

Abstract
Borrelia burgdorferi, the causative agent of Lyme disease, can be recovered from different organs of infected animals and patients, indicating that the spirochete is very invasive. Motility and chemotaxis contribute to the invasiveness of B. burgdorferi and play important roles in the process of the disease. Recent reports have shown that motility is required for establishing infection in mammals. However, the role of chemotaxis in virulence remains elusive. Our previous studies showed that cheA2, a gene encoding a histidine kinase, is essential for the chemotaxis of B. burgdorferi. In this report, the cheA2 gene was inactivated in a low-passage-number virulent strain of B. burgdorferi. In vitro analyses (microscopic observations, computer-based bacterial tracking analysis, swarm plate assays, and capillary tube assays) showed that the cheA2 mutant failed to reverse and constantly ran in one direction; the mutant was nonchemotactic to attractants. Mouse needle infection studies showed that the cheA2 mutant failed to infect either immunocompetent or immunodeficient mice and was quickly eliminated from the initial inoculation sites. Tick-mouse infection studies revealed that although the mutant was able to survive in ticks, it failed to establish a new infection in mice via tick bites. The altered phenotypes were completely restored when the mutant was complemented. Collectively, these data demonstrate that B. burgdorferi needs chemotaxis to establish mammalian infection and to accomplish its natural enzootic cycle.
INTRODUCTION
Lyme disease, the most commonly reported tick-borne disease in the United States (2, 55), is caused by the spirochete Borrelia burgdorferi (6, 26). In nature, B. burgdorferi is maintained via an enzootic cycle comprising both mammalian hosts and an Ixodes tick vector (6, 22). The enzootic cycle begins with feeding by uninfected tick larvae on an infected vertebrate. After the feeding, the spirochetes remain in the tick gut throughout the molting process. At the time that the infected nymph takes a blood meal on a mammal, the spirochetes begin to multiply and migrate from the tick gut to the salivary glands, from which they are transmitted to a new host, thereby completing the enzootic cycle. After being deposited into the skin of mammals following tick bites, the B. burgdorferi cells traverse the intracellular matrix, penetrate the vascular endothelial cell lining, enter the circulatory system of the host, and subsequently cause systemic infection (12, 13, 56, 65). The spirochetes can be recovered from different organs and tissues of infected animals (e.g., heart, joint, and bladder) (4) and patients (e.g., skin, blood, and endomyocardial and brain tissues) (18, 38, 54), demonstrating that B. burgdorferi is highly invasive.
Accumulating evidence has revealed that motility is an important virulence factor that is linked to the invasiveness and enzootic cycle of B. burgdorferi (11). First, B. burgdorferi has the ability to swim in highly viscous gel-like media (20, 24), including the connective tissue, and this ability is abrogated in aflagellated nonmotile mutants of B. burgdorferi (29, 35, 48). Second, real-time intravital microscopy analysis revealed that B. burgdorferi is able to penetrate the endothelium of blood vessels and quickly disseminate from the microvasculature in living mice and that the translational motility appears to be essential for transendothelial migration (34, 39). Third, a recent report indicates that B. burgdorferi transitions from a nonmotile phase to a motile phase during the period when the spirochete penetrates the tick gut basement membrane and migrates to the salivary glands of feeding ticks (14). Finally, our recent report describes a fliG1 mutant strain which is unable to translate in highly viscous media and fails to cause infection in a mouse model of the disease (29). Since bacterial motility is directed by chemotaxis (61), many Lyme disease researchers have long believed that chemotaxis may also be involved in the disease processes (11, 28, 44, 47, 52, 53) (e.g., facilitating spirochete migration from the tick gut to the salivary glands and initiating a new infection and/or dissemination from the site of deposition into the circulatory system of mammalian hosts). However, supporting evidence is very limited, owing primarily to the difficulty of constructing and complementing mutants in virulent strains of B. burgdorferi.
Bacterial chemotaxis is defined as a swimming behavior toward or away from chemical stimuli (61). The chemotaxis response is executed by a complex two-component phosphorelay system that is composed of a family of chemoreceptor proteins, a sensor histidine kinase (CheA), and a response regulator (CheY). The chemoreceptors sense a chemical stimulus and activate CheA via phosphorylation. The activated CheA then relays the phosphoryl group to CheY, yielding phosphorylated CheY (CheY-P). CheY-P in turn interacts with the motor switch proteins and controls the rotational direction of flagellar motors. In the well-studied model organisms Escherichia coli and Salmonella enterica serovar Typhimurium, there is only one copy each of cheA and cheY, and these genes are essential for chemotaxis. The null mutants of cheA or cheY run continuously and are nonchemotactic to attractants (42). In contrast, B. burgdorferi has multiple chemotaxis genes, e.g., two cheA genes (cheA1 and cheA2) and three cheY genes (cheY1, cheY2, and cheY3) (11, 17). We recently identified several genes that are essential for the chemotaxis of B. burgdorferi, including cheA2, cheY3, and cheX (an analogue of cheZ from E. coli) (28, 36, 37). The cheA2 and cheY3 mutants fail to reverse and constantly run, while the cheX mutant constantly flexes. All of these mutants are unable to sense and respond to attractants (3, 28, 36, 37). In this study, the cheA2 gene was chosen as a target to decipher the role of chemotaxis in the process of establishing disease. In vitro studies showed that the cheA2 gene is essential for chemotaxis. In vivo experiments revealed that chemotaxis is required for the spirochete to establish infection in mammals and for transmission from the tick vector to a mammalian host.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Infectious clone A3-68 Δbbe02 (wild type), a derivative strain from B. burgdorferi sensu stricto B31A3, was used in this study (45). This strain was a kind gift from P. Rosa (Rocky Mountain Laboratories, NIAID, NIH). Cells were grown in Barbour-Stoenner-Kelly II (BSK-II) medium as previously described (57), with an appropriate antibiotic(s) for selective pressure as needed, i.e., streptomycin (50 μg/ml), kanamycin (300 μg/ml), and/or gentamicin (40 μg/ml). To determine the expressional level of cheA2, 105 stationary-phase wild-type cells were inoculated into 10 ml of fresh BSK-II medium and then cultured under different conditions as previously documented (50, 63), including 23°C and pH 7.6 (unfed ticks [UF]), 34°C and pH 7.6 (routine laboratory cultural conditions), and 37°C and pH 6.8 (fed tick [FT] conditions). To mimic the FT conditions, 105 wild-type cells cultivated at 23°C and pH 7.6 were inoculated into 10 ml of BSK-II (pH 6.8) and grown at 37°C. To mimic a host adaptation condition, B31A3, the parental strain of A3-68 Δbbe02, was cultivated in dialysis membrane chambers (DMC) in rat peritonea as previously described (1).
