

Abstract
Concurrent with the global escalation of the AIDS pandemic, cryptococcal infections are increasing and are of significant medical importance. Furthermore, Cryptococcus neoformans has become a primary human pathogen, causing infection in seemingly healthy individuals. Although numerous studies have elucidated the virulence properties of C. neoformans, less is understood regarding lung host immune factors during early stages of fungal infection. Based on our previous studies documenting that pulmonary surfactant protein D (SP-D) protects C. neoformans cells against macrophage-mediated defense mechanisms in vitro (S. Geunes-Boyer et al., Infect. Immun. 77:2783–2794, 2009), we postulated that SP-D would facilitate fungal infection in vivo. To test this hypothesis, we examined the role of SP-D in response to C. neoformans using SP-D−/− mice. Here, we demonstrate that mice lacking SP-D were partially protected during C. neoformans infection; they displayed a longer mean time to death and decreased fungal burden at several time points postinfection than wild-type mice. This effect was reversed by the administration of exogenous SP-D. Furthermore, we show that SP-D bound to the surface of the yeast cells and protected the pathogenic microbes against macrophage-mediated defense mechanisms and hydrogen peroxide (H2O2)-induced oxidative stress in vitro and in vivo. These findings indicate that C. neoformans is capable of coopting host SP-D to increase host susceptibility to the yeast. This study establishes a new paradigm for the role played by SP-D during host responses to C. neoformans and consequently imparts insight into potential future preventive and/or treatment strategies for cryptococcosis.
INTRODUCTION
Cryptococcus neoformans is an opportunistic fungal pathogen that is endemic to many regions of the globe and is a leading cause of meningoencephalitis among immunocompromised individuals. Additionally, this fungus causes infection among seemingly immunocompetent persons (16), supporting its evolution to a primary human pathogen capable of initiating infection in seemingly immunocompetent people. C. neoformans cells are broadly subdivided into one of five serotype categories based on genetic properties and surface antigens: serotypes A (C. neoformans var. grubii), B (C. gattii), C (C. gattii), D (C. neoformans var. neoformans), and AD (the hybrid serotype). The majority of clinical isolates are serotype A. Infection is initiated when desiccated yeast cells or basidiospores are inhaled into the lungs, although the mechanisms of yeast cell survival and dissemination following inhalation remain incompletely understood. C. neoformans has been shown to be phagocytosed by alveolar macrophages (AMs), to proliferate intracellularly (15), and to undergo a phagosome extrusion event in which immune cells remain intact (3, 39). Furthermore, the capsule surrounding C. neoformans cells, an important virulence property of the yeast, has been shown to be protective against oxidative stresses (63), which play a critical role in the ability of the host to kill invading C. neoformans cells (2, 5).
Prior to encountering professional phagocytes in the lung, the fungal infectious propagule must interact with surfactant proteins (SPs), which have important functions during innate immune responses. Specifically, surfactant protein A (SP-A) and SP-D have been shown to opsonize and enhance the clearance of a number of microorganisms and allergens (14, 18, 23, 36, 43, 49, 50, 52) and thereby characteristically have protective functions in the lung. The roles of surfactant proteins during fungal infections remain unclear, as a number of studies have reported conflicting results, and few investigations have been performed to evaluate the role of SPs in response to C. neoformans infection in vivo. For example, Schelenz et al. have shown that SP-A binds to C. neoformans cells but does not enhance phagocytosis by macrophages (53); furthermore, we have demonstrated that SP-A does not play a significant role during C. neoformans infection in vivo (19). Moreover, SP-D has also been shown to bind and aggregate C. neoformans cells in vitro (58). An underlying variable likely affecting these results is the use of various C. neoformans strains in different studies. In addition, whether the yeast or basidiospore form of the fungus constitutes the infectious propagule remains unknown; however, despite variability in the size and the shape of basidiospores, together with differences in the associated immune responses, a rapid change to the yeast form is observed in the host (20, 60). Thus, we focused on the role played by SPs in response to the yeast form of serotype A, the most common clinical isolate, and consider the results presented here to be relevant to responses that occur during initial C. neoformans infection.
We have previously demonstrated that preopsonization with SP-D enhances phagocytosis of the acapsular cap59Δ mutant strain by macrophages and leads to the increased survival of both wild-type (H99) and cap59Δ cells (17) in vitro. Interestingly, those results are inconsistent with the paradigm that SP-D plays a host-protective role during innate immune responses, thus changing our hypothesized mechanism for the role of SP-D during C. neoformans infection. Consequently, the present study was undertaken to extend those studies by evaluating the role of SP-D during C. neoformans infection in vivo, utilizing a mouse model of C. neoformans infection. The data presented herein suggest that SP-D does indeed function to protect C. neoformans cells during infection and is exploited by the yeast cells to subvert host pulmonary immune mechanisms. To elucidate the mechanism(s) by which SP-D protects C. neoformans cells, we examined the role of SP-D during C. neoformans (strain H99, serotype A) infection using SP-D−/− and triple-transgenic inducible SP-D−/− mouse strains. Fungal burden and mouse survival were assessed, and in vitro assays were employed to examine the ability of SP-D to modulate C. neoformans growth in response to oxidative stress. We found that the presence of SP-D enhances the survival and proliferation of C. neoformans cells in vivo, as evidenced by the observation that SP-D−/− mice have decreased fungal burden and are partially protected during C. neoformans infection. Furthermore, AMs isolated from SP-D−/− mice demonstrated a greater ability to kill C. neoformans cells than did wild-type AMs, and preopsonization of the yeast cells with SP-D protected them against oxidative stress, an effect similar to that observed previously for the capsule on yeast cells (63), both in vitro and in vivo. Thus, we conclude that SP-D protects C. neoformans cells against host innate immune responses, in particular the activity of oxidants, and that this fungal protection is sufficient to partially overcome the enhanced antifungal activity of SP-D−/− murine macrophages, which may be related their enhanced oxidant production (62). These data suggest that C. neoformans cells have evolved a mechanism to subvert host innate immune defenses via the exploitation of host SP-D, a finding that could impact targeting strategies used to identify and manage patients presenting with cryptococcosis.
MATERIALS AND METHODS
Animals and reagents.
Experiments were conducted using pathogen-free C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME); SP-D−/− mice on the C57BL/6 background, as previously described (31); triple-transgenic [CCSP-rtTA (tetO)7-rSP-D SP-D−/−] inducible SP-D−/− mice, kindly provided by Jeffrey Whitsett (66) (designated plusdoxySP-D+/+ and offdoxySP-D−/− when fed doxycycline or not, respectively); and FVB/N controls for the triple-transgenic mice (Taconic, Hudson River Valley, NY). Inducible mice were raised on chow supplemented with 0.0625% doxycycline (625 ppm) to induce SP-D expression. Two weeks prior to infection with C. neoformans, half of the mice were switched to a diet of standard chow lacking doxycycline to obtain offdoxySP-D−/− mice, and the absence of SP-D in the lungs was validated by Western blot analysis of the bronchoalveolar lavage (BAL) fluid, which was tested following centrifugation to remove cells and cellular debris. All chow was provided ad libitum. For all experiments, the mice were age- and sex-matched and ranged between 8 and 10 weeks of age. Animals were anesthetized via intraperitoneal injection of ketamine (150 mg/kg of body weight) and xylazine (10 mg/kg) and euthanized by intraperitoneal injection of pentobarbital sodium (Nembutal; Abbot Laboratories, North Chicago, IL), followed by exsanguination. For isoflurane anesthesia, each mouse was exposed to isoflurane via inhalation in a closed container containing a Styrofoam platform atop gauze soaked in isoflurane. Mice were immediately removed from the container when their heads fell and rested on the platform.
Isolation of recombinant SP-D.
