

Abstract
We established a sensitive and accurate quantification system for Clostridium difficile in human intestines, based on rRNA-targeted reverse transcription-quantitative PCR (RT-qPCR). We newly developed a species-specific primer set for C. difficile targeting 23S rRNA gene sequences. Both the vegetative cells and the spores of C. difficile in human feces were quantified by RT-qPCR, with a lower detection limit of 102.4 cells/g of feces. In an analysis of the feces of residents (n = 83; age, 85 ± 8 years) and staff (n = 19; age, 36 ± 10 years) at a care facility for the elderly, C. difficile was detected by RT-qPCR in 43% of the residents (average count, log10 4.0 ± 2.0 cells/g of feces) and 16% of the staff (average count, log10 2.2 ± 0.1 cells/g of feces); these rates were far higher than those detected by qPCR (residents, 19%; staff, 0%) or selective cultivation (residents, 18%; staff, 5%). Another analysis of healthy adults (n = 63; age, 41 ± 11 years) also revealed the significant carriage rate of C. difficile in the intestines (detection rate, 13%; average count, log10 4.9 ± 1.2 cells/g of feces). From these results, it was suggested that rRNA-targeted RT-qPCR should be an effective tool for analyzing population levels of C. difficile in the human intestine.
INTRODUCTION
Clostridium difficile is a Gram-positive, anaerobic, spore-forming bacterium that has been identified as the major cause of nosocomial antibiotic-associated diarrhea, which in some cases leads to pseudomembranous colitis (4, 14, 34). This organism has been reported to be carried asymptomatically by 0% to 15% of healthy adults (10, 11, 20, 36, 37) and by 13% to 26% of hospitalized patients (30, 31, 42, 52). C. difficile forms metabolically dormant spores to survive in unfavorable environments, such as conditions of aerobiosis, nutrient deficiency, dryness, or high temperature (16, 22, 54). It is generally accepted that C. difficile spores are ingested from the environment into the human intestine; they then germinate and proliferate to initiate diseases when the normal intestinal microbiota is disrupted by the administration of broad-spectrum antibiotics (46, 56).
To investigate the population levels of C. difficile in the human intestinal tract, anaerobic culture techniques have conventionally been used (20, 37, 47, 51). This involves treatment of the feces with alcohol and culturing them on selective microbiological media containing sodium taurocholate to enhance spore germination (7, 26, 57), followed by the isolation of pure cultures and the application of confirmatory biochemical tests. This technique is effective for detecting spores, but it inevitably eliminates the vegetative cells. Moreover, classification and identification based on phenotypic traits do not always provide clear-cut results and are sometimes unreliable. Recently, PCR assays for detecting rRNA or toxin genes have become widely used for rapid and accurate testing for C. difficile (5, 35, 38, 45, 47, 50). The sensitivity of PCR, however, is about 105 to 106 cells/g of feces; this level seems to be insufficient for accurate quantification of this subdominant intestinal inhabitant in healthy subjects (36).
We have recently reported that reverse transcription-quantitative PCR (RT-qPCR) targeting of rRNA molecules enables the analysis of intestinal microbiota with 100 times the sensitivity of qPCR because of the high copy number of targeted rRNA molecules (27). This RT-qPCR method covers a wide variety of intestinal bacterial populations, including subdominant bacteria, and has several advantages, such as sensitivity, rapidity, and accuracy (23, 28). We have developed a specific primer set for C. difficile targeting the 23S rRNA gene, and here we examined the population levels of this bacterium in adult intestines by RT-qPCR.
MATERIALS AND METHODS
Clostridium difficile culture.
The bacterial strains listed in Table 1 were used. All C. difficile strains were routinely grown at 37°C for 24 h under anaerobic conditions in modified Gifu anaerobic broth (Nissui Pharmaceutical Co., Ltd., Tokyo, Japan) supplemented with 1% glucose (Glu-mGAM). The CFU counts of C. difficile vegetative cells were determined by culturing the specimens anaerobically on nonselective Glu-mGAM agar at 37°C for 1 day. To enumerate the CFU counts of C. difficile spores, specimens were mixed with an equal amount of 95% ethanol and kept at room temperature for 30 min and were then cultivated anaerobically on selective cycloserine-cefoxitin-mannitol agar (CCMA) at 37°C for 2 days. All anaerobic manipulations were performed in an anaerobic glove box (Coy Laboratory Products Inc., Grass Lake, MI). Total bacterial cell counts were determined by 4′,6-diamidino-2-phenylindole (DAPI) staining according to a method described previously (28).
Table 1.
Specificity tests with the newly developed primer set
| Taxon | Strain | Reactiona |
|---|---|---|
| Clostridium difficile | DSM 1296T | + |
| Clostridium difficile | 1470 | + |
| Clostridium difficile | KZ 1647 | + |
| Clostridium difficile | A5 | + |
| Clostridium difficile | A77 | + |
| Clostridium difficile | KZ 1678 | + |
| Clostridium difficile | A34 | + |
| Clostridium difficile | KZ 616 | + |
| Clostridium difficile | KZ 617 (= ATCC 17857) | + |
| Clostridium difficile | KZ 1858 (= VPI 10463) | + |
| Clostridium bifermentans | JCM 1386T | − |
| Clostridium ghonii | JCM 1400T | − |
| Clostridium glycolicum | JCM 1401T | − |
| Clostridium lituseburense | JCM 1404T | − |
| Clostridium sordellii | JCM 3814T | − |
| Clostridiumbutyricum | JCM 1391T | − |
| Clostridium paraputrificum | JCM 1293T | − |
| Clostridium perfringens | JCM 1290T | − |
| Faecalibacterium prausnitzii | ATCC 27768T | − |
| Clostridium orbiscindens | DSM 6740T | − |
| Veillonella parvula | ATCC 10790T | − |
| Blautia producta | JCM 1471T | − |
| Clostridium indolis | JCM 1380T | − |
| Clostridium aminovalericum | JCM 11016T | − |
| Clostridium symbiosum | JCM 1297T | − |
| Ruminococcus obeum | ATCC 29174T | − |
| Clostridium innocuum | DSM 1286T | − |
| Eubacterium bioforme | ATCC 27806T | − |
| Eubacteriumcylindroides | DSM 3983T | − |
| Eubacterium dolichum | DSM 3991T | − |
| Clostridium cocleatum | JCM 1397T | − |
| Clostridium ramosum | JCM 1298T | − |
| Clostridium spiroforme | JCM 1432T | − |
The specificity of the RT-qPCR assay for target bacteria performed with the Cd-lsu-F/Cd-lsu-R primer set was investigated by using RNA extracts corresponding to 105 cells from each strain described. Specificity was judged by using the criteria described in Materials and Methods. In addition, negative PCR results were obtained for the following bacterial strains: Bacteroides fragilis DSM 2151T, Bacteroides vulgatus ATCC 8482T, Bifidobacterium adolescentis ATCC 15703T, Bifidobacterium longum ATCC 15707T, Collinsella aerofaciens DSM 3979T, Eggerthella lenta ATCC 25559T, Prevotella melaninogenica ATCC 25845T, Fusobacterium varium ATCC 8501T, Escherichia coli JCM 1649T, Lactobacillus acidophilus ATCC 4356T, Enterococcus faecalis ATCC 19433T, Streptococcus salivarius subsp. salivarius JCM 5707T, Staphylococcus aureus ATCC 12600T, Lactococcus lactis subsp. lactis ATCC 19435T, and Pseudomonas aeruginosa IFO 12689T.
