up to 10-fold in some strains due to prophage induction. The low-SEA-producing group and the high-SEA-producing subgroup lacking phage-activated sea transcription showed no increase in SEA formation after the addition of MC. This study demonstrates that sea expression in enterotoxigenic strains is correlated with the clonal lineage of sea-carrying phages. The high-SEA-producing group, in particular the prophage-inducible sea1 group, may be more relevant to staphylococcal food poisoning than the low-SEA-producing group, harboring mainly sea2.
INTRODUCTION
Staphylococcus aureus produces a variety of heat-resistant enterotoxins causing staphylococcal food poisoning (SFP), which is one of the most prevalent food-borne intoxication diseases (1), and S. aureus is the fourth most common causative agent of food-borne illness in the European Union (7). Among the 21 recognized staphylococcal enterotoxins (SEs) or enterotoxin-like proteins (SEls), the bacteriophage-encoded staphylococcal enterotoxin A (SEA) (2) is the toxin most frequently reported to cause SFP in many countries worldwide, being detected in more than 50% of outbreaks (10, 21, 27). Differences in genetic backbones and regulatory systems of SE and SEl genes lead to diverse expression patterns and levels of toxins produced. The level of SEs produced may vary from a few nanograms to milligrams per milliliter of broth culture, depending on the bacterial strain and the SE type (6, 17). SEs usually accumulate extracellularly during bacterial growth in rich liquid broth media, up to the early stationary growth phase (6, 17, 25).
While the production of most SEs is regulated to some extent by the accessory gene regulator, the staphylococcal accessory regulator, and the repressor of toxin, the regulation mechanism of SEA is at least partially linked to the life cycle of the Siphoviridae family of temperate bacteriophages, which harbor the sea virulence region (2, 6, 8, 22, 23). This virulence region was recently shown to be connected to a distinct integrase group of Siphoviridae, Sa3int, associated with nasal isolates and other colonizing S. aureus strains (8). Using Northern blot analysis, Sumby and Waldor (22) demonstrated that activation of the prophage ΦSa3ms led to increased levels of mRNAs encoding SEA, SElG, SElK, and the fibrinolytic enzyme staphylokinase. In addition, we recently demonstrated that the expression and formation of SEA were enhanced by activating ΦMu50A in S. aureus Mu50 (25). Using acetic acid, a common preservative in the food industry, it was found that increased transcription of sea was correlated with larger numbers of intracellular sea gene copies and higher extracellular sea-containing phage titers. Using enzyme-linked immunosorbent assay (ELISA) to quantify the specific extracellular SEA levels, experiments with mitomycin C (MC), a known prophage inducer, confirmed that phage-mediated replication was responsible for the increased release of SEA. Furthermore, Borst and Betley (3) suggested that genetic differences in the prophage region upstream of sea may explain the variations in SEA level observed in S. aureus. Recently, genetic comparison of six completely sequenced enterotoxigenic strains showed the presence of two main versions of the sea gene, sea1 and sea2 (3, 25). In addition, sea genes with a few nucleotide differences have been reported (3, 9, 12).
The objectives of the present study were to systematically investigate the amounts of SEA produced by different S. aureus strains harboring sea, the genetic diversity of sea genes in relation to the levels of SEA produced, and the relationship between sea expression and the life cycle of the sea-encoding prophage. Twenty-one S. aureus strains, harboring sea1 or sea2 and including sequenced (whole genome) strains and food isolates, were investigated. Differences in the amounts of SEA produced by the different strains are discussed in relation to the genetic backbone of sea.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The S. aureus strains used in this study are listed in Table 1. Mu50 (LGC Promochem, London, United Kingdom), MW2 (donated from T. Baba, Juntendo University, Tokyo, Japan), Newman (donated by H. Ingmer, Copenhagen University, Copenhagen, Denmark), and MRSA252 (LGC Standards, London, United Kingdom) are completely sequenced. The remaining strains (donated by the Swedish Institute for Food and Biotechnology, SIK, Göteborg, Sweden), i.e., Sa17, Sa21, Sa41, Sa43, Sa44, Sa45, Sa46, Sa47, Sa48, Sa49, Sa51, Sa52, Sa53, Sa54, Sa55, Sa56, and Sa113, are food isolates. Four receiver strains, S. aureus RN27, RN450, RN451, and C104, were donated by J. R. Penades, Instituto Valenciano de Investigaciones Agrarias, Castellon, Spain. In order to obtain consistency and allow comparisons with previous data (9), all cultivation was performed in brain heart infusion (BHI) broth (Difco Laboratories, BD Diagnostic Systems, Le Point de Claix, France) or on BHI agar at 37°C. S. aureus was transferred from a glycerol stock to BHI agar for 1 day of cultivation, after which one colony was transferred to 15 ml of broth for overnight culture prior to the experiments. Broth was inoculated with a sufficient volume of S. aureus for overnight culture to give an optical density at 620 nm (OD620) of 0.1 at the start of cultivation. Batch cultivations were then performed in flasks on a shaker at 200 rpm (Innova 40/40R incubator shaker; New Brunswick Scientific). MC (Duchefa Biochemie, Haarlem, The Netherlands) was used at a concentration of 0.5 μg ml−1. Samples for quantitative reverse transcription-PCR (qRT-PCR) analysis, ELISA analysis, and PFU analysis were collected at a few time points, generally after 3 h, 6 h, and 24 h of growth, and each sample was analyzed three times. At least two independent cultivation replicates were performed using new culture medium and analytical reagents in order to confirm the results. Bacterial growth was monitored by measuring the OD620. All OD measurements were performed using a model U-1800 spectrophotometer (Hitachi High Technologies Inc., Pleasanton, CA).
