

Abstract
The NIH Human Microbiome Project (HMP) has produced several hundred metagenomic data sets, allowing studies of the many functional elements in human-associated microbial communities. Here, we survey the distribution of oral spirochetes implicated in dental diseases in normal human individuals, using recombination sites associated with the chromosomal integron in Treponema genomes, taking advantage of the multiple copies of the integron recombination sites (repeats) in the genomes, and using a targeted assembly approach that we have developed. We find that integron-containing Treponema species are present in ∼80% of the normal human subjects included in the HMP. Further, we are able to de novo assemble the integron gene cassettes using our constrained assembly approach, which employs a unique application of the de Bruijn graph assembly information; most of these cassette genes were not assembled in whole-metagenome assemblies and could not be identified by mapping sequencing reads onto the known reference Treponema genomes due to the dynamic nature of integron gene cassettes. Our study significantly enriches the gene pool known to be carried by Treponema chromosomal integrons, totaling 826 (598 97% nonredundant) genes. We characterize the functions of these gene cassettes: many of these genes have unknown functions. The integron gene cassette arrays found in the human microbiome are extraordinarily dynamic, with different microbial communities sharing only a small number of common genes.
INTRODUCTION
Integrons are genetic elements that acquire and excise gene cassettes from their locus via site-specific recombination. The type of integron first discovered in the 1980s as the source of antibiotic resistance determinants (37) has been named the resistance integron, or the mobile integron, as it is often found in plasmids or associated with transposons. Another type of integron, the chromosomal integron, was discovered in 1998 from examination of the Vibrio cholerae genome (22). Although they have similar structures, the two types of integrons (mobile integrons and chromosomal integrons) have different evolutionary histories and differ in that the mobile integrons usually carry relatively few genes, which are predominantly antibiotic resistance genes, while chromosomal integrons often carry far more genes, of very diverse functions (4). Chromosomal integrons are the ancestors of resistance integrons (34).
Integrons consist of a site-specific tyrosine recombinase (intI) gene, the primary recombination site attI immediately adjacent to the intI gene, and an array of captured gene cassettes encoding accessory functions (5). Gene cassettes are the minimal units that can be mobilized by the integrase, with each cassette containing one or a very small number of genes (6), and are separated by a recombination site, attC. Aggregation of different gene cassettes results in variable gene cassette arrays. The number of gene cassettes in integrons can reach several hundred; for example, the total length of the gene cassette pool from five Vibrio chromosomal integrons is equivalent to a small genome (34).
PCR with degenerate primers targeting the conserved regions of attC sites has recovered novel integrase genes and hundreds of diverse gene cassettes from various environments, including soil, sediment, biomass, and water habitats (24, 33, 38). Rowe-Magnus et al. employed a three-plasmid genetic strategy to recover integron genes, using the integrase to bind integron attC sites (34). These methods, which utilized the conserved nature of integron recombination sites, revealed a very dynamic integron gene repertoire and suggested that the gene cassette pool is likely to be limitless (7), while at the same time we do not know of work identifying the sources of integron genes.
Here, we report a different approach to discovering chromosomal integrons in human-associated microbial communities, using shotgun metagenomic sequences of the human microbiomes. Human bodies are complex ecological systems, in which various microbial organisms and viruses interact with each other and with human hosts. The MetaHit project has established a human gut microbial gene catalogue (29) and defined three enterotypes of human gut microbiomes (2). The Human Microbiome Project (HMP) (25) has resulted in >700 data sets of shotgun metagenomic sequence (http://www.hmpdacc.org/), from which we can learn the compositions and functions of human-associated microbial communities.
Our approach to integron discovery builds upon two novel computational methods: a targeted assembly approach for identifying the attC sites associated with chromosomal integrons (the repeats) in reads and a constrained assembly approach for identifying the gene cassettes, which first avidly retrieves potential paths in the de Bruijn graph (8, 26) for a metagenomic data set, constrained to contigs containing the attC sites, and then selects the paths that most likely represent cassette genes, based on several criteria (see Materials and Methods). As we show in Results, such specialized computational tools are important for a comprehensive characterization of metagenomic functional elements that contain repeats (such as the attC sites in the integron gene cassettes), as these repetitive regions are extremely difficult to assemble using a whole-metagenome assembly strategy.
In this study, we focus on the identification and characterization of integrons associated with Treponema species implicated in periodontal disease (9, 36) in the HMP data sets, using our integron discovery system. The Treponema denticola genome contains a chromosomal integron with 45 gene cassettes (6), and it is the only human-associated bacterial species that harbors chromosomal integrons (5). We also discover that the draft assemblies of two HMP reference genomes, Treponema vincentii and Treponema phagedenis (http://www.hmpdacc.org/HMRGD/), contain integron attC sites similar to those in T. denticola and possibly harbor integrons. We do not find integrons in other Treponema species, including T. pallidum SS14 uid58977 (21), T. pallidum Nichols uid57585 (12), T. primitia ZAS-2, and T. azotonutricium ZAS-9 (13). From the HMP data sets, we identify 826 integron gene cassettes that are related to the Treponema species, providing a gene cassette pool with 598 nonredundant genes. With these newly identified gene cassettes, we are able to compare the gene cassettes from different human subjects and study the dynamics of the integron gene cassettes in their natural environments (i.e., human bodies), providing a first survey of integron-containing Treponema species and their integrons in a normal human population.