Construction of CheA2/pBBE22G for complementing the cheA2 mutant.
pGA2kan, a previously constructed vector (28), was used to inactivate the cheA2 gene in the A3-68 Δbbe02 strain via allelic exchange mutagenesis. To complement the cheA2 mutant, pFlgBA2com, a previously constructed complementation vector (3), was digested with PstI and SphI to excise the PflgB-cheA2 fragment. The obtained fragment was then subcloned into the shuttle vector pBBE22G (27), producing CheA2/pBBE22G (see Fig. 2A).
Fig 2.
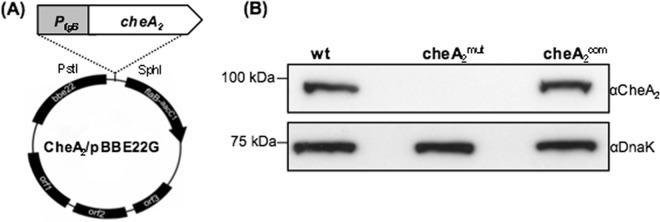
Detection of CheA2 in the cheA2mut strain and the complemented cheA2com strain. (A) Construction of CheA2/pBBE22G for complementation of cheA2mut. The fused PflgB-cheA2 fragment was released from pFlgBA2com, a previously constructed vector for complementing an avirulent cheA2 mutant (3), and cloned into pBBE22G (62), a shuttle vector of B. burgdorferi, yielding CheA2/pBBE22G. (B) Western blot analysis of cheA2mut and cheA2com strains. The same amounts of wild-type, cheA2mut, and cheA2com whole-cell lysates were analyzed by SDS-PAGE and then probed with CheA2 and DnaK antibodies as previously documented (28, 29).
SDS-PAGE and Western blots.
Equal amounts of B. burgdorferi whole-cell lysates (ranging from 10 to 20 μg) were separated in SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories, Hercules, CA). The immunoblots were probed with antibodies against B. burgdorferi CheA2, OspC, and DnaK (as an internal control) and developed using horseradish peroxidase-conjugated secondary antibody with an enhanced chemiluminescence (ECL) luminol assay as previously described (57). Signals were quantified using a Molecular Imager ChemiDoc XRS system with Image Lab software (Bio-Rad Laboratories).
Bacterial motion tracking analysis, swarm plate assays, and capillary tube-based chemotaxis assays.
The swimming velocity of B. burgdorferi cells was measured using a computer-based motion tracking system as previously described (3). Swarm plate analysis was conducted as previously described (28, 35). The diameters of swarm rings were measured and recorded in millimeters. The wild-type strain was used as a positive control. A previously constructed nonmotile flaB mutant (35) was used as a negative control to monitor the initial inoculum size. The capillary tube assay was carried out using N-acetyl-d-glucosamine as an attractant, as previously reported (3). The spirochete cells in the capillary tubes were enumerated using Petroff-Hausser counting chambers. The mean for five replicates was determined, and the data are expressed as the mean relative increase over a buffer control level containing no attractant. An increase in the number of spirochetes of ≥2-fold over that in the buffer control was considered significant.
qRT-PCR.
RNA samples for quantitative reverse transcription-PCR (qRT-PCR) analysis were prepared as previously described (25, 57, 66). Briefly, total RNAs from mouse tissues and ticks were isolated using TRIzol reagent (Invitrogen, Carlsbad, CA), and contaminating genomic DNA was removed using Turbo DNase (Ambion, Austin, TX). The DNase-treated RNAs were repurified and converted to cDNAs by use of an AffinityScript multiple-temperature cDNA synthesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions. The amounts of cDNAs were measured using iQ SYBR green Supermix (Bio-Rad). The spirochete burdens within infected mice and ticks were expressed as the flaB transcript levels relative the mouse or tick β-actin transcript level.
Mouse infection studies.
BALB/c, BALB/c SCID, and C3H mice at 4 to 6 weeks of age (Jackson Laboratory, Bar Harbor, ME) were used in this study. All animal experimentation was conducted following the NIH guidelines for housing and care of laboratory animals and was performed in accordance with the University of Buffalo and University of Maryland institutional regulations after review and approval by the Institutional Animal Care and Use Committees. The animal studies were carried out as previously described (29, 57, 66). Briefly, the mice were given a single subcutaneous injection of 105 spirochetes and sacrificed at 3 weeks postinfection. Tissues from the ear, skin, joints, and/or heart were harvested and placed into 1 ml BSK-II medium. The samples were incubated at 34°C for up to 2 weeks and monitored microscopically for the presence of spirochetes.
Experimental tick infection and tick-to-mouse infection.
Ixodes scapularis nymph ticks (Oklahoma State University, Stillwater, OK) were artificially infected by microinjection as previously described (23, 41). Injected ticks (3 ticks per pool; 3 pools for each B. burgdorferi strain) were allowed to rest in the incubator for 10 days, and the borrelial burdens in unfed ticks were analyzed by qRT-PCR as described earlier. For the analysis of spirochete levels in fed ticks and their ability to transmit to naïve hosts, unfed nymphs were infected via microinjection. Two days after the injection, the ticks were fed to repletion on naïve C3H mice (7 ticks/mouse; 3 mice for each B. burgdorferi strain) for 5 to 7 days and allowed to fall off. Engorged ticks were collected and subjected to qRT-PCR analysis to determine borrelial burdens (4 ticks from each mouse). At day 14 after the tick feeding, mice were sacrificed and tissues were subjected to either qRT-PCR analysis for determination of pathogen levels or spirochete culture in BSK-II medium (66).
Statistical analysis.
For the swarm plate, motion tracking, capillary tube assays, and the mouse and tick infection studies, the results are expressed as means ± standard errors of the means (SEM). The significance of differences between different experimental groups was evaluated with an unpaired Student t test (P values of <0.01 were considered significant).