Table 1.
SEA production in BHI broth without and with MC added after 3 h of growth
| Straina | SEA level (ng/ml) in BHI broth without MCb |
SEA level (ng/ml) in BHI broth with MCb |
Presence of sea genec |
||||
|---|---|---|---|---|---|---|---|
| 3 h | 6 h | 24 h | 6 h | 24 h | sea1 | sea2 | |
| Mu50 | 750.82 ± 166.87hA | 5,814.90 ± 458.61hC | 10,378.59 ± 1,842.13fD | 2,352.47 ± 547.11abB | 5,228.81 ± 935.77bcC | + | |
| Sa55 | 1,085.12 ± 142.77jA | 3,978.98 ± 207.22gB | 6,141.68 ± 529.34eC | 4,393.32 ± 699.72bB | 7,266.79 ± 1,839.55cdD | + | |
| Sa45 | 849.99 ± 59.16iA | 3,598.47 ± 404.40fB | 5,686.83 ± 416.37eC | 4,255.55 ± 263.70bB | 8,828.13 ± 1,379.18dD | + | |
| Sa17 | 633.03 ± 59.91gA | 3,108.70 ± 185.81eB | 4,671.77 ± 510.84dC | 7,403.17 ± 1,384.53cD | 13,650.30 ± 1,925.65eE | + | |
| Sa21 | 587.75 ± 64.04fgA | 3,544.74 ± 760.97fC | 4,900.44 ± 520.97dD | 1,735.17 ± 651.45aB | 3,649.56 ± 458.05abC | + | |
| Sa53 | 555.92 ± 50.97fA | 2,546.24 ± 209.22dA | 3,108.75 ± 197.63cA | 20,293.28 ± 4,193.16eB | 40,237.73 ± 5,415.26gC | + | |
| Sa43 | 355.27 ± 15.24cdA | 1,227.44 ± 107.09bcA | 2,699.43 ± 257.34cAB | 6,521.26 ± 3,972.53cB | 13,862.77 ± 8,222.78eC | + | |
| Sa48 | 441.98 ± 75.61eA | 1,520.24 ± 719.06cA | 2,863.48 ± 234.81cA | 15,185.01 ± 5,639.82dB | 35,181.77 ± 6,567.39fC | + | |
| Sa41 | 403.57 ± 85.68deA | 1,029.69 ± 83.43bAB | 1,940.13 ± 432.02bC | 1,460.00 ± 293.68aBC | 4,262.42 ± 1,494.25bcD | + | |
| MW2 | 287.33 0 ± 86.33cA | 947.72 ± 100.68bB | 1,470.25 ± 417.41bB | 1,301.72 ± 72.79aB | 3,108.66 ± 857.61abC | + | |
| MRSA252 | 133.40 ± 30.11bA | 340.80 ± 24.05aB | 473.31 ± 112.86aC | 280.90 ± 84.82aB | 535.46 ± 168.71aC | + | |
| Newman | 114.42 ± 16.39bA | 287.56 ± 39.24aC | 421.01 ± 69.18aD | 165.82 ± 9.98aB | 210.57 ± 47.23aB | + | |
| Sa56 | 0.38 ± 0.22aA | 4.01 ± 3.55aAB | 8.35 ± 8.25aAB | 6.52 ± 6.52aAB | 12.36 ± 12.47aB | + | |
| Sa54 | 0.49 ± 0.47aA | 2.50 ± 2.60aA | 4.58 ± 4.69aA | 2.42 ± 2.64aA | 4.39 ± 5.28aA | + | |
| Sa47 | 0.32 ± 0.12aA | 2.00 ± 0.78aA | 5.48 ± 2.37aB | 1.94 ± 0.77aA | 8.15 ± 2.86aC | + | |
| Sa52 | 0.53 ± 0.19aA | 2.42 ± 1.06aB | 2.63 ± 1.15aB | 1.76 ± 0.77aAB | 4.41 ± 2.34aC | + | |
| Sa51 | 0.37 ± 0.04aA | 0.72 ± 0.40aA | 0.98 ± 0.83aA | 0.97 ± 0.40aA | 1.17 ± 0.96aA | + | |
| Sa113 | 0.15 ± 0.08aA | 0.47 ± 0.45aAB | 0.58 ± 0.54aAB | 0.84 ± 0.68aAB | 1.12 ± 0.86aB | + | |
| Sa49 | 0.10 ± 0.05aA | 0.41 ± 0.27aB | 0.51 ± 0.34aBC | 0.40 ± 0.26aB | 0.71 ± 0.04aC | + | |
| Sa44 | 0.17 ± 0.06aA | 0.43 ± 0.09aB | 0.65 ± 0.17aC | 0.38 ± 0.04aB | 0.56 ± 0.04aC | + | |
| Sa46 | 0.09 ± 0.04aA | 0.34 ± 0.28aA | 0.41 ± 0.31aAB | 0.66 ± 0.65aAB | 1.13 ± 1.06aB | + | |
The strains are listed according to level of SEA production after 24 h under favorable conditions at 37°C.
