

Abstract
Neisseria meningitidis is a major cause of sepsis and bacterial meningitis worldwide. This bacterium expresses type IV pili (Tfp), which mediate important virulence traits such as the formation of bacterial aggregates, host cell adhesion, twitching motility, and DNA uptake. The meningococcal PilT protein is a hexameric ATPase that mediates pilus retraction. The PilU protein is produced from the pilT-pilU operon and shares a high degree of homology with PilT. The function of PilT in Tfp biology has been studied extensively, whereas the role of PilU remains poorly understood. Here we show that pilU mutants have delayed microcolony formation on host epithelial cells compared to the wild type, indicating that bacterium-bacterium interactions are affected. In normal human serum, the pilU mutant survived at a higher rate than that for wild-type bacteria. However, in a murine model of disease, mice infected with the pilT mutant demonstrated significantly reduced bacterial blood counts and survived at a higher rate than that for mice infected with the wild type. Infection of mice with the pilU mutant resulted in a trend of lower bacteremia, and still a significant increase in survival, than that of the wild type. In conclusion, these data suggest that PilU promotes timely microcolony formation and that both PilU and PilT are required for full bacterial virulence.
INTRODUCTION
Pathogenic Neisseria species, i.e., Neisseria gonorrhoeae and N. meningitidis, are human-specific bacterial species with a large set of virulence factors that facilitate the colonization of human mucosal surfaces. Type IV pili (Tfp) are essential for bacterial motility, the formation of bacterial aggregates, and the initial binding of bacteria to host epithelial cells (16, 22, 29). Furthermore, Tfp mediate natural competence for DNA transformation, which increases the probability of bacterial adaptation to the host environment (2, 25).
Tfp biogenesis in pathogenic Neisseria requires a principal set of 12 to 15 highly conserved proteins. Thousands of pilus subunits (PilE) are assembled into a helical structure that is 6 nm in diameter and several micrometers long. The prepilin peptidase, PilD, removes an N-terminal leader peptide from PilE before its incorporation into the pilus. Two ATPases, PilF and PilT, are associated with the cytoplasmic membrane and are at the core of pilus dynamics, with their actions contributing to Tfp elongation and retraction, respectively (21, 33). The pilus filament is secreted through a pore in the outer membrane formed by units of the PilQ protein (33). Adhesive properties of Tfp are modulated primarily through the adhesin PilC (18, 24, 26), extensive antigenic variation of PilE (19), and pilus posttranscriptional modification (5). However, minor pilins incorporated into the pilus filament also play an important role in fine-tuning Tfp function (1, 4, 17). Although the assembly of the pilus is relatively well understood, the maintenance and modulation of Tfp dynamics remain to be deciphered.
PilT shares a high degree of homology with two other ATPases, PilT2 and PilU, which have 60% amino acid conservation in the core region that contains the ATP-binding Walker A and B motifs (4, 6). In addition, PilT and PilU are also highly conserved among Tfp-expressing species, implicating a fundamentally conserved mechanism of Tfp biology (21). Previous studies of pathogenic Neisseria species have demonstrated that absence of pilus retraction by deletion of the pilT gene causes a hyperpiliated, hyperaggregative phenotype with increased adhesion to host cells, whereas twitching motility and natural competence are lost (23, 31). Despite increased adhesion, PilT mutants are unable to advance to intimate attachment (23). In comparison to PilT, PilU is slightly larger, with extended C- and N-terminal domains of unknown function. The pilT and pilU loci are organized into one operon, with the pilU gene downstream of pilT (20). In N. gonorrhoeae, PilU mutants retain piliation but fail to aggregate in liquid medium, whereas attachment to epithelial cells is increased 8-fold compared to that in the wild type and involves bacterial aggregation (20). In N. meningitidis, PilU-deficient mutants adhere better than the wild type to human umbilical vein endothelial cells (HUVEC) at 30 min and 90 min postinfection (4). However, no changes in aggregation or microcolony formation in liquid were reported.
Here we sought to further examine the role of PilU in meningococcal infection. We found that increased attachment of the pilU deletion mutant at 2 h postinfection is an inherent property of single bacteria and not an effect of increased autoagglutination. Using time-lapse imaging, we determined that pilU mutants were delayed in the formation and dispersal of microcolonies both during infection of host cells and in medium, suggesting that PilU modulates the dynamic regulation of Tfp-mediated autoagglutination. In a mouse model of disease, the pilU mutant was less virulent than the wild-type bacteria, suggesting that PilU is required for full virulence.
MATERIALS AND METHODS
Bacterial strains, cell culture, and growth conditions.
Neisseria meningitidis strains were derived from the serogroup C strain FAM20 (24) and grown at 37°C in a 5% CO2 atmosphere on GCB (GC medium base; Acumedia) supplemented with 1% Kellogg's supplement. A capsule mutant with a deletion of siaD was selected on kanamycin (Kan) (13), the pilT mutant was selected on Kan (13), the lpxA mutant, deficient in lipopolysaccharides (LPS), was selected on Kan, and the pilC1 and pilC2 mutants were selected on chloramphenicol (CM) (24); all of these mutants have been described previously. Escherichia coli strain DH5α was used for cloning and plasmid propagation. E. coli was grown on LB agar plates or in LB broth (Acumedia). Antibiotics were used at the following concentrations: ampicillin (Amp), 100 μg/ml; Kan, 50 μg/ml; tetracycline (Tet), 1 μg/ml; and CM, 4 μg/ml (Sigma). The human pharyngeal epithelial cell line FaDu (ATCC; HTB-43) was maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum (Sigma). Unless stated otherwise, all experiments were performed using 80% confluent cells cultivated in 24- or 48-well cell culture plates, with the cell culture medium freshly exchanged prior to the beginning of the experiment.
Generation of ΔpilU, ΔpilU_C, ΔpilT ΔpilU, and ΔpilTΔ pilU_C strains.