RESULTS
The level of CheA2 remains unchanged under various culture conditions.
During the enzootic cycle, B. burgdorferi robustly alters its gene expression, which allows the spirochete to adapt to and thrive in a new host (49). Whole-genome DNA microarray analyses have shown that a variety of genes change their expression under different cultural conditions that mimic the life cycle of B. burgdorferi in nature (5, 40, 46). Among these genes, some are involved in chemotaxis. For instance, Revel et al. reported that the level of cheA2 transcript under culture conditions mimicking unfed ticks (23°C and pH 7.6) was approximately 4.6-fold higher than that in fed ticks (37°C and pH 6.8) (46). We reasoned that the expression level of cheA2 may be variable during the enzootic cycle. To test this hypothesis, the wild-type strain was cultivated under different conditions as described in Materials and Methods. The level of CheA2 protein was monitored by quantitative Western blotting. There was no significant change evident under all examined conditions (Fig. 1), suggesting that the cheA2 gene is expressed constitutively during the enzootic cycle.
Fig 1.
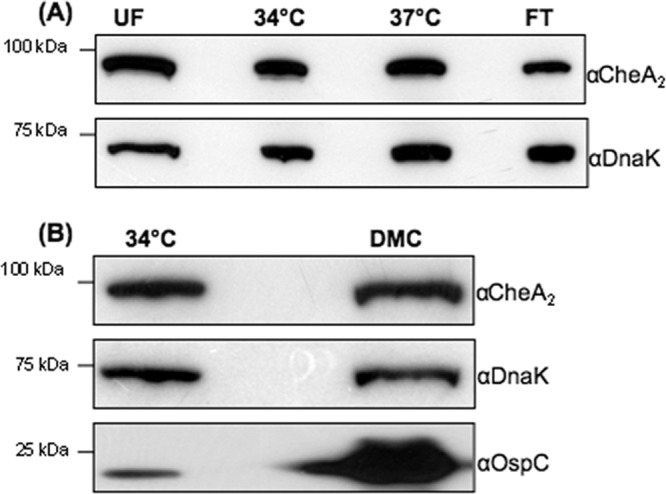
Detection of CheA2 under various culture conditions. The wild-type A3-68 Δbbe02 or B31A3 (15, 45) strain was cultivated under various conditions: UF conditions (23°C and pH 7.6), 34°C and pH 7.6, 37°C and pH 6.8, or FT conditions (switch from 23°C and pH 7.6 to 37°C and pH 6.8) (A) or in the presence of DMC (B) (1). Similar amounts of whole-cell lysates were analyzed by SDS-PAGE and then probed with CheA2, DnaK (a loading control), and OspC antibodies as previously described (57). Increased OspC expression was used as a marker for cells cultured in DMC.
Isolation of cheA2 mutant and its complemented strain.
To inactivate the cheA2 gene, pGA2kan, a previously constructed vector (28), was linearized and electrotransformed into wild-type competent cells. A previously described PCR analysis was first carried out to screen for clones with the desired targeted mutagenesis (28). B. burgdorferi contains 21 linear and circular plasmids (10). Because some of these plasmids are essential for infectivity and are easily lost during in vitro cultivations (43), a previously developed PCR method was used to determine if the obtained positive clones contained the same plasmids as their parental strain (15). One clone (referred to as cheA2mut) containing the same plasmid profile (see Fig. 3A) as the wild type was selected for further analysis. For complementation, the plasmid CheA2/pBBE22G (Fig. 2A) was electrotransformed into cheA2mut competent cells. The presence of CheA2/pBBE22G in antibiotic-resistant colonies was confirmed by PCR. One clone (designated cheA2com) containing an identical plasmid profile to that of the wild type (Fig. 3B) was selected for further analysis. As shown in Fig. 2B, a band of approximately 98 kDa was detected in the wild type and the cheA2com strain but absent in the cheA2mut strain, indicating that the cognate gene product was abrogated in the mutant and restored in the complemented strain.
Fig 3.
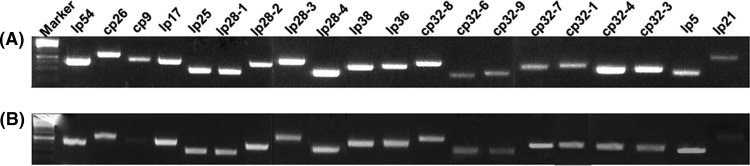
Detection of plasmid contents in the cheA2mut (A) and cheA2com (B) strains by PCR. The primers for PCR were described previously (15).
The cheA2mut mutant has an altered swimming behavior.
B. burgdorferi has three different swimming modes: run, flex, and reverse (28, 36). The cheA2 mutant fails to reverse or flex and runs constantly in one direction (28). However, all of these studies were carried out using a high-passage-number avirulent B31A strain. We hypothesized that a similar phenotype also applies in the virulent strain background. To test this hypothesis, the swimming behaviors of wild-type, cheA2mut, and cheA2com cells were analyzed using a computer-assisted cell tracker as previously described (3, 28). Similar to B31A, the A3-68 Δbbe02 strain also has three swimming modes, with runs interrupted by reverses or flexes approximately 2 times every 10 s (mean [± SEM] number of reversals/10 s = 2.2 ± 0.77; n = 30 cells) (Table 1; see Movie S1 in the supplemental material). In contrast to the wild type, cheA2mut failed to reverse and flex; it constantly ran in one direction (Table 1; see Movie S2). However, the swimming velocity of the mutant (15.9 ± 2.4 μm/s; n = 30 cells) was similar to that of the wild type (14.1 ± 1.9 μm/s; n = 30 cells). The altered swimming behaviors were completely restored in the complemented strain (cheA2com) (Table 1; see Movie S3). The phenotype observed here is similar to that previously reported for the avirulent background (3, 28). Collectively, these results indicate that cheA2mut has altered flagellar reversal frequency but not cell swimming velocity.
Table 1.
Swimming behavior of B. burgdorferi cheA2mut
| Strain | Velocity (μm/s)a | Reversal frequency (no. of reversals/10 s)a |
|---|---|---|
| Wild type | 14.1 ± 1.9 | 2.3 ± 0.77 |
| cheA2mut | 15.9 ± 2.4 | 0 |
| cheA2com | 15.3 ± 1.6 | 1.8 ± 0.63 |
Thirty swimming cells of each strain were tracked, and the data are expressed as means ± SEM.