Recombinant SP-D was isolated from Chinese hamster ovary cells expressing a clone of full-length rat SP-D, purified using maltose affinity chromatography, and stored at 4°C in 5 mM Tris buffer (pH 7.8) containing 2 mM EDTA, as previously described (37). Prior to the use of SP-D, EDTA was removed by dialysis against phosphate-buffered saline (PBS) lacking Ca2+ and Mg2+, to avoid protein precipitation.
Isolation of alveolar macrophages.
Mice were killed by intraperitoneal injection of pentobarbital sodium (Abbott Laboratories, North Chicago, IL), followed by exsanguination. The trachea was cannulated after a midline neck incision was made, and the lungs were lavaged five times with 1.0 ml of PBS solution containing EGTA (1 mM). The BAL fluid volume recovered from each mouse was documented for normalization purposes. The lavage fluid was centrifuged (600 × g for 10 min at 4°C), and the cells were washed with PBS and resuspended in Dulbecco's modified Eagle's medium (DMEM) (BioWhittaker, Inc., Walkersville, MD) containing 10% heat-inactivated fetal bovine serum, glutamine (300 μg/ml), penicillin (100 U/ml), and 20 mM HEPES (pH 7.2).
Culture and maintenance of C. neoformans strains.
Cells of C. neoformans serotype A wild-type strain H99 and the cap59Δ::HYG mutant, a mutant strain created from H99 (48), were maintained in glycerol stocks. Previously, we showed that SP-D increased the uptake of acapsular, avirulent cap59Δ::HYG cells by AMs and that the presence of capsule abrogated this effect. Consequently, H99 and cap59Δ::HYG cells were compared in the present in vivo study. Stocks of the respective strains were streaked out onto yeast extract-peptone-dextrose (YPD) agarose or YPD plus hygromycin agarose plates and incubated at 30°C. Liquid cultures were grown in YPD medium at 30°C for 16 to 18 h in a shaking incubator at 250 rpm.
Microscopy.
For all microscopy, a Zeiss (Thornwood, NY) Axioskop 2 Plus fluorescence microscope with an attached AxioCam MRM digital camera was utilized. A fixed exposure was used for all images, and the results represent data from experiments that were performed at least three times.
Preparation of C. neoformans for macrophage killing assays.
C. neoformans cells grown overnight in YPD liquid medium were collected by centrifugation at 2,000 × g, washed three times, and resuspended in sterile phosphate-buffered saline containing 0.9 μM CaCl2 at a concentration of 2 × 106 yeast cells/ml. Yeast cells were then incubated alone or with 1 μg/ml SP-D for 1 h at 37°C with shaking in an Eppendorf 5436 Thermomixer (Eppendorf, Westbury, NY). For macrophage infection, 10 μl of pretreated yeast cells (20,000 yeast cells) was inoculated into wells containing activated macrophages. This number of yeast cells was chosen to represent a multiplicity of infection (MOI) of 1:1, with the assumption that the macrophage numbers had doubled overnight.
Alveolar macrophage killing assays.
Alveolar macrophages were counted with a hemocytometer using trypan blue to identify dead cells and were resuspended at a concentration of 1 × 105 live cells/ml. One hundred microliters of this suspension (10,000 cells) was plated into a 96-well plate. Macrophages were allowed to adhere overnight, and the medium was subsequently replaced with 100 μl fresh complete medium containing 100 ng/ml lipopolysaccharide (LPS) (serotype 0111:B4) and 200 U/ml mouse gamma interferon for 3 h to activate the cells. An additional 100 μl of complete medium was then added to the cells, and C. neoformans cells pretreated with SP-D were added to the macrophages at an MOI of 1:1. Controls included wells containing macrophages alone and C. neoformans cells alone for each pretreatment condition. Killing was assessed after 16 h by removing the supernatant from each well and lysing the macrophages with 2 washes of 100 μl 0.01% SDS in phosphate-buffered saline. The supernatant and SDS washes were combined for each condition, serially diluted, and plated onto YPD agarose plates for colony forming unit (CFU) analysis. The results were assessed by comparing the CFUs from each macrophage and C. neoformans condition to those of the respective C. neoformans-alone controls, as follows: (CFUs [macrophage + C. neoformans]/CFUs [C. neoformans]) × 100. The resultant values were plotted by setting the yeast-only controls to zero to clearly indicate positive and negatives changes in C. neoformans growth in response to the different experimental conditions. This strategy allowed us to normalize for distinct growth rates of different strains and any effects caused by SP-D preopsonization. Experiments were performed in replicates of five, and the results represent data obtained from at least three independent experiments.
Animal infections.
Mice anesthetized by intraperitoneal injection of ketamine (150 mg/kg) and xylazine (10 mg/kg) were positioned on a taut string secured at either end, hanging from their incisors. C. neoformans cells in sterile phosphate-buffered saline at 1 × 106 yeast cells/ml or 2 × 105 yeast cells/ml were delivered by dropping the inoculum into the nares. Mice treated in the same way with sterile saline and uninfected mice were included as controls. For the delivery of exogenous SP-D, the C. neoformans inoculum was supplemented with 1 μg/ml (oxidative stress assays) or 2 μg/ml (rescue survival) of SP-D, as indicated. Anesthetized mice were kept warm with a heating blanket and monitored until full recovery from the procedure was achieved.
Protein measurement.
C. neoformans strain H99 or cap59Δ cells were intranasally instilled in anesthetized wild-type and SP-D−/− mice at a concentration of 5 × 104 cells/mouse. The mice were then euthanized at 12 h postinfection by a lethal injection of sodium pentobarbital followed by exsanguination. The lungs were lavaged using a 20-gauge cannula to inject phosphate-buffered saline containing 0.2 mM EGTA. The lungs were inflated to full capacity (approximately 1 ml of buffer), and the first 1 ml of lavage buffer was collected and stored at −80°C for later analysis of protein concentrations. The protein concentration in the BAL fluid was determined using the bicinchoninic acid (BCA) protein assay (Pierce), with known dilutions of bovine serum albumin as standards.
Histology.
Wild-type and SP-D−/− mice infected with C. neoformans cells as described above were euthanized by a lethal dose of pentobarbital sodium followed by exsanguination. The lungs were then perfused with warm sterile phosphate-buffered saline and fixed by the intratracheal instillation of 4% paraformaldehyde under gravity flow (27). The lungs were paraffin-embedded, sectioned at a thickness of 4 μm, and stained with hematoxylin and eosin to visualize lung morphology, cell infiltration, and fungal burden.
CFU analyses.
Wild-type and SP-D−/− mice infected with C. neoformans cells as described above were euthanized, and the lungs, spleens, and brains were harvested for CFU analyses at the indicated time points to determine fungal burden and dissemination. Whole organs were homogenized in 1 ml of sterile phosphate-buffered saline using a tissue homogenizer. Serial dilutions of the homogenized tissues were plated onto YPD agarose plates, and the colonies were counted following incubation at 30°C for 2 to 3 days.
Hydrogen peroxide assays.
C. neoformans cells (H99 and cap59Δ) were collected by centrifugation, washed three times with phosphate-buffered saline, and incubated with 1 μg/ml SP-A, 1 μg/ml SP-D, or buffer alone as a control for 1 h at 37°C with shaking. The yeast cells were then centrifuged to remove unbound protein and adjusted to a concentration of 1 × 106 yeast cells/ml in yeast nitrogen base (YNB) medium. The pretreated yeast cells were exposed to 1 mM hydrogen peroxide (H2O2) for 2 h at 37°C with shaking, followed by dilution and plating onto YPD agarose plates for CFU evaluation as described above.
XTT assays.