The primers used in this study are listed in Table 1. Upstream and downstream homology regions of pilU were amplified from FAM20 by using the primers uhs_pilU_KO_f and -r and dhs_pilU_KO_f and -r. A region containing the tetA gene, including its promoter, was amplified from plasmid pACYC184 with the primers tetA_pilU_KO_f and -r. All PCR fragments contained overlapping homologies, and the full-length construct was generated with a modified assembly PCR strategy utilizing high-fidelity Phusion DNA polymerase (Finnzymes). Plasmid pJET1.2-ΔpilU was generated by blunt-end ligation of the final PCR fragments into the cloning vector pJET1.2 according to the manufacturer's instructions (CloneJet; Fermentas). The plasmid pJET1.2-ΔpilU was then introduced into the chromosome of FAM20 by transformation. To complement the ΔpilU mutant, we followed a similar approach whereby the upstream and downstream homology regions were amplified from an area in a noncoding region between locus tags NMC0482 and NMC0483 from FAM20 genomic DNA to generate plasmid pJET1.2-ΔpilU_C, using the primers uhs_pilUC_f and -r and dhs_pilUC_f and -r. The pilU gene was amplified with the primers pilU_pilUC_f and -r, and the chloramphenicol resistance cassette CAT, including its promoter, was amplified from the plasmid pACYC184 (GenBank accession no. X06403) with the primers cat_pilUC_f and -r. The overlapping homologies between primers cat_pilUC_r and pilU_pilUC_f formed a synthetic σ70 promoter that drove the expression of pilU. The plasmid pJET1.2-ΔPilU_C was then introduced into the chromosome of the pilU mutant by transformation. To generate the pilT pilU double mutant strain, the FAM20 ΔPilU strain was transformed with genomic DNA extracted from a previously constructed pilT-negative strain (13). Because pilT-negative Neisseria meningitidis cells are nontransformable, the pilU-complemented pilT pilU double mutant was generated by transforming the ΔpilU_C strain in an identical manner. All mutants were confirmed by PCR, DNA sequencing (Eurofins), and Western blotting using unpurified polyclonal anti-PilT serum that also cross-reacts with PilU, which confirmed that the pilT mutants still expressed PilU (data not shown).
Table 1.
Primers used for cloning
| Primer | Sequence (5′–3′) |
|---|---|
| uhs_pilU_KO_f | ATGCCGTCTGAAATGCTGTCCGAATCGCTGACC |
| uhs_pilU_KO_r | GTATGGGGCTGCGTTAGCTTCTTTTCGGTTT |
| dhs_pilU_KO_f | GACCTGAATGGAATGATCCGCAAAACCCAATGCC |
| dhs_pilU_KO_r | ACCGCGCCTAAAATGGTAAAGC |
| tetA_pilU_KO_f | GAAGCTAACGCAGCCCCATACGATATAAGTTG |
| tetA_pilU_KO_r | GCGGATCATTCCATTCAGGTCGAGGTGGC |
| uhs_pilUC_f | TTGCATCCTGCTGTATAAAACC |
| uhs_pilUC_r | TCTTCCGTCAGCTATGGATGGCGGCGTTT |
| dhs_pilUC_f | CATTTCCTGAGCATTGGTAAAGCACGCGGGGAACGGAAA |
| dhs_PilUC_r | AGGTAGGCGGAAACGAGGGTA |
| pilU_pilUC_f | TAATGTGTGGAATTGTGAGCGGATAACAATTTCACACAGGAGGATCCTATGAATACCGATAACCTGCACG |
| pilU_pilUC_r | TTACCAATGCTCAGGAAATGAGGTTGAGACCG |
| cat_pilUC_f | CATCCATAGCTGACGGAAGATCACTTCGCA |
| cat_pilUC_r | GCTCACAATTCCACACATTATACGAGCCGATGATTAATTTTCAACAGCTC |
Competitive adhesion assays.
Bacterial colonies grown for 16 to 18 h on GC agar plates were suspended in DMEM and filtered through a 5-μm-pore-size filter to break apart preexisting bacterial aggregates. FaDu cells were infected with bacteria at a multiplicity of infection (MOI) of 100 and gently centrifuged at 200 × g for 5 min. After 2 h of incubation at 37°C in a 5% CO2 atmosphere, unbound bacteria were removed with 5 washes of phosphate-buffered saline (PBS), and the cells were then lysed with 1% saponin in GC liquid medium for 5 min. Lysates were plated onto nonselective and selective GC agar plates and incubated overnight, after which the CFU were counted. The competitive index (CI) was calculated as the ratio of the total CFU for the strains involved, meaning that in the competition of X versus Y, the CFU of X was divided by the CFU of Y to give the CI. Competitive adhesion assays were performed in triplicate and repeated on three independent occasions. Single-strain adhesion assays were performed in an identical manner, with antibiotics omitted from the plating medium (data not shown).
Western blot procedure and analysis.
Bacteria were harvested from GCB agar plates and thoroughly resuspended in PBS to an optical density at 600 nm (OD600) of 1.0. Bacterial suspensions were normalized according to the total protein content, as determined by the Bradford total protein assay (Bio-Rad), and equal amounts of total protein were analyzed by 12% SDS-PAGE. All samples were diluted in 4× sample buffer containing 1% β-mercaptoethanol and incubated at 95°C for 5 min before electrophoresis. Proteins were transferred from the gel onto polyvinylidene difluoride (PVDF) sheets (Immobilon-FL) and overlaid with any of the following primary antibodies: anti-PilT, anti-PilC, anti-pilus (27), anti-NafA (14), anti-PilQ (28), anti-all-Opa (4B12/C11), anti-OpaA and -D (H22.1), anti-OpaB (H21.1), and anti-OpaD (7-21-D9) (27) antibodies. The monoclonal EF-Tu antibody (dilution factor, 1:2,000: Hycult Biotech) was used as the loading/normalization control. Primary antibodies were followed by infrared (IR)-reactive dye-conjugated goat anti-mouse 680LT (dilution factor, 1:20,000) (Li-Cor) and/or goat anti-rabbit 800CW (dilution factor, 1:10,000) secondary antibody. Membranes were visualized using an Odyssey IR scanner (Li-Cor) at standardized 700- and 800-nm intensity settings of 3.0 and 6.5, respectively. The EF-Tu band was chosen as a normalizing band because it is likely to be expressed stably and is present in two identical copies in the genomes of both N. gonorrhoeae and N. meningitidis (tufA and tufB). Analysis of raw image data files was performed in ImageJ analysis software (version 1.43). Integrated pixel intensity areas for bands were calculated, with a manual correction for background intensity.
Outer membrane preparation.
Outer membrane preparations were obtained by utilizing a previously described protocol (8). In brief, one heavily streaked plate of bacteria was suspended in 2 ml LiCl buffer (0.2 M LiCl, 0.1 M sodium acetate, pH 5.8) and vortexed 3 times for 30 s each with 3-mm glass beads. After 6 1-min centrifugations at 20,000 × g, supernatants were subjected to ultracentrifugation at 100,000 × g for 2 h. The pellets were then dissolved in the SDS-PAGE sample buffer.