MATERIALS AND METHODS
Choosing representative repeat sequences for Treponema chromosomal integrons.
Eight distinct sequences were selected to represent the integron attC repeats in the T. denticola genome (the complete genomes of the other two integron-containing Treponema species, T. vincentii and T. phagedenis, are not available), given that not all the repeats are identical (Fig. 1). The pairwise sequence similarity between these eight sequences ranges from 77% to 44%, and all the attC sites in T. denticola can be aligned with at least one of the representative sequences with >85% sequence identity. Once the representative sequences are selected, we are able to identify new attC sites using similarity searches, instead of looking for features of integron recombination sites as in reference 39. One advantage of using similarity searches is that we can recover degenerate sites that may lack some typical characteristics of integron recombination sites.
Fig 1.

(A) The neighbor-joining tree of the eight representative sequences of the T. denticola chromosomal integron recombination sites. The sequences are named by the starting position of the sites in the genome. The multiple alignment was prepared using ClustalW, and the neighbor-joining tree was prepared using the jalview tool. (B) Predicted structure of one of the representative sequences, attC1870410, which has the typical structure of an integron recombination site, with two stems and one conserved unpaired G. The structure was predicted by RNAscf (3), software that performs simultaneous alignment and folding of RNAs, using the eight representative sequences as input.
Targeted assembly to identify integron attC sites.
The targeted assembly approach was developed to characterize CRISPR arrays from shotgun metagenomic sequences (31) and was employed here to identify and assemble the integron attC sites. The steps were (i) searching for reads that contain attC sites (with identity of >70% and covering >50% of at least one of the representative attC sequences) using BLAST (1); for paired-end reads, if one of a pair qualifies, both reads for the pair are included; and (ii) assembling the retrieved short reads using SOAPdenovo (18); note that we used k-mers of 31 bp, which were sufficiently long to assemble reads with the repetitive sequences found in the integrons; by contrast, whole-metagenome assembly generally uses shorter k-mers (29).
Constrained assembly to retrieve integron gene cassettes.
A second method, constrained assembly, was used to assemble integron gene cassettes from metagenomic shotgun reads. Since integron cassettes consist of genes that are much longer than the read length (∼100 bp for the current Illumina technology), and the attC sites behave like repeats that confuse (meta-)genome assemblers, it is extremely difficult to obtain gene sequences using either a whole-genome assembly method or the targeted-assembly approach (which is good for assembly of repeats but does not assemble very far beyond the repeats). As the integron cassettes are bounded by two attC repeats, we took advantage of this structure and devised a novel way to retrieve the cassette genes by traversing the assembly graph, constrained by the edges (contigs) that contain the attC sites. To avoid introducing artificial integron genes, we further applied several criteria to select paths that are most likely to present genuine gene cassettes. The constrained assembly approach consists of the following steps (Fig. 2): (i) assembling all shotgun reads in a metagenomic sequence data set—along with the contigs constructed by the targeted assembly approach, which may contain more complete attC sites than do shotgun reads—using SOAPdenovo (18) with k = 39 (see below for the selection of k-mer parameter), producing both contigs and the assembly graph (a de Bruijn graph) (26) (Fig. 2B); (ii) searching for attC sites in contigs using BLAST (with an identity threshold of 70% and coverage threshold of 50%) and tagging contigs with attC sites to be used as constraints to constrain the next step; (iii) extracting paths that start from one tagged contig and end at another tagged contig using a depth-first search algorithm and assembling the sequences for each path; the maximum length from one integron attC site to another attC site is set to 5,000 bp (Fig. 2C); (iv) checking the support of each assembled sequence by mapping the reads and read pairs onto the assembled sequences using BWA (17); we consider that a traverse between two contigs is valid if the flanking regions of the connection (of l bp at both sides; l is set to 15) are supported by at least one read or read pair, and an assembled sequence is considered to be supported only if all the traverses involved are supported by reads (Fig. 2D); (v) predicting the genes in each assembled sequence using FragGeneScan (30), with error model turned off; we require that the maximum gene number between any two integron attC sites be 3, considering that most integron cassettes contain 1 to 3 genes (6) (Fig. 2E).
Fig 2.