The cheA2mut mutant is defective in chemotaxis.
The swarm plate assay was first used to determine if the cheA2 gene is required for B. burgdorferi chemotaxis. The results showed that cheA2mut exhibited a poor swarming phenotype (Fig. 4A): the diameter of swarm rings formed by the mutant (mean ± SEM = 8.3 ± 0.38 mm; n = 4 plates) was substantially smaller than that for the wild type (11.4 ± 1.4 mm; n = 4 plates). The swarming ability was restored in the cheA2com strain (11.8 ± 1.4 mm; n = 4 plates). The chemotactic properties of cheA2mut were further analyzed using capillary tube assays with N-acetyl-d-glucosamine, a defined chemoattractant of B. burgdorferi (3). As shown in Fig. 4B, the wild-type and cheA2com strains exhibited a strong response to the attractant (the number of cells in the attractant-filled tubes was at least 2-fold higher than that in the negative control), whereas the cheA2mut strain was unable to accumulate in the attractant-filled tubes. In sum, the results from these two assays indicate that cheA2 is essential for the chemotaxis of B. burgdorferi.
Fig 4.
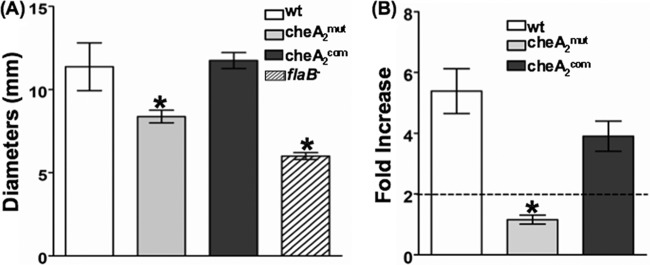
The B. burgdorferi cheA2 mutant is nonchemotactic. (A) Swarm plate assay. The assay was carried out on 0.35% agarose containing 1:10-diluted BSK-II medium as described previously (35). The flaB mutant, a previously constructed nonmotile mutant (35), was included to determine the sizes of the inocula. The data are presented as mean diameters (in millimeters) of rings ± SEM for four plates. (B) Capillary tube chemotaxis assay. Assays were carried out using 100 mM N-acetyl-d-glucosamine as an attractant according to a previous report (3). The results are expressed as fold increases in cell number entering the capillary tubes containing the attractant relative to the number entering control tubes without attractant (buffer alone). Results are expressed as means ± SEM for five tubes. A 2-fold increase is considered the threshold for a positive response. *, significant difference (P < 0.01).
The cheA2mut mutant fails to establish infection in mice by needle inoculation.
A previously developed mouse model of disease was carried out to determine if chemotaxis is essential for the virulence of B. burgdorferi (4). BALB/c mice were infected subcutaneously with equal numbers of the wild-type, cheA2mut, and cheA2com strains (4 mice per strain). Tissues from the ear, skin, and joints were harvested at 3 weeks postinfection and transferred to BSK-II medium. The wild-type and cheA2com strains were recovered successfully from all of the examined biopsy specimens, but the mutant was not (Table 2), which indicates that the cheA2 gene is essential for B. burgdorferi to establish an infection in mice. To further confirm this proposition, a similar experiment was implemented using immunodeficient SCID mice (3 mice per group). Similar to the case with immunocompetent mice, the wild-type and cheA2com strains were successfully reisolated from all tissue specimens. However, no living spirochetes were detected in the tissues from mice infected by cheA2mut (Table 2), suggesting that the failure of infection cannot be ascribed to adaptive immunity. Taken together, the results indicate that the chemotaxis mediated by CheA2 is essential for the infectivity of B. burgdorferi in mammals.
Table 2.
The cheA2mut mutant fails to infect micea
| Mouse strain | B. burgdorferi strain | No. of positive cultures/total no. of specimens examined |
No. of mice infected/total no. of mice used | |||
|---|---|---|---|---|---|---|
| Ear | Skin | Joint | All sites | |||
| BALB/c | Wild type | 4/4 | 4/4 | 4/4 | 12/12 | 4/4 |
| cheA2mut | 0/4 | 0/4 | 0/4 | 0/12 | 0/4 | |
| cheA2com | 4/4 | 4/4 | 4/4 | 12/12 | 4/4 | |
| SCID | Wild type | 3/3 | 3/3 | 3/3 | 9/9 | 3/3 |
| cheA2mut | 0/3 | 0/3 | 0/3 | 0/9 | 0/3 | |
| cheA2com | 3/3 | 3/3 | 3/3 | 9/9 | 3/3 | |
Groups of four BALB/c or three SCID mice were inoculated with 105 spirochetes of the wild-type, cheA2mut, or cheA2com strain. Mice were sacrificed at 3 weeks postinoculation, and ear, skin, and tibiotarsal joint specimens were harvested for spirochete culture in BSK-II medium.
Chemotaxis may facilitate rapid dissemination of B. burgdorferi cells from the initial deposition sites on the skin, consequently helping the spirochetes escape from host innate immunity and further establishing the systemic infection (11, 28). In this regard, the failure of cheA2mut to establish infection in SCID mice (Table 2) could be due to the loss of chemotaxis, which may be required for dissemination and/or evasion from innate immune attacks. To investigate these issues, the three strains were inoculated into SCID mice. After the injections, the infected mice were sacrificed at 24, 48, and 72 h. The skin specimens around the injection sites were harvested. The specimens were then subjected either to spirochete culture or to measurements of the borrelial burdens by qRT-PCR, using the flaB transcript as a surrogate indicator (41). At 24 and 48 h, the spirochete cells were successfully reisolated from all of the specimens infected by all three strains. However, at 72 h, while the wild-type and cheA2com strains were readily reisolated from the skin specimens, no living spirochetes were evident in the samples infected by the cheA2mut strain. Consistently, qRT-PCR analysis showed that the flaB transcripts were detectable in the cheA2mut specimens at 24 and 48 h, but there was no trace of the transcript detected in the sample isolated at 72 h (Fig. 5), indicating that the mutant was cleared from all of the inoculation sites after a critical window of 48 h. The obtained results strongly support the hypothesis that the chemotaxis of B. burgdorferi is involved in the process of bacterial dissemination and possibly in innate immune evasion that occurs at the initial infection sites.