Examination of LOS expression.
Tricine SDS-PAGE was used to examine the lipooligosaccharide (LOS) expression of bacterial mutants. Crude LOS preparations were made from whole-cell lysates of meningococci suspended in distilled water. A previously generated mutant strain lacking LOS, the ΔlxpA strain, was used as a negative control (3). The protein concentration was approximated using the Bradford assay (Bio-Rad) and adjusted to 2 μg/μl. A preparation of 4 μg of cell lysate in TE buffer (10 mM Tris [pH 8], 1 mM EDTA) containing 2% SDS was mixed with 2 μl of proteinase K (>20 mg/ml) (Sigma-Aldrich) and incubated for 30 min at 55°C. Another aliquot of proteinase K was added, and the incubation was repeated. Following digestion, loading buffer (0.5 M Tris-HCl [pH 6.8], 25% glycerol, 2% SDS, 0.5% β-mercaptoethanol, 0.5% bromophenol blue) was added, and the samples were heated for 5 min at 95°C. Separation was performed with a 16.5% acrylamide gel, and LOS was detected by silver staining according to the manufacturer's protocol (Invitrogen).
Live-cell amino labeling of bacteria and time-lapse microscopy.
Bacteria were carefully resuspended in 100 μl of sterile PBS (OD600 = 1.0) to which 0.25 μl of a 10-mg/ml DyLight488 N-hydroxysuccinimide ester (Thermo Scientific) suspension in dimethylformamide (Sigma) was added. After a 15-min incubation at 37°C, 100 μl of prewarmed Tris-buffered saline (TBS) was added to the bacterial suspension to stop the labeling reaction, after which bacteria were centrifuged at 20,000 × g for 1 min. The bacterial pellet was washed once in prewarmed DMEM supplemented with 10% fetal bovine serum (DMEM-F), resuspended in 100 μl prewarmed DMEM-F, and used to infect 80% confluent FaDu cells grown in 12-well poly-d-lysine-coated glass-bottom dishes (MatTek) containing 3 ml of DMEM-F. After a gentle centrifugation at 200 × g for 5 min, the plates were incubated at 37°C in 5% CO2 for 2 h, washed with DMEM-F three times, and observed in a live-cell microscope (Axiovert Z1; Zeiss) at 37°C and 5% CO2. Phase-contrast and fluorescence (488 nm) images were acquired with a 40× objective (numerical aperture [NA] = 1.36) every 10 min at four positions per well for 12 h. The experiment was repeated three times.
Capsule ELISA.
The total capsule was measured with a whole-cell enzyme-linked immunosorbent assay (ELISA) using a monoclonal antibody (National Institute for Biological Standards and Control). As a positive control, 200 ng of purified serogroup C polysaccharide was used to coat the plates (National Institute for Biological Standards and Control). Plates were coated with 100 μl of a bacterial suspension (OD600 = 0.032) and allowed to dry under laminar airflow overnight. To ensure equal loading, the total protein concentration was measured using the Bradford assay (Bio-Rad). The plates were then blocked with 200 μl of 2% skim milk powder (SMP) (Bio-Rad) in PBS (1 h) and incubated with 50 μl of capsule monoclonal antibody (1:400 in 0.2% SMP) for 2 h at room temperature. For detection, plates were incubated with horseradish peroxidase–goat anti-mouse immunoglobulin G (1:5,000) for 1 h. Peroxidase was detected using TMB (3,3′,5,5′-tetramethylbenzidine) (Invitrogen), and the absorbance was measured at 490 nm in a plate reader. The assay was performed three times in triplicate.
IL-6 and IL-8 ELISAs.
Bacteria were harvested from GCB agar plates and thoroughly suspended in DMEM to an OD600 of 0.32; 200 μl of this suspension was added to FaDu cells grown to 80% confluence in 24-well cell culture plates containing 800 μl of DMEM-F per well. After 2 and 8 h, cell culture supernatants were collected and centrifuged at 20,000 × g for 3 min to remove bacteria and cellular debris. One hundred microliters of supernatant was analyzed using commercially available interleukin-6 (IL-6) and IL-8 human antibody pairs (Invitrogen) according to the manufacturer's instructions. For detection, plates were incubated with horseradish peroxidase–goat anti-mouse immunoglobulin G (1:5,000) for 1 h. Plate-bound peroxidase was detected using TMB (Invitrogen), and the absorbance was measured at 490 nm in a plate reader. The assay was performed two times in triplicate.
Serum bactericidal and growth assays.
Bacterial colonies grown for 16 to 18 h on GC agar plates were suspended in DMEM, filtered through a 5-μm-pore-size filter to break apart bacterial aggregates, and diluted to an OD600 of 0.32 (corresponding to 1 × 108 CFU/ml, as confirmed by plating). Pooled normal human serum (NHS) was purchased from Sigma (H4522). One hundred microliters of bacterial suspension was added to the wells of a 24-well cell culture plate containing 900 μl of prewarmed DMEM supplemented with NHS to a final serum concentration of 35%. After incubation in NHS, the bacteria were serially diluted and plated. Bacterial growth in cell culture supernatants was performed in 12-well cell culture plates. To measure the optical density, bacteria were gently mixed by pipetting at the indicated time points. The assay was performed three times in triplicate. For live-cell microscopy, bacteria were added to an uncoated 24-well glass-bottom dish (MatTek) in an atmosphere of 5% CO2, with images acquired every 6 min for 12 h.
Mouse model of systemic meningococcal infection.
The CD46 transgenic BL6 mouse strain used for in vivo studies expresses human CD46 in a human-like pattern and has been described previously (11, 12). Six- to 8-week-old mice were challenged intraperitoneally with 108 CFU of wild-type FAM20, the ΔpilU mutant, or the ΔpilT mutant (n = 7 to 9) in 100 μl of GC broth. Bacteremia was determined at 2, 6, 24, and 48 h postchallenge by serial dilution and plating of 5 μl of blood sampled from the tail vein. Mice were monitored over a period of 3 days and euthanized upon signs of lethal infection. Animal care and experiments were performed in accordance with institutional guidelines and have been approved by national ethical committees.
Statistical analysis.
Student's t test was used to analyze differences between two groups. Differences between multiple groups were analyzed using analysis of variance (ANOVA) followed by a post hoc Tukey's honestly significantly different (HSD) test. Data from animal experiments were analyzed with the Kruskal-Wallis nonparametric one-way ANOVA. P values of <0.05 were considered statistically significant. Statistical analysis was performed using GraphPad Prism 4 software and the R statistical package.