C. neoformans cells (H99 and cap59Δ) were pretreated and exposed to H2O2 as described above for the H2O2 assays. The samples were then plated onto a 96-well plate in triplicate, and XTT {sodium 3-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro)benzene sulfonic acid hydrate} reagent (Sigma-Aldrich, St. Louis, MO) together with the electron donor phenazine methosulfate (Sigma-Aldrich, St. Louis, MO) were added to each well as previously described (51). This assay is based on a color change as the XXT reagent is metabolized by the organism under study to a formazan product, thus indicating the viability of the sample. Plates were evaluated with a plate reader at an absorbance wavelength of 450 nm to compare differences in cell metabolism under the different conditions.
Survival studies.
Mice were intranasally instilled with 5 × 104 or 1 × 104 yeast cells delivered in 50 μl of sterile phosphate-buffered saline as described above. For the rescue survival studies, 50 μl of exogenous SP-D at a concentration of 2 μg/ml was delivered into the lungs on days 0, 1, 3, 5, 10, and 15 postinfection. SP-D administered on day 0 was delivered together with the infectious inoculum via intranasal instillation. All subsequent doses were delivered via intratracheal instillation under isoflurane anesthesia. Mice were monitored until they regained consciousness and then twice daily for signs of morbidity (weight loss equal to or greater than 15% of their original body weight, symptoms of central nervous system [CNS] infection, respiratory distress, and any other signs of illness), at which point they were euthanized by carbon dioxide inhalation followed by bilateral thoracotomy.
Statistics.
The two-tailed Student t test was used for all comparisons between two groups. For comparisons of multiple groups, a one-way or two-way analysis of variance (ANOVA) followed by Tukey's multiple-comparison or Bonferroni's posttest, respectively, was employed. Survival analyses were conducted by a Kaplan-Meier survival curve analysis using the log-rank (Mantel-Cox) test. All statistical analyses were performed using Graphpad Prism, version 5.0, software. A P value of <0.05 was considered statistically significant.
RESULTS
SP-D−/− mice are protected during C. neoformans infection.
To assess whether SP-D affects the overall disease outcome in C. neoformans-infected mice, age- and weight-matched SP-D−/− and wild-type-background control mice were intranasally instilled with 5 × 104 C. neoformans cells, and survival studies were performed. The SP-D−/− mice displayed a significantly prolonged mean time to death compared to the wild-type mice, as assessed over 35 days (P < 0.0001) (Fig. 1A). To confirm that this observation was not due solely to the heightened inflammatory state associated with the SP-D−/− phenotype (62), survival studies were performed by employing a triple-transgenic SP-D−/− mouse strain containing tet-inducible recombinant rat SP-D (rrSP-D) driven by the CCSP (Clara cell-specific) promoter (66). Mice fed doxycycline express SP-D in the lungs (doxySP-D+/+), whereas those removed from the doxycycline diet lack SP-D (offdoxySP-D−/−). The presence and absence of SP-D in the lungs of doxySP-D+/+ and offdoxySP-D−/− mice were confirmed by Western blot analysis of the bronchoalveolar lavage (BAL) fluid (Fig. 1C). Of note, doxycycline has been observed to have no direct antifungal activity against H99 in vitro. As observed with the SP-D−/− mice, a significant delay in the mean time to death was documented for transgenic mice lacking SP-D (Fig. 1B), which suggested that the presence of SP-D was important for C. neoformans infection and enhanced disease progression (P < 0.0001 for doxySP-D+/+ compared to offdoxySP-D−/− mice).
Fig 1.

SP-D−/− mice are partially protected during C. neoformans H99 infection. (A) Survival curves for wild-type (WT) versus SP-D−/− mice following intranasal instillation of 5 × 104 yeast cells/mouse (P < 0.0001). (B) Survival curves for the inducible SP-D−/− mouse strain compared to controls following infection with 5 × 104 yeast cells/mouse. (C) Western blot showing the presence and absence of SP-D in the BAL fluid of the inducible mice in the absence (SP-D−/−) and in the presence (SP-D+/+) of doxycycline (P < 0.0001 for SP-D−/− groups compared to SP-D+/+ groups; P < 0.005 for the SP-D−/− group compared to the SP-D−/− group treated with SP-D). (D) Survival curves for WT versus SP-D−/− mice with and without administration of exogenous SP-D following intranasal instillation of 1 × 104 yeast cells/mouse. Each experimental group contained at least 10 mice (P < 0.0001).
Based on our previous in vitro results demonstrating that SP-D bound to the surface of C. neoformans cells provides protection against macrophage-mediated defense mechanisms, we tested whether exogenous application of SP-D could reverse the phenotype and improve the survival of SP-D−/− mice during C. neoformans infection. To answer this question, we performed survival analyses of SP-D−/− versus wild-type mice as described above, using a lower inoculum of C. neoformans cells (1 × 104 yeast cells) to obtain a greater difference between the survival curves and thus facilitate the detection of differences in the SP-D-treated and untreated groups. For this experiment, four groups of C. neoformans-infected mice were evaluated: (i) wild-type mice, (ii) wild-type mice with SP-D treatment, (iii) SP-D−/− mice, and (iv) SP-D−/− mice with SP-D treatment. During the course of the survival experiment, exogenous rrSP-D (50 μl at 2 μg/ml) was administered to the mice in the groups treated with SP-D at six time points (days 0, 1, 3, 5, 10, and 15). Although a complete reversal of the SP-D−/− phenotype was not observed, the SP-D−/− group treated with SP-D demonstrated a significantly faster mean time to death than the SP-D−/− group (Fig. 1D) (P < 0.005), consistent with our in vitro data providing evidence that SP-D facilitates the development of cryptococcosis. It is noteworthy that no significant differences were detected between the wild-type group and the wild-type group treated with SP-D, which indicated that the administration of SP-D above the levels present in wild-type control mice did not affect the host immune response to C. neoformans infection.
SP-D−/− mice demonstrate decreased fungal burden compared to wild-type mice at early time points postinfection.
To assess whether the capability of SP-D to promote C. neoformans infection was associated with innate immune responses, we examined the fungal burden in and histology of SP-D−/− and wild-type control mouse lungs at 12 h, 3 days, and 7 days postinfection. The SP-D−/− and wild-type mice displayed comparable fungal burdens in the lung tissue at 3 days postinfection, as confirmed by histology, which revealed similar levels of disease progression in the lung tissues of both strains (Fig. 2). Remarkably, a significantly decreased fungal burden was apparent in SP-D−/− BAL fluid compared to that in wild-type BAL fluid (P < 0.01) (Fig. 2A). Furthermore, in comparison to saline-instilled control mice, SP-D−/− mice infected with H99 or cap59Δ cells demonstrated increased protein levels in the BAL fluid at 12 h postinfection (data not shown). Because the alveolar compartment sampled by lavage represents an initial port of pathogen entry, these results suggest that SP-D plays an important role during initial infection by augmenting the survival and/or proliferation of C. neoformans cells, which may be critical for disease outcomes. In support of this hypothesis, at 7 days postinfection, SP-D−/− mice again displayed a decreased fungal burden in the BAL fluid (P = 0.053) and a significantly diminished fungal burden in the lung tissue (P < 0.01), with considerably less infection apparent by lung histology than for wild-type mice (Fig. 2B).
Fig 2.

SP-D−/− mice display decreased fungal burden in the BAL fluid from the lungs, but not the lung tissue, 3 days after instillation of H99 cells (A) and decreased fungal burden in both compartments at 7 days postinstillation (B). The results for fungal burden in the BAL fluid are presented as CFU/ml of the collected BAL fluid, and those for fungal burden in the lung tissue are presented as CFU/g of collected lung tissue. Each experimental group contained at least 10 mice. Scale bar, 100 μm. *, P = 0.053; **, P < 0.01.
SP-D promotes C. neoformans growth in the lung and increases dissemination to the spleen and brain in vivo.