Fig 5.
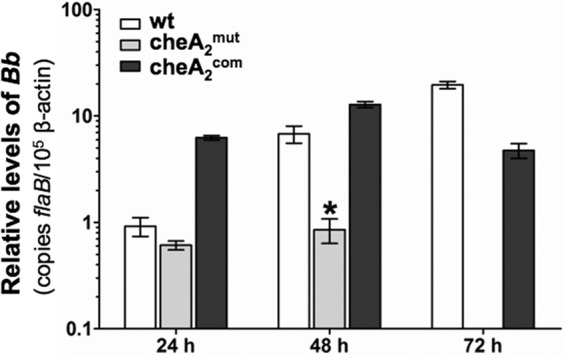
Detection of spirochete burdens in needle-infected SCID mice by qRT-PCR. Groups of three mice were needle inoculated with 105 cells of the wild-type, cheA2mut, and cheA2com strains. Infected animals were sacrificed at 24, 48, and 72 h. Skin samples from areas around the inoculation sites were collected and subjected to qRT-PCR. The bacterial burdens in these samples were measured by determining the number of copies of flaB mRNA compared to the number of copies of mouse β-actin transcript as previously documented (41, 66). The data are expressed as the means of relative levels of flaB transcript ± SEM for three independent experiments. At 72 h, the flaB mRNA was undetectable in the skin tissues infected by cheA2mut. *, significant difference (P < 0.01). Bb, B. burgdorferi.
The cheA2 gene is not required for artificial infection of I. scapularis ticks.
Since cheA2mut is noninfectious in mice, we used a microinjection-based infection procedure (23, 41) to generate infected ticks. I. scapularis nymphs were microinjected with equal amounts of the wild type, cheA2mut, and cheA2com strains. After the infection, one group of ticks (unfed) remained in the unfed condition for 10 days, while a parallel group of ticks was allowed to feed on naïve C3H mice (7 ticks/mouse; 3 mice for each strain) to repletion. The spirochete burdens in unfed ticks and engorged ticks were measured by qRT-PCR (66). As shown in Fig. 6, the bacterial burdens of the three strains in unfed ticks were very similar (P > 0.05). For the replete ticks, the bacterial burden of cheA2mut was slightly less than that of the wild-type (P > 0.1) and cheA2com (P > 0.05) strains, but the difference is not significant. Taken together, these results indicate that chemotaxis is not required for B. burgdorferi colonization and survival in the tick vector.
Fig 6.
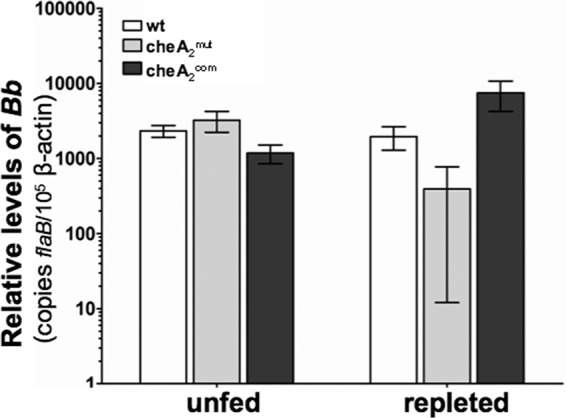
Detection of spirochete burdens in microinjected nymphal ticks before and after feeding, using qRT-PCR. RNA samples were extracted from whole unfed ticks (10 days after injection) and fed ticks (after repletion; 5 to 7 days) and subjected to PCR analysis. The bacterial burdens in ticks were measured by the number of copies of flaB mRNA compared to the number of copies of tick β-actin transcript as previously described (23, 41). The data are presented as the means of relative levels of flaB transcript ± SEM for three groups (each group containing 3 unfed ticks or 4 replete ticks) for each strain (wild type, cheA2mut, and cheA2com).
The cheA2mut mutant fails to establish infection in mice via tick bites.
The infectivity of cheA2mut in mice was evaluated via needle inoculation. However, this method is different from the natural route of mammalian infection. Because cheA2mut is still able to establish tick infection, we examined whether this mutant is able to be transmitted to mice that are fed upon by infected ticks. Groups of naïve mice were allowed to be parasitized by artificially infected ticks as detailed above. Fourteen days after the feeding, the mice were sacrificed. Skin, heart, joint, and bladder tissues were subjected either to qRT-PCR analysis for determination of pathogen levels or to spirochete isolation as described above. Consistent with the results obtained via needle inoculation, only the tissue specimens from mice fed upon by the wild-type- and cheA2com-infected ticks were positive for spirochetes in culture (data not shown). Results for the qRT-PCR analysis of pathogen burdens were consistent with the data from medium reisolation of spirochetes. In Fig. 7, representative qRT-PCR data are shown for skin and heart tissues, in which flaB transcripts could be detected only in the biopsy specimens from the mice infected by the wild-type and cheA2com strains. No flaB transcript was detected in the specimens from mice infected by cheA2mut, indicating that the mutant failed to be transmitted to mice. Collectively, these results suggest that chemotaxis is required for natural infections during the enzootic cycle of B. burgdorferi.
Fig 7.
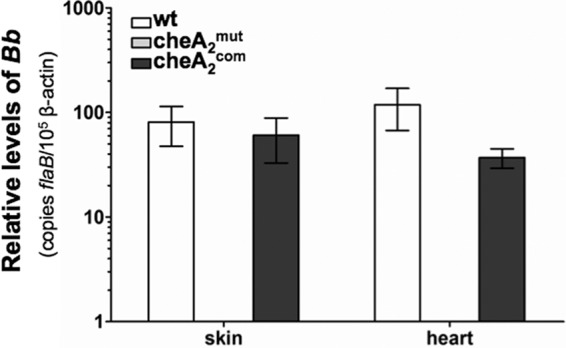
Detection of spirochete burdens in C3H mice infected via tick bite, using qRT-PCR. At day 14 after tick feeding, mice were sacrificed and tissues (skin, heart, joint, and bladder) collected for RNA isolations and then subjected to qRT-PCR analysis as described in the legend to Fig. 4. The data shown here are the results for skin and heart tissues. No trace of flaB message was detected in the mouse tissues infected by cheA2mut; since its PCR cycle thresholds (CT) are higher than that of the negative controls (no transcriptase added), it is considered negative.