Preparation of C. difficile vegetative cells.
Starter C. difficile cultures were prepared by overnight growth of the bacterial strains in Glu-mGAM broth, as described above. Thirty milliliters of another Glu-mGAM broth was inoculated with 3 μl of starter culture and then incubated anaerobically at 37°C for 1 day. The bacterial cells were washed twice with Dulbecco's PBS (−) (D-PBS) (Nissui Pharmaceutical Co., Ltd.) by centrifugation (5,000 × g at 4°C for 10 min) to remove medium. The resultant cell pellet was suspended in 1.0 ml of ice-cold D-PBS. The vegetative cell suspension was observed by using phase-contrast microscopy to confirm that the vegetative cell suspension did not include spores. The vegetative cell counts were determined by using DAPI staining, as described above.
Preparation of C. difficile spores.
Starter C. difficile cultures were prepared by overnight growth of the bacterial strains in Glu-mGAM broth, as described above. Thirty milliliters of sporulation medium (57) was inoculated with 1.5 ml of starter culture and then incubated anaerobically at 37°C for 3, 7, and 10 days to induce sporulation of C. difficile. For purification of spores, the bacterial cells were washed twice with ice-cold water by centrifugation (5,000 × g at 4°C for 10 min) and then sonicated twice by ultrasonic disrupter (catalog no. UD-201; Tomy Digital Biology Co., Ltd., Tokyo, Japan) (output level, 4.0; interval, 60% of intermittent output) for 5 min. The sonicated cells were suspended in 1% sodium dodecyl sulfate solution and then washed three times with ice-cold 2% Trypticase peptone water by centrifugation (1,000 × g at 4°C for 10 min) to remove cell debris. The resultant cell pellet was suspended in 1.0 ml of ice-cold 2% Trypticase peptone water (Becton, Dickinson, Sparks, MD). The spore suspension was observed by using phase-contrast microscopy to determine the spore count and to confirm that the spore suspension did not include cell debris or germinated spores.
Development of 23S rRNA gene-targeted primers.
Multiple alignment of the target groups and reference organisms was performed with the CLUSTAL_X program (49) by using 23S rRNA gene sequence information obtained from DDBJ/GenBank/EMBL databases. After comparison of the sequences in silico, target sites for C. difficile species-specific detection were identified and the primer set, Cd-lsu-F (5′-GGG AGC TTC CCA TAC GGG TTG-3′) and Cd-lsu-R (5′-TTG ACT GCC TCA ATG CTT GGG C-3′), was designed. The positions of the target sites for Cd-lsu-F and Cd-lsu-R are nucleotides 1,095 to 1,115 and 1,374 to 1,395 on the C. difficile 23S rRNA gene sequence (GenBank accession no. HM007603), respectively. The specificity of the designed primer was checked by submitting its sequence to the BLAST program of National Center for Biotechnology Information (NCBI) (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Total RNA extraction.
For RNA stabilization, each bacterial suspension (200 μl) was added to 2 volumes of RNAlater (Ambion, Inc., Austin, TX). After centrifugation of the mixture at 12,000 × g for 5 min, the supernatant was discarded and the pellets were stored at −80°C until they were used to extract RNA. RNA extraction were performed by using methods described previously (28) with minor modifications. Briefly, the thawed sample was resuspended in a solution containing 346.5 μl of RLT buffer (Qiagen Sciences, Germantown, MD), 3.5 μl of β-mercaptoethanol (Sigma-Aldrich Co., St. Louis, MO), and 100 μl of Tris-EDTA buffer. Glass beads (BioSpec Products, Inc., Bartlesville, OK) (300 mg; diameter, 0.1 mm) were added to the suspension, and the mixture was subjected to a vigorous vortex procedure for 5 min using a ShakeMaster Auto apparatus (catalog no. BMS-A15; Bio Medical Science Inc., Tokyo, Japan). Acid phenol (Wako Pure Chemical Industries, Ltd., Osaka, Japan) (500 μl) was added, and the mixture was incubated for 10 min at 60°C. After phenol-chloroform purification and isopropanol precipitation, the nucleic acid fraction was suspended in 0.2 ml of nuclease-free water (Ambion, Inc.).
RT-qPCR.
RT-qPCR was performed by using methods described previously (28) with a minor modification. Briefly, RT-qPCR was conducted in a one-step reaction using a Qiagen OneStep RT-PCR kit (Qiagen GmbH, Hilden, Germany), 10 μl of reaction mixture containing 5 μl of template RNA, and each specific primer at a concentration of 0.6 μM. The reaction mixture was dispensed into 384-well optical plates by using a Microlab STARlet liquid handling workstation (Hamilton Robotics, Inc., Reno, NV). The reaction mixture was incubated at 55°C for 30 min for reverse transcription. The continuous amplification program consisted of one cycle at 95°C for 15 min and 40 cycles at 94°C for 20 s, 60°C for 20 s, and 72°C for 50 s. Amplification and detection were performed by using an ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA).
DNA extraction and qPCR.
The bacterial suspension (200 μl) was subjected to DNA extraction. DNA extraction and qPCR were performed according to the method described by Matsuki et al. (29). Briefly, the thawed sample was mixed with 250 μl of extraction buffer (100 mM Tris-HCl, 40 mM EDTA; pH 9.0) and 50 μl of 10% sodium dodecyl sulfate. Glass beads (300 mg; diameter, 0.1 mm) and 500 μl of Tris-EDTA (TE)-saturated phenol were added to the suspension, and the mixture was subjected to a vigorous vortex procedure for 30 s using a FastPrep FP120 cell disruptor (Bio 101, Vista, CA) at a power level of 5.0. After phenol-chloroform purification and isopropanol precipitation, the nucleic acid fraction was suspended in 0.2 ml of nuclease-free water. qPCR was conducted using TaKaRa Taq (TaKaRa Bio Inc., Shiga, Japan), 10 μl of reaction mixture containing 5 μl of template DNA, and each specific primer at a concentration of 0.2 μM. The amplification program consisted of 1 cycle at 94°C for 5 min and 40 cycles at 94°C for 20 s, 60°C for 20 s, and 72°C for 50 s.