To ascertain the time course of C. neoformans infection and dissemination, CFU analyses of the lungs, spleen, and brain were performed to compare wild-type and SP-D−/− mouse strains at various time points postinfection. Not only did the SP-D−/− mice display decreased fungal burdens compared to those of wild-type mice in all organs at all time points evaluated, they also revealed a delay in the dissemination of the fungus from the lungs to the spleen and brain; dissemination occurred between 10 and 12 days postinfection in wild-type mice versus 12 to 15 days in SP-D−/− mice (Fig. 3). These studies were also performed using the triple-transgenic mouse strain, and similar results were obtained (data not shown). Of interest, experiments identical to the H99 studies were performed with the cap59Δ mutant strain, which does not disseminate from the lungs, and the results revealed significantly increased fungal burdens in the lungs of wild-type compared to SP-D−/− mice. In fact, infection with this mutant strain appeared to be effectively cleared in the SP-D−/− mice by 80 days postinfection (Fig. 4). These results are consistent with the delayed mean time to death observed for H99-infected SP-D−/− mice compared with SP-D-replete mice and suggest that SP-D interferes with innate immune responses to C. neoformans infection.
Fig 3.
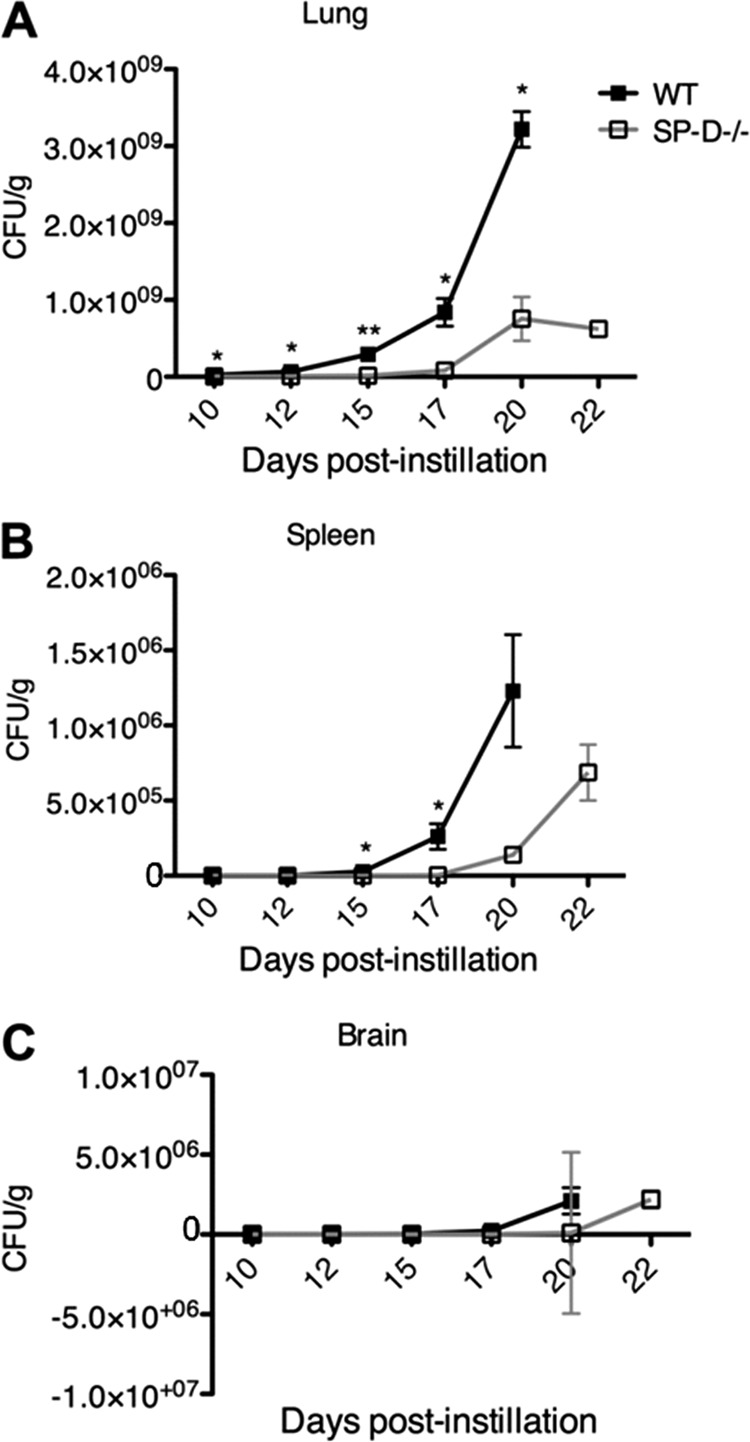
SP-D−/− mice display decreased fungal burden in the lung (A), spleen (B), and brain (C) at days 10, 12, 15, 17, 20, and 22 after instillation of C. neoformans (strain H99). The x axis indicates days postinstillation, and the y axis shows the calculated CFU/g of collected lung, spleen, and brain tissue. Each experimental group contained 4 mice. *, P < 0.05; **, P < 0.001.
Fig 4.
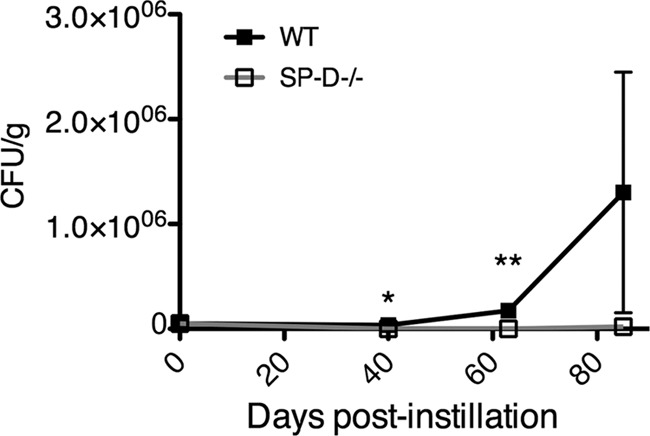
SP-D−/− mice display decreased fungal burden in the lung at days 40, 63, and 85 after instillation of cap59Δ cells. Each experimental group contained 4 mice. *, P < 0.05; **, P < 0.005.
SP-D−/− AMs demonstrate an enhanced ability to kill C. neoformans cells.
To assess whether the heightened inflammatory state of SP-D−/− macrophages contributes to the delay in disease progression observed for SP-D−/− mice, we performed killing assays with alveolar macrophages (AMs) lavaged from SP-D−/− and wild-type mice. We have previously shown that SP-D opsonization protects C. neoformans cells against macrophage-mediated defense mechanisms in vitro using the J774A.1 macrophage cell line (17). Because the differences in survival were greater for the SP-D−/− mice than for both the wild-type mice and the triple-transgenic mice expressing SP-D, we assessed whether SP-D−/− AMs kill C. neoformans cells with greater efficacy than do wild-type AMs. We tested both H99 and cap59Δ cells because we previously observed a relatively poor phagocytosis of H99 cells by AMs in vitro in comparison to AMs in vivo and to cap59Δ cells. The results demonstrated that, indeed, SP-D−/− AMs had an augmented capacity to kill C. neoformans cells, as evidenced by the significantly increased killing of mutant cap59Δ cells (P < 0.005) and a trend toward increased fungal killing of wild-type H99 cells in the presence of SP-D−/− AMs. Furthermore, precoating of the yeast cells with SP-D prior to macrophage infection reversed the enhanced killing ability of SP-D−/− AMs (Fig. 5).
Fig 5.