DISCUSSION
The role of chemotaxis in bacterial pathogenicity has been studied in several pathogens, such as Helicobacter pylori, Campylobacter jejuni, and Vibrio cholerae (7, 16, 32, 58, 59, 64). In H. pylori, chemotaxis plays a role in both colonization and persistence in the gastric mucosa during infection (16, 32, 51, 59). Mutants defective in chemotaxis are unable to establish an infection or cause only a localized infection and are later displaced by the wild type in a subsequent challenge, highlighting that chemotaxis is essential for the full virulence of the bacterium. In V. cholerae, the role of chemotaxis in the disease appears to be more complicated. In this bacterium, the direction of flagellar rotation seems to determine the infectivity of the cells, e.g., mutants with biased clockwise rotation show attenuated infection, while mutants with a biased counterclockwise rotation have a hyperinfectious phenotype (7–9, 33).
In pathogenic spirochetes, the linkage between chemotaxis and virulence was first established when Lux et al. showed that a nonchemotactic cheA mutant of Treponema denticola had impaired tissue penetration ability (30). Chemotaxis may be responsible for tissue identification and for guiding the spirochetes toward a specific tissue within the host (31). B. burgdorferi is unique in its ability to swim and penetrate into highly viscous environments, such as the host connective tissue, during infection (11, 14, 24, 56). The spirochete is also chemotactic to several host components (3, 52, 53), such as the extract of tick salivary glands and host serum. The nature of its complicated life cycle strongly suggests the requirement for a sensory guided movement in response to changes in stimuli during transmission. Furthermore, B. burgdorferi devotes approximately 6% of its genome to motility and chemotaxis genes, emphasizing the importance of motility and chemotaxis in its life cycle (11, 17). Thus, chemotaxis has been considered a virulence factor of B. burgdorferi. In this report, a comprehensive study was carried out to elucidate the role of chemotaxis in the pathogenesis of B. burgdorferi, using the virulent A3-68 Δbbe02 strain (45). The results clearly demonstrate that chemotaxis is an important virulence factor of B. burgdorferi.
In E. coli, the histidine kinase CheA is a key component of the chemotaxis signaling pathway (60, 61). B. burgdorferi has two cheA genes (cheA1 and cheA2). Our previous study in an avirulent B31A background showed that cheA2 is essential for chemotaxis, while disruption of cheA1 did not alter the chemotactic behavior of the cells (28). In this report, the in vitro experiments indicated that the cheA2mut mutant has a similar phenotype to that of its avirulent counterpart derived from B31A, i.e., the mutant fails to reverse, runs constantly in one direction, and is nonchemotactic to attractant (Table 1 and Fig. 4), which further highlights that CheA2 is a key component in the chemotaxis pathway of B. burgdorferi. The in vivo studies of the cheA2mut mutant demonstrated that chemotaxis is not essential for the survival of B. burgdorferi within the tick vector but is required for mammalian infection and/or transmission of spirochetes from the tick vector to a mammalian host. For instance, the cheA2mut mutant was able to colonize ticks but failed to establish an infection in mice via both needle inoculation and tick bites (Fig. 7 and Table 2).
One intriguing question still stands. The tick-to-mouse infection study showed that the cheA2mut strain failed to establish a natural infection in mice via tick bite (Fig. 7). This could be due to either the failure of the mutant to be transmitted from the tick vector to mice or the failure of the mutant to survive and establish an infection in mice after the completion of transmission. Bacterial chemotaxis is likely required for the early dissemination and invasion after initial deposition to evade incoming host innate immune cells, such as neutrophils (21). Consistently, the kinetics study of infection using needle inoculation revealed that the mutant cells were removed from the initial inoculation site within 72 h (Fig. 5), implying that chemotaxis is required for the early establishment of infection and/or immune evasion. During transmission, spirochetes migrate from the tick gut toward the salivary glands in order to be passed on to the host (14). Bacterial chemotaxis may guide the migration of spirochetes from the tick gut toward the salivary glands. An extract of tick salivary glands is a strong attractant to B. burgdorferi (53). Thus, the failure to recognize and swim toward attractants may block the early migration and transmission process. At this point of our studies, both events seem likely, and we are unable to rule out either of these two possibilities.
The other interesting question is the role of CheA1 in the chemotaxis and pathogenicity of B. burgdorferi. As mentioned earlier, the genome of B. burgdorferi carries multiple copies of putative chemotaxis genes, such as two cheA genes, three cheY genes, and three cheW genes (11, 17). The majority of these genes are located within two gene clusters: cheA2-cheW3-cheX-cheY3 and cheW2-bb0566-cheA1-cheB2-bb0569-cheY2 (17, 19, 28). The currently standing evidence has revealed that all of the chemotaxis genes in the cheA2 cluster are required for chemotaxis (28, 36, 37). In contrast, the genes in the cheW2 cluster that have been studied so far are not involved in chemotaxis, e.g., cheA1 and cheY2 mutants have a similar in vitro phenotype to that of the wild type (28, 37). It has been speculated that B. burgdorferi may possess two chemotaxis pathways that function in different hosts during the infection cycle (11, 28, 37), i.e., CheA2, CheW3, and CheY3 form a pathway that executes chemotaxis in mammalian hosts, whereas CheA1, CheW2, and CheY2 constitute another pathway that guides chemotactic responses in the tick host. Consistent with this speculation, the studies in this report have shown that inactivation of cheA2 affects the ability of spirochetes to establish infection in mice only, not in ticks. In future studies, a similar approach can be adopted to elucidate the roles of other genes, particularly the genes in the cheA1 cluster, in the chemotaxis and virulence of B. burgdorferi.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Rosa for providing the A3-68 Δbbe02 strain and M. Caimano for providing the DMC-cultured B31A3 sample.
This research was supported by Public Health Service grants (AI073354 and AI078958) to C. Li.