The amount of SEA was measured using six samples from each time point. SEA values in a column sharing a common lowercase letter or SEA values in a row sharing a common uppercase letter are not statistically different (P > 0.05).
Results from conventional PCR analysis of 21 S. aureus strains.
PCR mapping.
The PCR protocol used to map sequence differences between the two sea gene versions, i.e., sea1 and sea2, and accompanying DNA regions involved eight primer pairs and has been described previously (25).
sea expression analysis.
Primers and hybridization probes for the sea gene and the reference gene rrn were designed according to the method of Wallin-Carlquist et al. (25). The primers for the long sea-specific transcript were designed from the nucleotide sequence of the upstream region of sea and purchased from TIB Molbiol GmbH (Berlin, Germany). The forward primer was 5′-ATGAGTTGGGCAAGATGGTT-3′, and the reverse primer was 5′-GGACTTGTTGTCCACGTTAGG-3′. A fluorogenic TaqMan probe was obtained from TIB Molbiol GmbH, with the following sequence: 6-carboxyfluorescein (FAM)-CATACTGCAAGTGAAGTTGGGAAGTGT-BBQ.
Total RNA was extracted using phenol and chloroform as described by Lövenklev et al. (13), except that the RNA was resuspended in 100 μl RNA storage solution (Applied Biosystems, Foster City, CA). First-strand cDNA was synthesized in two separate reverse transcription assays using reverse primers specific to SEA, the reference gene 16S rRNA, and the long transcript described above, with 0.1 μg RNA in the reference gene assay and 0.5 μg RNA in the toxin gene assay.
Real-time PCR amplification was carried out on a LightCycler 2.0 instrument (Roche Diagnostics GmbH). The total volume of each PCR mixture was 20 μl, including 4 μl of template cDNA. The sea and rrn PCR conditions were the same as those described previously (25). The PCR assay for the long transcript was the same as the sea and rrn PCR assays, apart from the use of a TaqMan probe instead of hybridization probes. All reagents except the primers and probes were obtained from Roche Diagnostics GmbH. The water used was autoclaved ultrapure water. In order to detect the amplification of possible contaminants, a negative control consisting of water instead of DNA was included in the PCR analysis. Genomic DNA was used as a positive control. The following PCR protocol was used: initial denaturation at 95°C for 1 min followed by 45 cycles of denaturation at 95°C for 0 s (i.e., no hold at 95°C), primer annealing at 46°C (sea), 48°C (rrn), or 51°C (long sea transcript) for 5 s, and extension at 72°C for 25 s. A single fluorescence measurement was made at the end of the extension step for protocols using hybridization probes; for the protocol using the TaqMan probe, the measurement was made at the end of the annealing step. The crossing-point cycle for each transcript was determined using the second derivative maximum model in LightCycler software (ver. 3.5) (Roche Diagnostics GmbH), and the amplification efficiency in the exponential phase was calculated using the equation of Klein et al. (11).
The relative expression of the sea gene and the long sea transcript was calculated by relating the toxin gene expression to the constant expression of a reference gene, the 16S rRNA gene (18). To determine the amplification efficiency and the log-linear range of amplification for each real-time PCR assay, the total RNA was serially diluted. The dilutions were reverse transcribed and amplified in a LightCycler instrument three times to obtain standard curves. Samples were also amplified three times. Equal amounts of total RNA from each sample were reverse transcribed to quantify the transcript levels of sea and the long sea transcript. The relative expression levels were calculated from the amplification efficiency of each PCR assay and the crossing-point deviation of the unknown sample versus a calibration sample, as described previously (18).
ELISA.
An ELISA protocol (14, 25, 26) was used to quantify the extracellular concentration of SEA, using affinity-purified polyclonal antibodies from sheep (Toxin Technology, Inc., Sarasota, FL).
Statistical analysis.
SEA levels in all six independent samples for each strain and incubation time (with or without MC) were evaluated using a 21 × 5 (strain × incubation time) factorial design. Individual fixed effects and up to two-way interactions were evaluated with analysis of variance (ANOVA), using the model y = x1 + x2 + x1x2 in the GLM procedures of SPSS 10.0.1 for Windows, where x1 represents the strain and x2 represents the incubation time. Least-squares means were separated by post hoc comparisons using the Duncan test. All differences were reported at a significance level (alpha) of 0.05.
Phage plaque assay.