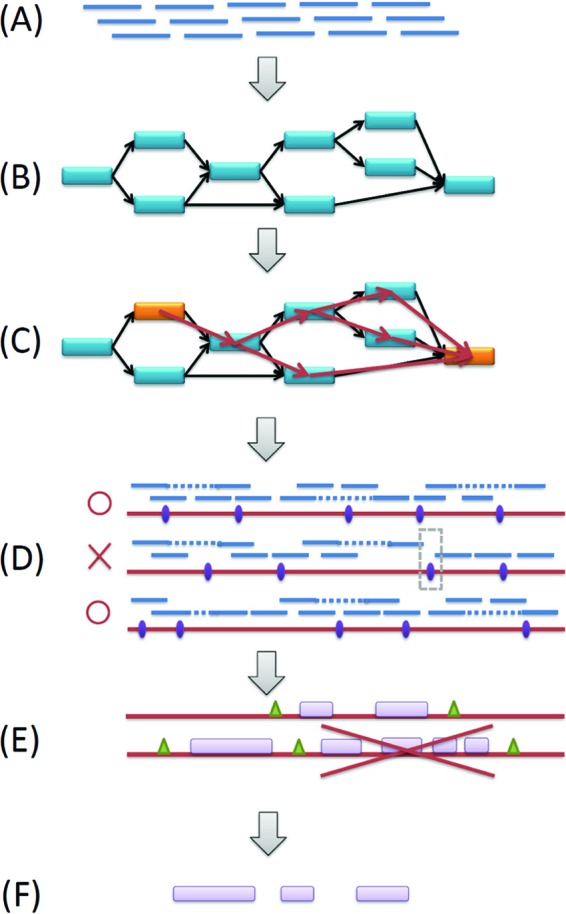
A diagram of the constrained assembly approach. (A) Paired-end and singleton reads from a metagenomic data set. (B) Assembly of all reads using SOAPdenovo, to generate contigs and a de Bruijn graph that connects the contigs. (C) Identification of contigs that consist of integron recombination repeats (shown as orange bars) and search for paths that start and end at a contig with repeats, using a depth-first search algorithm. At any intermediate node, the process will sort the coverage of all contigs connected by its outgoing edges and begin searching from the highest one. The starting and ending contig could be the same contig. (D) Validation of the assembled sequences (the paths) by read mapping and discarding of the paths that are not supported by reads (e.g., the middle sequence in the figure is discarded). (E) Identification of the integron repeats and their exact locations in the assembled sequences. Prediction of genes using FragGeneScan. Output sequences are between two repeats (attC sites) and consist of three or fewer genes. (F) Retrieval of the genes from sequences that pass all criteria.
Validation of constrained assembly using simulation.
We simulated three metagenomic data sets by sampling reads at different coverage (10×, 20×, and 31×) from nine Treponema genomes (or genome drafts) using MetaSim (32) with the Illumina 80-bp error model with an error rate of ∼1% provided by the authors (http://ab.inf.uni-tuebingen.de/software/metasim/errormodel-80bp.mconf). The species include T. denticola ATTC 35405 (NC_002967), T. azotonutricium ZAS-9 (NC_015577), T. primitia ZAS-2 (NC_015578), Treponema pallidum subsp. pallidum SS14 (NC_010741), T. pallidum subsp. pallidum strain Nichols (NC_000919), T. succinifaciens DSM 2489 (NC_015385), T. denticola strain F0402 (downloaded from http://www.broadinstitute.org/), T. vincentii (http://hmpdacc.org), and T. phagedenis (http://hmpdacc.org). We tested different k-mer parameters for the constrained assembly approach using these simulated data sets, and the results show that k = 39 resulted in the most integron genes for all the data sets (Fig. 3). The 31× data set contains 4,499,532 paired-end reads and 500,468 singleton reads. Seventy-three integron genes were identified from this data set by our constrained assembly approach: 37 genes from T. denticola ATCC 35405, 27 genes from T. denticola strain F0402, seven genes from T. vincentii, and two genes from T. phagedenis. We mapped these genes back to the genomes and confirmed that (i) all the genes were correctly assembled (error rate is 0%) and (ii) all the genes were mapped to the big integron or the degenerate, small integron region in the genomes (see Results). In addition, we did not find any genes in the Treponema species that do not harbor integrons. All suggest that our constrained approach is reliable even when reads from closely related species are present.
Fig 3.

The number of integron genes discovered from simulated metagenomic data sets using different k-mer settings. The x axis lists the k-mers, while the y axis shows the total number of genes assembled. We generated three data sets with different coverages (10×, 20×, and 31×) and applied our constrained assembly method to these data sets. Lines indicate the gene numbers found, and dashed lines are the numbers of genes that were identified solely at the contig level (i.e., genes on the contigs that are bounded between two integron recombination sites).
Functional annotation of identified gene cassettes.
We downloaded all protein sequences from the eggNOG v2.0 database (23) and retrieved the sequences with COG annotation (40). MUSCLE (11) was used to generate a multiple alignment for each COG family, and the HMM builder from the HMMER3 package (10) was then applied to build an HMM for each COG. HMMER searches (by hmmscan from the HMMER3 package) were used to annotate the predicted integron gene cassettes, with an E value cutoff of 0.001. For a gene with COG hits, we recorded the best nonoverlapped results, so that if a gene encodes multiple domains with distinct functions, all the functions will be reported.
Identification of potential source species of gene cassettes.
We used MEGAN (15) to identify the possible source species of the identified gene cassettes. We searched the genes against the NCBI NR database (as of September 2011) using BLASTP and applied the MEGAN software to analyze the similarity search results. Since the average length of the genes is 506 bp, we set the minimum score threshold to 100, as suggested by MEGAN′s authors for longer reads.
HMP data sets.