Determination of RT-PCR sensitivity.
C. difficile DSM 1296T, 1470, KZ 617, KZ 616, KZ 1858, KZ 1647, KZ 1678, A5, A34, and A77 were cultivated separately in Glu-mGAM broth. RNA and DNA fractions were extracted from culture samples in the early stationary phase (24 h), and bacterial counts were determined microscopically by DAPI staining. Serial RNA and DNA dilutions corresponding to bacterial counts ranging from 10−3 to 105 cells were assessed by RT-qPCR and qPCR assays, respectively. The range of RNA and DNA concentrations at which there was linearity with the threshold cycle (CT) value was confirmed (R2 > 0.99).
Determination of primer specificity.
Total RNA fractions extracted from the bacterial cells of each strain shown in Table 1 at a dose corresponding to 105 cells were assessed for RT-qPCR by using the primer set of Cd-lsu-F and Cd-lsu-R. Using the standard curve for C. difficile DSM 1296T, obtained as described above, the amplified signal was judged to be positive (+) when it was more than that of 104 standard cells and negative (−) when it was less than that of 10−1 standard cells. The amplified signal was defined as negative (−) when the corresponding melting curve had a peak different from that of the standard strain.
Determination of bacterial number by RT-qPCR.
A standard curve was generated with the RT-qPCR data (CT value) and the cell counts (DAPI staining) of dilution series of the following standard strains: C. difficile DSM 1296T (Table 2 and Table 3), 1470 (see Fig. 2 and 3), and KZ 617 (see Fig. 2). For determination of the bacterial counts in fecal samples, three serial 10-fold dilutions of the extracted RNA sample (corresponding to amounts of 1/2,000, 1/20,000, and 1/200,000 of the extracted RNA from 20 mg of feces) were used for RT-qPCR, and the CT values in the linear range of the assay were normalized to the standard curve generated in the same experiment to obtain the bacterial count. The amplified signal of the fecal sample was judged to be positive when it was more than that of 10−3 standard cells and the corresponding melting curve had a peak identical to that of the standard strain. Moreover, the amplification products were subjected to electrophoresis in 1.5% agarose and sequence analysis to confirm further the validity of the quantification of C. difficile by RT-qPCR.
Table 2.
Detection of C. difficile in healthy human adults by RT-qPCR, qPCR, culture, and EIAa
| Group (category) | n(by gender) | Age range (yr) (mean ± SD) | RT-qPCR |
qPCR |
Culture |
EIA DR (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| Log10 cells/g of fecesb | DR (%) | Log10 cells/g of fecesb | DR (%) | Log10 CFU/g of fecesb | DR (%) | ||||
| A-1 (facility residents) | 83 (M 24, F 59) | 68–106 (85 ± 8) | 4.0 ± 2.0 | 43 | 6.7 ± 1.0 | 19 | 4.9 ± 0.9 | 18 | 0 |
| A-2 (facility staff) | 19 (M 5, F 14) | 23–56 (36 ± 10) | 2.2 ± 0.1 | 16 | ND | 0 | 2.9 | 5 | 0 |
| B (healthy adults living at home) | 63 (M 40, F 23) | 20–65 (41 ± 11) | 4.9 ± 1.2 | 13 | NT | NT | NT | ||
Fecal samples were collected by the procedure described in Materials and Methods. DR, detection ratio; M, male; F, female; NT, not tested; ND, not detected.
Data are expressed as means and standard deviations.
Table 3.
Comparison of C. difficile counts by RT-qPCR, qPCR, and the anaerobic culture method
| Group and subjecta | Log10 cell count or CFU/g of feces |
Δ value |
|||
|---|---|---|---|---|---|
| RT-qPCR | qPCR | Culture | RT-qPCR versus qPCR | RT-qPCR versus culture | |
| A-1 (facility residents) | |||||
| SO-5 | 6.4 | 6.7 | 5.0 | −0.3 | 1.3 |
| SO-13 | 2.0 | ||||
| SO-14 | 2.2 | ||||
| SO-15 | 2.4 | ||||
| SO-16 | 6.3 | 6.6 | 4.0 | −0.2 | 2.3 |
| SO-19 | 5.3 | 5.5 | 4.4 | −0.3 | 0.9 |
| SO-21 | 5.8 | 6.5 | 4.8 | −0.7 | 1.0 |
| SO-22 | 2.4 | ||||
| SO-23 | 5.8 | 6.4 | 5.5 | −0.6 | 0.3 |
| SO-24 | 2.4 | ||||
| SO-25 | 6.4 | 6.9 | 6.0 | −0.5 | 0.4 |
| SO-26 | 5.2 | 5.9 | 3.4 | −0.7 | 1.8 |
| SO-28 | 4.4 | 5.1 | 3.8 | −0.7 | 0.6 |
| SO-29 | 2.0 | ||||
| SO-31 | 5.8 | 5.9 | 4.8 | −0.1 | 1.0 |
| SO-32 | 2.5 | ||||
| SO-37 | 7.8 | 8.8 | 4.6 | −0.9 | 3.2 |
| SO-45 | 6.0 | 6.5 | 5.8 | −0.4 | 0.3 |
| SO-48 | 2.0 | ||||
| SO-49 | 2.6 | ||||
| SO-50 | 2.0 | ||||
| SO-53 | 2.0 | ||||
| SO-55 | 5.5 | 6.0 | −0.5 | ||
| SO-57 | 2.1 | ||||
| SO-58 | 7.0 | 7.8 | 4.4 | −0.8 | 2.6 |
| SO-60 | 6.9 | 7.6 | 4.7 | −0.6 | 2.2 |
| SO-62 | 3.5 | ||||
| SO-64 | 2.2 | ||||
| SO-69 | 3.1 | ||||
| SO-70 | 2.5 | ||||
| SO-73 | 2.3 | ||||
| SO-75 | 2.1 | ||||
| SO-87 | 7.2 | 7.9 | 6.3 | −0.7 | 0.9 |
| SO-93 | 2.2 | ||||
| SO-98 | 2.0 | ||||
| SO-99 | 5.9 | 6.4 | 6.6 | −0.4 | −0.6 |
| A-2 (facility staff) | |||||
| SO-100 | 2.3 | 2.9 | −0.6 | ||
| SO-113 | 2.1 | ||||
| SO-120 | 2.3 | ||||
Subjects who tested positive for C. difficile are listed.
Fig 2.