RESULTS
Deletion of pilU promotes bacterial adherence to human pharyngeal epithelial cells.
To study the role of PilU in meningococcal infection, we generated a set of mutants of the N. meningitidis serogroup C strain FAM20. First, we constructed a pilU deletion mutant (ΔpilU) by replacing the pilU open reading frame with the tetracycline resistance gene tetA. We then complemented the mutant by reintroducing pilU driven from a synthetic σ70 promoter integrated at a noncoding region of the chromosome (ΔpilU_C strain). Finally, we deleted pilT from both the ΔpilU and ΔpilU_C strains by transforming these strains with genomic DNA from a previously constructed pilT-negative mutant (ΔpilT), generating the ΔpilT ΔpilU and ΔpilT ΔpilU_C strains. To evaluate bacterial adherence to pharyngeal epithelial FaDu cells, we infected cells for 2 h with the mutant and wild-type strains at a 1:1 ratio and then plated adherent bacteria on selective and nonselective media. The CI was defined as the ratio of the number of adherent mutant bacteria to the number of adherent wild-type bacteria. The pilU mutant adhered 5-fold better than the wild-type strain, and this effect was abolished in the complemented ΔpilU_C strain (Fig. 1). As expected, the ΔpilT mutant bound much more strongly than the wild type, with a CI of 15. In comparing the ΔpilT ΔpilU double mutant strain with the ΔpilT mutant, no additional increase in adhesion was detected, demonstrating that the increase in adhesion resulting from the removal of the PilU protein was dependent on PilT (Fig. 1). We also performed single-strain adhesion assays, which confirmed these results (data not shown). To exclude changes in adhesion-associated components, the mutants and the wild type were compared in terms of their expression of several virulence factors. The ΔpilU and complemented ΔpilU_C mutants retained expression of virulence-linked proteins such as PilC1/PilC2, PilT, PilE, and Opa (Fig. 2), as well as PilQ and NafA (data not shown). There were no detectable differences in either outer membrane protein profiles or LOS expression between the ΔpilU mutant and the wild type or the complemented pilU mutant (see Fig. S1 in the supplemental material). Electron microscopy studies did not reveal any differences in piliation level or pilus morphology between the mutants and the parental strain (data not shown). In a recent study, pilU mutants were found to attach 3-fold more strongly than the wild type to HUVEC at 90 min postinfection (4). In conclusion, the stronger binding of the pilU mutant suggests that PilU also has antiadhesion properties that are activated upon the interaction of bacteria and epithelial host cells and that this effect is masked by a concurrent mutation in pilT.
Fig 1.

A pilU mutant outcompetes wild-type FAM20 in a competitive adhesion assay. The assay was performed using wild-type FAM20 (WT), a pilU mutant (ΔpilU), a complemented pilU mutant (ΔpilU_C), a pilT mutant (ΔpilT), a pilU pilT double mutant (ΔpilT ΔpilU), and the double mutant complemented with pilU (ΔpilT ΔpilU_C). Mutant and wild-type bacteria were mixed at a 1:1 ratio as determined by the OD600 and viable counts. The bacterial mixture was immediately added to FaDu cells (total MOI = 100) and allowed to adhere for 2 h in a standard adhesion assay. The competitive index denotes the relative amounts of mutants adherent to cells as determined by plating on both selective and nonselective media. The dashed line indicates a competitive index of 1, i.e., no difference in adhesion. Error bars indicate standard deviations, and asterisks denote statistically significant differences from a CI of 1 (P < 0.001; paired t test).
Fig 2.

Characterization of adhesion factors in the pilU mutant. Immunoblots show the expression levels of PilC, PilT, PilE, and Opa in the wild-type FAM20 (WT), pilU mutant (ΔpilU), and complemented (ΔpilU_C) strains. Controls for the PilC immunoblot were FAM20 mutants expressing only one defined PilC variant, i.e., lanes PilC1+/PilC2− and PilC1−/PilC2+. The control for the PilT immunoblot was the PilT-deficient mutant (ΔpilT). Whole-cell lysates were analyzed in a 2-color Western blot and visualized with anti-PilC, anti-PilT, anti-pili, and anti-Opa antibodies in combination with a monoclonal anti-EF-Tu antibody. The EF-Tu band intensity was used as a loading control and used for normalization. R.I. denotes the relative EF-Tu-normalized band intensity for at least three separate experiments, set to 1.0 for the wild type. Asterisks denote statistically significant differences relative to the wild type (P < 0.05; Student's t test).
PilU- and PilT-deficient mutants induce proinflammatory responses similar to those in wild-type bacteria.
Induction of proinflammatory cytokines upon bacterial infection is a critical defense mechanism and constitutes the first step toward elimination of pathogenic microorganisms. Cytokines are stimulated when the host cells recognize bacterial pathogen-associated molecular patterns. To study whether PilU and PilT can modulate cytokine expression, we investigated the cellular response to infection of the wild type, the ΔpilU mutant, and the complemented ΔpilU_C mutant by measuring the release of the proinflammatory cytokines IL-6 and IL-8 from the human pharyngeal epithelial cell line FaDu at 2 and 8 h postinfection. In these studies, we did not observe any measurable differences between the strains, indicating that PilU and PilT are not involved in mediating cytokine stimulation in host cells (Fig. 3).
Fig 3.

pilU and pilT mutants stimulate IL-6 and IL-8 similarly to the stimulation by the wild type. The graphs show the release of IL-6 (A) and IL-8 (B) from FaDu cells following infection with bacteria (MOI = 100) for 2 h (black bars) and 8 h (white bars), as quantified by ELISA of the cell culture supernatant. The following strains were used: wild-type FAM20 (WT), pilU mutant (ΔpilU), complemented pilU mutant (ΔpilU_C), pilT mutant (ΔpilT), pilU pilT double mutant (ΔpilT ΔpilU), and the double mutant complemented with pilU (ΔpilT ΔpilU_C). Values are normalized to those for uninfected controls at each time point. N.C. (negative control) indicates uninfected cells. Error bars show standard deviations.
PilU affects microcolony formation and dispersal both on epithelial cells and in cell-free medium.