We used the Human Microbiome Illumina WGS Reads (HMIGWS) Build 1.0 and the whole-metagenome assemblies (PGAs) from the HMP consortium (http://www.hmpdacc.org/). There are 757 total metagenomic samples from 103 subjects (individuals). The reference genomes were also downloaded from this website.
Availability of software and predicted integron genes.
We implemented the integron discovery pipeline in C++ and Perl. Software and predicted integron genes can be downloaded from our website http://omics.informatics.indiana.edu/integron.
RESULTS
The T. denticola integron attC sites are unique to Treponema species.
BLAST searches using the eight representative attC sequences against the NCBI nucleotide collection (NT) and the genome database (chromosomes) with default settings hit only Treponema genomes. Using an identity threshold of 70% and coverage threshold of 50%, 64 attC sites were found in the T. denticola ATCC 35405 genome, of which 45 are located within the chromosomal integron (positions 1817049 to 1874294) identified by reference 6. We also found two additional attC sites downstream of the integron region, suggesting that the integron may be even larger and contain more genes. The attC site located immediately downstream of the previously reported integron location is more degenerative (barely passes the coverage threshold), but the site further downstream is more complete, and we believe that these two attC sites are genuine. In addition, we found 7 attC sites outside the big integron region (for example, there is an attC site located between positions 300167 and 300227, which shares 98% sequential identity with the attC site within the integron array between positions 1870410 and 1870474). Furthermore, a degraded intI gene exists between positions 302289 and 302350, suggesting that a degraded, small integron may exist in this region of the genome. We also discovered integron sequences in T. denticola F0402 (sequence downloaded from http://www.broadinstitute.org/). While the integrase genes (intI) are very similar between these two strains (with 95% identity), the integron gene cassettes are quite different—only 10 integron genes are shared between these two strains (see Fig. S1 in the supplemental material).
Instances of the T. denticola attC sites were also found in the draft assemblies of two human microbiome reference genomes (as of July 2011): T. vincentii ATCC 35580 and T. phagedenis F0421. A total of 16 attC sites were found in five contigs of T. vincentii ATCC 35580, and 6 attC sites were found in three contigs of T. phagedenis F0421. We further checked the T. vincentii and T. phagedenis genomes for features indicative of integrons. In both genomes, there are gene cassettes flanked by attC sequences: we identified one gene in a T. phagedenis contig and 12 genes from three contigs of T. vincentii. One of the T. vincentii contigs exhibits a very clear integron structure, as shown in Fig. S2 in the supplemental material. None of the 12 genes identified in T. vincentii share significant similarity with the integron genes of the T. denticola integron, suggesting that the gene cassettes of the two integron loci have undergone substantial changes since these two species diverged. We also searched the T. vincentii and T. phagedenis genomes using the T. denticola intI gene and detected a significant (sequence similarity = 86%) and long(953-bp) intI gene on the T. vincentii contig ACYH1000073, which is shown in Fig. S2. Together with the recombination sites and the gene cassettes, this region contains all elements required for an integron.
Integron-containing Treponema species are found in 80% of a normal human population, using searches for integron attC sites.
We identified integron attC sites in 300 of >700 HMP samples, using targeted assembly. The body sites that have identified integrons are summarized in Table 1. Most samples with integrons are orally related (including hard palate, supragingival plaque, saliva, tongue dorsum, subgingival plaque, throat, buccal mucosa, and attached/keratinized gingiva sites), whereas non-oral samples, including stool and vaginal samples, do not contain integron attC sites (repeats). This evidence suggests that a high proportion of oral samples contain the Treponema species implicated in dental diseases, implying that these pathogens are ubiquitous among people. The existence of Treponema species implicated in dental diseases in most normal human individuals (though of low abundance) is also supported by mapping the sequencing reads onto the available complete genomes (or drafts) of the three integron-containing Treponema species (T. denticola, T. vincentii, and T. phagedenis) (see mapping results in Fig. S3 in the supplemental material). We found only rare samples from nose (anterior nares) and ear (retroauricular crease) with integron repeats.
Table 1.
Summary of the HMP samples with identified T. denticola integron attC sites
| Location | No. of samples with attC sites | Total no. of samples | % of samples with attC sites |
|---|---|---|---|
| Hard palate | 1 | 1 | 100 |
| Supragingival plaque | 98 | 128 | 77 |
| Saliva | 5 | 5 | 100 |
| Tongue dorsum | 109 | 136 | 80 |
| Vaginal introitus | 0 | 3 | 0 |
| Stool | 0 | 150 | 0 |
| Midvagina | 0 | 2 | 0 |
| Subgingival plaque | 8 | 8 | 100 |
| Throat | 6 | 7 | 86 |
| Posterior fornix | 0 | 62 | 0 |
| Anterior nares | 2 | 94 | 2 |
| Buccal mucosa | 60 | 122 | 49 |
| Right retroauricular crease | 2 | 18 | 11 |
| Left retroauricular crease | 0 | 9 | 0 |
| Palatine tonsils | 6 | 6 | 100 |
| Attached/keratinized gingiva | 3 | 6 | 50 |
The 300 samples containing attC sites resulted in 85 out of 103 individuals having an identified infection of Treponema species (82.5%; between 1 and 15 samples per individual). This number is consistent with a previous report that disease associated with T. denticola occurs in 80% of adults, at some time in their lives (36).