Footnotes
Published ahead of print 16 April 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Akins DR, Bourell KW, Caimano MJ, Norgard MV, Radolf JD. 1998. New animal model for studying Lyme disease spirochetes in a mammalian host-adapted state. J. Clin. Invest. 101:2240–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bacon RM, Kugeler KJ, Mead PS. 2008. Surveillance for Lyme disease—United States, 1992–2006. MMWR Surveill. Summ. 57:1–9 [PubMed] [Google Scholar]
- 3. Bakker RG, Li C, Miller MR, Cunningham C, Charon NW. 2007. Identification of specific chemoattractants and genetic complementation of a Borrelia burgdorferi chemotaxis mutant: flow cytometry-based capillary tube chemotaxis assay. Appl. Environ. Microbiol. 73:1180–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barthold SW. 1995. Animal models for Lyme disease. Lab. Invest. 72:127–130 [PubMed] [Google Scholar]
- 5. Brooks CS, Hefty PS, Jolliff SE, Akins DR. 2003. Global analysis of Borrelia burgdorferi genes regulated by mammalian host-specific signals. Infect. Immun. 71:3371–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burgdorfer W, et al. 1982. Lyme disease, a tick-borne spirochetosis? Science 216:1317–1319 [DOI] [PubMed] [Google Scholar]
- 7. Butler SM, Camilli A. 2004. Both chemotaxis and net motility greatly influence the infectivity of Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 101:5018–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Butler SM, Camilli A. 2005. Going against the grain: chemotaxis and infection in Vibrio cholerae. Nat. Rev. Microbiol. 3:611–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Butler SM, et al. 2006. Cholera stool bacteria repress chemotaxis to increase infectivity. Mol. Microbiol. 60:417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Casjens S, et al. 2000. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 35:490–516 [DOI] [PubMed] [Google Scholar]
- 11. Charon NW, Goldstein SF. 2002. Genetics of motility and chemotaxis of a fascinating group of bacteria: the spirochetes. Annu. Rev. Genet. 36:47–73 [DOI] [PubMed] [Google Scholar]
- 12. Coleman JL, et al. 1997. Plasminogen is required for efficient dissemination of B. burgdorferi in ticks and for enhancement of spirochetemia in mice. Cell 89:1111–1119 [DOI] [PubMed] [Google Scholar]
- 13. Comstock LE, Thomas DD. 1991. Characterization of Borrelia burgdorferi invasion of cultured endothelial cells. Microb. Pathog. 10:137–148 [DOI] [PubMed] [Google Scholar]
- 14. Dunham-Ems SM, et al. 2009. Live imaging reveals a biphasic mode of dissemination of Borrelia burgdorferi within ticks. J. Clin. Invest. 119:3652–3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elias AF, et al. 2002. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect. Immun. 70:2139–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foynes S, et al. 2000. Helicobacter pylori possesses two CheY response regulators and a histidine kinase sensor, CheA, which are essential for chemotaxis and colonization of the gastric mucosa. Infect. Immun. 68:2016–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fraser CM, et al. 1997. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580–586 [DOI] [PubMed] [Google Scholar]
- 18. Garcia-Monco JC, Fernandez Villar B, Calvo Alen J, Benach JL. 1990. Borrelia burgdorferi in the central nervous system: experimental and clinical evidence for early invasion. J. Infect. Dis. 161:1187–1193 [DOI] [PubMed] [Google Scholar]
- 19. Ge Y, Charon NW. 1997. Molecular characterization of a flagellar/chemotaxis operon in the spirochete Borrelia burgdorferi. FEMS Microbiol. Lett. 153:425–431 [DOI] [PubMed] [Google Scholar]
- 20. Goldstein SF, Charon NW, Kreiling JA. 1994. Borrelia burgdorferi swims with a planar waveform similar to that of eukaryotic flagella. Proc. Natl. Acad. Sci. U. S. A. 91:3433–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hartiala P, et al. 2008. Borrelia burgdorferi inhibits human neutrophil functions. Microbes Infect. 10:60–68 [DOI] [PubMed] [Google Scholar]
- 22. Hayes EB, Piesman J. 2003. How can we prevent Lyme disease? N. Engl. J. Med. 348:2424–2430 [DOI] [PubMed] [Google Scholar]
- 23. Kariu T, Coleman AS, Anderson JF, Pal U. 2011. Methods for rapid transfer and localization of Lyme disease pathogens within the tick gut. J. Vis. Exp. 2011:2544 doi:10.3791/2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kimsey RB, Spielman A. 1990. Motility of Lyme disease spirochetes in fluids as viscous as the extracellular matrix. J. Infect. Dis. 162:1205–1208 [DOI] [PubMed] [Google Scholar]
- 25. Kumar M, Yang X, Coleman AS, Pal U. 2010. BBA52 facilitates Borrelia burgdorferi transmission from feeding ticks to murine hosts. J. Infect. Dis. 201:1084–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lane RS, Piesman J, Burgdorfer W. 1991. Lyme borreliosis: relation of its causative agent to its vectors and hosts in North America and Europe. Annu. Rev. Entomol. 36:587–609 [DOI] [PubMed] [Google Scholar]
- 27. Lawrenz MB, Wooten RM, Norris SJ. 2004. Effects of vlsE complementation on the infectivity of Borrelia burgdorferi lacking the linear plasmid lp28-1. Infect. Immun. 72:6577–6585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li C, et al. 2002. Asymmetrical flagellar rotation in Borrelia burgdorferi nonchemotactic mutants. Proc. Natl. Acad. Sci. U. S. A. 99:6169–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li C, Xu H, Zhang K, Liang FT. 2010. Inactivation of a putative flagellar motor switch protein FliG1 prevents Borrelia burgdorferi from swimming in highly viscous media and blocks its infectivity. Mol. Microbiol. 75:1563–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lux R, Miller JN, Park NH, Shi W. 2001. Motility and chemotaxis in tissue penetration of oral epithelial cell layers by Treponema denticola. Infect. Immun. 69:6276–6283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lux R, Moter A, Shi W. 2000. Chemotaxis in pathogenic spirochetes: directed movement toward targeting tissues? J. Mol. Microbiol. Biotechnol. 2:355–364 [PubMed] [Google Scholar]