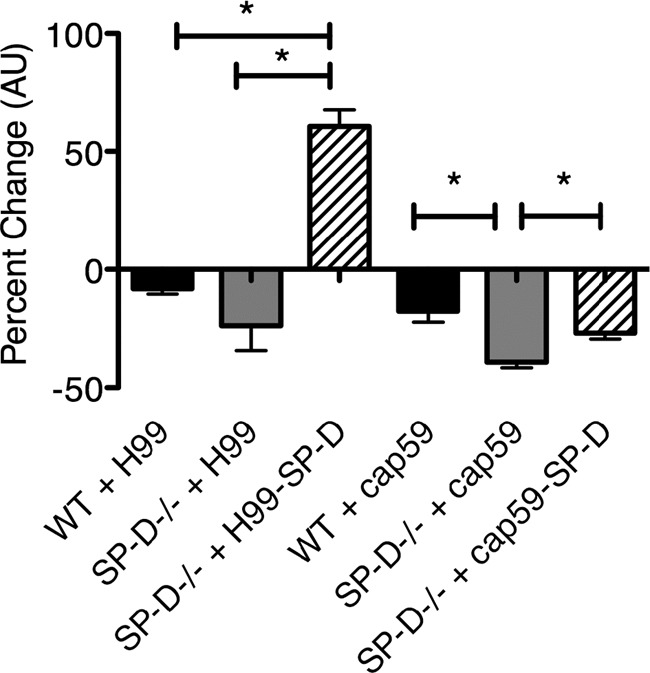
Precoating of C. neoformans cells with SP-D reverses the enhanced ability of SP-D−/− macrophages to diminish C. neoformans cell growth in vitro, compared to wild-type AMs. Alveolar macrophages from wild-type and SP-D−/− mice were isolated from the BAL fluid and incubated with H99 and cap59Δ cells for 16 h to compare the killing abilities of these 2 macrophage populations. *, P < 0.005. AMs isolated from SP-D−/− mice displayed an enhanced ability to kill C. neoformans H99 and cap59Δ cells, and this effect was reversed when the C. neoformans cells were precoated with 5 μg/ml SP-D (n = 5). AU, arbitrary units.
SP-D protects C. neoformans cells against oxidative stress, and yeast cells recovered from infected SP-D−/− mice display a “leaky-capsule” morphology.
To further assess the mechanism(s) of SP-D's pathogen-protective effect, we evaluated H2O2 both in vitro and in vivo (Fig. 6A and B, respectively). Wild-type yeast cells obtained from the BAL fluid of SP-D−/− mice at 3 days postinfection were more susceptible to oxidative insult than those obtained from wild-type mice, as indicated by CFU analysis following exposure to H2O2. Furthermore, precoating of the yeast cells with SP-D prior to instillation reversed this effect, which demonstrated that SP-D had a functional role on the surface of C. neoformans cells: it protected the yeast cells against high levels of oxidative stress, such as those potentially encountered following the immune cell respiratory burst in vivo (P < 0.05) (25, 26, 47) (Fig. 6A). A direct examination of yeast cells from the host via India ink staining revealed an abrogated capsular morphology among those yeast cells recovered from previously infected SP-D−/− mice, as evidenced by the inclusion of ink within the capsular matrix, thus suggesting a less dense or leaky capsule (Fig. 6C).
Fig 6.

H99 cells retrieved from SP-D−/− mice at 3 days postinstillation demonstrate decreased resistance to oxidative stress.(A) H99 or SP-D-coated H99 cells instilled into WT and SP-D−/− mice, lavaged 3 days postinstillation, and exposed to 1 mM H2O2 for 1 h at 37°C. Yeast cells retrieved from SP-D−/− BAL fluid showed a significantly increased susceptibility to oxidative stress (n = 5). (B) H99 cells in SP-D−/− BAL fluid with increasing concentrations of SP-D correlate with increasing resistance to oxidative stress. NT, no protein control (n = 5). *, P < 0.05; **, P < 0.01. (C) C. neoformans cells recovered from SP-D−/− mice display a “leaky-capsule” morphology. Representative yeast cells were recovered from the BAL fluid of SP-D−/− or wild-type mice at 3 days postinfection and stained with India ink. In the absence of SP-D, the India ink penetrates the capsule matrix. Scale bar, 10 μm.
Next, we investigated the protective effect of SP-D in vitro by adding increasing amounts of SP-D to wild-type yeast cells suspended in SP-D−/− BAL fluid prior to H2O2 exposure. Consistent with the in vivo results, SP-D dose-dependently protected C. neoformans cells against oxidative stressors (P < 0.05 and P < 0.01) (Fig. 6B). We further noted that the capsules of C. neoformans cells incubated in the presence of SP-D were larger than those of C. neoformans cells incubated in the absence of SP-D (Fig. 7), and therefore, we considered whether the protective effect of SP-D on yeast cells might be attributable to a facilitation of capsule enlargement, to SP-D itself, or, potentially, to both mechanisms.
Fig 7.
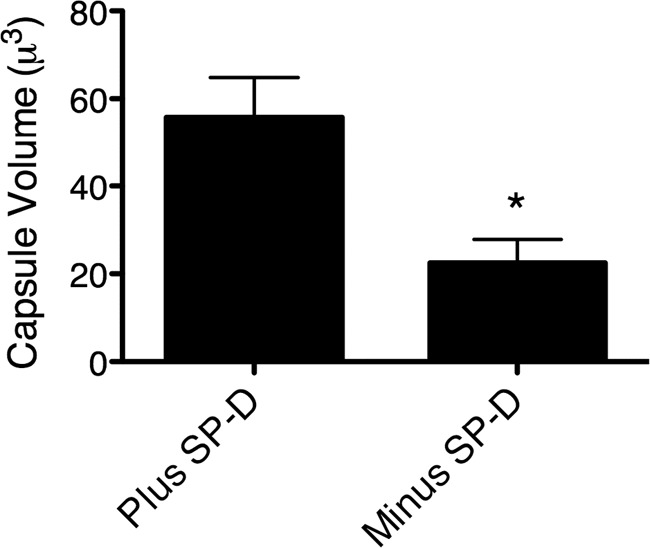
C. neoformans cells incubated in the presence of SP-D exhibit larger capsules than those of C. neoformans cells incubated in the absence of SP-D. Yeast cells were incubated in bronchoalveolar lavage fluid of SP-D−/− with or without 1 μg/ml of rrSP-D for 3 days at 37°C. The yeast cells were then stained with India ink and viewed under a microscope, and micrographs were obtained for capsule measurements. Capsule was measured using Adobe Photoshop CS2. The diameters of the whole cell (DWC) and the cell body (DCB) were measured, the volume was calculated, and the capsule portion of each yeast cell was defined as the difference between the whole cell (plus capsule) and the cell body (no capsule). The capsule volumes were determined using the equation for the volume of a sphere [4/3 × π × (D/2)3], as previously described (64). The capsule volumes of 15 to 20 yeast cells were determined per group. *, P < 0.05.
SP-D is sufficient to protect C. neoformans cells against oxidative stress.
To evaluate whether SP-D functions to reinforce and/or to physically stabilize the protective effect of the capsule or is sufficient alone to protect C. neoformans cells against oxidative stress, we utilized the acapsular cap59Δ mutant and assessed yeast viability following exposure to 1 mM H2O2. The results were validated by using two techniques: CFU analysis and a sodium 3-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro)benzene sulfonic acid hydrate (XTT) assay. SP-D, but not SP-A, protected cap59Δ cells against oxidative stress (Fig. 8), which demonstrated that SP-D itself could shield this yeast against critical host-mediated defense mechanisms, even in the absence of capsule (P < 0.05 and P < 0.01).
Fig 8.

SP-D protects against oxidative stress in the presence and absence of capsule. Shown are data for H99 and cap59Δ cells precoated with SP-D (unbound SP-D was washed away) and then exposed to 1 mM H2O2. The respective unexposed cap59Δ and SP-D-coated cap59Δ controls were incubated under the same conditions, but without H2O2, to calculate the percent change compared to the control. (A) CFU assay. (B) XTT assay (n = 4). *, P < 0.05; **, P < 0.01.