To study aggregation and microcolony formation, we next monitored bacterial adherence to host epithelial cells over a period of 12 h by time-lapse microscopy. Bacteria were labeled with a nontoxic N-hydroxysuccinimide ester-based dye and allowed to adhere to FaDu cells in glass-bottom cell culture dishes at an MOI of 100. At 2 h, nonadhered bacteria were removed by careful washing to prevent bacterial overgrowth, and incubation was continued in fresh cell medium. Observation in the microscope was performed with four fields of view per sample in duplicate on three independent occasions. The wild type, the ΔpilU mutant, and the ΔpilU_C complemented mutant all adhered primarily as single bacteria, with no detectable clumping, whereas a control pilT mutant adhered in large aggregates (Fig. 4A, top row). These data demonstrate that the stronger binding of the pilU mutant than the wild type (Fig. 1) was not due to the aggregation or clumping of bacteria on host cells. At later time points, bacterial microcolonies started to build up on the host cell surface. We defined microcolony formation as the time point when at least one persistent bacterial aggregate with a diameter that exceeded 10 μm became visible. For the wild-type strain, microcolonies became visible 4 h ± 30 min (mean ± standard deviation [SD]) after infection and reached their maximum size, with an average diameter of 12 μm, at 5.5 h ± 10 min, after which they quickly collapsed into single bacteria over a period of 20 min. The microcolonies of the ΔpilU mutant started to form after 6 h ± 65 min (SD) and reached an average size of 14.5 μm at 9 h ± 10 min, after which they also collapsed into single bacteria over a 20-min time frame (Fig. 4A). The starts of microcolony formation (Fig. 4B) and microcolony dispersal (Fig. 4C) were significantly different between the wild-type strain and the ΔpilU mutant. At 4 h, the ΔpilU_C strain had an average microcolony size similar to that of the wild type, supporting the complemented phenotype. As a control, the experiment was also performed with unlabeled bacteria, with similar results (data not shown).
Fig 4.

The pilU mutant is delayed in microcolony formation on human epithelial cells. (A) Live-cell phase-contrast and fluorescence imaging of pharyngeal epithelial FaDu cells infected with wild-type FAM20 and the ΔpilU, ΔpilU_C, and ΔpilT mutants during a total time of 12 h. Representative time points were chosen to show when the wild type and the pilU mutant reached their respective maximal microcolony sizes. Prior to infection, bacteria were passed through a 5-μm-pore-size filter to break apart bacterial aggregates. The initial inoculum of bacteria was stained with the fluorescent DyLight488 N-hydroxysuccinimide ester, visible in red. Microcolonies are highlighted with a yellow dashed line around the periphery. The ΔpilT mutant was included as a positive control for aggregation. Bar = 10 μm. Quantification of microcolony formation (B) and microcolony dispersal (C) was done for the wild type and the ΔpilU mutant. Microcolony formation was determined as the time point when one persistent microcolony with a size of 10 μm first became visible. Microcolony dispersal was defined as the time point when tight bacterial aggregates visibly started to dissolve into single bacteria. Values are means ± standard deviations for the experiment depicted in panel A and are representative of all assays performed. Significant differences from the wild type are indicated with asterisks (P < 0.05; two-tailed Student's t test).
We next performed the same type of experiment in liquid medium in the absence of host cells and obtained similar results. Bacteria suspended in DMEM supplemented with fetal calf serum were monitored by time-lapse microscopy for 12 h. The wild-type strain and complemented ΔpilU_C strain reached their maximum microcolony size at around 3 h, whereas the ΔpilU mutant displayed delayed microcolony formation that peaked at 9 h (Fig. 5). Because bacterial growth rates might influence the formation of microcolonies, we performed bacterial growth assays with cell culture supernatants. There was no detectable difference in the growth rates of the wild type, the ΔpilU mutant, and the ΔpilU_C complemented strain, excluding the possibility that variation in growth rates caused delayed microcolony formation (data not shown). Taken together, these data demonstrate that pilU mutants have delayed microcolony formation relative to that of the wild type. Thus, the presence of PilU facilitates the formation of microcolonies both in liquid medium and on host epithelial cells.
Fig 5.

Microcolony formation is delayed in a meningococcal pilU mutant in cell-free medium. Live-cell phase-contrast microscopy was performed on bacteria growing in cell-free medium. Representative images were selected based on when the wild type and the ΔpilU mutant reached their respective maximal microcolony sizes. Microcolonies are highlighted with a yellow dashed line around the periphery. The ΔpilT mutant was included as a positive control for aggregation. Bar = 10 μm.
In a time-lapse study, the initial inoculum is fluorescently labeled, but the surface labeling of bacteria becomes diluted as they grow and divide, meaning that actively dividing bacteria and their offspring quickly lose their fluorescence. Because the inoculum was fluorescently visible, we were able to follow the fate of these bacteria. We determined that all microcolonies, except for those of the ΔpilT and ΔpilT ΔpilU strains (data not shown, but identical to those for the ΔpilT strain), but even small microcolonies that formed very early, were composed almost exclusively of nonfluorescent bacteria (Fig. 4). These data indicate that microcolonies are formed primarily by actively dividing bacteria for both the wild type and the pilU mutant and are not a result of an association of bacteria present in the initial inoculum. As a control, we included and observed heat-inactivated labeled bacteria and were unable to detect a significant reduction in the fluorescence signal during the course of the experiment, suggesting that the loss of fluorescence was not due to bleaching of the dye (data not shown).
Loss of PilU significantly increases bacterial survival in normal human serum.
N. meningitidis has the capacity to colonize the nasopharyngeal mucosa, pass the epithelial barrier, and reach subepithelial tissues and the bloodstream. An essential feature of meningococcal pathogenesis is resistance to human complement activation. To investigate whether PilU and PilT contribute to complement resistance, we performed a serum-mediated killing assay. Wild-type N. meningitidis FAM20 and its ΔpilU mutant were incubated in 35% NHS for 60 min and then spread on plates for viable counts. The ΔpilU mutant survived significantly better than the wild type, with 51% and 7% of the inoculum recovered, respectively (Fig. 6A). The serum resistance of the complemented ΔpilU_C strain was reduced almost to the wild-type level, supporting a complemented phenotype. The serum resistance of the ΔpilT and ΔpilT ΔpilU mutants did not differ significantly from that of the wild-type strain, again indicating a function of PilU dependent on PilT, or possibly a function masked by ΔpilT mutant-mediated hyperpiliation (Fig. 6A). The same assay was performed with heat-inactivated NHS, and no difference in growth between the mutant and wild-type strains was observed (data not shown). Further examination of serum-mediated killing via microscopy showed that the serum-resistant phenotype of the ΔpilU mutant at 1 h was not mediated by bacterial aggregates present in the inoculum (data not shown). To exclude the possibility that capsule expression levels differed between the pilU mutant and the wild type, we measured capsule levels by ELISA. There was no difference in capsule level between the mutant and wild-type strains (Fig. 6B). Additionally, the pilU mutant retained the LOS and Opa expression of the wild type (Fig. 2; see Fig. S1 in the supplemental material). In conclusion, we determined that a pilU mutant was 8-fold more resistant to NHS than the wild-type strain, in a pilT-dependent manner.