Effect of growth phase on C. difficile counts determined by RT-qPCR, qPCR, and the culture method. Throughout the growth phase in sporulation medium culture, the numbers of C. difficile 1470 (A) and KZ 617 (B) were determined by RT-qPCR (●), qPCR (○), and the culture method. Standard curves generated with the RNA and DNA dilution series for each target strain in the stationary phase (24 h) were used to quantify the bacteria by RT-qPCR and qPCR, respectively. CFU counts of vegetative cells were determined by culturing samples on Glu-mGAM agar plates (◇), and those of spores were determined by a method that involved treatment of samples with ethanol and culturing them on CCMA plates (⧫). Results are expressed as the means and standard deviations of the results from triplicate samples.
Fig 3.

Quantitative detection of C. difficile spiked in human feces by RT-qPCR and the culture method. Vegetative cells (Vc) and spores (Sp) were prepared from C. difficile 1470 and observed by using phase-contrast microscopy. (A) Fecal samples collected from three individuals were spiked with serial dilutions of purified vegetative cells or spores to final concentrations ranging from 102.4 to 107.0 cells/g and then assessed by RT-qPCR (B) or the culture method (C). RT-qPCR counts were obtained by using a standard curve generated with the RNA dilution series for the vegetative cells of C. difficile 1470. CFU counts were determined by culturing the same fecal samples treated with ethanol on CCMA. Bacterial counts of spiked vegetative cells and spores were determined by DAPI staining and by spore counting with phase-contrast microscopy, respectively, and then plotted against the corresponding RT-qPCR or CFU counts.
Sequencing of the RT-PCR-amplified rRNA.
RT-PCR products generated with the primer set of Cd-lsu-F and Cd-lsu-R were purified by a High Pure PCR product purification kit (Roche Diagnostics GmbH, Mannheim, Germany) and used for the sequence analysis of 23S rRNA gene fragments. Cycle sequencing reactions were performed with a BigDye Terminator version 3.1 cycle sequencing kit (Applied Biosystems) according to the manufacturer's specifications. Sequences were automatically analyzed on an ABI Prism 3130 Genetic Analyzer (Applied Biosystems). Comparison of rRNA gene sequences obtained was performed by using the BLAST program of the NCBI for assignment of a strain to a particular species.
Quantification of C. difficile spiked in human feces by RT-qPCR and culture.
Fecal samples were collected from three healthy adult males (ages, 31, 45, and 46 years) who had been confirmed in advance by RT-qPCR not to include C. difficile in their indigenous intestinal populations. Each fecal sample was weighed and suspended in 9 volumes of sterilized anaerobic transfer medium (28) in an anaerobic glove box. Vegetative cells (1 day of culture) and spores (3 days of culture) of C. difficile 1470 prepared as described above were serially diluted and were spiked into the fecal homogenates to make final concentrations ranging from 102.4 to 107 cells/g of feces. RNA fractions extracted from 200 μl of each sample were assessed by RT-qPCR assay. The CT values obtained were applied to the standard curve generated with the RNA dilution series for the vegetative cells of C. difficile 1470 to determine the RT-qPCR counts.
Collection and preparation of fecal samples.
A spoonful of feces (0.3 to 0.5 g) was collected into a fecal collection tube (catalog no. 80.734.001; Sarstedt AG & Co., Nümbrecht, Germany) containing 2 ml of RNAlater (for total RNA and DNA extraction) and into an identical tube without liquid (for culture and enzyme immunoassay [EIA]); the tubes were stored at 4°C and −20°C, respectively.
Primary treatment of fecal samples.
Primary treatment of feces for total RNA and DNA extraction was done as follows. Briefly, fecal samples were weighed and suspended in 9 volumes of RNAlater to make a fecal homogenate (100 mg feces/ml). In preparation for total RNA extraction, 200 μl of the fecal homogenate was added to 1 ml of D-PBS. After centrifugation of the mixture at 12,000 × g for 5 min, the supernatant was discarded by decantation, and the pellet was stored at −80°C until it was used to extract RNA. For DNA extraction, 200 μl of the fecal homogenate was added to 1 ml of D-PBS and, after centrifugation of the mixture at 12,000 × g for 5 min, 1 ml of the supernatant was discarded. After another wash with 1 ml of D-PBS, the pellets were stored at −30°C until they were used to extract DNA.
Isolation of C. difficile.
Fecal samples for culture and enzyme immunoassay were weighed and suspended in 9 volumes of sterilized anaerobic transfer medium (28) to make a fecal homogenate (100 mg feces/ml). Fifty microliters of the fecal homogenate was mixed with an equal amount of 95% ethanol and kept at room temperature for 30 min. After serial dilution of the fecal homogenates with anaerobic diluting solution, 100-μl portions of the appropriate diluents were spread onto CCMA, which was then cultured at 37°C for 2 days. Colonies on the agar plates were screened by colony morphology and cell morphology after Gram staining. The screened colonies were subjected to colony PCR with the primer set of Cd-lsu-F and Cd-lsu-R to confirm the identification. Finally, the number of colonies identified was counted to calculate CFU counts for target bacteria per gram of feces (wet weight). The lower limit of bacterial detection with this procedure was 400 CFU/g of feces.
EIA.
An enzyme immunoassay (EIA) was performed with C. Diff TOX-A/B Quik Chek (Techlab Inc., Blacksburg, VA) to detect toxin A and/or toxin B in fecal samples and bacterial culture. The fecal homogenate (0.1 ml) prepared for C. difficile isolation and bacterial culture was subjected to the assay in accordance with the manufacturer's specifications.
Typing of C. difficile.
PCR ribotyping was performed according to the method described by Stubbs et al. (48) with a minor modification. Briefly, 50 μl of PCR mixture was concentrated by heating at 95°C for 15 min and separated by electrophoresis in MetaPhor agarose (Lonza Rockland, Inc., Rockland, ME) (2.0%) at a constant voltage of 100 V for 2.5 h. C. difficile isolates with patterns that differed by at least one major band were assigned to different PCR ribotypes. To investigate toxigenicity of C. difficile isolates, PCR assay was performed to detect toxin A and toxin B genes (tcdA and tcdB) according to the method described by Kato et al. (20).
Statistical analysis.
SPSS 14.0 software (SPSS Japan Inc., Tokyo, Japan) and the program R (http://www.r-project.org) were used. Regression analysis was performed to determine the statistical correlations of the results, and Pearson's product-moment correlation coefficient was calculated. Fischer's exact test and Tukey's honestly significant difference (HSD) test were performed to assess the statistical differences between proportions. P < 0.05 was regarded as significant.
RESULTS
Specific quantification of C. difficile by RT-qPCR in comparison with qPCR.