DISCUSSION
Surfactant proteins A and D have been found to play critical host-protective roles in the lung during innate immune responses to infectious and allergenic materials (8, 9, 49). Intriguingly, and in contrast to the host-protective prototype proposed for these proteins against invading pathogens, we found that SP-D in fact functions to facilitate infection with the pathogenic fungus C. neoformans by protecting the fungus against host-produced reactive oxygen species (ROS) and facilitating its dissemination from the lungs to the CNS. Consistent with our previous studies showing that SP-D is protective against macrophage-mediated defense mechanisms (17), we found here that SP-D shielded C. neoformans cells against H2O2-induced oxidative stress, a component of the immune cell respiratory burst, in vitro and on yeast cells recovered directly from the alveolar compartment. We further showed that in the presence of endogenous SP-D in wild-type mice, the progression of C. neoformans infection was facilitated, leading to greater fungal burdens in the lung, spleen, and brain and faster dissemination from the lungs to the CNS than in SP-D−/− mice. These findings support our hypothesis that, in contrast to its well-accepted host-protective roles, SP-D promotes the course of infection of C. neoformans cells and thus may function as a risk factor.
SP-D has been found to facilitate the clearance of numerous bacterial, viral, and fungal pathogens via a number of different mechanisms, including aggregation, increased phagocytosis, and the elicitation of various cytokines/chemokines. The mechanisms of SP-D's host-protective role have been well enumerated in the context of bacteria and viruses (18, 22, 50), yet its function during fungal infections is less well characterized, with the most comprehensive fungal studies being performed with Aspergillus fumigatus. The pathogenic fungus A. fumigatus can cause both hypersensitivity reactions and invasive disease, and SP-D has been shown to be host protective in both of these disease models, as evidenced by robust in vitro and in vivo studies (43, 44). For example, Strong et al. demonstrated that the administration of recombinant SP-D to sensitized SP-D−/− mice rescues the elevated pulmonary hypersensitivity of SP-D−/− mice to A. fumigatus allergen challenge (57). Haczku et al. showed that SP-D levels are increased in the pulmonary space following A. fumigatus challenge of sensitized mice in an interleukin-4 (IL-4)- and IL-13-dependent manner and that these increased levels of endogenous SP-D protect the host against excessive inflammatory damage (21). In invasive aspergillosis, SP-D was shown to increase the uptake and killing of A. fumigatus conidia in vitro (1) and to decrease the fungal burden and increase the survival of mice, in part by increasing the levels of proinflammatory cytokines (44, 55). SP-D was also shown to protect against infection with Candida albicans (59), although its host protective functions during infection with other fungi such as Blastomyces dermatitidis, Pneumocystis jiroveci, Pneumocystis carinii, Coccidioides immitis, Coccidioides posadasii, C. neoformans, and C. gattii remain unclear. Studies investigating the latter fungal pathogens have hinted at the possibility of the respective pathogens' abilities to subvert host innate immune responses via SP-D, directly via the downregulation of SP-D expression and indirectly via the SP-D inhibition of the proinflammatory cytokine tumor necrosis factor alpha (TNF-α) (6, 9, 17, 30, 34, 61). Furthermore, AIDS patients, who are at a particularly high risk of developing opportunistic fungal pulmonary infections, have been shown to possess elevated levels of SP-D in their lungs (28). Based on the present findings, we speculate that following yeast cell inhalation, increased levels of SP-D in the pulmonary space might participate in promoting C. neoformans infection.
In the present study, we demonstrated that C. neoformans yeast cells subverted host pulmonary innate immune responses via SP-D bound to their surface for protection against oxidative stress, which has been shown to be an important host-protective mechanism during C. neoformans infection (2, 46, 63). Furthermore, we demonstrated that SP-D−/− mice survived longer than did SP-D-replete mice following intranasal infection with C. neoformans cells; SP-D−/− mice exhibited a decreased fungal burden and delayed yeast dissemination compared with wild-type mice. Similar results were obtained with both SP-D−/− and the triple-transgenic inducible SP-D−/− mouse strains, which confirmed that the observed effect was dependent on the absence of SP-D and was not due solely to the heightened inflammation characterized for SP-D−/− mice (62, 65). In addition, the administration of exogenous SP-D partially reversed the phenotype of delayed disease progression in SP-D−/− mice, as assessed by survival studies, which further demonstrated the ability of SP-D to facilitate the progression of C. neoformans infection. We speculate that the inability to completely restore the SP-D−/− phenotype during C. neoformans infection via the administration of exogenous SP-D may be due to a partial contribution of the heightened inflammatory state present in these mice. It is also possible that the application of exogenous SP-D reduces the elevated capacity of SP-D−/− macrophages to kill C. neoformans cells. These hypotheses are further supported by the slightly greater difference in survival observed between SP-D−/− and wild-type mice than that between offdoxySP-D−/− and plusdoxySP-D+/+ mice. Alternatively, higher concentrations of SP-D and/or a more frequent dosing schedule than those utilized in the present study may be necessary to achieve a complete rescue of the SP-D−/− phenotype. To discriminate between the two hypotheses, survival experiments may be performed in inducible mice using different periods of doxycycline removal, as previous analyses have shown that the number of activated macrophages increases with time after doxycycline has been removed from the diet.
An ability to subvert the host-protective functions of SP-D has been reported previously for some fungal pathogens. A host-detrimental effect of SP-D was described for P. carinii, although in that study, mice overexpressing SP-D were compared to those expressing physiological levels of SP-D (61). In addition, SP-D has been shown to abrogate TNF1-α production by alveolar macrophages in response to B. dermatitidis infection (34), potentially facilitating disease progression. These findings suggest that some fungal pathogens are equipped to utilize the host-protective properties of SP-D to their advantage; however, further investigations using in vivo models of disease are needed to confirm this hypothesis, as disparities between in vitro and in vivo models are often encountered (17). It is intriguing to speculate that the innate host immune functions of SP-D established for bacterial and viral infections are not generally applicable to certain fungal infections. One might postulate an evolutionary explanation for this observation; for example, eukaryotic fungal cells may possess a honed ability to recognize, respond to, and subvert host machinery compared with prokaryotic bacteria and viruses.
C. neoformans cells exploit a number of strategies to promote their survival in the host. We have previously shown that SP-D on the surface of C. neoformans cells functions as an opsonin by increasing their phagocytosis by macrophages. However, we have also observed that SP-D-bound fungi are in fact protected against macrophage-mediated defense mechanisms; they display increased survival compared with uncoated yeast cells. Other studies have shown that C. neoformans cells can survive intracellularly and then extrude from host immune cells via a process known as vomocytosis (3, 4, 38, 39, 41). In support of these properties being beneficial to the fungus, a recent study employing a panel of C. gattii strains with various virulence patterns revealed that increased intracellular survival correlates directly with increased fungal virulence (40). This finding is consistent with hypotheses that C. neoformans cells disseminate from the pulmonary space via intracellular parasitism and that the ability to survive intracellularly enables the fungus to evade host immune responses. From an evolutionary perspective, the ability of C. neoformans cells to survive in amoebae may have provided this fungus the opportunity to evolve robust intracellular survival strategies in nature (56). In the present study, we provide further evidence that C. neoformans cells have evolved sophisticated tactics to promote their survival during infection by using host innate immune factors like SP-D.