Fig 6.

Loss of PilU increases serum resistance. (A) Survival of the wild-type strain (WT), the ΔpilU mutant, the ΔpilU_C mutant, the ΔpilT mutant, the ΔpilT ΔpilU double mutant, and the ΔpilT ΔpilU_C mutant after 60 min of incubation in 35% NHS. Bacterial survival is expressed as the percentage of CFU remaining from the initial inoculum of 107 CFU in 1 ml of medium, which was set to 100%. Error bars show standard deviations. Statistically significant differences from the wild type are indicated with asterisks (P < 0.001; Tukey's range test). (B) Measurement of capsular polysaccharide by ELISA. A capsule-negative mutant (SiaD−) was used as the negative control. Purified capsule (P.C.) was used as the positive control.
ΔpilT and ΔpilU mutants are not as virulent as the wild-type strain in a mouse model of meningococcal disease.
Next, we aimed to study the role of PilT and PilU in vivo. We used a mouse model of meningococcal infection with the possibility to follow bacteremia and disease outcomes for 3 days (12). CD46 transgenic mice were infected intraperitoneally (i.p.) with 108 CFU of wild-type FAM20 or the ΔpilT or ΔpilU mutant. Levels of bacteremia were determined at 2, 6, 24, and 48 h postinfection by the serial dilution and plating of blood samples. The status of the mice was monitored regularly until 3 days postinfection, after which mice had either developed signs of lethal disease and been euthanized or recovered from the infection. At 6 h and later, bacteremia levels in mice infected with the ΔpilT mutant were significantly lower than those in mice infected with the wild-type strain (Fig. 7A). Consequently, mice infected with the ΔpilT mutant demonstrated 50% survival, whereas mice infected with the wild-type strain demonstrated 0% survival, which represented a significant difference (Fig. 7B). Interestingly, mice infected with the ΔpilU mutant also had a significant increase in survival compared to mice challenged with the wild-type strain (Fig. 7B). However, blood counts were not significantly different between the wild type and the ΔpilU mutant at any time point. Still, meningococcal blood titers from mice challenged with the ΔpilU mutant showed a trend of lower values than those for mice challenged with the wild-type strain, as the bacteremia seemed to decline more rapidly at the 24- and 48-h time points after being initially identical at 2 and 6 h postchallenge (Fig. 7A). It is also interesting that mice infected with the ΔpilU or ΔpilT mutant had a delayed initial onset of sickness (36 h versus 24 h) and displayed a more constant rate of onset of terminal symptoms throughout the course of the experiment than the sharp drop-off seen for the wild-type strain (Fig. 7B). Survival of the wild-type and mutant strains in 35% normal mouse serum obtained from the CD46 transgenic mice did not differ (data not shown). In fact, the mouse serum did not have a bactericidal effect on the meningococci. Taken together, these results show for the first time that expression of PilU and PilT contributes extensively to meningococcal virulence in an in vivo model.
Fig 7.
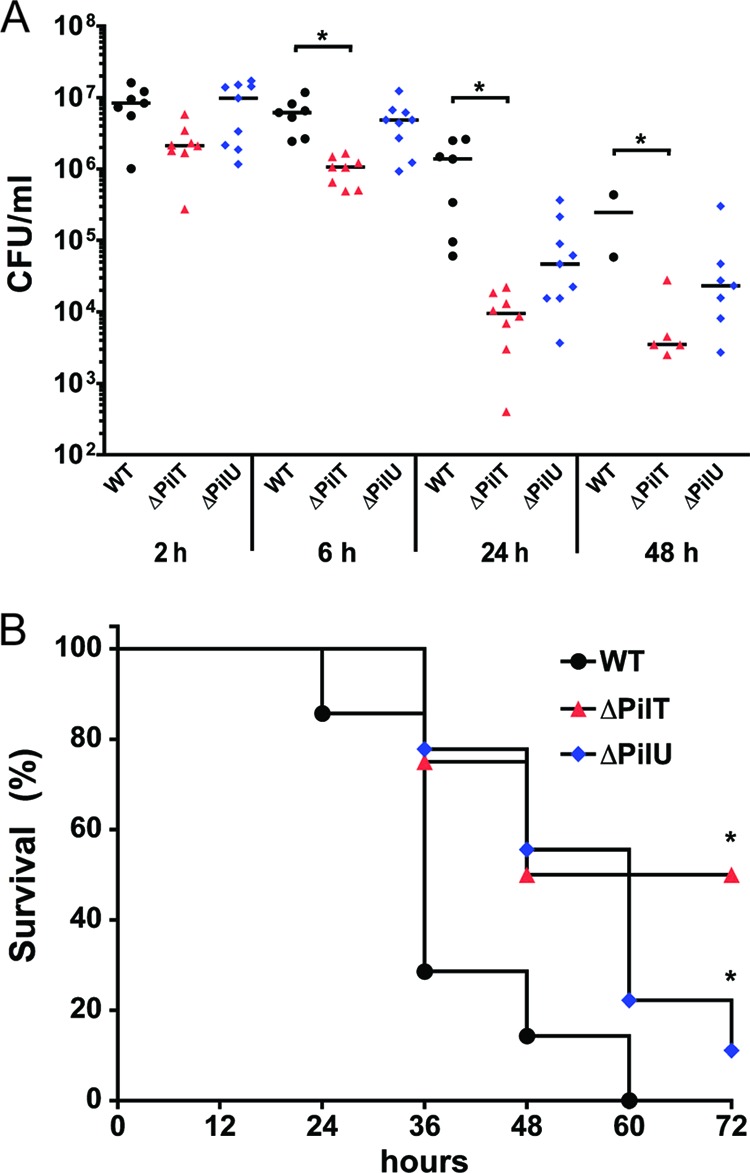
Meningococcal ΔpilT and ΔpilU mutants are less virulent than the wild type in a mouse model of meningococcal disease. Human CD46 transgenic mice were infected i.p. with the wild-type strain, the ΔpilT mutant, or the ΔpilU mutant (108 CFU/mouse). (A) Bacterial counts in blood (CFU/ml) were determined 2, 6, 24, and 48 h after the start of infection. Statistically significant values are indicated with asterisks (P < 0.05; Kruskal-Wallis nonparametric one-way ANOVA). (B) Survival of mice (n = 7 to 9 mice per group) after i.p. challenge with the wild-type strain, the ΔpilT mutant, or the ΔpilU mutant (*, P < 0.05; Kaplan-Meier survival analysis).