The counts of C. difficile in the pure culture obtained by DAPI staining (x axis) and the corresponding CT values obtained by RT-qPCR (y axis) showed good correlation for the 10 different C. difficile strains tested (Fig. 1; R2 > 0.99). There were no significant differences between RT-qPCR and qPCR in the slopes of the fitted curves, indicating that the two reactions gave similar amplification efficiencies, whereas the y-axis intercept of the RT-qPCR curves was 8.9 cycles less than that of qPCR, indicating that the RT-qPCR assay was some 400 times more sensitive than the qPCR assay. Total RNA fractions extracted from 48 bacterial strains corresponding to 105 cells were assessed by RT-qPCR with the primer set of Cd-lsu-F and Cd-lsu-R (Table 1): The primer set was specific for C. difficile strains and did not amplify any of the RNAs extracted from nontarget microorganisms tested.
Fig 1.
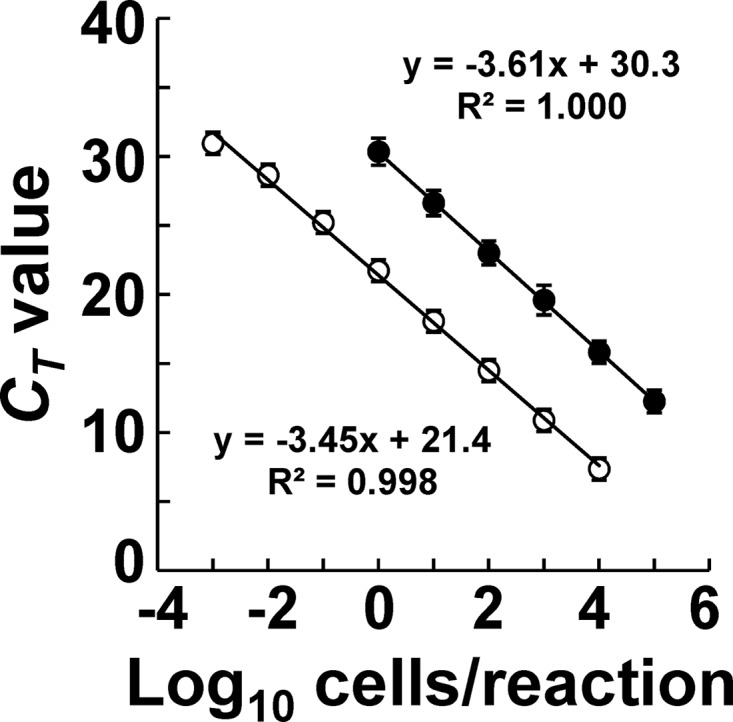
Quantitative detection of C. difficile strains by RT-qPCR in comparison with qPCR. C. difficile DSM 1296T, 1470, KZ 617, KZ 616, KZ 1858, KZ 1647, KZ 1678, A5, A34, and A77 were cultivated separately in Glu-mGAM broth. RNA and DNA fractions were extracted from culture samples in the early stationary phase (24 h), and the bacterial counts were determined microscopically with DAPI staining. On the basis of the bacterial counts, 10-fold serial dilutions of RNA or DNA from 10−3 to 105 cells were assessed by RT-qPCR (○) and qPCR (●) assays. The CT values obtained were plotted against the log10 number of bacterial cells subjected to each reaction; data are expressed as means and standard deviations of the results from 10 strains.
Effect of growth phase on RT-qPCR counts.
The counts of C. difficile 1470 and KZ 617 in in vitro culture were evaluated periodically for 10 days by RT-qPCR, qPCR, and the culture method, starting with a concentration of 105 CFU/ml (Fig. 2). The qPCR counts were always higher than those determined by the other methods; this did not change significantly from day 1 to day 10 after rapid growth to the level of 108 cells/ml after 0.5 days of culture. Like the qPCR counts, the RT-qPCR counts increased dramatically after 0.5 days, but they subsequently decreased from day 1 to day 5 to the level of 107 cells/ml, which was in good agreement with the changes in the CFU counts on Glu-mGAM. From day 7 to day 10 of C. difficile KZ 617 culture, the CFU counts on CCMA overtook those on the Glu-mGAM plates, showing a pattern similar to those of the RT-qPCR counts.
To investigate further the detection of C. difficile spores by RT-qPCR, spores were purified from 3, 7, and 10 days of culture of C. difficile 1470 and KZ 617 and subjected to the RT-qPCR assay (see Fig. S1 in the supplemental material). There were no significant differences in the fitted curves according to the cultivation period, indicating the sensitivity for detection of C. difficile spores by RT-qPCR.
Quantitative detection of C. difficile spiked in human feces.
Vegetative cells and spores were prepared from C. difficile 1470 (Fig. 3A), and the bacterial counts were determined by DAPI staining and by spore counting with phase-contrast microscopy, respectively. Serial dilutions of vegetative cells (1-day culture) or spores (3-day culture) were spiked into the feces of volunteers who had been confirmed in advance by RT-qPCR not to have any form of indigenous C. difficile. Both vegetative cells and spores were detected by RT-qPCR, even at a concentration of 102.4 cells/g of feces, and the counts of spiked bacteria (x axis) and those determined by RT-qPCR (y axis) were well correlated over bacterial concentrations ranging from 102.4 to 107.0 cells/g of feces (R2 > 0.95) (Fig. 3B). In contrast, use of the selective cultivation method underestimated the vegetative cell counts compared with the counts by DAPI staining and RT-qPCR (Fig. 3C).
Quantitative detection of C. difficile in healthy human adults by RT-qPCR.
RT-qPCR analysis was performed to enumerate the intestinal populations of C. difficile in healthy adults from three study groups (A-1, A-2, and B) (Table 2). A-1 consisted of residents at a care facility for the elderly (24 males and 59 females aged 68 to 106 years [average, 85 ± 8 years]); A-2 was staff at the same facility (5 males and 14 females aged 23 to 56 years [average, 36 ± 10 years]); and B consisted of healthy adults living at home (40 males and 23 females aged 20 to 65 years [average, 41 ± 11 years]). The rates of the subjects taking antibiotics before the sampling were 6% of group A-1, 0% of group A-2, and 7% of group B, respectively.
RT-qPCR amplification by the Cd-lsu-F/Cd-lsu-R primer set was observed in 38 subjects in group A-1, 4 subjects in group A-2, and 21 subjects in group B. In the gel electrophoresis analysis of the RT-qPCR products, 2 of 38 subjects in group A-1, 1 of 4 subjects in group A-2, and 13 of 21 subjects in group B had a fragment with a size different from that of the standard strain and judged to be negative. To confirm further the validity of the RT-qPCR amplification, sequence analysis of the RT-qPCR products was performed. The dominant 0.30-kb fragments generated by the Cd-lsu-F/Cd-lsu-R primer set from 36 subjects of group A-1, 3 subjects of group A-2, and 8 subjects of group B resulted in sequences most similar to the 23S rRNA gene of the target C. difficile, with identities exceeding 99% (see Table S1 in the supplemental material). Finally, C. difficile was detected by RT-qPCR in 43% of group A-1 at an average count of 104.0 cells/g of feces, 16% of group A-2 at 102.2, and 13% of group B at 104.9, respectively.