We checked the size of each oral sample (as measured by the total bases) and found that oral samples with identified integron attC sites are significantly larger than samples without attC sites (Welch's t test, Z = 4.63, degrees of freedom = 230, P < 0.001). This is expected, as the Treponema species implicated in dental disease are not abundant in oral sites of normal individuals (see Fig. S3 in the supplemental material) and will be difficult to detect when sequencing is shallow. Thus, the 80% prevalence may be a conservative estimate.
Integron gene cassettes identified in HMP whole-metagenome assemblies.
We first identified integrons in the contigs from the whole-metagenome assemblies of human metagenomes by looking for genes flanked by attC sites. Seven hundred forty-one attC sites were detected in the whole-metagenome assemblies, but most contigs carry only one attC site. As a result, we only found 66 nonredundant (at a 97% identity cutoff) genes from 25 samples: 17 are from supragingival plaque, six are from tongue dorsum, and two samples are from subgingival plaque. The sample distribution shows that we can indeed find integron genes associated with Treponema species (and hence demonstrate the existence of these oral pathogens) in mouth-related samples.
Figure 4 shows an example from contig SRS049318_LANL_scaffold_118938, with two attC sites at 176 to 226 and 817 to 877 bp. FragGeneScan predicted one protein-coding gene between the two sites, and a similarity search of this predicted protein against the NCBI NR database revealed similarity to a hypothetical protein in the T. denticola genome and to an HNH nuclease domain (superfamily ID, cl00083) (42). HNH endonuclease features 11 conserved residues, and all are conserved in the predicted protein.
Fig 4.
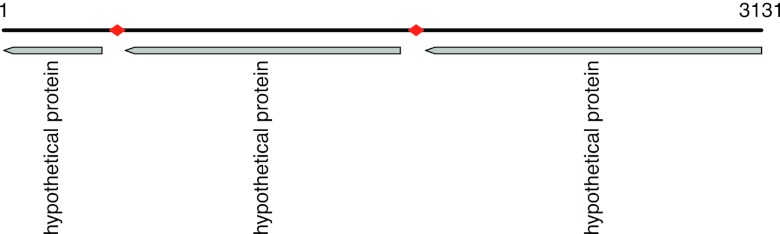
Annotation of a contig from sample SRS022602 (SRS022602_Baylor_scaffold_118781) of 3,131 bp. Red diamonds indicate the two repeats identified in this contig with similarity to the attC sites in the T. denticola chromosomal integron, and the three gray boxes indicate the predicted genes. The first gene (1–407) shares 46% sequence identity and 66% similarity along 97% of the gene with a protein (YP_001868417.1) from the Nostoc punctiforme PCC 73102 genome (a nitrogen-fixing cyanobacterium). The second gene (503–1639) shares 31% identity (53% similarity) along 99% of the gene with a protein (ADE86468.1) from Rhodobacter capsulatus SB 1003 (a purple, nonsulfur photosynthetic bacterium). The third gene (1743–3131) shares 24% identity and 45% similarity, covering 88% of the gene, with a protein (ZP_04160697.1) from Bacillus mycoides Rock3-17 (a Gram-positive, nonmotile soil bacterium); this gene also shares 24% sequence identity and 46% similarity (covering 65% of the gene) with a protein (YP_002158281.1, nuclease-related domain family protein, NERD) from Vibrio fischeri MJ11 (20).
Many more integron gene cassettes can be identified in the HMP data sets using constrained assembly.
Using the whole-metagenome assemblies, we were able to retrieve only 66 integron-associated genes (see above). Application of our constrained assembly approach (see Materials and Methods for details and a validation of the method using simulated data sets) to the HMP data sets led to the identification of 794 genes in 47 samples. After combining both predictions and keeping only unique genes for each sample, we derived a total of 826 unique genes (598 97% nonredundant). Table S1 in the supplemental material shows the comparison of the results by the constrained assembly approach results and the whole-metagenome assembly for individual HMP samples. The distribution of sample locations and the number of genes in each location are listed in Table 2. We identified genes in 24 supragingival plaque samples, 19 tongue dorsum samples, and 4 subgingival samples. The proportion of samples with gene cassettes identified using the constrained assembly approach is still low—compared with samples with identified attC sites (300)—due to the low abundance of the Treponema species in many samples (see Fig. S3). But we can still utilize the attC sites (taking advantage of the multiple copies of the attC sites) to identify T. denticola or related species in those samples, demonstrating the power of using unique repeats to trace rare species. We note that mapping shotgun sequences onto the known reference genomes (or drafts) of Treponema species can be used to identify the existence of these species in the HMP samples, but such a mapping cannot be effectively used to identify the integron gene cassettes due to the dynamic nature of the integron genes (e.g., the two T. denticola strains share only 10 cassette genes; see above).
Table 2.