Potential lysogenic phages were induced using a protocol described by Resch et al. (15). Briefly, bacterial cells were grown at 37°C in BHI medium until the late exponential phase. MC was added to the culture to a final concentration of 0.5 μg ml−1, followed by further incubation for 30 min. The cells were resuspended in fresh BHI medium, incubated again for 4 h at 37°C, and thereafter washed once with BHI medium. The cultures were centrifuged and filtered through a 0.2-μm-pore-size filter to remove cell debris, and the resultant supernatants were spotted onto agar and the plates incubated overnight (15). Fresh cultures of four receiver strains, S. aureus RN27, RN450, RN451, and C104, were prepared as described above. Receiver strains were grown to the exponential phase and then diluted to an OD620 of 0.1 and spread on BHI agar (15% agar with 4 mM Ca2+). Ten microliters of each phage supernatant was dropped onto a lawn of a receiver strain. The production of lytic spots was assessed after 24 h of incubation at 37°C. Phage DNA for PCR analysis was purified using zinc chloride as previously described by Santos (20), without previous DNase or RNase treatments.
PFU determination.
The receiver strain RN450 was used for PFU determination. S. aureus Sa17 and Sa51 were cultivated in 200 ml BHI medium, and after 3 h of growth, the cultures were divided equally between two flasks, one of which received MC. Samples were taken after 3 h, 4 h, 6 h, and 8 h of incubation. The receiver cultures were diluted to an OD620 of 0.1 (around 5 × 107 cells ml−1). Diluted phage supernatant suspension (dilution range of 1 to 10−6) was mixed with 0.2 ml RN450 and 4 ml soft BHI agar (7.5% agar with 4 mM Ca2+); the mixture was incubated for 1 min and poured onto BHI agar (15% agar with 4 mM Ca2+). The number of PFU was counted after 1 day of incubation at 37°C (19).
RESULTS
High- and low-SEA-producing S. aureus strains.
Twenty-one S. aureus strains harboring sea, including 4 sequenced (whole genome) strains and 17 food isolates, were examined systematically during batch growth at 37°C to determine the levels of SEA produced. The results are given in Table 1, in which the strains are arranged according to SEA production. The extracellular SEA concentration was determined by ELISA after 3 h, 6 h, and 24 h of growth, using six samples from each time point. The average maximum specific growth rate (μmax) of the 21 S. aureus strains was 2.25 ± 0.28 h−1, with growth rates ranging from 1.52 to 2.70 h−1. Ten of the 21 enterotoxigenic strains, namely, Mu50, Sa55, Sa45, Sa17, Sa21, Sa53, Sa43, Sa48, Sa41, and MW2, produced large amounts of SEA, with levels of >1,000 ng/ml after 24 h of growth. Nine strains, namely, Sa56, Sa54, Sa47, Sa52, Sa51, Sa113, Sa49, Sa44, and Sa46, produced small amounts of SEA, with levels of <10 ng/ml after 24 h. The differences in the amounts of accumulated extracellular SEA between high- and low-SEA-producing strains were up to 4 log units. Two clinical type strains, MRSA252 and Newman, produced intermediate levels of SEA (roughly 450 ng/ml). More than half of the SEA was produced after 6 h of growth, indicating that sea expression is linked mainly to the growth phase when bacteria are cultivated under favorable batch conditions. PCR mapping was performed to determine which version of the sea gene, sea1 or sea2, was present. Interestingly, all 10 high-SEA-producing strains harbored sea1, and 7 of the 9 low-SEA-producing strains, as well as MRSA252 and Newman, harbored the sea2 version.
SEA gene expression pattern of Mu50.
Detailed studies of relative sea expression and SEA production were carried out at 37°C without pH control in order to determine the expression pattern of SEA during the whole growth period (Fig. 1). The strain Mu50, producing the largest amount of SEA, was batch cultivated 10 times independently, and samples for sea mRNA and SEA determinations were withdrawn every hour for the first 24 h and after 48 h of growth. The relative expression quantity of the sea transcript was determined by referring to the housekeeping gene encoding the 16S rRNA (25). As expected, the relative level of sea mRNA was found to increase in the exponential growth phase and to peak in the transition to the stationary growth phase. The sea mRNA level remained high for several hours and then decreased. After 24 h of growth, sea mRNA was hardly detectable. Extracellular SEA accumulated in the culture medium during early cultivation. As the relative sea mRNA concentration declined, the amount of SEA stopped increasing. Notably, the relative levels of sea mRNA varied between the cultivation replicates, reflecting batch-to-batch variations. The time difference between the build-up of sea mRNA and the extracellular build-up of SEA was approximately 6 h.
Fig 1.

Growth, SEA production, and relative sea expression of S. aureus Mu50. The values were obtained from 10 independent batch cultivations grown in duplicate (duplicates A [filled symbols and bars] and B [open symbols and bars]). Circles denote the average growth, expressed as ln OD620, and squares denote the average extracellular SEA levels of duplicates A and B. Bars show the average relative sea expression (RE).
Phage-activated sea expression.