Comparison of RT-qPCR with qPCR, anaerobic culture, and EIA.
The RT-qPCR results were compared to those obtained by qPCR, anaerobic culture, and EIA. The mean incidences obtained by using qPCR (group A-1, 19%; group A-2, 0%) and the culture method (group A-1, 18%; group A-2, 5%) were far lower than those obtained by RT-qPCR (P < 0.01) (Table 2). Table 3 shows the differences of the C. difficile counts between the 3 methods in groups A-1 and A-2: the RT-qPCR counts were always lower than the qPCR counts (the delta values ranging from −0.9 to −0.1) and were in most cases greater than the CFU counts (the delta values ranging from −0.6 to 3.2). The EIA analysis performed with C. Diff TOX-A/B Quik Chek showed that all the fecal samples tested were negative for C. difficile toxins A and B (Table 2).
Typing of C. difficile isolates obtained from the residents and staff at the facility.
PCR ribotypes and toxigenicity of C. difficile isolates were examined with a maximum of eight colonies for all the isolates obtained from 15 subjects in group A-1 and 1 subject in group A-2 (see Table S2 in the supplemental material [“First test” column]). All the isolates were classified into seven different ribotypes, srt 1 to 7, and two different toxin gene types, those positive for toxin A and B genes (tcdA+ tcdB+) (50%) and those lacking toxin genes (50%). The EIA analysis of the isolates showed the results to be concordant with those determined by the PCR assay for tcdA and tcdB (data not shown).
DISCUSSION
The PCR method has thus far been the major molecular technique used to detect C. difficile, especially in the clinical setting (5, 35, 45, 47, 50). The detection limit of PCR, however, has been reported to be about 105 to 106 cells/g of feces, which seems to be insufficient for the detection of indigenous intestinal C. difficile, which may be present at low levels. A major advantage of quantifying C. difficile by using this 23S rRNA-targeted RT-qPCR assay is the method's high sensitivity, with a lower detection limit of 102 to 103 cells/g of feces. Because of the differences between the methods in the copy numbers of targeted molecules, the sensitivity of RT-qPCR is 400 times that of conventional qPCR (Fig. 1) (23, 27, 28). Moreover, recently, the RNA molecule has been reported to be positively correlated with viability in some bacterial killing regimens (6, 15) and has been used as an indicator of bacterial cell viability as an alternative to analyses of colony-forming ability or the DNA molecule (9). In in vitro culture of C. difficile, RT-qPCR counts were in good agreement with the total CFU counts at different growth phases, whereas qPCR counts were largely dissociated from the results obtained by the other methods, especially in the bacterial death phase (Fig. 2). This might have been because the C. difficile DNA persisted in a PCR-detectable form even in a culture-negative environment, whereas rRNA molecules were more susceptible than DNA molecules in the death phase (21). These data, taken together with previous findings, suggested that RT-qPCR should be more suitable than qPCR for use in enumerating viable populations of C. difficile.
C. difficile is detected in the feces and in hospital environments in a metabolically dormant spore form (22, 54). The spores possess thick layers of highly cross-linked coat proteins, a modified peptidoglycan spore cortex, and abundant intracellular constituents, such as calcium chelates of dipicolinic acid and small, acid-soluble spore proteins (17), making it difficult to extract the nucleic acid efficiently. Additionally, in the case of Bacillus subtilis, the total RNA content has been reported to vary by up to 700% when the spores germinate and begin growing (33). We detected C. difficile spores in the feces by RT-qPCR with sensitivity comparable to that of detection of vegetative cells (Fig. 3). Moreover, the sensitivity for detection of C. difficile spores by RT-qPCR was same among different cultivation periods of spores (see Fig. S1 in the supplemental material). These results indicate that the rRNA-targeted RT-qPCR method is suitable for quantification of C. difficile irrespective of the growth phase. Moreover, the RT-qPCR counts of C. difficile in healthy adults were significantly higher than the CFU counts (Table 3, groups A-1 and A-2); this can be explained by the lower sensitivity of the culture method compared to that of RT-qPCR. The culture method might underestimate the total C. difficile count because of elimination of vegetative cells during selective quantification (Fig. 3).
Several risk factors for acquisition of C. difficile and development of its infection have been described, including age, treatment with antibiotics, and hospital admission (40); those factors are considered to relate to changes in the intestinal microbiota, immunosenescence, or the presence of underlying diseases (44). The present results clearly showed that the carriage rate of C. difficile in the facility residents (43%) was significantly higher than those in the facility staff (16%) and the healthy adults living at home (13%) (Table 2) (P < 0.05, as determined by Tukey's HSD test), which supports the findings in the previous reports. It has been well documented that C. difficile spreads nosocomially and causes hospital outbreaks of C. difficile-associated diarrhea in various clinical settings (4, 34), where infected and colonized patients and contaminated environments have been implicated as the potential sources of C. difficile (30, 35). In the present study, we identified clusters of individuals carrying the same ribotype of C. difficile (see Table S2 in the supplemental material). And the periodical examinations conducted for 6 months demonstrated that identical strains were maintained in 4 (57%) of 7 individuals and that different strains were acquired by 3 subjects (43%), which are findings in accordance with the previous results indicating the occurrence of C. difficile cross-transmission in hospital settings (3, 55). On the other hand, the occurrence of cross-transmission from the residents to the staff seems to be low from the results indicating the lower carriage rate of C. difficile by the staff. However, to ascertain the occurrence of C. difficile cross-transmission in the residents as well as the staff at the care facility, further studies to compare multiple occasions are needed.