Further studies should be aimed at confirming whether SP-D functions to subvert the innate immune response and enhance cryptococcal infection in humans, particularly in light of ongoing diagnoses of C. gattii infection in apparently immunocompetent individuals in the Pacific Northwest (7, 10–13, 29, 42). Such studies might consider whether the various SP-D polymorphisms described in the literature (24, 32, 33, 35, 54) affect susceptibility to C. neoformans infection and/or C. gattii infection and, if so, whether genetic screening may facilitate infection risk stratification for prevention in individuals possessing other risk factors for cryptococcal disease. The present study provides evidence that SP-D is subverted by C. neoformans cells for increased uptake by immune cells and protection against oxidative stress, thus aiding disease progression. Further investigations in our laboratory are aimed at elucidating whether SP-D protects C. neoformans cells against oxidative stress by acting as a scavenger of highly reactive oxidant molecules or by upregulating the antioxidant capabilities of the yeast itself. Precedent for the latter is supported by recent studies demonstrating the capacity of mouse anticapsular antibodies to induce changes in the gene expression of C. neoformans cells (45). Because the containment of infection during its early stages is critical for avoiding disease, the present findings elucidate important early host-pathogen interactions that could be targeted for clinical intervention, with the goal of both risk stratification and, ultimately, the development of a target(s) for a drug(s) or vaccine(s) to prevent infection and/or dissemination.
ACKNOWLEDGMENTS
We thank Kathy Evans for preparation and purification of SP-D.
These studies were supported by NIH grants HL-30923 (J.R.W.), HL-51134 (J.R.W.), AI73896 (J.R.P.), AI28388 (J.R.P.), and AI42159 (J.H.).
We have no conflicts of interest.
We remember and are grateful for the extraordinary insights and leadership provided by Jo Rae Wright for this science. She was a truly an inspirational person and scientist, and her legacy will continue to inspire others in the pursuit of scientific answers and the truth.
Footnotes
Published ahead of print 30 April 2012
REFERENCES
- 1. Allen M, Harbeck R, Smith B, Voelker D, Mason R. 1999. Binding of rat and human surfactant proteins A and D to Aspergillus fumigatus conidia. Infect. Immun. 67:4563–4569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alspaugh J, Granger D. 1991. Inhibition of Cryptococcus neoformans replication by nitrogen oxides supports the role of these molecules as effectors of macrophage-mediated cytostasis. Infect. Immun. 59:2291–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alvarez M, Casadevall A. 2006. Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr. Biol. 16:2161–2165 [DOI] [PubMed] [Google Scholar]
- 4. Alvarez M, Saylor C, Casadevall A. 2008. Antibody action after phagocytosis promotes Cryptococcus neoformans and Cryptococcus gattii macrophage exocytosis with biofilm-like microcolony formation. Cell. Microbiol. 10:1622–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aratani Y, et al. 2006. Contribution of the myeloperoxidase-dependent oxidative system to host defence against Cryptococcus neoformans. J. Med. Microbiol. 55:1291–1299 [DOI] [PubMed] [Google Scholar]
- 6. Atochina E, et al. 2004. Delayed clearance of Pneumocystis carinii infection, increased inflammation, and altered nitric oxide metabolism in lungs of surfactant protein-D knockout mice. J. Infect. Dis. 189:1528–1539 [DOI] [PubMed] [Google Scholar]
- 7. Bartlett K, Kidd S, Kronstad J. 2008. The emergence of Cryptococcus gattii in British Columbia and the Pacific Northwest. Curr. Infect. Dis. Rep. 10:58–65 [DOI] [PubMed] [Google Scholar]
- 8. Brinker K, et al. 2001. Surfactant protein D enhances bacterial antigen presentation by bone marrow-derived dendritic cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 281:L1453–L1463 [DOI] [PubMed] [Google Scholar]
- 9. Brummer E, Stevens D. 2010. Collectins and fungal pathogens: roles of surfactant proteins and mannose binding lectin in host resistance. Med. Mycol. 48:16–28 [DOI] [PubMed] [Google Scholar]
- 10. Byrnes EJ, III, Bildfell R, Dearing P, Valentine B, Heitman J. 2009. Cryptococcus gattii with bimorphic colony types in a dog in western Oregon: additional evidence for expansion of the Vancouver Island outbreak. J. Vet. Diagn. Invest. 21:133–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Byrnes EJ, III, et al. 2009. Molecular evidence that the range of the Vancouver Island outbreak of Cryptococcus gattii infection has expanded into the Pacific Northwest in the United States. J. Infect. Dis. 199:1081–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Datta K, Bartlett K, Marr K. 2009. Cryptococcus gattii: emergence in Western North America: exploitation of a novel ecological niche. Interdiscip. Perspect. Infect. Dis. 2009:176532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dixit A, Carroll S, Qureshi S. 2009. Cryptococcus gattii: an emerging cause of fungal disease in North America. Interdiscip. Perspect. Infect. Dis. 2009:840452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Erpenbeck V, et al. 2005. Surfactant protein D increases phagocytosis and aggregation of pollen-allergen starch granules. Am. J. Physiol. Lung Cell. Mol. Physiol. 288:L692–L698 [DOI] [PubMed] [Google Scholar]
- 15. Feldmesser M, Kress Y, Novikoff P, Casadevall A. 2000. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect. Immun. 68:4225–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fraser J, et al. 2005. Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 437:1360–1364 [DOI] [PubMed] [Google Scholar]
- 17. Geunes-Boyer S, et al. 2009. Surfactant protein D increases phagocytosis of hypocapsular Cryptococcus neoformans by murine macrophages and enhances fungal survival. Infect. Immun. 77:2783–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giannoni E, Sawa T, Allen L, Wiener-Kronish J, Hawgood S. 2006. Surfactant proteins A and D enhance pulmonary clearance of Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 34:704–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giles S, Zaas A, Reidy M, Perfect J, Wright J. 2007. Cryptococcus neoformans is resistant to surfactant protein A mediated host defense mechanisms. PLoS One 2:e1370 doi:10.1371/journal.pone.0001370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giles SS, Dagenais TR, Botts MR, Keller NP, Hull CM. 2009. Elucidating the pathogenesis of spores from the human fungal pathogen Cryptococcus neoformans. Infect. Immun. 77:3491–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haczku A, et al. 2006. IL-4 and IL-13 form a negative feedback circuit with surfactant protein-D in the allergic airway response. J. Immunol. 176:3557–3565 [DOI] [PubMed] [Google Scholar]
- 22. Hartshorn K, et al. 1996. Interactions of recombinant human pulmonary surfactant protein D and SP-D multimers with influenza A. Am. J. Physiol. 271:L753–L762 [DOI] [PubMed] [Google Scholar]
- 23. Hartshorn KL, et al. 2010. Viral aggregating and opsonizing activity in collectin trimers. Am. J. Physiol. Lung Cell. Mol. Physiol. 298:L79–L88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heidinger K, et al. 2005. Polymorphisms in the human surfactant protein-D (SFTPD) gene: strong evidence that serum levels of surfactant protein-D (SP-D) are genetically influenced. Immunogenetics 57:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hidalgo H, Helmke R, German V, Mangos J. 1992. Pneumocystis carinii induces an oxidative burst in alveolar macrophages. Infect. Immun. 60:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hidalgo H, Helmke R, German V, Mangos J. 1991. Role of the zymolyase-sensitive cyst wall of Pneumocystis carinii in the oxidative burst of macrophages. J. Protozool. 38:30S–31S [PubMed] [Google Scholar]