- 32. McGee DJ, et al. 2005. Colonization and inflammation deficiencies in Mongolian gerbils infected by Helicobacter pylori chemotaxis mutants. Infect. Immun. 73:1820–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Merrell DS, et al. 2002. Host-induced epidemic spread of the cholera bacterium. Nature 417:642–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moriarty TJ, et al. 2008. Real-time high resolution 3D imaging of the Lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS Pathog. 4:e1000090 doi:10.1371/journal.ppat.1000090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Motaleb MA, et al. 2000. Borrelia burgdorferi periplasmic flagella have both skeletal and motility functions. Proc. Natl. Acad. Sci. U. S. A. 97:10899–10904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Motaleb MA, et al. 2005. CheX is a phosphorylated CheY phosphatase essential for Borrelia burgdorferi chemotaxis. J. Bacteriol. 187:7963–7969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Motaleb MA, Sultan SZ, Miller MR, Li C, Charon NW. 2011. CheY3 of Borrelia burgdorferi is the key response regulator essential for chemotaxis and forms a long-lived phosphorylated intermediate. J. Bacteriol. 193:3332–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nocton JJ, et al. 1994. Detection of Borrelia burgdorferi DNA by polymerase chain reaction in synovial fluid from patients with Lyme arthritis. N. Engl. J. Med. 330:229–234 [DOI] [PubMed] [Google Scholar]
- 39. Norman MU, et al. 2008. Molecular mechanisms involved in vascular interactions of the Lyme disease pathogen in a living host. PLoS Pathog. 4:e1000169 doi:10.1371/journal.ppat.1000169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ojaimi C, et al. 2003. Profiling of temperature-induced changes in Borrelia burgdorferi gene expression by using whole genome arrays. Infect. Immun. 71:1689–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pal U, et al. 2004. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J. Clin. Invest. 113:220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parkinson JS. 1977. Behavioral genetics in bacteria. Annu. Rev. Genet. 11:397–414 [DOI] [PubMed] [Google Scholar]
- 43. Purser JE, Norris SJ. 2000. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. U. S. A. 97:13865–13870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Radolf JD, Caimano MJ, Stevenson B, Hu LT. 2012. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat. Rev. Microbiol. 10:87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rego RO, Bestor A, Rosa PA. 2011. Defining the plasmid-borne restriction-modification systems of the Lyme disease spirochete Borrelia burgdorferi. J. Bacteriol. 193:1161–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Revel AT, Talaat AM, Norgard MV. 2002. DNA microarray analysis of differential gene expression in Borrelia burgdorferi, the Lyme disease spirochete. Proc. Natl. Acad. Sci. U. S. A. 99:1562–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosa PA, Tilly K, Stewart PE. 2005. The burgeoning molecular genetics of the Lyme disease spirochaete. Nat. Rev. Microbiol. 3:129–143 [DOI] [PubMed] [Google Scholar]
- 48. Sal MS, et al. 2008. Borrelia burgdorferi uniquely regulates its motility genes and has an intricate flagellar hook-basal body structure. J. Bacteriol. 190:1912–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Samuels DS. 2011. Gene regulation in Borrelia burgdorferi. Annu. Rev. Microbiol. 65:479–499 [DOI] [PubMed] [Google Scholar]
- 50. Schwan TG, Piesman J, Golde WT, Dolan MC, Rosa PA. 1995. Induction of an outer surface protein of Borrelia burgdorferi during tick feeding. Proc. Natl. Acad. Sci. U. S. A. 92:2909–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schweinitzer T, Josenhans C. 2010. Bacterial energy taxis: a global strategy? Arch. Microbiol. 192:507–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shi W, Yang ZM, Geng Y, Wolinsky LE, Lovett MA. 1998. Chemotaxis in Borrelia burgdorferi. J. Bacteriol. 180:231–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shih CM, Chao LL, Yu CP. 2002. Chemotactic migration of the Lyme disease spirochete (Borrelia burgdorferi) to salivary gland extracts of vector ticks. Am. J. Trop. Med. Hyg. 66:616–621 [DOI] [PubMed] [Google Scholar]
- 54. Stanek G, Klein J, Bittner R, Glogar D. 1990. Isolation of Borrelia burgdorferi from the myocardium of a patient with longstanding cardiomyopathy. N. Engl. J. Med. 322:249–252 [DOI] [PubMed] [Google Scholar]
- 55. Steere AC, Coburn J, Glickstein L. 2004. The emergence of Lyme disease. J. Clin. Invest. 113:1093–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Szczepanski A, Furie MB, Benach JL, Lane BP, Fleit HB. 1990. The interaction between Borrelia burgdorferi and endothelium in vitro. J. Clin. Invest. 85:1637–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sze CW, Li C. 2011. Inactivation of bb0184, which encodes carbon storage regulator A, represses the infectivity of Borrelia burgdorferi. Infect. Immun. 79:1270–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takata T, Fujimoto S, Amako K. 1992. Isolation of nonchemotactic mutants of Campylobacter jejuni and their colonization of the mouse intestinal tract. Infect. Immun. 60:3596–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Terry K, Williams SM, Connolly L, Ottemann KM. 2005. Chemotaxis plays multiple roles during Helicobacter pylori animal infection. Infect. Immun. 73:803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vladimirov N, Sourjik V. 2009. Chemotaxis: how bacteria use memory. Biol. Chem. 390:1097–1104 [DOI] [PubMed] [Google Scholar]
- 61. Wadhams GH, Armitage JP. 2004. Making sense of it all: bacterial chemotaxis. Nat. Rev. Mol. Cell. Biol. 5:1024–1037 [DOI] [PubMed] [Google Scholar]
- 62. Xu Q, McShan K, Liang FT. 2008. Essential protective role attributed to the surface lipoproteins of Borrelia burgdorferi against innate defences. Mol. Microbiol. 69:15–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang X, et al. 2000. Interdependence of environmental factors influencing reciprocal patterns of gene expression in virulent Borrelia burgdorferi. Mol. Microbiol. 37:1470–1479 [DOI] [PubMed] [Google Scholar]
- 64. Yao RJ, Burr DH, Guerry P. 1997. CheY-mediated modulation of Campylobacter jejuni virulence. Mol. Microbiol. 23:1021–1031 [DOI] [PubMed] [Google Scholar]
- 65. Zambrano MC, Beklemisheva AA, Bryksin AV, Newman SA, Cabello FC. 2004. Borrelia burgdorferi binds to, invades, and colonizes native type I collagen lattices. Infect. Immun. 72:3138–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang X, Yang X, Kumar M, Pal U. 2009. BB0323 function is essential for Borrelia burgdorferi virulence and persistence through tick-rodent transmission cycle. J. Infect. Dis. 200:1318–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.