The mean carriage rate of C. difficile in the facility residents determined by RT-qPCR (43%) (Table 2) was found to be significantly higher than those reported previously (0% to 20%) (2, 44). This result suggests that there is a higher potential risk of acquiring C. difficile infection in the elderly. Recently, several outbreaks of C. difficile infection in the elderly at the care facilities have been reported (8, 13). Moreover, Riggs et al. showed asymptomatic colonization of more than half of the residents of a facility in the midst of an epidemic (39). Therefore, it may be prudent to monitor the C. difficile colonization levels of patients undergoing treatment with antibiotics. The pathogenicity of C. difficile is associated with the production of two large toxins, toxin A and toxin B, both of which are implicated in mucosal damage (53). Nontoxigenic strains are not pathogenic, and their colonization in humans and a hamster model was found to be protective against C. difficile infection by preventing colonization of toxigenic strains (32, 41, 43). Most strains produce both toxins, but pathogenic strains of C. difficile producing toxin B only have previously been reported (1, 19). Analysis of the toxin gene type showed that 8 (50%) of the 16 isolates were positive for tcdA and tcdB and that the other half lacked tcdA and tcdB (see Table S2 in the supplemental material [“First test” column]), although all the specimens were negative in the EIA (Table 2). Because the sensitivity of the EIA was not as high as 106 CFU/g of feces, the incidence of toxigenic strains might have been underestimated. The precise environmental signals modulating toxin expression remain unclear, but in vitro studies indicate that toxin expression may be enhanced by stressors, including antibiotics, catabolite repression, and sporulation (18, 53). To further characterize such opportunistic pathogens in the intestines, it is essential to evaluate toxin production. Therefore, identification of expression of the mRNA of these toxin genes by the use of the same total RNA fractions should be the next objective after quantification of exact population levels by RT-qPCR; this step would enable a more precise recognition of the pathogenic activity of C. difficile in particular environments.
In conclusion, we have developed a sensitive, culture-independent system for quantification for C. difficile in the human intestine by using rRNA-targeted RT-qPCR. It should prove to be an effective tool for analyzing subdominant populations of C. difficile in the human intestine.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kikuo Takano and all of the staff and volunteers at Sousen Hospital for their participation in this study. We also thank Yukiko Kado, Akira Takahashi, Norikatsu Yuki, Kiyohito Ogata, and Toshihiko Takada for their assistance in this research.
Footnotes
Published ahead of print 11 May 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Alfa MJ, et al. 2000. Characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile responsible for a nosocomial outbreak of Clostridium difficile-associated diarrhea. J. Clin. Microbiol. 38:2706–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arvand M, et al. 2012. High prevalence of Clostridium difficile colonization among nursing home residents in Hesse, Germany. PLoS One 7:e30183 doi: 10.1371/journal.pone.0030183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barbut F, et al. 2000. Epidemiology of recurrences or reinfections of Clostridium difficile-associated diarrhea. J. Clin. Microbiol. 38:2386–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartlett JG. 2006. Narrative review: the new epidemic of Clostridium difficile-associated enteric disease. Ann. Intern. Med. 145:758–764 [DOI] [PubMed] [Google Scholar]
- 5. Bélanger SD, Boissinot M, Clairoux N, Picard FJ, Bergeron MG. 2003. Rapid detection of Clostridium difficile in feces by real-time PCR. J. Clin. Microbiol. 41:730–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bleve G, Rizzotti L, Dellaglio F, Torriani S. 2003. Development of reverse transcription (RT)-PCR and real-time RT-PCR assays for rapid detection and quantification of viable yeasts and molds contaminating yogurts and pasteurized food products. Appl. Environ. Microbiol. 69:4116–4122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borriello SP, Honour P. 1981. Simplified procedure for the routine isolation of Clostridium difficile from faeces. J. Clin. Pathol. 34:1124–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burns K, et al. 2010. Infection due to C. difficile ribotype 078: first report of cases in the Republic of Ireland. J. Hosp. Infect. 75:287–291 [DOI] [PubMed] [Google Scholar]
- 9. Cenciarini-Borde C, Courtois S, La Scola B. 2009. Nucleic acids as viability markers for bacteria detection using molecular tools. Future Microbiol. 4:45–64 [DOI] [PubMed] [Google Scholar]
- 10. Chachaty E, Bourneix C, Renard S, Bonnay M, Andremont A. 1993. Shedding of Clostridium difficile, fecal beta-lactamase activity, and gastrointestinal symptoms in 51 volunteers treated with oral cefixime. Antimicrob. Agents Chemother. 37:1432–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chachaty E, et al. 1992. Presence of Clostridium difficile and antibiotic and beta-lactamase activities in feces of volunteers treated with oral cefixime, oral cefpodoxime proxetil, or placebo. Antimicrob. Agents Chemother. 36:2009–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reference deleted. [Google Scholar]
- 13. Crogan NL, Evans BC. 2007. Clostridium difficile: an emerging epidemic in nursing homes. Geriatr. Nurs. 28:161–164 [DOI] [PubMed] [Google Scholar]
- 14. Dolgin E. 2011. ‘Game changer’ antibiotic and others in works for superbug. Nat. Med. 17:10 doi: 10.1038/nm0111-10 [DOI] [PubMed] [Google Scholar]
- 15. Dreier J, Stormer M, Kleesiek K. 2004. Two novel real-time reverse transcriptase PCR assays for rapid detection of bacterial contamination in platelet concentrates. J. Clin. Microbiol. 42:4759–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Freeman J, Wilcox MH. 2003. The effects of storage conditions on viability of Clostridium difficile vegetative cells and spores and toxin activity in human faeces. J. Clin. Pathol. 56:126–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henriques AO, Moran CP., Jr 2007. Structure, assembly, and function of the spore surface layers. Annu. Rev. Microbiol. 61:555–588 [DOI] [PubMed] [Google Scholar]
- 18. Kamiya S, Ogura H, Meng XQ, Nakamura S. 1992. Correlation between cytotoxin production and sporulation in Clostridium difficile. J. Med. Microbiol. 37:206–210 [DOI] [PubMed] [Google Scholar]
- 19. Kato H, et al. 1998. Identification of toxin A-negative, toxin B-positive Clostridium difficile by PCR. J. Clin. Microbiol. 36:2178–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kato H, et al. 2001. Colonisation and transmission of Clostridium difficile in healthy individuals examined by PCR ribotyping and pulsed-field gel electrophoresis. J. Med. Microbiol. 50:720–727 [DOI] [PubMed] [Google Scholar]
- 21. Keer JT, Birch L. 2003. Molecular methods for the assessment of bacterial viability. J. Microbiol. Methods 53:175–183 [DOI] [PubMed] [Google Scholar]