DISCUSSION
In this work, we investigated the roles of PilU and PilT in meningococcal virulence. Earlier studies have shown that inactivation of pilU in both N. gonorrhoeae and N. meningitidis results in increased binding to host cells compared to that of wild-type strains (4, 20). We showed that PilU affects meningococcal microcolony formation both during infection of cells and in medium alone. The presence of PilU enabled meningococci to form microcolonies at earlier time points than those observed in the absence of PilU. In addition, a meningococcal pilU deletion mutant became much more resistant to human serum than the wild type, indicating that the ability to control PilU may be important during bloodstream invasion. However, in investigating the ability of meningococci to cause systemic disease in mice, we found that the lack of PilU or PilT reduces virulence in terms of bacterial blood counts and disease development, suggesting that these proteins are necessary for full virulence of N. meningitidis.
Ever since the PilU protein was originally described for Neisseria gonorrhoeae, its function has remained enigmatic, with the conflicting phenotypes of increased host cell adhesion and reduced autoagglutination (20). Infection of epithelial cells with gonococci has been reported to result in extensive microcolony formation at 2 h postinfection (20), indicating variations in Tfp biogenesis and dynamics between species. In this work, we found that microcolony formation of the meningococcal ΔpilU mutant was delayed 3 to 4 h compared to that of the wild-type strain both during attachment to epithelial cells and in cell culture medium (Fig. 4 and 5). However, the ΔpilU meningococcal mutant showed a 5-fold increase in the initial bacterial adhesion to FaDu epithelial cells compared to the wild type (Fig. 1), which closely matches the previously observed 8-fold increase of PilU-deficient gonococci adhering to human ME180 cells (20) and the 3-fold increase of N. meningitidis gonococci adhering to HUVEC (4). Increased binding of pilU mutants has been speculated to be attributable to bacterial binding in aggregates to host cells. By utilizing nondestructive fluorescence labeling of bacteria in a live-cell infection assay, we are confident in concluding that both the wild-type strain and the ΔpilU mutant primarily bind epithelial cells as single diplococci under standard assay conditions (Fig. 4). This observation implies that individual bacterial cells of the ΔpilU strain display an inherent increase in cellular adhesion that is not coupled to the ability to form microcolonies.
Microcolony formation has been linked to multiple pilus-associated proteins, such as the positive regulators PilX (10) and PilZ (4) and the negative regulators NafA (14), PilV, PilT2 (4), and PilT (32). Therefore, it is extremely difficult to speculate about the cause of the dysregulation observed for the pilU mutant. However, because pilU mutant microcolonies do not display gross differences in morphology compared to those of the wild type, aside from a slight increase in average size, it does not seem likely that there are any absolute deficiencies in the above-mentioned proteins. Instead, PilU may be involved in the interaction, proper localization, and dynamics of surface components.
The serum-resistant phenotype of the ΔpilU mutant is intriguing. Certain strains of N. meningitidis express a 29-kDa protein, factor H binding protein (fHBP), that binds the complement regulator factor H. Expression levels of fHBP have been shown to correlate with the level of fH binding and thus with increasing serum resistance (15). We cannot at this point exclude a change in expression or localization of fHBP as the cause of the increase in serum resistance in the pilU-negative mutant. How the absence of PilU makes bacteria more serum resistant in a PilT-dependent manner remains to be determined.
The importance of PilT and PilU during disease in vivo has been investigated previously in the Tfp-expressing species Pseudomonas aeruginosa and Dichelobacter nodosus (7, 9, 34). In Pseudomonas, both PilU and PilT are required for corneal colonization and for bacterial spread from the lung to the liver in a mouse model of acute pneumonia, whereas the absence of PilT and PilU does not significantly affect mortality. PilU-deficient mutants of P. aeruginosa are hyperpiliated and have lost twitching motility, supporting a difference in PilU phenotypes between P. aeruginosa and N. meningitidis (30). In this study, we examined for the first time the impact of PilT and PilU on disease caused by N. meningitidis. In an in vivo murine model of meningococcal disease, we found a 50% reduced mortality in mice challenged with the ΔpilT mutant and a smaller but still significant reduction of mortality in mice infected with the ΔpilU mutant. The increased resistance to human serum observed for the pilU mutant does not confer any advantage in mouse serum, most likely because of an effect of species-specific serum components. The reduced virulence of the pilU mutant observed in the mouse model may instead be due to increased cellular clearance of bacteria. It is of special interest that we were able to recover bacteria from the blood of mice injected i.p. with PilT-deficient bacteria, meaning that even though this mutant is hyperpiliated and lacks retracting pili, it can still cross over from the peritoneal cavity, enter the bloodstream, and cause disease, albeit at a drastically reduced rate. In addition, the data presented here argue that the roles of PilU and PilT in disease may differ in N. meningitidis and P. aeruginosa.
To summarize, we have shown that microcolony formation is delayed in bacteria lacking pilU, implying that PilU facilitates the formation and subsequent dispersal of microcolonies on epithelial cells. Furthermore, PilU renders bacteria more sensitive to normal human serum, in a PilT-dependent manner. In a mouse model of disease, both PilT and PilU are essential for full virulence of N. meningitidis. Although the results described in this paper answer some open questions posed in the literature, they also open exciting new avenues of research in this field.