In order to study whether the induction of the presumed sea-carrying prophage influenced the amount of SEA produced, MC was added to all 21 growing strains. The time for prophage induction was predicted to be in the middle of the exponential growth phase, during the build-up of sea mRNA, and MC was therefore added after 3 h of batch cultivation. Samples for ELISA were withdrawn after 6 h and 24 h of cultivation. The results are presented in Table 1. Eight of 21 strains, all harboring the sea1 gene, showed an increase in SEA formation after MC addition. In comparison to controls without MC, the increase in the food isolates was more than 12 times for Sa48 and Sa53, 5 times for Sa43, and 2 times for Sa17, Sa41, and MW2, while a slight but significant increase was seen in Sa45 and Sa55 after 24 h of cultivation. Two high-SEA-producing strains, Sa21 and Mu50, showed a slight decrease in absolute levels of accumulated extracellular SEA after 24 h compared to cultures without MC. In contrast, SEA levels were not significantly affected when MC was added to cultures of the low-SEA-producing strains.
To further investigate whether this increase in SEA levels in some enterotoxigenic strains was due to induction of the prophage by MC, the possibility of detecting two types of sea transcript (long and short) was investigated. A long transcript, associated with a presumed latent phage promoter located upstream of the endogenous sea promoter, as demonstrated by Sumby and Waldor (22), plus a short transcript, i.e., the sum of the short and long transcripts (23), was quantified using qRT-PCR. The results are given in Table 2. Overall, sea mRNA was detected in 12 strains, including all of the high-SEA-producing strains, MRSA252, and Newman. The relative levels of sea mRNA varied between the biological replicates of each sample, as also observed in Mu50 (Fig. 1). The long transcript was detected in 8 of the 12 strains containing measurable amounts of sea mRNA. These eight strains were all high-SEA-producing strains carrying sea1, showing an increase in SEA production after the addition of MC. No sea mRNA could be detected in the low-SEA-producing strains.
Table 2.
Relative expression of total and long sea transcripts without and with MC present during growtha
| Strain | Total transcript level |
Long transcript levelb | ||
|---|---|---|---|---|
| 3 h without MC | 6 h without MC | 6 h with MC | ||
| Mu50 | 0.37 ± 0.05 | 0.52 ± 0.25 | 0.0055 ± 0.00 | — |
| Sa55 | 0.54 ± 0.13 | 0.031 ± 0.00 | 1.26 ± 0.28 | 1.03 ± 0.14 |
| Sa45 | 0.57 ± 0.04 | 0.088 ± 0.02 | 19.40 ± 2.23 | 7.99 ± 0.99 |
| Sa17 | 0.20 ± 0.03 | 0.36 ± 0.09 | 121.65 ± 34.57 | 100.74 ± 15.15 |
| Sa21 | 0.23 ± 0.01 | 0.77 ± 0.06 | 0.23 ± 0.04 | — |
| Sa53 | 0.31 ± 0.08 | 0.097 ± 0.04 | 7.61 ± 0.80 | 12.01 ± 2.94 |
| Sa43 | 0.71 ± 0.05 | 0.12 ± 0.03 | 40.92 ± 6.63 | 21.35 ± 2.85 |
| Sa48 | 0.37 ± 0.03 | 0.049 ± 0.00 | 122.12 ± 27.27 | 94.37 ± 13.76 |
| Sa41 | 0.39 ± 0.03 | 0.067 ± 0.01 | 3.92 ± 1.42 | 3.89 ± 0.22 |
| MW2 | 0.29 ± 0.04 | 0.0036 ± 0.00 | 1.16 ± 0.38 | 0.97 ± 0.43 |
| MRSA252 | 0.14 ± 0.02 | 0.036 ± 0.01 | 0.20 ± 0.03 | — |
| Newman | 1.03 ± 0.13 | 0.0041 ± 0.00 | 0.025 ± 0.00 | — |
The amount of sea mRNA was too small to determine in the low-SEA-producing strains.
—, no detectable long transcript.
The number of viable phage particles, i.e., PFU, was investigated to explore the relationship between the formation of phages and sea expression. To investigate strains liberating bacteriophages, a conventional phage plaque assay was performed using four different receiver strains of S. aureus: RN27, RN450, RN451, and C104 (Table 3). Based on the previous results for both mRNA and toxin levels, two representative strains, Sa17 (a high-SEA-producing strain) and Sa51 (a low-SEA-producing strain), were selected. Both strains were cultivated in the presence and absence of MC in order to determine the relationship between PFU and sea expression (Fig. 2). MC was added after 3 h of cultivation. Phage numbers in supernatants were counted after 3 h, 4 h, and 6 h of growth. A pronounced increase in PFU was observed for Sa17 (from 7 × 103/ml to 8 × 106/ml), with a more modest increase for Sa51 (from 7 × 102/ml to 103/ml), within 3 h of MC addition. The presence of the sea gene in the phage released into the culture supernatant and subsequently isolated was confirmed by PCR. Sa17 showed an increase in both short and long sea1 mRNA levels in the cultures containing MC, which followed the PFU pattern. The long transcript was not detected in the Sa51 culture, as expected, nor was it possible to detect sea2 mRNA.