- 27. Hsia CC, Hyde DM, Ochs M, Weibel ER. 2010. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am. J. Respir. Crit. Care Med. 181:394–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jambo K, French N, Zijlstra E, Gordon S. 2007. AIDS patients have increased surfactant protein D but normal mannose binding lectin levels in lung fluid. Respir. Res. 8:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kidd S, et al. 2007. Cryptococcus gattii dispersal mechanisms, British Columbia, Canada. Emerg. Infect. Dis. 13:51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koneti A, Linke M, Brummer E, Stevens D. 2008. Evasion of innate immune responses: evidence for mannose binding lectin inhibition of tumor necrosis factor alpha production by macrophages in response to Blastomyces dermatitidis. Infect. Immun. 76:994–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Korfhagen TR, et al. 1998. Surfactant protein-D regulates surfactant phospholipid homeostasis in vivo. J. Biol. Chem. 273:28438–28443 [DOI] [PubMed] [Google Scholar]
- 32. Lahti M, et al. 2002. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr. Res. 51:696–699 [DOI] [PubMed] [Google Scholar]
- 33. Lawson P, Perkins V, Holmskov U, Reid K. 1999. Genomic organization of the mouse gene for lung surfactant protein D. Am. J. Respir. Cell Mol. Biol. 20:953–963 [DOI] [PubMed] [Google Scholar]
- 34. Lekkala M, et al. 2006. Effect of lung surfactant collectins on bronchoalveolar macrophage interaction with Blastomyces dermatitidis: inhibition of tumor necrosis factor alpha production by surfactant protein D. Infect. Immun. 74:4549–4556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leth-Larsen R, et al. 2005. A common polymorphism in the SFTPD gene influences assembly, function, and concentration of surfactant protein D. J. Immunol. 174:1532–1538 [DOI] [PubMed] [Google Scholar]
- 36. LeVine A, et al. 2004. Surfactant protein-d enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 31:193–199 [DOI] [PubMed] [Google Scholar]
- 37. Lu J, Willis A, Reid K. 1992. Purification, characterization and cDNA cloning of human lung surfactant protein D. Biochem. J. 284(Pt 3):795–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma H, Croudace J, Lammas D, May R. 2007. Direct cell-to-cell spread of a pathogenic yeast. BMC Immunol. 8:15 doi:10.1186/1471-2172-8-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma H, Croudace J, Lammas D, May R. 2006. Expulsion of live pathogenic yeast by macrophages. Curr. Biol. 16:2156–2160 [DOI] [PubMed] [Google Scholar]
- 40. Ma H, et al. 2009. The fatal fungal outbreak on Vancouver Island is characterized by enhanced intracellular parasitism driven by mitochondrial regulation. Proc. Natl. Acad. Sci. U. S. A. 106:12980–12985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma H, May R. 2009. Virulence in Cryptococcus species. Adv. Appl. Microbiol. 67:131–190 [DOI] [PubMed] [Google Scholar]
- 42. MacDougall L, et al. 2007. Spread of Cryptococcus gattii in British Columbia, Canada, and detection in the Pacific Northwest, USA. Emerg. Infect. Dis. 13:42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Madan T, et al. 2001. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J. Clin. Invest. 107:467–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Madan T, et al. 2001. Protective role of lung surfactant protein D in a murine model of invasive pulmonary aspergillosis. Infect. Immun. 69:2728–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McClelland EE, Nicola AM, Prados-Rosales R, Casadevall A. 2010. Ab binding alters gene expression in Cryptococcus neoformans and directly modulates fungal metabolism. J. Clin. Invest. 120:1355–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Missall T, Pusateri M, Lodge J. 2004. Thiol peroxidase is critical for virulence and resistance to nitric oxide and peroxide in the fungal pathogen, Cryptococcus neoformans. Mol. Microbiol. 51:1447–1458 [DOI] [PubMed] [Google Scholar]
- 47. Murayama T, Suzuki K, Yamamoto K, Kuze F. 1990. Oxygen radical generation of murine alveolar macrophages. Nihon Kyobu Shikkan Gakkai Zasshi 28:741–749 (In Japanese.) [PubMed] [Google Scholar]
- 48. Nelson R, Pryor B, Lodge J. 2003. Sequence length required for homologous recombination in Cryptococcus neoformans. Fungal Genet. Biol. 38:1–9 [DOI] [PubMed] [Google Scholar]
- 49. Pikaar J, et al. 1995. Opsonic activities of surfactant proteins A and D in phagocytosis of gram-negative bacteria by alveolar macrophages. J. Infect. Dis. 172:481–489 [DOI] [PubMed] [Google Scholar]
- 50. Restrepo C, Dong Q, Savov J, Mariencheck W, Wright J. 1999. Surfactant protein D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 21:576–585 [DOI] [PubMed] [Google Scholar]
- 51. Roehm N, Rodgers G, Hatfield S, Glasebrook A. 1991. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Methods 142:257–265 [DOI] [PubMed] [Google Scholar]
- 52. Sawada K, et al. 2010. Pulmonary collectins protect macrophages against pore-forming activity of Legionella pneumophila and suppress its intracellular growth. J. Biol. Chem. 285:8434–8443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schelenz S, Malhotra R, Sim RB, Holmskov U, Bancroft GJ. 1995. Binding of host collectins to the pathogenic yeast Cryptococcus neoformans: human surfactant protein D acts as an agglutinin for acapsular yeast cells. Infect. Immun. 63:3360–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Selman M, et al. 2003. Surfactant protein A and B genetic variants predispose to idiopathic pulmonary fibrosis. Hum. Genet. 113:542–550 [DOI] [PubMed] [Google Scholar]
- 55. Singh M, et al. 2009. Therapeutic effects of recombinant forms of full-length and truncated human surfactant protein D in a murine model of invasive pulmonary aspergillosis. Mol. Immunol. 46:2363–2369 [DOI] [PubMed] [Google Scholar]
- 56. Steenbergen J, Shuman H, Casadevall A. 2001. Cryptococcus neoformans interactions with amoebae suggest an explanation for its virulence and intracellular pathogenic strategy in macrophages. Proc. Natl. Acad. Sci. U. S. A. 98:15245–15250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Strong P, Reid K, Clark H. 2002. Intranasal delivery of a truncated recombinant human SP-D is effective at down-regulating allergic hypersensitivity in mice sensitized to allergens of Aspergillus fumigatus. Clin. Exp. Immunol. 130:19–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van de Wetering JK, Coenjaerts FE, Vaandrager AB, van Golde LM, Batenburg JJ. 2004. Aggregation of Cryptococcus neoformans by surfactant protein D is inhibited by its capsular component glucuronoxylomannan. Infect. Immun. 72:145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van Rozendaal B, van Spriel A, van De Winkel J, Haagsman H. 2000. Role of pulmonary surfactant protein D in innate defense against Candida albicans. J. Infect. Dis. 182:917–922 [DOI] [PubMed] [Google Scholar]
- 60. Velagapudi R, Hsueh YP, Geunes-Boyer S, Wright JR, Heitman J. 2009. Spores as infectious propagules of Cryptococcus neoformans. Infect. Immun. 77:4345–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vuk-Pavlovic Z, et al. 2006. Surfactant protein D enhances Pneumocystis infection in immune-suppressed mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 290:L442–L449 [DOI] [PubMed] [Google Scholar]
- 62. Wert S, et al. 2000. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc. Natl. Acad. Sci. U. S. A. 97:5972–5977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zaragoza O, et al. 2008. Capsule enlargement in Cryptococcus neoformans confers resistance to oxidative stress suggesting a mechanism for intracellular survival. Cell. Microbiol. 10:2043–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zaragoza O, Fries B, Casadevall A. 2003. Induction of capsule growth in Cryptococcus neoformans by mammalian serum and CO(2). Infect. Immun. 71:6155–6164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang L, Hartshorn K, Crouch E, Ikegami M, Whitsett J. 2002. Complementation of pulmonary abnormalities in SP-D(−/−) mice with an SP-D/conglutinin fusion protein. J. Biol. Chem. 277:22453–22459 [DOI] [PubMed] [Google Scholar]
- 66. Zhang L, Ikegami M, Dey C, Korfhagen T, Whitsett J. 2002. Reversibility of pulmonary abnormalities by conditional replacement of surfactant protein D (SP-D) in vivo. J. Biol. Chem. 277:38709–38713 [DOI] [PubMed] [Google Scholar]