Breakdown by body location of the samples that have identified Treponema chromosomal integron gene cassettes
| Body location | No. of samples | No. of genes | No. of genes without COG hits |
|---|---|---|---|
| Supragingival plaque | 24 | 457 | 252 |
| Tongue dorsum | 19 | 283 | 203 |
| Subgingival plaque | 4 | 86 | 46 |
Similarly, among the 300 samples with detected attC sites, the samples with gene cassettes assembled by constrained assembly were significantly larger than those with no identified genes (Welch's t test, Z = 4.42, degrees of freedom = 68, P < 0.001). This can also explain why we did not find gene cassettes in samples from buccal mucosa—the buccal mucosa samples are significantly smaller than other oral data sets (Welch's t test, Z = 25.28, degrees of freedom = 388, P ≪ 0.001), partially caused by a large contamination of human DNAs in the buccal mucosa samples.
Many cassettes are of unknown function.
We annotated the predicted cassette genes using similarity searches. Among the 826 genes, 501 cannot be assigned to a COG family (Table 2): ∼60% are unassigned. Of the remaining genes, ∼60% are assigned to COG categories R (general function prediction only) and S (function unknown). Combining these two categories, 85% of the 826 genes are of unknown function: the proportion is even higher than reported for other integrons (it was reported that 75% of the cassette pool associated with Vibrionales genomes corresponds to genes with undefined functions [4, 5]).
To analyze genes with identified functions, we clustered the genes within each location (at 97% identity) to see how many genes are unique to distinct locations. The functional category L (replication, recombination, and repair) is the majority among all functional categories (25%); genes associated with categories D (cell cycle control, cell division, and chromosome partitioning), K (transcription), N (cell motility), and T (signal transduction mechanisms) are also elevated among all functional categories, with 12%, 11%, 13%, and 13% of the genes with known functions, respectively (see Table S2 in the supplemental material). Integron genes with these functions have been reported previously: for example, category L and category T are among the most prevalent functions reported by reference 5. Genes in other categories, such as genes predicted to be part of the toxin/antitoxin system in category D, DNA-methyltransferase in category K, and methyl-accepting chemotaxis protein in category N, were also reported by reference 5. This again demonstrates that our results are consistent with the previous findings of gene functions encoded by chromosomal integrons.
We further compared the predicted genes found in the HMP data sets with the genes in the T. denticola chromosomal integron (located at positions 1817049 to 1874294 on NC_002967, as reported by reference 6) using BLAST with an E value cutoff of 0.001. We found that of the 826 genes, 192 (23%) hit to the genome's integron genes. We also found that of the 70 integron genes identified in the T. denticola genome, 39 (56%) genes had homologs in the 826 genes retrieved from the human samples. In other words, about 44% of integron genes in the complete genome were missing from our broad survey of human samples. This clearly demonstrates that the T. denticola integron is undergoing an active process of cassette insertion and excision.
Integron genes can be traced to a variety of sources.
To infer the potential origins of the integron gene cassettes associated with Treponema species, we applied MEGAN (15) to analyze all the gene cassettes identified in the HMP samples. The MEGAN taxonomic assignments of the gene cassettes are summarized in Fig. 5. A total of 365 (44%) genes cannot be assigned to any taxon. Among the genes (461) assigned to a taxon, 152 (18%) are assigned to T. denticola (at the species level), 47 (6%) genes are assigned to T. vincentii, and 262 genes likely originate from other species: 117 (14%) genes from other spirochete species and 145 (17%) genes from nonspirochete species. Table S3 in the supplemental material lists the candidate donor species, and the annotations of the potential donor genes in these species. Here, we show two examples: the first example is 14 genes assigned to the order Clostridiales, which were first discovered in soil but also appear in human microbiomes (14, 35), and the other example is 25 genes assigned to Spirochaeta caldaria, a thermophilic bacterium (27).
Fig 5.

Taxonomic assignments of the integron genes by MEGAN. The numbers following clade names are the numbers of genes assigned to that taxonomic rank, not including the genes assigned to the taxa below that rank (for example, there are 63 genes assigned to T. denticola species, 49 genes assigned to strain ATCC 35405, and 40 genes assigned to strain F0402; in total, 138 genes can be assigned to the T. denticola species).
Most integron genes are unique to samples and individuals.
In order to characterize the cassette genes shared among different samples, we clustered genes from different samples using CD-HIT (19), with an identity cutoff of 70% at the amino acid level, and then mapped the clustered genes to samples. Figure 6 clearly shows that gene sharing among samples is minimal. Most of the genes uniquely belong to only one sample—only 84 genes are shared between exactly two samples and 63 genes are shared among three or more samples. This finding is consistent with the findings from reference 16 that integron genes from 12 Vibrio isolates share only a very small number (<10%) of genes. The HMP cohort contains individuals who were sampled at multiple body sites and visits, enabling us to compare the sharing of the integron cassette genes within and across individuals. We calculated the proportion of shared genes between any two samples and found that samples from the same individual tend to share more genes than do samples from different individuals: the average proportion of genes shared between samples from the same individual is 13%, and the average proportion of genes shared between samples from different individuals is slightly lower, 8%. Note again, it was reported previously that Vibrio isolates share <10% of their integron genes (16). Our results indicate that even within an individual, there is strong population subdivision between Treponema species collected at different sites.