Table 3.
Receiver strains used in spot tests of 21 tested S. aureus strains
| Strain | Receiver strain(s) in spot testsa |
|---|---|
| Mu50 | — |
| Sa55 | RN450, RN451 |
| Sa45 | RN450, RN451 |
| Sa17 | RN27, RN450, RN451, C104 |
| Sa21 | RN450, RN451 |
| Sa53 | RN450, RN451 |
| Sa43 | — |
| Sa48 | RN450, RN451 |
| Sa41 | — |
| MW2 | — |
| MRSA252 | — |
| Newman | — |
| Sa56 | RN27, RN450, RN451 |
| Sa54 | — |
| Sa47 | — |
| Sa52 | C104 |
| Sa51 | RN27, RN450, RN451, C104 |
| Sa113 | RN27, RN450, RN451, C104 |
| Sa49 | — |
| Sa44 | — |
| Sa46 | — |
—, no receiver strains were observed.
Fig 2.

Comparison of relative expression, PFU, and growth (OD620) of S. aureus Sa17 (a) and PFU and growth of Sa51 (b) in noninduced and induced cultures. The relative expression of short and long sea transcripts, together with PFU, was analyzed at 3 h, 4 h, and 6 h of cultivation. Filled symbols denote the results for noninduced (without MC) cultures, and open symbols denote the results for induced (with MC) cultures. Solid lines show the relative sea expression levels, with circles representing the short transcript and triangles the long transcript. Dashed lines show PFU, and dotted lines show growth. No relative sea expression and long transcript levels were detectable in Sa51.
DISCUSSION
It has been shown for many bacterial species that bacteriophages contribute to controlling the expression of virulence and the evolution of pathogenesis. For example, increased virulence expression due to prophage induction has been proved for Panton-Valentine leukocidin in S. aureus (28), streptococcal pyrogenic exotoxin C and a DNase in Streptococcus pyogenes (5), and the Shiga toxins in Escherichia coli (16, 24). In this study, we showed that SEA formation is closely linked to the Siphoviridae family of temperate bacteriophages. Based solely on the SEA production level of each S. aureus strain, 19 of the 21 enterotoxigenic strains were distinctly identified as being either high-SEA-producing or low-SEA-producing strains (Table 1). The observation that enterotoxigenic strains may produce either high or low levels of SEA was first reported by Borst and Betley in 1994 (3), using Western blot analysis. They observed that 4 strains among the 10 investigated showed larger amounts of SEA than the other 6 strains. In addition, we previously divided SEA-producing strains into two groups based on genetic diversity analysis of the Siphoviridae prophage region harboring sea, with one group containing sea1 and the other containing sea2 (25). Figure 3 shows that both sea1 and sea2 share the common endogenous promoter region P1, characterized by Borst and Betley (4), apart from a number of nucleotide sequence differences. The sea1 group was thereafter divided into two lines, with one group representing transcriptional prophage-activated sea1 strains and the other group harboring the sea1-producing strains Mu50, Sa21, Sa54, and Sa56, lacking phage-activated sea transcription. These findings were also in agreement with the results in Tables 1 and 2.
Fig 3.
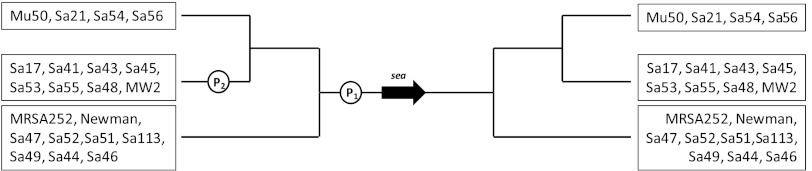
Assumed gene map of sea promoter regions in 21 S. aureus strains. P1 is the endogenous sea gene promoter, and P2 represents the latent promoter activated by phage induction in some sea1 strains. The black arrow indicates the sea gene. Nucleotide sequence analysis of the sea promoter region was performed on the sequenced (whole genome) S. aureus strains MRSA252 (GenBank accession no. BX571856), MSSA476 (GenBank accession no. BX571857), Mu3 (GenBank accession no. AP009324), Mu50 (GenBank accession no. BA000017), MW2 (GenBank accession no. BA000033), and Newman (GenBank accession no. AP009315).
Sumby and Waldor (22) showed that there is a linkage between prophage induction and increased sea expression in S. aureus. They proved that induction of the prophage ΦSa3ms in strain MSSA476 resulted in two long phage-activated sea transcripts, of 3.0 kb and 6.8 kb, apart from a sea-specific 1.7-kb transcript originating from the endogenous promoter, P1. The two long sea transcripts seemed to be initiated from the same latent phage promoter but differed regarding termination. The 6.8-kb transcript was found to contain sak located downstream of sea, indicating a transcriptional readthrough of both virulence genes. These findings appear to confirm the assumption that sea expression may be controlled by at least two promoters: an endogenous sea promoter and a latent phage promoter that is repressed under lysogenic conditions. Interestingly, Borst and Betley (3) wrote upon discovering the endogenous promoter in 1994 that “it is conceivable that alternative promoter regions might be active under different physiological conditions.” We therefore tested this hypothesis by using MC induction conditions to analyze sea transcripts and SEA levels in 21 genetically different S. aureus clones.