Supplementary Material
Footnotes
Published ahead of print 16 April 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Aas FE, et al. 2006. Neisseria gonorrhoeae type IV pili undergo multisite, hierarchical modifications with phosphoethanolamine and phosphocholine requiring an enzyme structurally related to lipopolysaccharide phosphoethanolamine transferases. J. Biol. Chem. 281:27712–27723 [DOI] [PubMed] [Google Scholar]
- 2. Aas FE, Lovold C, Koomey M. 2002. An inhibitor of DNA binding and uptake events dictates the proficiency of genetic transformation in Neisseria gonorrhoeae: mechanism of action and links to type IV pilus expression. Mol. Microbiol. 46:1441–1450 [DOI] [PubMed] [Google Scholar]
- 3. Albiger B, Johansson L, Jonsson AB. 2003. Lipooligosaccharide-deficient Neisseria meningitidis shows altered pilus-associated characteristics. Infect. Immun. 71:155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown DR, Helaine S, Carbonnelle E, Pelicic V. 2010. Systematic functional analysis reveals that a set of seven genes is involved in fine-tuning of the multiple functions mediated by type IV pili in Neisseria meningitidis. Infect. Immun. 78:3053–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chamot-Rooke J, et al. 2011. Posttranslational modification of pili upon cell contact triggers N. meningitidis dissemination. Science 331:778–782 [DOI] [PubMed] [Google Scholar]
- 6. Chiang P, et al. 2008. Functional role of conserved residues in the characteristic secretion NTPase motifs of the Pseudomonas aeruginosa type IV pilus motor proteins PilB, PilT and PilU. Microbiology 154:114–126 [DOI] [PubMed] [Google Scholar]
- 7. Comolli JC, et al. 1999. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect. Immun. 67:3625–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frasch CE, Mocca LF. 1978. Heat-modifiable outer membrane proteins of Neisseria meningitidis and their organization within the membrane. J. Bacteriol. 136:1127–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han X, et al. 2008. Twitching motility is essential for virulence in Dichelobacter nodosus. J. Bacteriol. 190:3323–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Helaine S, et al. 2005. PilX, a pilus-associated protein essential for bacterial aggregation, is a key to pilus-facilitated attachment of Neisseria meningitidis to human cells. Mol. Microbiol. 55:65–77 [DOI] [PubMed] [Google Scholar]
- 11. Johansson L, et al. 2003. CD46 in meningococcal disease. Science 301:373–375 [DOI] [PubMed] [Google Scholar]
- 12. Johansson L, et al. 2005. Human-like immune responses in CD46 transgenic mice. J. Immunol. 175:433–440 [DOI] [PubMed] [Google Scholar]
- 13. Jones A, Georg M, Maudsdotter L, Jonsson AB. 2009. Endotoxin, capsule, and bacterial attachment contribute to Neisseria meningitidis resistance to the human antimicrobial peptide LL-37. J. Bacteriol. 191:3861–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuwae A, et al. 2011. NafA negatively controls Neisseria meningitidis piliation. PLoS One 6:e21749 doi:10.1371/journal.pone.0021749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Madico G, et al. 2006. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J. Immunol. 177:501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Merz AJ, So M, Sheetz MP. 2000. Pilus retraction powers bacterial twitching motility. Nature 407:98–102 [DOI] [PubMed] [Google Scholar]
- 17. Naessan CL, et al. 2008. Genetic and functional analyses of PptA, a phospho-form transferase targeting type IV pili in Neisseria gonorrhoeae. J. Bacteriol. 190:387–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nassif X, et al. 1994. Roles of pilin and PilC in adhesion of Neisseria meningitidis to human epithelial and endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 91:3769–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nassif X, et al. 1993. Antigenic variation of pilin regulates adhesion of Neisseria meningitidis to human epithelial cells. Mol. Microbiol. 8:719–725 [DOI] [PubMed] [Google Scholar]
- 20. Park HS, Wolfgang M, Koomey M. 2002. Modification of type IV pilus-associated epithelial cell adherence and multicellular behavior by the PilU protein of Neisseria gonorrhoeae. Infect. Immun. 70:3891–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pelicic V. 2008. Type IV pili: e pluribus unum? Mol. Microbiol. 68:827–837 [DOI] [PubMed] [Google Scholar]
- 22. Pujol C, Eugene E, de Saint Martin L, Nassif X. 1997. Interaction of Neisseria meningitidis with a polarized monolayer of epithelial cells. Infect. Immun. 65:4836–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pujol C, Eugene E, Marceau M, Nassif X. 1999. The meningococcal PilT protein is required for induction of intimate attachment to epithelial cells following pilus-mediated adhesion. Proc. Natl. Acad. Sci. U. S. A. 96:4017–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rahman M, Kallstrom H, Normark S, Jonsson AB. 1997. PilC of pathogenic Neisseria is associated with the bacterial cell surface. Mol. Microbiol. 25:11–25 [DOI] [PubMed] [Google Scholar]
- 25. Rudel T, et al. 1995. Role of pili and the phase-variable PilC protein in natural competence for transformation of Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U. S. A. 92:7986–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rudel T, Scheurerpflug I, Meyer TF. 1995. Neisseria PilC protein identified as type-4 pilus tip-located adhesin. Nature 373:357–359 [DOI] [PubMed] [Google Scholar]
- 27. Sjolinder H, Jonsson AB. 2007. Imaging of disease dynamics during meningococcal sepsis. PLoS One 2:e241 doi:10.1371/journal.pone.0000241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tonjum T, Caugant DA, Dunham SA, Koomey M. 1998. Structure and function of repetitive sequence elements associated with a highly polymorphic domain of the Neisseria meningitidis PilQ protein. Mol. Microbiol. 29:111–124 [DOI] [PubMed] [Google Scholar]
- 29. Virji M, et al. 1991. The role of pili in the interactions of pathogenic Neisseria with cultured human endothelial cells. Mol. Microbiol. 5:1831–1841 [DOI] [PubMed] [Google Scholar]
- 30. Whitchurch CB, Mattick JS. 1994. Characterization of a gene, pilU, required for twitching motility but not phage sensitivity in Pseudomonas aeruginosa. Mol. Microbiol. 13:1079–1091 [DOI] [PubMed] [Google Scholar]
- 31. Wolfgang M, et al. 1998. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol. Microbiol. 29:321–330 [DOI] [PubMed] [Google Scholar]
- 32. Wolfgang M, Park HS, Hayes SF, van Putten JP, Koomey M. 1998. Suppression of an absolute defect in type IV pilus biogenesis by loss-of-function mutations in pilT, a twitching motility gene in Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U. S. A. 95:14973–14978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wolfgang M, van Putten JP, Hayes SF, Dorward D, Koomey M. 2000. Components and dynamics of fiber formation define a ubiquitous biogenesis pathway for bacterial pili. EMBO J. 19:6408–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zolfaghar I, Evans DJ, Fleiszig SM. 2003. Twitching motility contributes to the role of pili in corneal infection caused by Pseudomonas aeruginosa. Infect. Immun. 71:5389–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.