Fig 6.

Sharing of gene cassettes among the samples. In this map, rows are the samples and columns are the genes found in the integron gene cassettes, clustered at 70% sequence identity at the amino acid level (by CD-HIT). A red cell means that the corresponding gene exists in the corresponding sample. The naming convention for the samples is SRS ID_individual ID_female/male_body site_location. Note that some samples are from the same individual (e.g., two samples collected from a female with an individual ID of 761143397 are highlighted with blue triangles in the figure, and another two samples from individual 160158126 are highlighted in orange diamonds).
The functions of the shared genes also vary, and the majority of them are still of unknown function: for the 84 genes shared between two samples, 56 genes cannot be assigned to any COG function, and 19 are assigned to unknown function (category R or S). Similarly, for the 63 genes shared by three or more samples, 30 genes do not hit to any COG function and 17 genes hit to unknown functions. (See Table S4 in the supplemental material for detailed numbers of hits to COG functions.) Overall, the percentage of shared genes with an unknown function is 83%. This number is similar to the proportion for all 826 genes. Furthermore, the number of genes in category L (replication, recombination, and repair) is again the highest among all categories with known functions. These numbers hint that the genes shared among two or more samples are sampled from all integron genes, without any preference for genes of certain functions.
DISCUSSION
To assemble integron gene cassettes, we designed a novel method to trace the de Bruijn assembly graph and then extract sequences bounded by contigs that contain attC sites. Assembly approaches based on de Bruijn graphs typically report the sequences of the edges (i.e., contigs) while discarding the connections between contigs embedded in the graph—the ambiguous connections between contigs may be difficult to resolve if no further information can be applied (28). Our novel constrained assembly approach to integron gene cassettes enables us to traverse between the contigs in the de Bruijn graph by applying further information learned from the integron structures. The effect is enormous, as we obtained 826 genes de novo using this approach, compared to only 66 genes in the whole-metagenome assembly contigs.
Our integron gene discovery pipeline (see Materials and Methods) includes two validation steps (step 4 and step 5): only genes included in the sequences that are supported by read mapping (step 4) and in sequences that contain 1 to 3 genes (step 5) will be reported as candidate integron genes. For the HMP data sets, only 22% of sequences passed the first validation process and 56% of genes passed the second. We did not observe any misassembled integron genes when we applied the pipeline to the simulated data sets. We cannot completely exclude the possibility of having misassemblies in the real HMP data sets, considering that the prediction of the integron genes may be affected by reads from unknown species. Also, our method may miss some integron genes due to the heterogeneity of attC sites of the Treponema species in the real samples.
Our targeted assembly and constrained assembly approaches can in principle be applied to any metagenome containing an integron system. Given the attC sites, we are able to detect species with the corresponding integrons and generate integron gene cassettes. For example, the coral-mucus-associated Vibrio integrons (16) can be used to detect this coral pathogen in ocean samples, such as the Sargasso Sea metagenomic samples (41). By analyzing integron genes, we can help to understand how this species evolves and coexists with coral. We can also analyze genes from different sites (or depths) of the ocean and understand how bacteria in these sites interact with the outer environment. Even if species with integrons are of low abundance, we can still detect their existence in metagenomic samples, as in the case of T. denticola.
Note that our targeted assembly (used in this work to characterize the integron attC sites) was developed to characterize CRISPR arrays in metagenomic samples (31). CRISPR/Cas systems are a widespread class of adaptive immunity systems that bacteria and archaea mobilize against foreign nucleic acids; the CRISPR arrays contain repeats, and short spacers that are likely derived from viral genomes or plasmids. Because the spacers in CRISPR arrays are significantly shorter than Illumina reads, we could easily assemble CRISPR arrays using targeted assembly alone, by first collecting reads containing repeats and then assembling the reads using optimized parameters. In contrast, integron spacers (cassettes) contain 1 to 3 genes between the attC sites, so it is hard to assemble the gene cassettes using targeted assembly alone. The constrained assembly approach was developed to overcome this limitation and allows the assembly and characterization of integron gene cassettes. Both applications (the identification of the CRISPR arrays using the targeted assembly approach and the identification of integron gene cassettes) demonstrate the importance of directed computational approaches for studies of important functional elements—which are poorly analyzed using generalized computational approaches (such as whole-metagenome assembly)—and show that they are essential for the analysis of metagenomic sequences.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Human Microbiome Project (HMP) consortium for providing the sequencing data and the whole-metagenome assemblies of the HMP data sets and thank Haixu Tang for his expertise on assembly and helpful discussions.