Comparing the SEA levels in cultures with and without MC showed that 8 of the 10 high-SEA-producing strains (all except for Mu50 and Sa21) indeed exhibited increased SEA levels in the presence of MC, while none of the low- and intermediate-SEA-producing strains showed any increase. In addition, the eight inducible strains showed enhanced transcription of sea and the occurrence of long sea transcripts after the addition of MC. Remarkably, some high-SEA-producing strains showed increases in SEA levels of up to 10-fold, with a hundred times increase of sea mRNA. Based on the relative levels of sea mRNAs of both types of transcripts, the phage-activated transcription dominated sea expression in most of the eight strains (Table 2). In addition, MC prophage induction most likely boosts phage DNA replication, resulting in increased sea gene copy numbers, and contributes to some extent to increased sea expression. This was also demonstrated by Sumby and Waldor (22) by construction of a replication-deficient ΦSa3ms phage lacking the pronounced increase in sea transcription. Additional support for the sea gene dose effect was observed in a previous study using Mu50 (23). That study showed that acetic acid increased SEA expression and formation in Mu50 by activating the prophage ΦMu50A. However, the expected boost in SEA production accompanying the increased intracellular sea gene copy levels and extracellular sea-containing phage copy numbers was not observed. The absence of the long sea transcript in Mu50 observed in this study (Table 2) may explain the modest enhancement of SEA expression and formation in the previous study.
When the numbers of viable phage particles liberated from a high-SEA-producing strain, Sa17, and a low-SEA-producing strain, Sa51, were investigated, we found that the PFU increased by around 103 times in Sa17 and 1.5 times in Sa51 within 3 h after MC addition. This means that both types of prophages can be induced, but to different extents. The reason that one strain, Sa17, exhibited increased SEA formation with an apparent boost of phage-activated transcription, while the other did not, could be that in Sa51 a latent phage promoter (such as P2) able to read through the weakly expressed sea2 gene is either lacking or not activated. Furthermore, the high phage titer of Sa17, i.e., phage replication and cell lysis, will also contribute to the massive release of SEA. This means that sea expression is closely associated with the phage life cycle and the clonal lineage of Siphoviridae. It is unlikely to be a coincidence that all strains with phage-induced transcription belong to a specific group harboring sea1. Betley and Mekalanos (2) reported already in 1985 that the sea gene was carried by a family of prophages showing polymorphism. This polymorphism was suggested by Borst and Betley (3) to affect the amount of SEA produced by the bacterial strains carrying the prophages. Today, six different sea-carrying prophages have been sequenced: Φ252B, ΦMu3, ΦMu50A, ΦNM3, ΦSa3ms, and ΦSa3mw (for a recent review, see reference 21). They all carry the genes for SEA, staphylokinase, and the complement inhibitor. The integration of sea-carrying prophages into the S. aureus genome mediates negative conversion of the β-hemolysin gene, a gene required for lysis of red blood cells. Recently, it was shown that these prophages are connected to a distinct integrase group of Siphoviridae, Sa3int, associated with nasal isolates and other colonizing S. aureus strains (8).
Overall, these data highlight the significance of connecting variations in SEA formation with the biology of the different Sa3int bacteriophages. By knowing which SEA-encoding prophage an S. aureus strain harbors, risk assessment and predictive microbiology to control SFP in foods may be improved. The finding that the formation of SEA is partially regulated by stress-regulated prophage induction in S. aureus addresses the importance of studying the behavior of enterotoxigenic bacteria in food environments. Current data have shown that there are significant differences in the behavior of S. aureus enterotoxin expression and formation in food compared to planktonic bacteria in cultivation broth (26). Instead of a short time of growth-associated enterotoxin expression, prolonged expression and formation of SEA were observed in processed pork meat, indicating a more complex behavior of phage-mediated sea expression in food.
ACKNOWLEDGMENT
We acknowledge the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS) for financial support.