- 22. Kramer A, Schwebke I, Kampf G. 2006. How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect. Dis. 6:130 doi: 10.1186/1471-2334-6-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kubota H, et al. 2010. Detection of human intestinal catalase-negative, Gram-positive cocci by rRNA-targeted reverse transcription-PCR. Appl. Environ. Microbiol. 76:5440–5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reference deleted. [Google Scholar]
- 25.Reference deleted. [Google Scholar]
- 26. Marler LM, et al. 1992. Comparison of five cultural procedures for isolation of Clostridium difficile from stools. J. Clin. Microbiol. 30:514–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsuda K, Tsuji H, Asahara T, Kado Y, Nomoto K. 2007. Sensitive quantitative detection of commensal bacteria by rRNA-targeted reverse transcription-PCR. Appl. Environ. Microbiol. 73:32–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsuda K, et al. 2009. Establishment of an analytical system for the human fecal microbiota, based on reverse transcription-quantitative PCR targeting of multicopy rRNA molecules. Appl. Environ. Microbiol. 75:1961–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuki T, et al. 2004. Quantitative PCR with 16S rRNA-gene-targeted species-specific primers for analysis of human intestinal bifidobacteria. Appl. Environ. Microbiol. 70:167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McFarland LV, Mulligan ME, Kwok RY, Stamm WE. 1989. Nosocomial acquisition of Clostridium difficile infection. N. Engl. J. Med. 320:204–210 [DOI] [PubMed] [Google Scholar]
- 31. McFarland LV, Surawicz CM, Stamm WE. 1990. Risk factors for Clostridium difficile carriage and C. difficile-associated diarrhea in a cohort of hospitalized patients. J. Infect. Dis. 162:678–684 [DOI] [PubMed] [Google Scholar]
- 32. Merrigan MM, Sambol SP, Johnson S, Gerding DN. 2009. New approach to the management of Clostridium difficile infection: colonisation with non-toxigenic C. difficileduring daily ampicillin or ceftriaxone administration. Int. J. Antimicrob. Agents 33 (Suppl. 1): S46–S50 [DOI] [PubMed] [Google Scholar]
- 33. Moeller R, et al. 2006. A method for extracting RNA from dormant and germinating Bacillus subtilis strain 168 endospores. Curr. Microbiol. 53:227–231 [DOI] [PubMed] [Google Scholar]
- 34. Monaghan T, Boswell T, Mahida YR. 2009. Recent advances in Clostridium difficile-associated disease. Postgrad Med. J. 85:152–162 [DOI] [PubMed] [Google Scholar]
- 35. Mutters R, et al. 2009. Quantitative detection of Clostridium difficile in hospital environmental samples by real-time polymerase chain reaction. J. Hosp. Infect. 71:43–48 [DOI] [PubMed] [Google Scholar]
- 36. Nakamura S, et al. 1981. Isolation of Clostridium difficile from the feces and the antibody in sera of young and elderly adults. Microbiol. Immunol. 25:345–351 [DOI] [PubMed] [Google Scholar]
- 37. Ozaki E, et al. 2004. Clostridium difficile colonization in healthy adults: transient colonization and correlation with enterococcal colonization. J. Med. Microbiol. 53:167–172 [DOI] [PubMed] [Google Scholar]
- 38. Penders J, et al. 2005. Quantification of Bifidobacterium spp., Escherichia coli and Clostridium difficile in faecal samples of breast-fed and formula-fed infants by real-time PCR. FEMS Microbiol. Lett. 243:141–147 [DOI] [PubMed] [Google Scholar]
- 39. Riggs MM, et al. 2007. Asymptomatic carriers are a potential source for transmission of epidemic and nonepidemic Clostridium difficile strains among long-term care facility residents. Clin. Infect. Dis. 45:992–998 [DOI] [PubMed] [Google Scholar]
- 40. Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7:526–536 [DOI] [PubMed] [Google Scholar]
- 41. Sambol SP, Merrigan MM, Tang JK, Johnson S, Gerding DN. 2002. Colonization for the prevention of Clostridium difficile disease in hamsters. J. Infect. Dis. 186:1781–1789 [DOI] [PubMed] [Google Scholar]
- 42. Samore MH, et al. 1994. Clostridium difficile colonization and diarrhea at a tertiary care hospital. Clin. Infect. Dis. 18:181–187 [DOI] [PubMed] [Google Scholar]
- 43. Shim JK, Johnson S, Samore MH, Bliss DZ, Gerding DN. 1998. Primary symptomless colonisation by Clostridium difficile and decreased risk of subsequent diarrhoea. Lancet 351:633–636 [DOI] [PubMed] [Google Scholar]
- 44. Simor AE, Bradley SF, Strausbaugh LJ, Crossley K, Nicolle LE. 2002. Clostridium difficile in long-term-care facilities for the elderly. Infect. Control Hosp. Epidemiol. 23:696–703 [DOI] [PubMed] [Google Scholar]
- 45. Sloan LM, Duresko BJ, Gustafson DR, Rosenblatt JE. 2008. Comparison of real-time PCR for detection of the tcdC gene with four toxin immunoassays and culture in diagnosis of Clostridium difficile infection. J. Clin. Microbiol. 46:1996–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sorg JA, Sonenshein AL. 2008. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 190:2505–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stamper PD, et al. 2009. Comparison of a commercial real-time PCR assay for tcdB detection to a cell culture cytotoxicity assay and toxigenic culture for direct detection of toxin-producing Clostridium difficile in clinical samples. J. Clin. Microbiol. 47:373–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stubbs SL, Brazier JS, O'Neill GL, Duerden BI. 1999. PCR targeted to the 16S-23S rRNA gene intergenic spacer region of Clostridium difficile and construction of a library consisting of 116 different PCR ribotypes. J. Clin. Microbiol. 37:461–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van den Berg RJ, et al. 2005. Prospective multicenter evaluation of a new immunoassay and real-time PCR for rapid diagnosis of Clostridium difficile-associated diarrhea in hospitalized patients. J. Clin. Microbiol. 43:5338–5340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vanpoucke H, De Baere T, Claeys G, Vaneechoutte M, Verschraegen G. 2001. Evaluation of six commercial assays for the rapid detection of Clostridium difficile toxin and/or antigen in stool specimens. Clin. Microbiol. Infect. 7:55–64 [DOI] [PubMed] [Google Scholar]
- 52. Viscidi R, Willey S, Bartlett JG. 1981. Isolation rates and toxigenic potential of Clostridium difficile isolates from various patient populations. Gastroenterology 81:5–9 [PubMed] [Google Scholar]
- 53. Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18:247–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wilcox MH, Fawley WN. 2000. Hospital disinfectants and spore formation by Clostridium difficile. Lancet 356:1324. [DOI] [PubMed] [Google Scholar]
- 55. Wilcox MH, Fawley WN, Settle CD, Davidson A. 1998. Recurrence of symptoms in Clostridium difficile infection—relapse or reinfection? J. Hosp. Infect. 38:93–100 [DOI] [PubMed] [Google Scholar]
- 56. Wilson KH. 1983. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J. Clin. Microbiol. 18:1017–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wilson KH, Kennedy MJ, Fekety FR. 1982. Use of sodium taurocholate to enhance spore recovery on a medium selective for Clostridium difficile. J. Clin. Microbiol. 15:443–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.