Footnotes
Published ahead of print 25 May 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arumugam M, et al. 2011. Enterotypes of the human gut microbiome. Nature 473:174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bafna V, Tang H, Zhang S. 2006. Consensus folding of unaligned RNA sequences revisited. J. Comput. Biol. 13:283–295 [DOI] [PubMed] [Google Scholar]
- 4. Boucher Y, Labbate M, Koenig JE, Stokes HW. 2007. Integrons: mobilizable platforms that promote genetic diversity in bacteria. Trends Microbiol. 15:301–309 [DOI] [PubMed] [Google Scholar]
- 5. Cambray G, Guerout AM, Mazel D. 2010. Integrons. Annu. Rev. Genet. 44:141–166 [DOI] [PubMed] [Google Scholar]
- 6. Coleman N, Tetu S, Wilson N, Holmes A. 2004. An unusual integron in Treponema denticola. Microbiology 150:3524–3526 [DOI] [PubMed] [Google Scholar]
- 7. Collis CM, Recchia GD, Kim MJ, Stokes HW, Hall RM. 2001. Efficiency of recombination reactions catalyzed by class 1 integron integrase IntI1. J. Bacteriol. 183:2535–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Compeau PE, Pevzner PA, Tesler G. 2011. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 29:987–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dashper SG, Seers CA, Tan KH, Reynolds EC. 2011. Virulence factors of the oral spirochete Treponema denticola. J. Dent. Res. 90:691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eddy SR. 2009. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23:205–211 [PubMed] [Google Scholar]
- 11. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fraser CM, et al. 1998. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 281:375–388 [DOI] [PubMed] [Google Scholar]
- 13. Graber JR, Leadbetter JR, Breznak JA. 2004. Description of Treponema azotonutricium sp. nov. and Treponema primitia sp. nov., the first spirochetes isolated from termite guts. Appl. Environ. Microbiol. 70:1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hattori M, Taylor TD. 2009. The human intestinal microbiome: a new frontier of human biology. DNA Res. 16:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res. 17:377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koenig JE, et al. 2011. Coral-mucus-associated Vibrio integrons in the Great Barrier Reef: genomic hotspots for environmental adaptation. ISME J. 5:962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li R, et al. 2010. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20:265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659 [DOI] [PubMed] [Google Scholar]
- 20. Mandel MJ, Wollenberg MS, Stabb EV, Visick KL, Ruby EG. 2009. A single regulatory gene is sufficient to alter bacterial host range. Nature 458:215–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matejkova P, et al. 2008. Complete genome sequence of Treponema pallidum ssp. pallidum strain SS14 determined with oligonucleotide arrays. BMC Microbiol. 8:76 doi:10.1186/1471-2180-8-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mazel D, Dychinco B, Webb VA, Davies J. 1998. A distinctive class of integron in the Vibrio cholerae genome. Science 280:605–608 [DOI] [PubMed] [Google Scholar]
- 23. Muller J, et al. 2010. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 38:D190–D195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nield BS, et al. 2001. Recovery of new integron classes from environmental DNA. FEMS Microbiol. Lett. 195:59–65 [DOI] [PubMed] [Google Scholar]
- 25. Peterson J, et al. 2009. The NIH human microbiome project. Genome Res. 19:2317–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pevzner PA, Tang H, Waterman MS. 2001. An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. U. S. A. 98:9748–9753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pohlschroeder M, Leschine S, Canale-Parola E. 1994. Spirochaeta caldaria sp. nov., a thermophilic bacterium that enhances cellulose degradation by Clostridium thermocellum. Arch. Microbiol. 161:17–24 [Google Scholar]
- 28. Pop M. 2009. Genome assembly reborn: recent computational challenges. Brief. Bioinform. 10:354–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qin J, et al. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rho M, Tang H, Ye Y. 2010. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38:e191 doi:10.1093/nar/gkq747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rho M, Wu Y-W, Doak T, Ye Y. 2012. Diverse CRISPRs evolving in human microbiomes. PLoS Genet. 8:e1002441 doi:10.1371/journal.pgen.1002441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Richter DC, Ott F, Auch AF, Schmid R, Huson DH. 2008. MetaSim: a sequencing simulator for genomics and metagenomics. PLoS One 3:e3373 doi:10.1371/journal.pone.0003373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodriguez-Minguela CM, Apajalahti JH, Chai B, Cole JR, Tiedje JM. 2009. Worldwide prevalence of class 2 integrases outside the clinical setting is associated with human impact. Appl. Environ. Microbiol. 75:5100–5110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rowe-Magnus DA. 2009. Integrase-directed recovery of functional genes from genomic libraries. Nucleic Acids Res. 37:e118 doi:10.1093/nar/gkp561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanada I, Nishida S. 1965. Isolation of Clostridium tetani from soil. J. Bacteriol. 89:626–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seshadri R, et al. 2004. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc. Natl. Acad. Sci. U. S. A. 101:5646–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stokes HW, Hall RM. 1989. A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: integrons. Mol. Microbiol. 3:1669–1683 [DOI] [PubMed] [Google Scholar]
- 38. Stokes HW, et al. 2001. Gene cassette PCR: sequence-independent recovery of entire genes from environmental DNA. Appl. Environ. Microbiol. 67:5240–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stokes HW, O'Gorman DB, Recchia GD, Parsekhian M, Hall RM. 1997. Structure and function of 59-base element recombination sites associated with mobile gene cassettes. Mol. Microbiol. 26:731–745 [DOI] [PubMed] [Google Scholar]
- 40. Tatusov RL, et al. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Venter JC, et al. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304:66–74 [DOI] [PubMed] [Google Scholar]
- 42. Wilson D, et al. 2009. SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37:D380–D386 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.