Footnotes
Published ahead of print 27 April 2012
REFERENCES
- 1.Bergdoll MS. 1989. Staphylococcus aureus, p 463– 523 In Doyle MP. (ed), Foodborne bacterial pathogens. Marcel Dekker Inc, New York, NY. [Google Scholar]
- 2.Betley MJ, Mekalanos JJ. 1985. Staphylococcal enterotoxin A is encoded by phage. Science 229: 185– 187 [DOI] [PubMed] [Google Scholar]
- 3.Borst DW, Betley MJ. 1994. Phage-associated differences in staphylococcal enterotoxin A gene (sea) expression correlate with sea allele class. Infect. Immun. 62: 113– 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borst DW, Betley MJ. 1994. Promoter analysis of the staphylococcal enterotoxin A gene. J. Biol. Chem. 269: 1883– 1888 [PubMed] [Google Scholar]
- 5.Broudy TB, Pancholi V, Fischetti VA. 2001. Induction of lysogenic bacteriophage and phage-associated toxin from group A streptococci during coculture with human pharyngeal cells. Infect. Immun. 69: 1440– 1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czop JK, Bergdoll MS. 1974. Staphylococcal enterotoxin synthesis during the exponential, transitional, and stationary growth phases. Infect. Immun. 9: 229– 235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eurosurveillance Editorial Team 2012. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2010. Euro Surveill. 17: 20113 doi:10.2903/j.efsa.2012.2597 [PubMed] [Google Scholar]
- 8.Goerke C, et al. 2009. Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 191: 3462– 3468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jang HJ, Nde C, Toghrol F, Bentley WE. 2008. Microarray analysis of toxicogenomic effects of ortho-phenylphenol in Staphylococcus aureus. BMC Genomics 9: 411 doi:10.1186/1471-2164-9-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kérouanton A, et al. 2007. Characterization of Staphylococcus aureus strains associated with food poisoning outbreaks in France. Int. J. Food Microbiol. 115: 369– 375 [DOI] [PubMed] [Google Scholar]
- 11.Klein D, et al. 1999. Proviral load determination of different feline immunodeficiency virus isolates using real-time polymerase chain reaction: influence of mismatches on quantification. Electrophoresis 20: 291– 299 [DOI] [PubMed] [Google Scholar]
- 12.Letertre C, Perelle S, Dilasser F, Fach P. 2003. A strategy based on 5′ nuclease multiplex PCR to detect enterotoxin genes sea to sej of Staphylococcus aureus. Mol. Cell. Probes 17: 227– 235 [DOI] [PubMed] [Google Scholar]
- 13.Lövenklev M, Holst E, Borch E, Rådström P. 2004. Relative neurotoxin gene expression in Clostridium botulinum type B, determined using quantitative reverse transcription-PCR. Appl. Environ. Microbiol. 70: 2919– 2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marta D, Wallin-Carlquist N, Schelin J, Borch E, Rådström P. 2011. Extended staphylococcal enterotoxin D expression in ham products. Food Microbiol. 28: 617– 620 [DOI] [PubMed] [Google Scholar]
- 15.Matsuzaki S, et al. 2003. Experimental protection of mice against lethal Staphylococcus aureus infection by novel bacteriophage phi MR11. J. Infect. Dis. 187: 613– 624 [DOI] [PubMed] [Google Scholar]
- 16.Mühldorfer I, et al. 1996. Regulation of the Shiga-like toxin II operon in Escherichia coli. Infect. Immun. 64: 495– 502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otero A, Garcia ML, Garcia MC, Moreno B, Bergdoll MS. 1990. Production of staphylococcal enterotoxins C1 and C2 and thermonuclease throughout the growth cycle. Appl. Environ. Microbiol. 56: 555– 559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29: e45 doi:10.1903/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Resch A, Fehrenbacher B, Eisele K, Schaller M, Götz F. 2005. Phage release from biofilm and planktonic Staphylococcus aureus cells. FEMS Microbiol. Lett. 252: 89– 96 [DOI] [PubMed] [Google Scholar]
- 20.Santos MA. 1991. An improved method for the small scale preparation of bacteriophage DNA based on phage precipitation by zinc chloride. Nucleic Acids Res. 19: 5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schelin J, et al. 2011. The formation of Staphylococcus aureus enterotoxin in food environments and advances in risk assessment. Virulence 2: 580– 592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sumby P, Waldor MK. 2003. Transcription of the toxin genes present within the staphylococcal phage phiSa3ms is intimately linked with the phage's life cycle. J. Bacteriol. 185: 6841– 6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tremaine MT, Brockman DK, Betley MJ. 1993. Staphylococcal enterotoxin A gene (sea) expression is not affected by the accessory gene regulator (agr). Infect. Immun. 61: 356– 359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner PL, et al. 2001. Role for a phage promoter in Shiga toxin 2 expression from a pathogenic Escherichia coli strain. J. Bacteriol. 183: 2081– 2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallin-Carlquist N, et al. 2010. Acetic acid increases the phage-encoded enterotoxin A expression in Staphylococcus aureus. BMC Microbiol. 10: 147 doi:10.1186/1471-2180-10-147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wallin-Carlquist N, Marta D, Borch E, Rådström P. 2010. Prolonged expression and production of Staphylococcus aureus enterotoxin A in processed pork meat. Int. J. Food Microbiol. 141 (Suppl 1): S69– S74 [DOI] [PubMed] [Google Scholar]
- 27.Wieneke AA, Roberts D, Gilbert RJ. 1993. Staphylococcal food poisoning in the United Kingdom, 1969–90. Epidemiol. Infect. 110: 519– 531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wirtz C, Witte W, Wolz C, Goerke C. 2009. Transcription of the phage-encoded Panton-Valentine leukocidin of Staphylococcus aureus is dependent on the phage life-cycle and on the host background. Microbiology 155: 3491– 3499 [DOI] [PubMed] [Google Scholar]